

Abstract
To understand the molecular mechanisms of parasitism in vivo, it is essential to elucidate how the transcriptomes of the human hosts and the infecting parasites affect one another. Here we report the RNA-seq analysis of 116 Indonesian patients infected with the malaria parasite Plasmodium falciparum (Pf). We extracted RNAs from their peripheral blood as a mixture of host and parasite transcripts and mapped the RNA-seq tags to the human and Pf reference genomes to separate the respective tags. We were thus able to simultaneously analyze expression patterns in both humans and parasites. We identified human and parasite genes and pathways that correlated with various clinical data, which may serve as primary targets for drug developments. Of particular importance, we revealed characteristic expression changes in the human innate immune response pathway genes including TLR2 and TICAM2 that correlated with the severity of the malaria infection. We also found a group of transcription regulatory factors, JUND, for example, and signaling molecules, TNFAIP3, for example, that were strongly correlated in the expression patterns of humans and parasites. We also identified several genetic variations in important anti-malaria drug resistance-related genes. Furthermore, we identified the genetic variations which are potentially associated with severe malaria symptoms both in humans and parasites. The newly generated data should collectively lay a unique foundation for understanding variable behaviors of the field malaria parasites, which are far more complex than those observed under laboratory conditions.
Plasmodium species, including P. falciparum (Pf), cause worldwide health problems that require immediate action (Aregawi et al. 2011). Intensive international efforts have been made to analyze their genomes and transcriptomes (Daily et al. 2007; Volkman et al. 2007; Mu et al. 2010; Otto et al. 2010). For various Plasmodium species, including human malaria parasites Pf and P. vivax (Pv), entire genome sequence data are now available (Aurrecoechea et al. 2009). Genetic variations of Pf in different regions of the world have also been analyzed (Volkman et al. 2007). Manske et al. (2012) generated a data set of 86,158 exonic single nucleotide polymorphisms in 227 Pf samples from Africa, Asia, and Oceania. Considerable efforts were also made to enrich the genome annotations with transcriptome information (Daily et al. 2007; Otto et al. 2010; Tuda et al. 2011). In PlasmoDB, a representative database of Plasmodium species, a wide variety of expression data, such as those collected at several time points during the intra-erythrocytic developmental cycle, were archived and made freely available (Aurrecoechea et al. 2009).
On the other hand, several pathways or genes that play pivotal roles in the host defense system in humans have been also identified. Particularly, innate immune response genes are well characterized as first line defense sensors. Toll-like receptors (TLRs) and several other pattern recognition receptors recognize pathogens and related components and trigger downstream signaling cascades (Kawai and Akira 2010; Takeuchi and Akira 2010). Especially in a malaria infection, hemozoin, a degradation product of heme, appears after heme is scavenged by parasites. TLR9 specifically recognizes hemozoin (Coban et al. 2005, 2010; Parroche et al. 2007). Upon recognition, the downstream TLR pathway is activated, eventually leading to the activation of two major groups of transcription factors: the NF-kB (such as the NFKB1:RELA complex) and AP-1 (such as the FOS:JUN complex) group and the IRFs. Through these transcription factors, a series of proinflammatory cytokines, such as TNF, IL1, and IL6, are subsequently induced (Pahl 1999; Dinarello 2000; Gilmore 2006; Hoffmann et al. 2006; Kishimoto 2006). Particularly, the IRF group of transcription factors induces type I interferon (IFN) responses (Sato et al. 2009). They further modulate the inflammatory responses and invoke acquired immune responses (Ito et al. 2002; Palm and Medzhitov 2009).
In spite of rapid progress in both parasite genomics and analysis of host immune responses in humans, current knowledge is mostly limited to that obtained from either rodent models or laboratory infection systems. Little is known about the in situ gene expression patterns in humans and parasites in the field. Indeed, clinical features, including the severity of malaria symptoms and drug resistance, are highly variable depending on parasites and patients. To address this issue, we used RNA-seq analysis. To simultaneously analyze gene expressions from both the human hosts and infecting parasites, we used a mixture of host and parasite RNA isolated from infected patients for the RNA sequencing. In this way, we also hoped to avoid the technical difficulties associated with isolating parasites at field hospitals, which is frequently the largest barrier to retaining the quality of the materials, or other manipulations that might introduce bias to the expression information. In addition, by utilizing the RNA sequences for calling single nucleotide polymorphisms (SNPs), we were also able to analyze genetic variation among the Pf samples at the same time. Here we describe the interactive transcriptome analysis of clinical malaria patients.
Results
RNA sequencing of human-parasite mixed mRNA populations
To analyze the interactions between transcriptomes of human host cells and infecting parasites, we carried out RNA-seq analysis of peripheral blood samples of malaria patients (Fig. 1A). We generated an average of 30 million RNA-seq tags per sample from each of 116 patients (Table 1). When the RNA-seq tags were mapped onto the reference genomes of humans and parasites, we found that ∼90% were uniquely mapped to the human genome and the rest were mapped to the Pf genome. There were essentially no RNA-seq tags mapped to both the human and Pf genomes (Supplemental Table 1). We also collected control samples from 25 healthy people and 28 people with other infectious diseases. From these control samples, essentially no Pf tags were identified (Supplemental Table 1; Supplemental Fig. 1). Based on these results, we concluded that we were able to use the mixed RNAs for the RNA-seq analysis and to separate the RNA-seq tags by mapping them to the respective genomes.
Figure 1.
