

Abstract
Microbiota regulate intestinal physiology by modifying host gene expression along the length of the intestine, but the underlying regulatory mechanisms remain unresolved. Transcriptional specificity occurs through interactions between transcription factors (TFs) and cis-regulatory regions (CRRs) characterized by nucleosome-depleted accessible chromatin. We profiled transcriptome and accessible chromatin landscapes in intestinal epithelial cells (IECs) from mice reared in the presence or absence of microbiota. We show that regional differences in gene transcription along the intestinal tract were accompanied by major alterations in chromatin accessibility. Surprisingly, we discovered that microbiota modify host gene transcription in IECs without significantly impacting the accessible chromatin landscape. Instead, microbiota regulation of host gene transcription might be achieved by differential expression of specific TFs and enrichment of their binding sites in nucleosome-depleted CRRs near target genes. Our results suggest that the chromatin landscape in IECs is preprogrammed by the host in a region-specific manner to permit responses to microbiota through binding of open CRRs by specific TFs.
Animal physiology is directed by interactions between factors encoded in the animal’s genome and those encountered in its environment. The impact of these interactions on animal health is most evident in the intestine, where digestion and absorption of dietary nutrients occur in the presence of complex communities of microorganisms (intestinal microbiota). The identification of intestinal microbiota as prominent environmental factors shaping diverse aspects of intestinal and extraintestinal health and disease has fueled intense interest in defining the mechanisms underlying host-microbiota interactions (Sommer and Bäckhed 2013). The primary interface between animal hosts and their microbiota is the intestinal epithelium, which encounters dynamic environmental stimuli from microbiota along the length of the gut (Camp et al. 2009; Pott and Hornef 2012). As with other tissues, intestinal epithelial function is predicated on the ability to produce and maintain multiple cell types while also retaining the ability to respond to environmental stimuli, all using the same genome. Accordingly, the intestinal epithelium exhibits extensive functional specialization along its proximal-distal axis characterized by distinct gene expression programs and differences in cell-type abundance (van der Flier and Clevers 2009). Comparisons of mice reared in the absence of microorganisms (germ-free or GF) to those colonized with a normal microbiota have revealed that gene expression in the intestine is profoundly altered by the presence of a microbiota (Rawls et al. 2006; El Aidy et al. 2012; Larsson et al. 2012; Pott et al. 2012). Furthermore, comparisons of GF mice to those colonized by microbiota for variable lengths of time revealed that microbiota-induced alterations to host gene expression are temporally dynamic and require several weeks to reach homeostasis (El Aidy et al. 2012, 2013). Proper orchestration of these microbiota-induced gene expression programs in a tissue-specific context is essential for establishing host-microbe commensalism and sustaining host health. However, the regulatory mechanisms through which microbiota modify host gene expression in the intestinal epithelium remain unresolved.
Specification and tuning of gene transcription proceeds in part through coordinate interactions between transcription factors (TFs) and cis-regulatory DNA. Cis-regulatory regions (CRRs) harbor binding sites for multiple activating or repressing TFs and can be located proximal to the transcription start site (TSS), within gene bodies, as well as in intergenic regions distal to the TSS (Bulger and Groudine 2011). CRRs are generally distinguished by the low occupancy of nucleosomes on genomic DNA, which can be experimentally captured by hypersensitivity to DNase I cleavage (Boyle et al. 2008). DNase-seq is a high-throughput, quantitative method that generates genome-wide accessible chromatin profiles which strongly correlate with in vivo transcription factor occupancy and gene expression levels (Thurman et al. 2012). We reasoned that DNase-seq could be used to discover CRRs of various types (e.g., promoters, enhancers, silencers, locus control regions) that mediate host transcriptional responses to microbiota in epithelial cells along the length of the intestinal tract. We found that regional differences in gene transcription along the length of the intestine were accompanied by major alterations in the accessible chromatin landscape. Surprisingly, we discovered that commensal microbiota modify the transcriptional landscape in the intestinal epithelium without significantly impacting the accessible chromatin landscape. Instead, we find that open intestinal CRRs linked to microbiota-responsive genes are enriched with binding motifs for microbiota-responsive TFs. Our results suggest that the chromatin landscape in intestinal epithelial cells is “preprogrammed” by the host in a region-specific manner to permit transcriptional responses to environmentally acquired intestinal microbiota likely through differential binding of CRRs by specific TFs. This data extends support for the model that cell fate specification is associated with acquisition of a specific accessible chromatin architecture, which is subsequently utilized by cells to respond to a perpetually dynamic environment (John et al. 2011; Samstein et al. 2012). Cumulatively, this work provides a foundational approach and essential resource for understanding the role of the cis-regulatory genome in mediating host-microbe commensalism in the intestine.
Results
RNA-seq reveals acute and chronic transcriptome alterations in response to microbiota in the mouse ileal and colonic epithelium
To determine the genome-wide impact of microbiota on host gene transcription in the gut epithelium, we measured the messenger RNA transcriptome in intestinal epithelial cells (IECs) isolated from the ileum and colon of mice reared in the presence and absence of microbiota (Fig. 1; Supplemental Table S1). We compared three distinct microbial states in order to determine acute and chronic effects of microbiota on host transcription (Fig. 1A). Germ-free (GF) mice were reared for 10–12 wk in the absence of any microbes. Conventionally raised (CR) mice were reared since birth in the presence of microbiota for 10–12 wk (chronic colonization). Conventionalized (CV) mice were reared under GF conditions for 8–10 wk and then colonized for two weeks with microbiota (acute colonization). Isolated IECs display uniform expression of the pan-epithelial cell surface marker EpCAM (Bjerknes and Cheng 1981; von Furstenberg et al. 2011) and lack the CD31 endothelial and immune cell surface marker (Fig. 1B). As expected, we observed robust differences between ileal and colonic IEC transcriptomes, supporting the significant physiological differences between these distinct tissues (Fig. 1C,D; Supplemental Figs. S1, S2; Supplemental Table S2). Biological replicates from each microbial state clustered together, a result consistent in both the ileal and colonic epithelium (Fig. 1C). We found that acute colonization (CV) has a larger impact on IEC gene expression than lifelong presence of microbiota (CR) in comparison to GF IEC transcriptomes, a finding supported by previous studies of temporal responses to microbiota (Fig. 1C,D; El Aidy et al. 2012, 2013). We determined a set of genes from CR and CV mice that were significantly different than GF in either the ileum or colon (Fig. 1E; Supplemental Table S3). Hierarchical clustering of these genes followed by Gene Ontology (GO) functional categorization revealed the impact of microbiota on distinct intestinal epithelial biological processes in each tissue (Supplemental Table S4). Consistent with previous studies (Rawls et al. 2006; El Aidy et al. 2012; Larsson et al. 2012; Pott et al. 2012), our RNA-seq data reveal that microbiota induce various aspects of immune response in both ileal and colonic IECs under both CR and CV conditions (Fig. 1F). Gene clusters involved in transport and metabolism of lipids and other nutrients were generally down-regulated by microbiota in both the ileum and colon. Together, our RNA-seq data revealed that gut microbiota elicit genome-wide alterations to host gene transcription in the intestinal epithelium, a response that varies depending on intestinal region and time post-colonization.