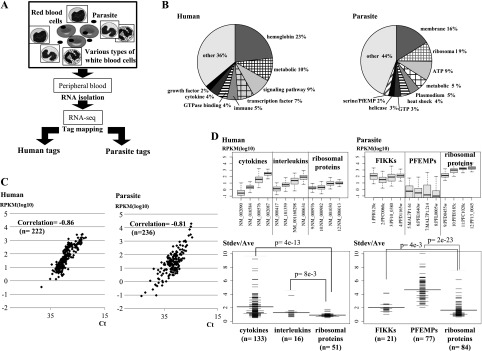
RNA-seq analysis of human-parasite mixed transcriptomes. (A) Schematic representation of the RNA-seq analysis using mixed human-parasite RNA. Note that in each sample, different cell types from both humans and parasites are represented. (B) Distribution of the RNA-seq tags assigned to the indicated functional categories of genes in humans (left) and parasites (right). Definitions of the gene categories are shown in Supplemental Table 3. (C) Real time RT-PCR validation of the RNA-seq analysis. A total of 458 cases ([left panel] 222 cases in humans; [right panel] 236 cases for parasites) were examined. Overall correlation between RNA-seq and real time RT-PCR-based gene expression was r = −0.83 (r = −0.86 in humans; r = −0.81 in parasites). (D) (Top) Distributions of the relative deviations in gene expression patterns in humans (left) and parasites (right). Relative deviation was calculated as the standard deviation divided by the average of the expression levels for each gene. (Bottom) Distribution of the variance in gene expressions for the indicated categories of genes. The y-axis represents the standard deviation of the gene expression level (Stdev) divided by the average gene expression level (Ave) for the corresponding gene in the 116 patients. Horizontal bar represents the average of the Stdev/Ave value for the category. The number of genes (n) included in the analysis for each category is shown in parentheses. All of the data for the genes binned under each category are presented in Supplemental Table 4.
Table 1.
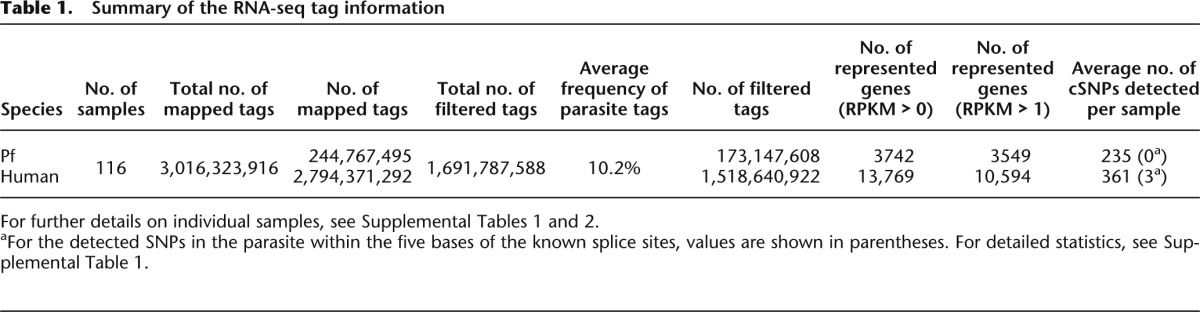
Summary of the RNA-seq tag information
Based on the generated RNA-seq tag information, we analyzed gene expression patterns in human and Pf (Supplemental Fig. 2A,B). Both human and parasite tags were normalized against the expression of GAPDH in the respective organisms (see Supplemental Table 2 for gene expression information for each gene). Figure 1B shows the average breakdown of the tags in a patient. In humans, a considerable portion of the tags was derived from immune-related genes, such as cytokine genes and their receptor genes (2%–10%, depending on the category), although many of them were from the beta globin gene (23%), as expected. In parasites, the largest portion of the tags was derived from functionally uncharacterized genes (44%) (for details, see Supplemental Table 3), perhaps due to still inadequate genome annotations in Pf.
For the validation analysis, we conducted real-time RT-PCR assays for a total of 458 cases (Fig. 1C). A reasonable correlation existed between the RNA-seq and the RT-PCR data, with an overall Pearson’s correlation r = −0.83 (r = −0.86; n = 222 in humans and r = −0.81; n = 236 in Pf) (for examples, see Supplemental Fig. 3).
Variable gene expression patterns in humans and parasites
For a group of genes, expression patterns were widely varied among samples, and this variation had little correlation with their total expression levels (Supplemental Fig. 4A). For example, in humans, the cytokine and interleukin genes showed a more diverse expression, regardless of their expression levels, among patients than did the ribosomal protein genes (P = 6 × 10−2 and P = 1 × 10−9, respectively) (Fig. 1D, left panels; see Supplemental Table 4 for a comprehensive list of genes). Similarly, in parasites, the expressions of the FIKK and PfEMP gene families, which encode representative surface proteins, were highly variable compared to the ribosomal protein genes (P = 4 × 10−3 and P = 2 × 10−23, respectively) (Fig. 1D, right panels). These genes are known to encode proteins used by parasites to escape from host immune systems. Considering that different patients have different malaria symptoms, correspondingly, different expression patterns in human and parasite may represent different modes of host-parasite interactions (also see Supplemental Fig. 4B).
Correlation of gene expression patterns with clinical information
We compared the clinically observed parasitemia of the parasites with the frequency of the Pf tags (%Pf tags). We found that the infectious rate (as diagnosed by microscopic analysis of thin blood films which is believed to be the most quantitative) and the Pf read counts are reasonably well correlated (Pearson’s correlation r = 0.85; n = 14) (Fig. 2A,B). We also validated whether they are correlated in a wider dynamic range in an even more quantitative manner. Using in vitro samples of a Pf strain, 3D7, for which parasitemia can be flexibly controlled and more precisely evaluated, we analyzed and observed that the correlation was almost perfect under this experimental condition (r = 0.97; n = 8 of diverse parasitemia, ranging from 0.5% to 10%) (Fig. 2C). In addition, for 40 samples correlation between the rapid diagnosis, which is a simple but qualitative and less accurate method, and is more frequently used in the field, and the Pf read counts is also reasonable (Spearman’s r = 0.53; n = 40) (Fig. 2D). Taken together, we concluded that RNA read counts correlate well with clinical statuses of malaria at a sufficient level, at least for the present study.
Figure 2.