Figure 1.

RNA-seq reveals transcriptome alterations in the presence and absence of microbiota in the mouse ileal and colonic epithelium. (A) Overview of experiments described in this study. Schematic of the mouse gastrointestinal tract showing the stomach (dark gray), duodenum (teal), jejunum (dark gray), ileum (blue), cecum (dark gray), and colon (red). Adapted from Stevens (1977). © 1933 by H.H. Dukes; © 1977 by Cornell University. Used by permission of the publisher, Cornell University Press. Approximately 6-cm sections of the duodenum, ileum, or colon were used for intestinal epithelial cell (IEC) isolation (see Methods). DNase-seq and RNA-seq were performed on intestinal epithelial cells (IECs, ∼90% purity) isolated from the ileum and colon of germ-free (GF), conventionally raised (CR), and ex-GF conventionalized (CV) mice. DNase-seq was also performed on IECs isolated from the duodenum of CR mice. (B) Fluorescence-activated cell sorting of pooled duodenal and ileal IECs labeled with antibodies marking either epithelial cells (EpCAM) or endothelial cells/leukocytes/platelets (CD31) reveal that ∼90% of cells were epithelial (EpCAM positive and CD31 negative). Similar results were obtained from colonic IEC preparations (data not shown). (C) Dendrogram of Jensen-Shannon divergence shows that RNA-seq replicates from GF, CR, or CV ileal or colonic IECs cluster. Note that anatomical location and environmental condition, rather than sibling relationship, drives the clustering. (D) Principal component analysis (PCA) confirms tissue type (PC1) and colonization state (PC2 and PC3) explains much of the variance observed in the RNA-seq data. Arrow tips denote sample position in PCA coordinates. (E) Volcano plot showing pairwise comparisons of RNA expression between GF versus CR and GF versus CV conditions for each tissue. Green dots represent genes that are significantly different (FDR < 0.05). (F) Hierarchical clustering of FPKM values for all genes that exhibited differential expression in the pairwise comparisons in D. Gene clusters were submitted to DAVID to determine Gene Ontology functional enrichment. Shown are top enrichments for each gene cluster. See also Supplemental Tables S1, S3, and S4.
Chromatin accessibility displays regional variation along the length of the GI tract and correlates with gene expression
We next sought to determine the feasibility of using DNase-seq to discover cis-regulatory regions (CRRs) that control epithelial transcriptional response to gut microbiota along the length of the intestine (Fig. 2). Because there is high endogenous DNase activity in the intestine (Fig. 2A; Lacks 1981), we developed a modified DNase-seq protocol (Song and Crawford 2010) using endogenous DNases to digest IEC chromatin (Fig. 2B). Using CR mice, we found that endogenous DNase activity identified duodenal, ileal, and colonic DNase hypersensitive sites (DHSs) that are highly reproducible (Supplemental Fig. S3), often evolutionarily conserved (Fig. 2C; Supplemental Fig. S4B), demarcate transcription start sites in promoter regions (Fig. 2D,E), and overlap both novel and previously described intestinal enhancers (Fig. 2E; Supplemental Fig. S4C–F; Madison 2002; Shen et al. 2012). In addition, DNase-seq in IECs identified accessible chromatin at biomarker genes associated with abundant and rare epithelial cell types including enterocytes, enteroendocrine cells, goblet cells, Paneth cells, and stem cells (Supplemental Fig. S5). These results confirm that our modified DNase-seq strategy effectively captures the IEC accessible chromatin landscape in the duodenum, ileum, and colon and exhibited hallmarks of previously described DNase-seq data sets that used exogenous DNase to digest chromatin.
Figure 2.

Endogenous DNase activity distinguishes open chromatin in mouse intestinal epithelial cells. (A) Pulse-field gel image of nuclei digested for 15 min at 37°C with increasing concentrations of exogenous DNase I. Note that high-molecular-weight (HMW) DNA is stable at 0°C; however, there is significant DNA digestion even with no addition of exogenous DNase when nuclei are incubated for 15 min at 37°C. (m) Yeast chromosome marker. (B) Endogenous DNase activity is detected within 30 sec after moving nuclei to 37°C, and by 8 min, most HMW DNA is digested. Patterns were consistent for duodenum, ileum, and colon (see Supplemental Fig. S4). The observed digestion pattern is similar to reported digestion patterns using exogenous DNase I (Song and Crawford 2010). For DNase-seq library preparation, nuclei digested for 2, 4, and 8 min were pooled to capture a range of DNase hypersensitivities. Libraries were prepared for duodenal, ileal, and colonic IECs. (C) Average phastCons scores plotted for the top 100,000 DHSs from duodenal, ileal, and colonic IECs centered at the peak maximum. Nongenic DNA flanking ileal DNase hypersensitive sites (DHSs) was used to assess background conservation (control). (D) Feature distribution of the top 100,000 and 25,000 DHSs from each tissue. Note the increased representation of promoter-associated sites (<2 kb from annotated transcription start sites) in the 25,000 DHSs with the highest signal intensity. (E) DNase-seq signal tracks from conventionally raised (CR) duodenal, ileal, and colonic IECs at the villin 1 (Vil1) locus. Note strong peaks at the transcription start site (DHS 1) and within the first intron (DHS 2). A 12.4-kb region including both DHS 1 and DHS 2 drives IEC-specific crypt and villous expression in the duodenum, ileum, and colon (Madison 2002); however, DHS 2 is required for crypt expression. For comparison, DNase-seq signal from the liver is also shown. (F) Venn diagram enumerating differential DHSs along the length of the GI tract. (G) Hierarchical clustering of differential DHSs across replicates of CR duodenal, ileal, and colonic IECs reveals open chromatin sites specific to each tissue. (cs) Colon-specific; (i:c) ileum and colon; (ds) duodenum specific; (d:c) duodenum and colon; (is) ileum specific; (d:i) duodenum and ileum. (H) Feature distribution showing that the majority of segment-specific DHSs are located in intergenic (>2 kb away from a gene body) or intronic regions of the genome. See also Supplemental Figures S2–S4 and Supplemental Tables S4 and S6.
Proximal-distal functional specialization along the intestinal tract is associated with widespread alterations in gene expression (Fig. 1; Supplemental Fig. S2), but the relationship with the accessible chromatin landscape was unknown. We compared DNase-seq in IECs isolated from the duodenum, ileum, and colon of CR mice (see Methods) in order to discover segment-specific cis-regulatory regions (CRRs) along the length of the intestine (Fig. 2E; Supplemental Table S5). We identified 131,073 accessible chromatin regions that are shared between each segment of the intestinal tract (Fig. 2F). These “pan-intestine” DHSs are associated with a wide variety of genes that have known functions in intestinal epithelial cell biology including an enrichment near genes involved in nutrient transporter activity, adherens junctions, and intestinal morphogenesis (Supplemental Table S6). We identified 7211 DHSs that are common to both the epithelium of duodenum and ileum but absent from the colon (small intestine-specific). These DHSs are near genes enriched in GO Biological Process categories characteristic for small intestinal activities including metabolism of steroids, peptides, lipids, and lipoproteins (Supplemental Table S6). In addition, we identified 2361, 2554, and 21,724 DHSs that are specific to the duodenum, ileum, or colon, respectively, with a false discovery rate (FDR) < 0.0001 (Fig. 2F,G). These segment-specific DHSs are generally located in intergenic or intronic DNA (Fig. 2H, D), are enriched for whole intestine-specific H3K4me1 histone marks (enhancers) (Supplemental Fig. S4D–F), and are near genes enriched in diverse molecular functions and biological processes specific to each intestinal segment (Supplemental Table S6). These data provide a genome-wide atlas of accessible chromatin in the intestinal epithelium of conventionally raised mice and indicate significant regional specialization of gene regulatory activity in IECs along the length of the intestinal tract.
We next compared the mRNA and accessible chromatin landscapes in CR ileal or colonic IECs to determine the correlation of segment-specific DHSs with gene expression. We identified 2773 transcripts that are differentially expressed between ileal and colonic IECs (Fig. 3A; Supplemental Fig. S2; Supplemental Table S2). We also identified numerous quantitative differences in DNase hypersensitivity between ileal and colonic IECs (Fig. 3B). Qualitatively, we found that many differentially expressed genes have one or more segment-specific DHSs nearby, which likely explains differences in gene expression observed between ileal and colonic IECs. For example, diacylglycerol O-acyltransferase 1 (Dgat1), an enzyme that catalyzes the formation of triglycerides in ileal enterocytes (Lee et al. 2010), is highly expressed in the ileum but not the colon (Fig. 3C). DNase-seq identified accessible chromatin in the first and third introns of Dgat1 specific to the ileal epithelium (Fig. 3C). In contrast, the aquaporin 8 (Aqp8) gene encodes a water channel protein highly expressed in the colonic epithelium (Yang et al. 2005) and has a colon-specific DHS ∼13 kb upstream of the transcription start site (Fig. 3C). Indeed, most genes (1897 out of 2175; 87.2%) that are differentially expressed between the ileal and colonic IECs have a segment-specific DHS within their gene regulatory domain (see Methods; Figs. 3D, 5D). Additionally, we find that increased DNase hypersensitivity at the proximal promoter is best associated with increased gene expression (Supplemental Fig. S6A). However, the greatest number of differential DHSs associated with differential gene expression are within the gene body, and we speculate that many of these tissue-restricted DHSs are facilitating enhancer activity to promote nearby gene expression (Supplemental Fig. S6B). Collectively, these results integrate genome-wide RNA-seq and DNase-seq data to identify putative CRRs controlling segment-specific patterns of gene transcription in IECs underlying proximal-distal functional specialization along the intestinal tract.
Figure 3.

Differential open chromatin between ileal and colonic IECs correlates with differential gene expression. (A) Volcano plot showing pairwise comparison of RNA expression between conventionally raised (CR) ileal and colonic epithelium. Blue and orange dots represent genes more highly expressed in the ileum or colon, respectively (FDR < 0.05). (B) The fold difference in DNase signal intensity from CR ileal versus colonic IECs plotted against the average DNase signal observed in 250-bp windows. Significantly differential windows are highlighted in red and blue (FDR < 0.0001). (C) Representative signal track view highlighting two genes, diacylglycerol O-acyltransferase 1 (Dgat1) and aquaporin 8 (Aqp8), that exhibit differential open chromatin and transcript abundance in the ileum or colon. (D) Two-sided Kolmogorov-Smirnov goodness-of-fit test shows a positive relationship between the presence of a nearby tissue-specific DHS (within 2 kb of and including the gene body) and increased transcript abundance in that tissue. The y-axis shows the cumulative fraction of genes linked to a nearby tissue-specific DHS. Deviation from the null expectation that linked genes display a normal distribution centered on a fold change of 1 (black line) suggests that segment-specific DHSs are enriched near genes of higher expression in that tissue. See also Supplemental Figures S5 and S6.
Figure 5.
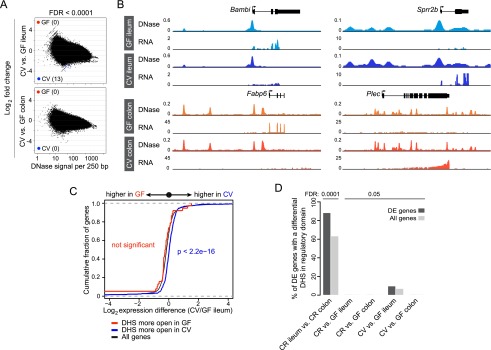
Microbiota do not substantially remodel the intestinal epithelial chromatin landscape upon acute colonization. (A) The fold difference in DNase signal intensity from conventionalized (CV) versus germ-free (GF) ileal or colonic IECs plotted against the average DNase signal observed in 250-bp windows. Significantly differential windows are highlighted in red and blue (FDR < 0.0001). (B) Representative signal track highlighting multiple genes in the ileum or colon that show differences in transcript abundance upon colonization with microbiota but no detectable change in the open chromatin landscape. (Bambi) BMP and activin membrane-bound inhibitor; (Sprr2b ) small proline-rich protein 2B; (Fabp6) fatty acid binding protein 6; (Plec) plectin. See also Supplemental Tables S3 and S7. (C) Two-sided Kolmogorov-Smirnov goodness-of-fit test shows a weak relationship between the presence of a nearby tissue-specific DHS (within 2 kb of the gene body) and increased transcript abundance in the GF versus CV ileum comparison at FDR < 0.05. The y-axis shows the cumulative fraction of genes linked to a nearby tissue-specific DHS. Deviation from the null expectation that linked genes display a normal distribution centered on a fold change of 1 (black line) suggests that CV-specific DHSs are enriched near genes of higher expression in CV ileal IECs. (D) Percent of differentially expressed genes that have a differential DNase hypersensitive site within their regulatory domain at two cutoffs (FDR < 0.0001 and FDR < 0.05). See also Supplemental Figure S7 and Supplemental Table S8.