Associations of RNA-seq read counts and clinical information of parasitemia. (A) Representative image of a thin blood film from a clinical sample (top) and 3D7 (bottom) from which infected red blood cells were counted and parasitemia was calculated. Arrow indicates the stained parasite. (B) Relation between the Pf read counts and the parasitemia, which was diagnosed by a microscopic view of the thin blood film (n = 14). Numbers of infected red blood cells were counted microscopically. (C) Results of a similar analysis from an in vitro cultured Pf strain, 3D7. Parasites were cultured at the indicated parasitemia, which was validated microscopically and used for the RNA-seq analysis, as in the case in the clinical samples (n = 8). Pf tag counts were normalized with those of the human beta globin gene. (D) Qualitative comparison between clinical parasitemia and Pf read counts (n = 40). For this analysis, parasitemia was qualitatively determined by rapid diagnosis according to the standard protocol. The correlation coefficient calculated by the indicated methods is shown in the respective graphs.
Gene expression variations were occasionally associated with the patients’ clinical data. The clinical data analyzed included the %Pf tags, which should represent severity of malaria, body temperature, age of the patient, and the duration since symptom onset (Table 2A). In this study, even though it is technically molecular information and not a direct indicator of malaria symptoms, we used %Pf tags as a clinical condition based on the results of the correlation analyses. We conducted an exhaustive statistical enrichment test using the Wilcoxon signed rank test and identified a number of genes whose expression patterns were significantly correlated with clinical data (Table 2B; see Supplemental Tables 5–8 for details).
Table 2.
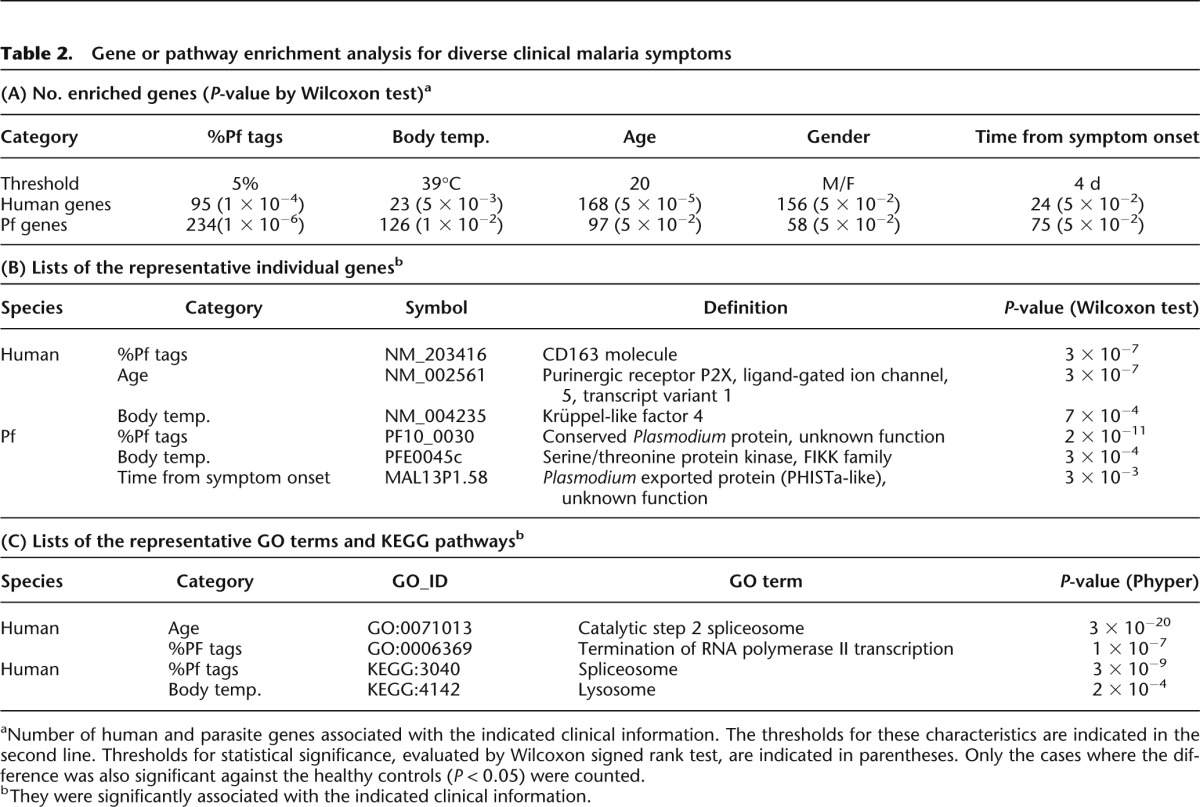
Gene or pathway enrichment analysis for diverse clinical malaria symptoms
Among the identified genes, several are potentially involved in the severity of malaria symptoms. As exemplified in Figure 3A, expression of the haptoglobulin receptor gene (CD163 molecule) was induced in patients with higher %Pf tags (P = 3 × 10−7). Observed expression changes were further validated by independent real-time RT-PCR analysis (Fig. 3A, middle panel) and by RNA-seq tag counts in finer bins (P = 2 × 10−7). We also validated the statistical significance of the differences, considering the control samples. We could show that the difference was significant between malaria patients and healthy controls (Supplemental Fig. 5A,B). Previous reports showed that this gene plays a role in clearance and endocytosis of hemoglobin complexes by macrophages (Kristiansen et al. 2001). Subsequently, accumulated hemozoin may induce further responses of the innate immune system.
Figure 3.
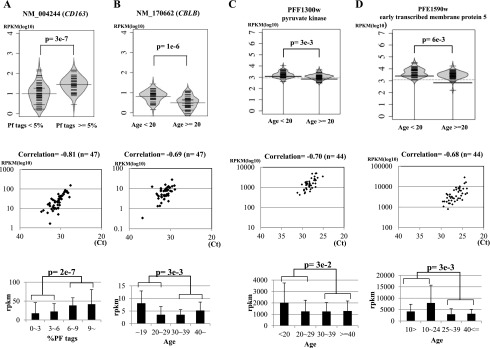
Association of gene expression patterns with clinical information. Examples of human genes (A,B) and parasite genes (C,D) associated with the indicated clinical information. (Top) Bean plots of the gene expression levels (y-axis in reads per kilobase per million, RKPM) are shown for the indicated populations. Statistical significances (P) of the differences are shown within the plots. (Middle) Validation analysis of the expression levels by real-time RT-PCR. (Bottom) Differential expressions, identified by both RNA-seq tag counts and real-time RT-PCR validations, are shown for further breakdown of the populations. Particularly for human genes (A,B), gene expression levels for the healthy controls are shown in Supplemental Figure 5. Detailed evaluation of the statistical significances in the differences against the control samples is also shown there. Data represent the means of three experiments. The number of samples used (n) is as indicated in the margin. Note that demographic data were not always available for all of the samples. Statistical significances of the indicated differences are shown above the plots.
Similarly, as exemplified in Figure 3B, expression levels of CBLB were higher in younger malaria patients (P = 1 × 10−6), which was also validated by real-time RT-PCR and tag counts in finer bins (P = 3 × 10−3). Again, we found that the difference was statistically significant against the age-matched controls (Supplemental Fig. 5, C and D, shows the statistical significance in the difference in the respective age range). Indeed, by also considering the control samples, we found that the difference was derived from insufficient down-regulation of CBLB expression from the normal level in young malaria patients (Supplemental Fig. 5D). Genes associated with young malaria patients are particularly important, as they present the most pressing problem. It is known that CBLB, an E3 ubiquitin ligase (Sawasdikosol et al. 2000), negatively controls the association between TLR4 and the intracellular adaptor MYD88 and thereby represses eventual mal-activations of T cells and inflammatory responses (Rao et al. 2002; Han et al. 2010). CBLB-deficient mice showed an autoimmune disease-like phenotype with enhanced T-cell activation, in which increased release of inflammatory cytokines was also observed. As opposed to the case in the CBLB-deficient mice, young patients occasionally show more severe malaria symptoms (Rogier et al. 1996), because the retained expression of the CBLB gene may result in insufficient activation of the cytokine and inflammatory responses.
In the parasite genome, expression of the Pf pyruvate kinase gene was enhanced in patients younger than 20 yr old (Fig. 3C). Because a high expression level of this gene indicates active metabolism in parasites, this observation may also reflect severe malarial symptoms in young patients. We also found that the Pf early transcribed membrane protein 5 gene is induced in young patients (Fig. 3D). This gene may serve as a drug target for vaccine development to block the parasite growth at an early stage, especially in young patients.
Correlation of a group of genes with clinical information
We also analyzed and identified several groups of genes belonging to functional GO categories (Ashburner et al. 2000) and KEGG pathways (Kanehisa et al. 2012) that showed statistically significant associations with the indicated clinical data (Table 2C; see Supplemental Tables 7 and 8 for further details). Among them, we particularly focused on the innate immune response pathway. Generally, genes in this pathway were up-regulated (Supplemental Table 9), which is partly consistent with previous results (Hartgers et al. 2008; Franklin et al. 2009; Fu et al. 2012). We also observed that patients with higher %Pf tags showed characteristic patterns of expression changes compared to patients with lower %Pf tags (Fig. 4). Also, by considering the healthy controls and the controls with other infectious diseases, we could further classify the gene expression patterns, namely, whether they are characteristic of malaria patients or common to patients of different infectious diseases. For example, the mRNA levels of the TLR2, which senses parasites’ glycosylphosphatidylinositol (GPI) anchors (Krishnegowda et al. 2005), was up-regulated, while those of TLR9, which senses hemozoin, a degradation product of heme, (Coban et al. 2005, 2007, 2010; Parroche et al. 2007), remained almost the same in given patients. Similarly, mRNA levels of TICAM2, but not TIRAP, were up-regulated. We also observed that activations of the representative downstream target genes involved in inflammatory cytokine and type I interferon responses showed characteristic patterns (Supplemental Table 9), depending on the patients. Thus, quite unexpectedly, gene up-regulations proportional to the %Pf tags occurred only for restricted members of the pathway.
Figure 4.

Gene expression patterns of the TLR4 pathway genes. (A) Examples of the gene expression patterns between healthy controls, malaria patients with %Pf tags of <5%, >5%, and patients with other infectious diseases. Left and right panels exemplify the cases where gene expression levels increased with increasing %Pf tags (the case of the JUN gene) and where they decreased (the case of the IKBKG), respectively. Also note that left and right panels exemplify the cases where the observed difference was malaria-specific and nonmalaria-specific, respectively, which was revealed by considering the control patients with other infectious diseases. Statistical significances in the differences were evaluated by Wilcoxon signed rank test and are shown in the margins. (B) Global patterns of gene expression in the TLR4 pathway. Significant differences between two populations (%Pf tags of >5% or <5%) are shown above the plots. Genes significantly up-regulated in the patients with high %Pf tags (P < 0.05) are enclosed in bold boxes. Also, by considering the controls (healthy controls and patients with other infectious diseases), “A” and “B” indicate genes where the increase and decrease in the gene expressions were observed for the increasing or decreasing %Pf tags, respectively, and such differences were statistically significant against healthy controls. Among them, “C” indicates the cases where the gene expression differences were significant against the control patients with other infectious diseases, thus, the difference appeared to be malaria-specific. For evaluating statistical significance in the differences, Wilcoxon signed rank test was used.