Microbiota modulate gene expression without remodeling the intestinal epithelial accessible chromatin landscape
Our comparative analysis of accessible chromatin across intestinal segments suggested that DNase-seq could be used to identify CRRs that mediate intestinal epithelial responses to microbiota. To test the hypothesis that commensal microbiota modify IEC transcription through modification of the accessible chromatin landscape, we generated DNase-seq data sets from IECs isolated from the ileum and colon of GF mice and compared them to CR animals that had been exposed to microbiota from birth (Fig. 1A). Surprisingly, we discovered that the accessible chromatin landscape in IECs of GF and CR mice is nearly identical for both the ileum and colon (Fig. 4). Hierarchical clustering did not identify a significant subpopulation of DHS specific to GF or CR conditions in either the colon or the ileum (Fig. 4A). In accord, DNase signal intensities within GF and CR accessible chromatin in the ileum and colon were highly correlated with Spearman’s rho of 0.938 and 0.919, respectively (Fig. 4B). This is in contrast to the correlation observed between CR ileum and CR colon (0.634), CR duodenum and CR colon (0.647), and CR duodenum and CR ileum (0.777) (Fig. 4B; Supplemental Fig. S7). We scanned the genome for differential DNase cleavage in GF and CR ileal or colonic IECs across 250-bp windows. Using the same FDR threshold (<0.0001) from our analysis that discovered thousands of differential DNase hypersensitive sites between intestinal segments in CR mice (Fig. 3B; Supplemental Fig. S7B), we found only one DHS that was significantly different between GF and CR conditions in either the ileum or colon (Fig. 4C). Loosening the FDR threshold 500-fold to FDR < 0.05, we identified only nine 250-bp windows (Supplemental Table S7) with significantly different DNase hypersensitivity between GF ileum and CR ileum and identified none in the colon (Supplemental Fig. S8). The nine DHSs reaching modest significance in the ileum were not near any gene known to be regulated by microbiota (Supplemental Table S7; Rawls et al. 2006; Donohoe et al. 2011; El Aidy et al. 2012; Larsson et al. 2012; Pott et al. 2012). Therefore, the differential transcript levels observed for many genes in GF and CR IECs (Fig. 1E; Supplemental Table S3) were not linked to any significant alteration in local chromatin accessibility. For example, angiopoietin-like 4 (Angptl4), known to be suppressed by microbiota in ileal IECs (Bäckhed et al. 2004; Camp et al. 2012), was corroborated by our RNA-seq analysis. However, the accessible chromatin landscape at this locus is identical in both the GF and CR ileum (Fig. 4D). A similar relationship was observed for other genes with known gene expression responses to microbiota, including fibroblast growth factor 15 (Fgf15) (Sayin et al. 2013), cytochrome P450, family 4, subfamily b, polypeptide 1 (Cyp4b1) (Larsson et al. 2012), and angiogenin, ribonuclease A family, member 4 (Ang4) (Hooper et al. 2003). These results revealed that mice, reared lifelong in the presence or absence of microbiota, have nearly identical IEC accessible chromatin landscapes.
Figure 4.
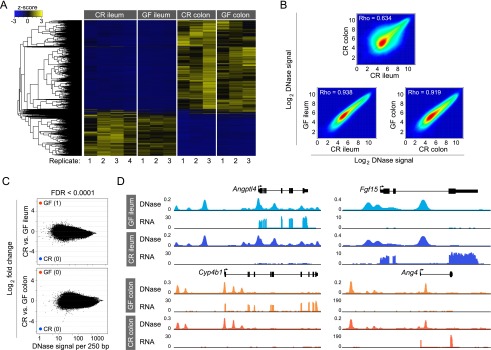
Life in the presence or absence of microbiota does not affect the intestinal epithelial accessible chromatin landscape. (A) Hierarchical clustering of differential DHSs across all replicates of conventionally raised (CR) versus germ-free (GF) ileal and colonic IECs. Note the similarity between GF and CR conditions for each tissue. (B) Density scatter plot showing the correlation of DNase-seq signal intensity for the top 100,000 DHSs for CR colon and CR ileum (top), GF ileum and CR ileum (bottom left), and GF colon and CR colon (bottom right). (C) The fold difference in DNase signal intensity plotted against the average DNase signal observed in 250-bp windows. Significantly differential windows are highlighted in red and blue (FDR < 0.0001). Comparing across tissues (CR colon vs. CR ileum) discovered thousands of differential DNase hypersensitive sites (see Fig. 3B). Comparing tissues in the presence or absence of microbiota reveals undetectable change in the open chromatin landscape in response to microbiota. (D) Representative signal track highlighting multiple genes in the ileum or colon that show differences in transcript abundance in the presence of microbiota but no change in the open chromatin landscape. (Angptl4) Angiopoietin-like 4; (Fgf15) fibroblast growth factor 15; (Cyp4b1) cytochrome P450, family 4, subfamily b, polypeptide 1; (Ang4) angiogenin, ribonuclease A family, member 4. See also Supplemental Tables S3 and S7.
Colonization of GF mice with microbiota is known to evoke dynamic temporal alterations in gene expression (El Aidy et al. 2012, 2013), raising the possibility that transcriptional responses to acute and lifelong colonization may utilize distinct regulatory mechanisms. We therefore tested whether acute colonization with microbiota would alter IEC accessible chromatin by generating DNase-seq data sets from IECs isolated from the ileum and colon of mice raised GF for 8 wk, then conventionalized (CV) for 2 wk with microbiota. Again, despite a robust effect on the gene expression landscape (Fig. 1C–F), conventionalization with microbiota had minimal impact on the accessible chromatin landscape in either the ileum or colon (Fig. 5A,B). Loosening the FDR threshold (FDR < 0.05), we were able to identify regions of differential accessible chromatin in the ileum that are near microbiota-regulated genes (Supplemental Fig. S8A,B; Supplemental Table S8). DHSs more open in CV had a weak but significant correlation with differential gene expression in the ileum (Fig. 5C); however, the vast majority (91%) of microbiota-regulated genes in CV versus GF ileum did not have a differential DHS nearby (Fig. 5D). Visual inspection of many of the putatively differential DHSs revealed qualitatively minimal alterations in accessible chromatin (Supplemental Fig. S8C,D). Notably, there was no significant functional enrichment of genes linked to nearby DHSs putatively differential in GF versus CV ileum (GREAT v2.0.2 default thresholds) (data not shown). In addition, we failed to identify any differential DHSs near microbiota-regulated genes in the colon (Fig. 5D). Indeed, we failed to observe any regions of substantial accessible chromatin differences in the presence or absence of microbiota in either the ileum or colon. This result was fundamentally different from results obtained in our between-tissue comparisons (Figs. 2E, 3B,C; Supplemental Figs. S4D–G, S7). Cumulatively, these data revealed that commensal microbiota modify the transcriptional landscape in the intestinal epithelium without remodeling the host’s accessible chromatin landscape.