Correlated patterns of gene expression between humans and parasites
We calculated the Spearman’s correlation for all pairs of human-Pf genes. We identified a total of 52,044 pairs of human and parasite genes with positive correlations and 188 pairs with negative correlations (correlation > 0.5 or < −0.5, respectively) (Fig. 5A). Permutation tests using randomly correlated human-Pf gene expression patterns detected essentially no gene with correlations in this range (Supplemental Fig. 6). For example, expression of the tumor necrosis factor alpha-induced protein 3 (TNFAIP3) gene in humans, which also belongs to the immune response or inflammatory pathway (Song et al. 1996; Vereecke et al. 2009), was positively correlated with the Pf putative polyadenylate-binding protein gene (r = 0.64) (Fig. 5B; also see Fig. 5C for the case of the human JUND and the Pf putative eukaryotic translation initiation factor 3 subunit 10 gene). On the other hand, negative correlation was observed between expression of the human TNFAIP8L2 and the parasite putative methyltransferase gene (r = −0.55) (Fig. 5D).
Figure 5.
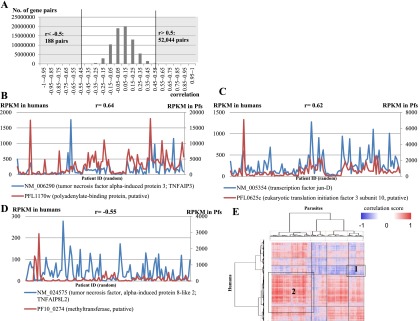
Positive and negative correlations between expression patterns of human and parasite genes in 116 patients. (A) Distribution of the Spearman’s correlation coefficients calculated for each pair of human-parasite genes. The number of gene pairs with correlation coefficients >0.5 (positive correlation) or < −0.5 (negative correlation) is given to the right and left, respectively. (B–D) Examples of positively (B,C) and negatively (D) correlated human (blue) and parasite (red) gene pairs. (B) The human tumor necrosis factor alpha-induced protein 3 (TNFAIP3) and the parasite putative polyadenylate-binding protein gene; (C) the human JUND and the parasite putative eukaryotic translation initiation factor 3 subunit 10 gene; (D) the human TNFAIP3 and the parasitic putative methyltransferase gene. Spearman’s correlation coefficients are shown above each plot. On the x-axis, patients are ordered by their patient ID numbers (random order). (E) Hierarchal clustering analysis. Vertical and horizontal axes represent the human and parasite genes, respectively, each of which was paired with at least one gene in the other organism with a Spearman’s coefficient of either >0.5 or < −0.5. The heat map represents the degree of correlation according to the color scale shown in the legend. Numbered boxes represent clusters where genes with the indicated functional categories described in the text are enriched.
A hierarchical clustering analysis of the identified correlations (Fig. 5E) showed that some of the positive or negative correlations were enriched in particular GO categories. Namely, GO terms’ “innate immune response (GO:0045087)” in humans and “metabolic process (GO:0008152)” in parasites were enriched (P = 3 × 10−4 and P = 1 × 10−4, respectively) in a cluster of negatively correlated genes (as indicated by Box 1), suggesting that active innate responses of human hosts are repressive for parasite metabolisms in general. In another cluster of positively associated genes (as indicated by Box 2), the GO terms’ “sequence-specific DNA binding transcription factor activity (GO:0003700)” in humans and “eukaryotic translation initiation factor 3 complex (GO:0005852)” in parasites were enriched (P = 0.04 and P = 1 × 10−4, respectively). Among these human transcriptional regulatory factors, the SUZ12 (NM_015355), which is a component of polycomb complex (Birve et al. 2001), and other histone modifiers, such as the histone-lysine N-methyltransferase (KMT2E) (Cosgrove and Patel 2010), were included. Although the general lack of gene function information for Pf genes imposes an obstacle for further interpretations of the observed associations, it would be interesting to know whether positive or negative correlation of these gene expression patterns represents gene expression programs that aggressively compete for mutually conflicting benefits between hosts and parasites.
Genetic variations of parasites
We were also able to collect information on SNPs. Although the available information was only for expressed genes, and sequence depth depends on their expression levels, we were still able to confidently call an average of 235 SNPs per sample for parasites (Supplemental Table 1).
We tentatively focused on the SNPs in previously characterized drug resistance genes. In the Pf chloroquine resistance transporter gene (PfCRT), whose mutation is reported to be responsible for chloroquine susceptibility (Fidock et al. 2000), we confidently called the T214A substitution in at least 17 cases and the G215C substitution in two additional cases (Fig. 6A; Supplemental Table 10). Both of these substitutions cause cysteine to serine amino acid changes at this position (C72S), which was reported to be one of the representative mutations involved in acquiring drug resistance. In addition, we detected the A227C substitution (K76T substitution in amino acids), which is another drug susceptibility mutation in 28 cases. Previously uncharacterized mutations were also scattered throughout the gene. We similarly analyzed the SNPs in the Pf multiple drug resistance gene 1 (PfMDR1) and the Pf calcium-transporting ATPase 6 gene (PfATP6), which are reportedly responsible for resistance to a wide variety of anti-malarial drugs, including quinine, halofantrine, mefloquine, and artemisinin (Reed et al. 2000; Price et al. 2004; Duraisingh and Cowman 2005). As shown in Figure 6, B and C, we found putative drug resistance-acquiring mutations in a considerable number of cases. These findings should sound an alarm that a significant population of parasites in this region may have acquired drug resistance.
Figure 6.

Identification of SNVs in parasite putative drug resistance-related genes. (A–C) SNVs detected in the Pf chloroquine resistance transporter gene (A), Pf multiple drug resistance gene 1 (PfMDR1) (B), and the Pf calcium-transporting ATPase 6 gene (PfATP6) (C). Patients are ordered by their sample ID numbers (randomly) on the y-axis. Base changes observed at the indicated positions for each patient are indicated according to the color scheme shown in the legend. Only the nonsynonymous SNPs are shown. The number of tags mapped at each genomic coordinate is represented by the grayscale shown in the legend. Positions of the SNPs that were previously reported (known) or newly identified in this study (novel) are indicated by gray and black vertical lines, respectively. For detailed information on SNPs in each sample, see Supplemental Table 10. Also note that SNPs located in overlapping regions of the splicing sites may have an increased error rate, which is derived from mapping of RNA-seq tags onto the genome sequence. Further intensive manual inspection may be necessary for these sites (Supplemental Table 1).
Finally, we conducted an association study between the presence of a particular SNP and %Pf read counts both for humans and parasites. As shown in Figure 7, we could identify several candidates of such associate genes both in humans and parasites. Although further validation studies should be necessary using a larger cohort, we believe this should lay the first important base to elucidate biological associations between host humans and infecting parasites occurring in vivo.
Figure 7.
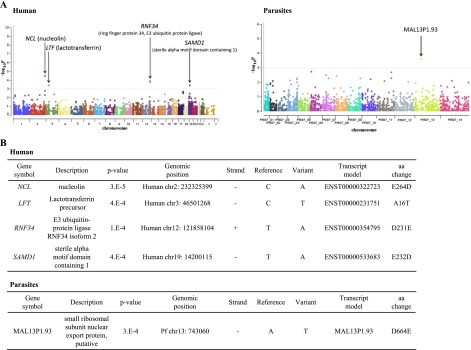
Association study to identify human and parasite genes related to severe malaria symptoms. (A) Results of the association study to identify genes that are associated with the severity of malaria. Results are shown as Manhattan plots for humans (left panel) and parasites (right panel). Calculated P-values are shown on the y-axis. Gene names are indicated by arrows. (B) Summarized information of the associated SNVs in humans (top) and parasites (bottom). Genomic coordinates and the deduced amino acid change are shown.
Discussion
In this study, we used a mixture of human and parasite RNA for the transcriptome analysis of malaria samples so that the expression profiles of both could be represented simultaneously. A similar approach could be applied to analyze any parasite in the field or to parasites that are impossible to be isolated from patient tissues.
An obvious drawback of this approach is that obtained expression profiles should represent those of all cell types mixed together. Consequently, it is difficult to precisely separate information from different types of human blood cells and different stages of parasitic life cycles. Also, we have set a very conservative threshold for statistical significance, considering the effects from diverse environmental factors inherent to clinical samples, which, in turn, may have caused false negative detection of otherwise significant associations (Table 2A). Therefore, some subtle but solid associations may have also been overlooked in this study. To address this concern, sample size should also be expanded. To accomplish this, one of the recently introduced RNA-seq methods, such as not so random (NSR) (Armour et al. 2009), might be useful.
It was also challenging to examine correlations between the abundance of individual genes based on a single time point. For example, Figure 5 shows some pairs of positively or negatively correlated genes across the patient samples. However, it is possible that these observations were obtained as a consequence of complex factors. For example, some of the “anti-correlated” genes may have appeared so because they are underexpressed in the parasite in the bloodstream. This has in fact been shown to be true because the circulating forms of Pf are mainly in the early stage ring form of the parasite that is not actively metabolizing. Later forms are sequestered in the vasculature, so that different expression patterns may represent different populations of the parasites.
Despite these drawbacks, our approach has enabled the first analyses of in situ samples in their intact statuses at the genome-wide level. In particular, we have demonstrated that activation of mRNA expression is not uniform throughout the TLR network (Fig. 4) in vivo. Interestingly, genes comprising the same signaling complex occasionally responded differently (Fig. 4). These genes might be differentially regulated at the translational or post-translational levels, such as via phosphorylation or ubiquitination (Chuang and Ulevitch 2004; Miggin and O’Neill 2006). By doing so, the host immune systems may realize a versatile network in terms of its dynamics and robustness, enabling flexible responses against various types of pathogens.
Indeed, our current analysis has brought numerous important clues that deserve future in-depth biological analyses. We fully acknowledge that intensive analyses of laboratory strains, such as 3D7, may be extremely useful; however, behaviors of the field malaria parasites are far more complex than those observed under laboratory conditions (Daily et al. 2007). Furthermore, it is now recognized that parasite genotypes are rapidly diversifying (Miotto et al. 2013). We believe complementary use of both in field and laboratory strains will eventually reveal a global view of malaria etiology occurring in patients.
Methods
Samples
Blood samples of 116 patients diagnosed with Pf infections by the rapid malaria paper test (Abbott) and occasionally by the smear test (according to the standard method [Moll et al. 2008]) were collected at several field hospitals in the surrounding area of Sam Ratulangi University Hospital in Manado, Indonesia from 2006 to 2010 (Supplemental Table 11). All of the samples were collected following informed consent of the patients, and the collections were approved by the local ethical committee of Sam Ratulangi University and that of the University of Tokyo. To estimate false positive detection rates of Pf tags, samples were similarly collected from 25 healthy people and 28 patients infected with other pathogens. Details of the sample descriptions are also shown in Supplemental Figure 1 and Supplemental Tables 1, 2, and 11.
RNA-seq
For the RNA-seq analysis, we used 2.5-mL samples of peripheral blood that were first isolated and stabilized with a PAXgene Blood RNA Tube (BD). From 2.5 mL of the PAXgene Blood RNA Tube sample, total RNA was extracted using the PAXgene Blood RNA Kit (BD). RNA-seq libraries were prepared following the manufacturers’ instructions using the TruSeq RNA-seq kit (Illumina). A single lane of 36-bp single-end sequencing was performed for each sample (Illumina GAIIx platform). The RNA-seq tags were then mapped to the reference genomes of human (hg19) (UCSC Genome Browser; http://genome.ucsc.edu/) and Pf (PlasmoDB Release 6.0; http://plasmodb.org/plasmo/), allowing two-base mismatches, using the BWA mapping software (Li and Durbin 2009). Details of the mapping procedure are described in the legend for Supplemental Figure 1. For the raw data for each gene used in this study, see Supplemental Table 2. Experimental conditions, results of the real time RT-PCR, and the primers used for the validation are shown in Supplemental Table 12.