Microbiota-regulated transcription factors have binding sites enriched in accessible chromatin near microbiota-responsive genes
Our results indicate that microbiota-induced modifications to the transcriptional landscape in the intestinal epithelium are achieved by a mechanism other than overt chromatin remodeling. We therefore tested the hypothesis that differential TF binding to sites within a tissue-restrictive accessible chromatin landscape could explain the observed differences in gene expression. First, we tested whether this hypothesis could explain the distinct transcriptional responses to acute (CV) and chronic (CR) microbiota exposure (Fig. 1; Supplemental Fig. S9A,C; Supplemental Table S3). Indeed, we found that many TFs that exhibit differential expression between CV and CR states (Supplemental Fig. S9C,D) have binding sites enriched within accessible chromatin near genes differentially expressed between CV and CR states (Supplemental Fig. S9E,F; Supplemental Tables S9, S10). For example, nuclear factor of activated T cells 5 (NFAT5) has previously been shown to regulate IEC differentiation (Wang et al. 2011, 2013), suggesting that a component of the initial response to microbes may be mediated through IEC turnover. Moreover, both JUN (also known as AP-1 in humans) (Hasselblatt et al. 2008) and early growth response 1 (EGR1) (Moon et al. 2007) have been implicated in the response to injury in the intestine and might mediate the initial response to microbiota during conventionalization (Mukherji et al. 2013).
When comparing our data with other published results, we did not find a robust set of genes that consistently discriminate CR and CV states (Supplemental Figs. S9, S10; Rawls et al. 2006; Donohoe et al. 2011; El Aidy et al. 2012; Larsson et al. 2012; Pott et al. 2012; data not shown). Differences related to experimental design (e.g., whole tissue vs. IEC), expression detection method (e.g., RNA-seq vs. microarray), mouse strain, microbial community composition, time post-colonization, diet, and tissue heterogeneity may explain differences between various CR and CV data sets. However, despite these differences, we were able to identify core sets of genes that were consistently regulated by the presence of microbiota in the ileum or colon (up- or down-regulated in both CR and CV compared with GF) (Supplemental Fig. S10; Supplemental Table S11). We combined these sets with our accessible chromatin data to identify TFs that might mediate a consistent response to microbiota in either the ileum (Fig. 6) or colon (Fig. 7). We found that genes consistently up-regulated in the ileum and colon are significantly enriched for immune and inflammatory response GO categories, whereas genes down-regulated in the ileum and colon are enriched for diverse metabolic processes (Figs. 6A, 7A). We queried these groups of up- or down-regulated genes to identify TFs that are consistently regulated by microbiota across multiple studies (Figs. 6B, 7B). Next, we searched for TF binding sites (TFBSs) in DHSs within the regulatory domains of up- or down-regulated genes in either the ileum or colon (Figs. 6C, 7C; Supplemental Tables S12, S13). Strikingly, we found TFBS enrichment of many of the TFs that are themselves differentially regulated by microbiota (Figs. 6B,C, 7B,C). For example, DHSs near genes up-regulated in the ileum are enriched for motifs matching Interferon regulatory factors (IRFs), signal transducer and activator of transcription (STATs), and E-twenty-six (ETS) family members. Consistent with these observations, the TFs Stat4, Stat1, Stat2, Irf1, Irf8, and Ets1 are all up-regulated in the colonized ileum. In contrast, TFBSs for many nuclear receptors are enriched in accessible chromatin near genes down-regulated in colonized versus GF ileum. In accord, we find that nuclear receptors Pparg, Ppara, Thra, Thrb, Nr1h3, Nr1i3, Nr1d1, Nr1d2, Nr2e3, Nr3c2, and coactivator Ppargc1a all display decreased expression in the colonized ileum. Similar relationships between enriched TFs and their TFBSs were observed in the colon data (Fig. 7B,C). Finally, several of these TF expression-TFBSs enrichment correlations were validated using Ingenuity Pathway Analysis (IPA) upstream regulator prediction (Figs. 6D, 7D) and ChIP-seq data (Figs. 6E, 7E). Though most of the ChIP-seq experiments were performed in nonintestinal tissues (Supplemental Table S14), this in vivo binding data provides strong support for these predicted TFs to regulate microbiota response through DHSs identified in our study. Collectively, this analysis integrates accessible chromatin and transcriptome data to suggest specific transcription factors and target cis-regulatory regions that likely mediate the impact of microbiota on IEC transcription and physiologic function.
Figure 6.

Integrating gene expression and open chromatin data identifies candidate transcription factors regulating response to microbiota colonization in the ileum. (A) Integration of our data set with published studies comparing ileum gene expression in the presence and absence of microbiota reveals a set of genes consistently up- or down-regulated by microbiota across at least four studies. Significant functional enrichments are shown for each gene set (see Supplemental Fig. S9; Supplemental Table S11). (B) Heat map of known transcription factors (TFs; including DNA binding transcription factors and transcription cofactors) that consistently display differential RNA expression levels in response to microbiota across multiple experimental studies in the ileum. Relative expression levels are indicated, where white represents no data. TFs are annotated with their predicted DNA binding domain family. Highlighted with blue or red circles are TFs with motif (C) or binding support (E). (C) Transcription factor binding site (TFBS) prediction in DHSs within the regulatory domain of genes consistently differentially regulated by microbiota in the ileum (see Supplemental Tables S12, S13). Fold enrichments were calculated relative to a GC matched background (Guturu et al. 2013). Motifs are colored based on fold enrichment ratios between down and up gene sets. (Teal) Enriched in DHSs near down genes; (brown) enriched in DHSs near up genes. Highlighted with blue or red circles are motifs matching TFs with differential expression (B) or binding support (E). (D) Scatter plot showing P-values for IPA upstream regulator analysis for the ileum up and ileum down gene lists identifies TFs and other factors that have previously been shown to influence expression of genes within these lists. (E) Plot showing the overlap of ChIP-seq peaks from multiple TFs (measured in various tissues) (see Supplemental Table S14) with DHSs within the regulatory domain of genes either consistently up-regulated (y-axis) or down-regulated (x-axis) by microbiota in the ileum. Fold enrichments were calculated relative to a uniformly distributed null model. Highlighted are the TFs where the up/down fold ratio is at least one standard deviation away from the mean of all fold ratios.