Computational procedures
Gene Ontology (GO) terms were obtained from the UCSC Genome Browser for human and from PlasmoDB for parasites. KEGG data were obtained directly from the KEGG database (http://www.genome.jp/kegg/). Statistical significance was calculated using the indicated methods. To identify the genes and pathways which are associated with the clinical information, only the cases where the difference was also statistically significant against the healthy controls were counted. In all cases, the statistical analysis software package R was used for the calculation (Gentleman et al. 2004).
For calling SNPs, a SNP caller GATK was used (McKenna et al. 2010). SNPs called with confidence scores greater than 50 were selected. SNPs supported by more than five independent RNA-seq tags were further selected and used for the analysis. Hierarchal clustering was performed using Bioconductor in R. A phylogenic tree for each gene was drawn using MEGA 4 (Tamura et al. 2007). The genetic distance between each genotype was calculated by considering the mutual Hamming distance of the detected SNPs (Isaev 2004). Only genes with at least five mutually comparable SNPs were considered. To identify SNVs which are associated with %PF tags, statistical bias of the occurrence in the patient group giving a larger number of Pf read counts was evaluated for each of the identified SNVs. For this purpose, Wilcoxon signed rank test was used, and the cases giving P-values of 1 × 10−3 were selected.
To provide a viewer, we constructed the Full-Malaria database (http://fullmal.hgc.jp). A search example is also shown in Supplemental Figure 7.
Data access
The sequencing data from this study have been submitted to the DNA Data Bank of Japan (DDBJ; http://www.ddbj.nig.ac.jp/) under accession number DRA000949.
Acknowledgments
We thank J. Watanabe for the many years of work on this project. We also thank T. Horiuchi and K. Toya for their excellent programming work, K. Abe for constructing the RNA-seq libraries, and K. Imamura, A. Kanai, and M. Tosaka for performing the RNA-seq sequencing. This work was supported by a Grant-in-Aid for Publication of Scientific Research Results from the Japan Society for Promotion of Science and JST and a Grant from the Asia-Africa S&T Strategic Cooperation Promotion Program by the Special Coordination Funds for Promoting Science & Technology from the Ministry of Education, Culture, Sports, Science and Technology of Japan. This research was also supported in part by the Cabinet Office, Government of Japan and the Japan Society for the Promotion of Science (JSPS) through the Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST Program).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.158980.113.
References
- Aregawi M, Cibulskis RE, Lynch M, Williams R, World Health Organization, Global Malaria Programme . 2011. World malaria report 2011. World Health Organization, Geneva. [Google Scholar]
- Armour CD, Castle JC, Chen R, Babak T, Loerch P, Jackson S, Shah JK, Dey J, Rohl CA, Johnson JM, et al. . 2009. Digital transcriptome profiling using selective hexamer priming for cDNA synthesis. Nat Methods 6: 647–649 [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. . 2000. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS, et al. . 2009. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res 37: D539–D543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birve A, Sengupta AK, Beuchle D, Larsson J, Kennison JA, Rasmuson-Lestander A, Muller J. 2001. Su(z)12, a novel Drosophila Polycomb group gene that is conserved in vertebrates and plants. Development 128: 3371–3379 [DOI] [PubMed] [Google Scholar]
- Chuang TH, Ulevitch RJ. 2004. Triad3A, an E3 ubiquitin-protein ligase regulating Toll-like receptors. Nat Immunol 5: 495–502 [DOI] [PubMed] [Google Scholar]
- Coban C, Ishii KJ, Kawai T, Hemmi H, Sato S, Uematsu S, Yamamoto M, Takeuchi O, Itagaki S, Kumar N, et al. . 2005. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J Exp Med 201: 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coban C, Ishii KJ, Horii T, Akira S. 2007. Manipulation of host innate immune responses by the malaria parasite. Trends Microbiol 15: 271–278 [DOI] [PubMed] [Google Scholar]
- Coban C, Igari Y, Yagi M, Reimer T, Koyama S, Aoshi T, Ohata K, Tsukui T, Takeshita F, Sakurai K, et al. . 2010. Immunogenicity of whole-parasite vaccines against Plasmodium falciparum involves malarial hemozoin and host TLR9. Cell Host Microbe 7: 50–61 [DOI] [PubMed] [Google Scholar]
- Cosgrove MS, Patel A. 2010. Mixed lineage leukemia: a structure-function perspective of the MLL1 protein. FEBS J 277: 1832–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daily JP, Scanfeld D, Pochet N, Le Roch K, Plouffe D, Kamal M, Sarr O, Mboup S, Ndir O, Wypij D, et al. . 2007. Distinct physiological states of Plasmodium falciparum in malaria-infected patients. Nature 450: 1091–1095 [DOI] [PubMed] [Google Scholar]
- Dinarello CA 2000. Proinflammatory cytokines. Chest 118: 503–508 [DOI] [PubMed] [Google Scholar]
- Duraisingh MT, Cowman AF. 2005. Contribution of the pfmdr1 gene to antimalarial drug-resistance. Acta Trop 94: 181–190 [DOI] [PubMed] [Google Scholar]
- Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, Ursos LM, Sidhu AB, Naude B, Deitsch KW, et al. . 2000. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell 6: 861–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin BS, Parroche P, Ataide MA, Lauw F, Ropert C, de Oliveira RB, Pereira D, Tada MS, Nogueira P, da Silva LH, et al. . 2009. Malaria primes the innate immune response due to interferon-γ induced enhancement of toll-like receptor expression and function. Proc Natl Acad Sci 106: 5789–5794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Ding Y, Zhou T, Fu X, Xu W. 2012. Plasmodium yoelii blood-stage primes macrophage-mediated innate immune response through modulation of toll-like receptor signalling. Malar J 11: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. . 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore TD 2006. Introduction to NF-κB: players, pathways, perspectives. Oncogene 25: 6680–6684 [DOI] [PubMed] [Google Scholar]
- Han C, Jin J, Xu S, Liu H, Li N, Cao X. 2010. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol 11: 734–742 [DOI] [PubMed] [Google Scholar]
- Hartgers FC, Obeng BB, Voskamp A, Larbi IA, Amoah AS, Luty AJ, Boakye D, Yazdanbakhsh M. 2008. Enhanced Toll-like receptor responsiveness associated with mitogen-activated protein kinase activation in Plasmodium falciparum-infected children. Infect Immun 76: 5149–5157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Natoli G, Ghosh G. 2006. Transcriptional regulation via the NF-κB signaling module. Oncogene 25: 6706–6716 [DOI] [PubMed] [Google Scholar]
- Isaev A. 2004. Introduction to mathematical methods in bioinformatics. Springer, New York. [Google Scholar]
- Ito T, Amakawa R, Fukuhara S. 2002. Roles of toll-like receptors in natural interferon-producing cells as sensors in immune surveillance. Hum Immunol 63: 1120–1125 [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. 2012. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40: D109–D114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11: 373–384 [DOI] [PubMed] [Google Scholar]
- Kishimoto T 2006. Interleukin-6: discovery of a pleiotropic cytokine. Arthritis Res Ther (Suppl 2) 8: S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnegowda G, Hajjar AM, Zhu J, Douglass EJ, Uematsu S, Akira S, Woods AS, Gowda DC. 2005. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J Biol Chem 280: 8606–8616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, Moestrup SK. 2001. Identification of the haemoglobin scavenger receptor. Nature 409: 198–201 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manske M, Miotto O, Campino S, Auburn S, Almagro-Garcia J, Maslen G, O’Brien J, Djimde A, Doumbo O, Zongo I, et al. . 2012. Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature 487: 375–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. . 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miggin SM, O’Neill LA. 2006. New insights into the regulation of TLR signaling. J Leukoc Biol 80: 220–226 [DOI] [PubMed] [Google Scholar]
- Miotto O, Almagro-Garcia J, Manske M, Macinnis B, Campino S, Rockett KA, Amaratunga C, Lim P, Suon S, Sreng S, et al. . 2013. Multiple populations of artemisinin-resistant Plasmodium falciparum in Cambodia. Nat Genet 45: 648–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll K, Ljungstrom I, Perlmann H, Scherf A, Wahlgren M. 2008. Methods in malaria research, 5th ed., pp. 18–19. Malaria Research and Reference Reagent Resource Center (MR4), American Type Culture Collection, Manassas, VA. [Google Scholar]
- Mu J, Myers RA, Jiang H, Liu S, Ricklefs S, Waisberg M, Chotivanich K, Wilairatana P, Krudsood S, White NJ, et al. . 2010. Plasmodium falciparum genome-wide scans for positive selection, recombination hot spots and resistance to antimalarial drugs. Nat Genet 42: 268–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto TD, Wilinski D, Assefa S, Keane TM, Sarry LR, Bohme U, Lemieux J, Barrell B, Pain A, Berriman M, et al. . 2010. New insights into the blood-stage transcriptome of Plasmodium falciparum using RNA-Seq. Mol Microbiol 76: 12–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl HL 1999. Activators and target genes of Rel/NF-κB transcription factors. Oncogene 18: 6853–6866 [DOI] [PubMed] [Google Scholar]
- Palm NW, Medzhitov R. 2009. Pattern recognition receptors and control of adaptive immunity. Immunol Rev 227: 221–233 [DOI] [PubMed] [Google Scholar]
- Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, Halmen KA, Lamphier M, Olivier M, Bartholomeu DC, et al. . 2007. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci 104: 1919–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price RN, Uhlemann AC, Brockman A, McGready R, Ashley E, Phaipun L, Patel R, Laing K, Looareesuwan S, White NJ, et al. . 2004. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet 364: 438–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao N, Dodge I, Band H. 2002. The Cbl family of ubiquitin ligases: critical negative regulators of tyrosine kinase signaling in the immune system. J Leukoc Biol 71: 753–763 [PubMed] [Google Scholar]
- Reed MB, Saliba KJ, Caruana SR, Kirk K, Cowman AF. 2000. Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature 403: 906–909 [DOI] [PubMed] [Google Scholar]
- Rogier C, Commenges D, Trape JF. 1996. Evidence for an age-dependent pyrogenic threshold of Plasmodium falciparum parasitemia in highly endemic populations. Am J Trop Med Hyg 54: 613–619 [DOI] [PubMed] [Google Scholar]
- Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T. 2009. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat Med 15: 696–700 [DOI] [PubMed] [Google Scholar]
- Sawasdikosol S, Pratt JC, Meng W, Eck MJ, Burakoff SJ. 2000. Adapting to multiple personalities: Cbl is also a RING finger ubiquitin ligase. Biochim Biophys Acta 1471: M1–M12 [DOI] [PubMed] [Google Scholar]
- Song HY, Rothe M, Goeddel DV. 1996. The tumor necrosis factor-inducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-κB activation. Proc Natl Acad Sci 93: 6721–6725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. 2010. Pattern recognition receptors and inflammation. Cell 140: 805–820 [DOI] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24: 1596–1599 [DOI] [PubMed] [Google Scholar]
- Tuda J, Mongan AE, Tolba ME, Imada M, Yamagishi J, Xuan X, Wakaguri H, Sugano S, Sugimoto C, Suzuki Y. 2011. Full-parasites: database of full-length cDNAs of apicomplexa parasites, 2010 update. Nucleic Acids Res 39: D625–D631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vereecke L, Beyaert R, van Loo G. 2009. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol 30: 383–391 [DOI] [PubMed] [Google Scholar]
- Volkman SK, Sabeti PC, DeCaprio D, Neafsey DE, Schaffner SF, Milner DA Jr, Daily JP, Sarr O, Ndiaye D, Ndir O, et al. . 2007. A genome-wide map of diversity in Plasmodium falciparum. Nat Genet 39: 113–119 [DOI] [PubMed] [Google Scholar]