Figure 7.

Integrating gene expression and open chromatin data identifies candidate transcription factors regulating response to microbiota colonization in the colon. (A) Integration of our data set with published studies comparing colon gene expression in the presence and absence of microbiota reveals a set of genes consistently up- or down-regulated by microbiota across at least four studies. Significant functional enrichments are shown for each gene set (see Supplemental Fig. S9; Supplemental Table S11). (B) Heat map of known transcription factors (TFs; including DNA-binding transcription factors and transcription cofactors) that consistently display differential RNA expression levels in response to microbiota across multiple experimental studies in the colon. Relative expression levels are indicated, where white represents no data. TFs are annotated with their predicted DNA binding domain family. Highlighted with blue or red circles are TFs with motif (C) or binding support (E). (C) Transcription factor binding site (TFBS) prediction in DHSs within the regulatory domain of genes consistently differentially regulated by microbiota in the colon (see Supplemental Tables S12, S13). Fold enrichments were calculated relative to a GC matched background. Motifs are colored based on fold enrichment ratios between down and up gene sets. (Teal) Enriched in DHSs near down genes; (brown) enriched in DHSs near up genes. Highlighted with blue or red circles are motifs matching TFs with differential expression (B) or binding support (E). (D) Scatter plot showing P-values for IPA upstream regulator analysis for the colon up and colon down gene lists identifies TFs and other factors that have previously been shown to influence expression of genes within these lists. (E) Plot showing the overlap of ChIP-seq peaks from multiple TFs (measured in various tissues) (see Supplemental Table S14) with DHSs within the regulatory domain of genes either consistently up-regulated (y-axis) or down-regulated (x-axis) by microbiota in the colon. Fold enrichments were calculated relative to a uniformly distributed null model. Highlighted are the TFs where the up/down fold ratio is at least one standard deviation away from the mean of all fold ratios.
Discussion
The ability of the intestinal epithelium to serve as an effective interface between animals and their microbial environment is achieved through orchestration of tissue-specific and microbiota-induced gene expression programs. This orchestration is fundamental to intestinal physiology and host-microbe commensalism, and the underlying mechanisms represent attractive therapeutic targets for promoting health. In order to understand how IECs interpret microbial inputs to regulate gene expression in a tissue-specific context, we generated a total of 20 DNase-seq and 18 RNA-seq data sets from primary IECs isolated from multiple intestinal segments from CR, CV, and GF mice. We developed a modified DNase hypersensitivity assay allowing for the identification of segment-specific CRRs covering a range of abundant and rare IEC types whose loci were distinguished by accessible chromatin distinct from other tissues. These atlases of the IEC accessible chromatin and gene expression landscapes should be a valuable resource for researchers interested in (1) discovering molecular mechanisms controlling cell type-specific and microbiota-regulated gene transcription in different segments of the intestine, (2) discovering differential splicing and novel transcripts regulated by microbiota in the intestine, and (3) generating cell type- or tissue-specific transgenic constructs.
Previous studies in gnotobiotic mice have established that the commensal microbiota modify host physiology through impacting gene expression in the intestinal epithelium along the length of the intestinal tract (Bäckhed et al. 2004; Hooper 2004; Rawls et al. 2006; Donohoe et al. 2011; Vaishnava et al. 2011; El Aidy et al. 2012, 2013; Pott and Hornef 2012; Alenghat et al. 2013; Sayin et al. 2013). Here we observed that microbiota, although potent manipulators of host transcription, have essentially no impact on the accessible chromatin landscape in the ileal and colonic intestinal epithelia of healthy mice (see Supplemental Material). These results suggest a model in which chromatin accessibility is organized during intestinal development in a region-specific manner and maintained similarly in the presence or absence of microbiota (Supplemental Fig. S11). In accord, adult rodents reared in the absence of microbiota develop crypt-villus units and do not display major alterations in the frequency of IEC types (Kandori et al. 1996; Falk et al. 1998). Our results imply that intestinal epithelial cells utilize a strategy other than large-scale chromatin remodeling to respond to the complex activities of the microbiota. This also suggests that the distinct accessible chromatin landscapes of differentiated cells are restricted in their range of response to environmental variables. This supports recently published data showing that TFs utilize pre-existing chromatin landscapes to respond to extracellular cues following terminal differentiation programs (John et al. 2011; Samstein et al. 2012). Interestingly, the accessible and histone-modified chromatin landscape in intestinal stem cells was recently found to be very similar to their differentiated epithelial cell lineages in CR mice (Kim et al. 2014). Together, these findings suggest that a significant component of intestinal epithelial specification is the establishment of a chromatin environment competent to maintain appropriate tissue-specific physiological function while also allowing appropriate tissue-specific responses to microbiota.
Based on our results, we predict that differential occupancy or activity of specific TFs within tissue-specific accessible chromatin may underlie much of the differential transcript abundance observed in GF versus CR or CV conditions (Supplemental Fig. S11). Some TF families implicated here, such as STAT and IRF, are known to integrate inflammatory stimuli to promote expression of immune response genes in the intestinal epithelium and mediate crosstalk with underlying mucosal immune cells (Jiang et al. 2009; Shulzhenko et al. 2011). Our results also identify TFs not previously implicated in microbiota responses. Strikingly, TFBSs for nuclear receptor TFs were enriched near down-regulated genes in both ileum and colon, with many nuclear receptor transcripts also being down-regulated by microbiota in these tissues. This association of nuclear receptors with microbiota-dependent reduction of host gene expression suggests an important role for this family of ligand-binding TFs (Markov and Laudet 2011). Furthermore, comparisons of GF animals to those raised under CV or CR conditions suggest specific TFs that might mediate acute or chronic responses to microbiota, respectively. Future studies will be needed to define the particular TF binding events that regulate gene expression though identified CRRs and to elucidate the upstream host-microbe signal transduction networks converging on these TFs and CRRs.
In this study, we focused on healthy mice reared GF or colonized with specific pathogen-free microbiota. Our results provide a framework for future exploration into how disease states, host genotype, microbiota composition, and other environmental challenges such as infection by pathogenic microbes, diet alterations, or drug exposures may impact the chromatin landscape in the intestinal epithelia. For example, human SNPs associated with inflammatory bowel diseases are enriched in putative cis-regulatory regions (Mokry et al. 2014), demanding improved understanding of how variation in the regulatory genome contributes to this and other human diseases. It will also be important to determine whether the hyporesponsiveness of the accessible chromatin landscape observed in IECs is shared by other cell populations, such as leukocytes, which may exhibit chromatin-based adaptations to particular microbial stimuli (Ganal et al. 2012). This work marks a significant step toward integrating transcriptional regulatory genomics with microbiota research to identify the mechanisms that underlie host-microbe commensalism in the intestine. Future investigations in appropriate gnotobiotic animal models will be required to interrogate the underlying regulatory logic that governs tissue-specific host transcriptional responses to intestinal microbiota.
Methods
Mouse husbandry
All mice used in this study were in the C57BL/6 strain originally sourced from Jackson Laboratories and maintained in the National Gnotobiotic Rodent Resource Center (NGRRC) at the University of North Carolina (UNC) at Chapel Hill. Mice were reared under specific pathogen-free (conventionally raised or CR) conditions, germ-free (GF) conditions, or reared GF and colonized with a conventional microbiota from SPF mice for 14 d (conventionalized or CV). Production, colonization, maintenance, feeding, and sterility testing of GF mice were performed using the standard procedures of the NGRRC. Animals were housed on Alpha-dri bedding (Shepherd) and fed 3500 Autoclavable Breeder Chow (Prolab) or Picolab mouse diet 5058 (LabDiet) ad libitum. All experiments using mice were performed according to established protocols approved by the Institutional Animal Care and Use Committee at UNC at Chapel Hill. For additional information, see Supplemental Table S1.
DNase hypersensitivity on IECs
IECs were isolated from the duodenum (anterior 5 cm of midgut), ileum (posterior 6 cm of midgut), and colon (6 cm of terminal hindgut) of 8- to 12-wk-old mice as described (Gracz et al. 2012). DNase hypersensitivity assays were performed as described (Song and Crawford 2010) with the following modifications using endogenous DNase activity to digest chromatin. Cells were gently lysed by adding 10 mL 0.1% Igepal in resuspension buffer (RSB; 10 mM Tris-Cl at pH 7.4, 10 mM NaCl, 3 mM MgCl2) containing 1× Complete Protease Inhibitors. Isolated nuclei were incubated for 30 sec, 1 min, 2 min, 4 min, or 8 min at 37°C; and endogenous DNase activity was stopped by addition of 0.33 mL cold 50 mM EDTA, and stored on ice. Stabilization of nuclei in agarose plugs, determination of appropriate DNase digestion patterns, library preparation, and sequencing were performed as described (Song and Crawford 2010). See Supplemental Material for additional information.
RNA preparation and sequencing
Total RNA was isolated using TRIzol Reagent (Invitrogen) and further purified using the Qiagen RNeasy (Qiagen) kit according to the manufacturer’s protocol. Two micrograms of total RNA were used for standard TruSeq library preparation with polyA selection (performed by the UNC High Throughput Sequencing Core) for mRNA Illumina sequencing using 2 × 50-bp paired-end reads.
Bioinformatic analysis of RNA-seq data sets
RNA-seq reads were aligned to the mouse genome (NCBI37/mm9) using TopHat v2.0.8b (Trapnell et al. 2012; Kim et al. 2013), allowing for up to two mismatches with UCSC gene transcriptome-guided mapping but permitting nonreference mapping. Normalized fragments per kilobase of transcript per million mapped reads (FPKM) expression values were obtained for reference and novel transcripts via Cufflinks, and pairwise differential gene expression tests were carried out with Cuffdiff v2.0.2 (Trapnell et al. 2012). The default significance threshold of FDR < 5% was used for each comparison. Principle components analysis for RNA-seq was performed with R package cummeRbund v2.0.0. Hierarchical clusterings of RNA-seq data (Fig. 1F; Supplemental Fig. S2A) were performed using heatmap.2 from the gplots package (http://CRAN.R-project.org/package=gplots). A two-sided Kolmogorov-Smirnov test was used to assess the global association of differential DHS and nearby gene expression differences between ileum and colon and in the presence or absence of microbiota. GO enrichments were performed using DAVID v6.7 (Huang et al. 2009a,b). For additional information, see the Supplemental Material.
Bioinformatic analysis of DNase-seq data sets
The top 100,000 DHS peaks in each DNase-seq biological replicate were merged and windowed to 250 bp (with 50bp overlaps) to establish a liberal search space for differential DNase hypersensitivity (signal). Raw base-pair resolution DH signal was summed for each sample in each window as input for the R package DESeq v1.8.3 (Anders and Huber 2010). Sequencing depth normalization, variance fitting, and pairwise differential analyses were performed via DESeq v1.8.3. Overlapping windows with significantly differential DHS signal at the desired FDR threshold (<0.01% for tissue comparisons and <5% for GF vs. CR comparisons) were subsequently merged to reconstitute differential DHS peaks for enumeration. Feature counts were obtained by an in-house script to annotate DHSs with mm9 UCSC gene elements. In all analyses, 2 kb upstream of reference or RNA-seq-derived TSS were considered proximal promoter regions. In the relatively rare cases where a DHS fell within 2 kb of a TSS at two different genes, we selected the gene with the nearest TSS to the midpoint of the DHS. Conservation of DHSs was assessed using the Cistrome conservation plots tool by computing the base-wise phastCons score in the 1000 bp surrounding the DHS peak center. Functional enrichments for sets of DHSs were computed using default parameters with GREAT v2.0.2 (McLean et al. 2010). Refer to http://bejerano.stanford.edu/great for a description of statistical outputs for each set of functional enrichments. For additional information, see the Supplemental Material.
Data access
DNase-seq and RNA-seq data sets have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE57919 and as a trackHub viewable at the UCSC Genome Browser (see http://rawlslab.duhs.duke.edu/data).
Acknowledgments
The authors are grateful to Chris Packey and Maureen Bower for assistance with gnotobiotic mice, Lingyun Song, Yoichiro Shibata, Alexias Safi, and Jeremy Simon for technical assistance with accessible chromatin data set generation and analysis, and Adam Gracz for help with IEC isolation. This work was supported by grants from the National Institutes of Health (P30-DK034987, P40-OD010995, R01-DK081426, R01-HD059862, P01-DK094779), the National Science Foundation (DGE-1147470), the PhRMA Foundation, and the Pew Scholars in the Biomedical Sciences Program.
Author contributions: The study was designed by J.G.C., J.F.R., C.L.F., and G.E.C. Experiments were performed by J.G.C. and C.L.F. DNase and RNA-seq analyses were conducted by C.L.F. and J.G.C., and integration of transcriptome data sets was performed by C.R.L. Motif prediction was performed by C.R.L., J.G.C., C.L.F., and H.G. Overlap enrichment analysis was conducted by T.R., H.G., and G.B. ChIP data sets were curated by A.M.W., J.C., and G.B. The manuscript was written by J.G.C., C.L.F., J.F.R., G.E.C., and C.R.L. with input from all authors.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.165845.113.
References
- Alenghat T, Osborne LC, Saenz SA, Kobuley D, Ziegler CGK, Mullican SE, Choi I, Grunberg S, Sinha R, Wynosky-Dolfi M, et al. . 2013. Histone deacetylase 3 coordinates commensal-bacteria-dependent intestinal homeostasis. Nature 504: 153–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. 2004. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci 101: 15718–15723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerknes M, Cheng H. 1981. Methods for the isolation of intact epithelium from the mouse intestine. Anat Rec 199: 565–574 [DOI] [PubMed] [Google Scholar]
- Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. 2008. High-resolution mapping and characterization of open chromatin across the genome. Cell 132: 311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulger M, Groudine M. 2011. Functional and mechanistic diversity of distal transcription enhancers. Cell 144: 327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp JG, Kanther M, Semova I, Rawls JF. 2009. Patterns and scales in gastrointestinal microbial ecology. Gastroenterology 136: 1989–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp JG, Jazwa AL, Trent CM, Rawls JF. 2012. Intronic cis-regulatory modules mediate tissue-specific and microbial control of angptl4/fiaf transcription. PLoS Genet 8: e1002585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe DR, Garge N, Zhang X, Sun W, O’Connell TM, Bunger MK, Bultman SJ. 2011. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab 13: 517–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Aidy S, van Baarlen P, Derrien M, Lindenbergh-Kortleve DJ, Hooiveld G, Levenez F, Doré J, Dekker J, Samsom JN, Nieuwenhuis EES, et al. . 2012. Temporal and spatial interplay of microbiota and intestinal mucosa drive establishment of immune homeostasis in conventionalized mice. Mucosal Immunol 5: 567–579 [DOI] [PubMed] [Google Scholar]
- El Aidy S, Merrifield CA, Derrien M, van Baarlen P, Hooiveld G, Levenez F, Doré J, Dekker J, Holmes E, Claus SP, et al. . 2013. The gut microbiota elicits a profound metabolic reorientation in the mouse jejunal mucosa during conventionalisation. Gut 62: 1306–1314 [DOI] [PubMed] [Google Scholar]
- Falk PG, Hooper LV, Midtvedt T, Gordon JI. 1998. Creating and maintaining the gastrointestinal ecosystem: what we know and need to know from gnotobiology. Microbiol Mol Biol Rev 62: 1157–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganal SC, Sanos SL, Kallfass C, Oberle K, Johner C, Kirschning C, Lienenklaus S, Weiss S, Staeheli P, Aichele P, et al. . 2012. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity 37: 171–186 [DOI] [PubMed] [Google Scholar]
- Gracz AD, Puthoff BJ, Magness ST. 2012. Identification, isolation, and culture of intestinal epithelial stem cells from murine intestine. Methods Mol Biol 879: 89–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guturu H, Doxey AC, Wenger AM, Bejerano G. 2013. Structure-aided prediction of mammalian transcription factor complexes in conserved non-coding elements. Philos Trans R Soc Lond B Biol Sci 368: 20130029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselblatt P, Gresh L, Kudo H, Guinea-Viniegra J, Wagner EF. 2008. The role of the transcription factor AP-1 in colitis-associated and β-catenin-dependent intestinal tumorigenesis in mice. Oncogene 27: 6102–6109 [DOI] [PubMed] [Google Scholar]
- Hooper LV 2004. Bacterial contributions to mammalian gut development. Trends Microbiol 12: 129–134 [DOI] [PubMed] [Google Scholar]
- Hooper LV, Stappenbeck TS, Hong CV, Gordon JI. 2003. Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nat Immunol 4: 269–273 [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. 2009a. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. 2009b. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Jiang H, Patel PH, Kohlmaier A, Grenley MO, McEwen DG, Edgar BA. 2009. Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell 137: 1343–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S, Sabo PJ, Thurman RE, Sung M-H, Biddie SC, Johnson TA, Hager GL, Stamatoyannopoulos JA. 2011. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet 43: 264–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandori H, Hirayama K, Takeda M, Doi K. 1996. Histochemical, lectin-histochemical and morphometrical characteristics of intestinal goblet cells of germfree and conventional mice. Exp Anim 45: 155–160 [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T-H, Li F, Ferreiro-Neira I, Ho L-L, Luyten A, Nalapareddy K, Long H, Verzi M, Shivdasani RA. 2014. Broadly permissive intestinal chromatin underlies lateral inhibition and cell plasticity. Nature 506: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacks SA 1981. Deoxyribonuclease I in mammalian tissues. Specificity of inhibition by actin. J Biol Chem 256: 2644–2648 [PubMed] [Google Scholar]
- Larsson E, Tremaroli V, Lee YS, Koren O, Nookaew I, Fricker A, Nielsen J, Ley RE, Bäckhed F. 2012. Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut 61: 1124–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Fast AM, Zhu J, Cheng J-X, Buhman KK. 2010. Intestine-specific expression of acyl CoA:diacylglycerol acyltransferase 1 reverses resistance to diet-induced hepatic steatosis and obesity in Dgat1−/− mice. J Lipid Res 51: 1770–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison BB 2002. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem 277: 33275–33283 [DOI] [PubMed] [Google Scholar]
- Markov GV, Laudet V. 2011. Origin and evolution of the ligand-binding ability of nuclear receptors. Mol Cell Endocrinol 334: 21–30 [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28: 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokry M, Middendorp S, Wiegerinck CL, Witte M, Teunissen H, Meddens CA, Cuppen E, Clevers H, Nieuwenhuis EES. 2014. Many inflammatory bowel disease risk loci include regions that regulate gene expression in immune cells and the intestinal epithelium. Gastroenterology 146: 1040–1047 [DOI] [PubMed] [Google Scholar]
- Moon Y, Yang H, Kim YB. 2007. Up-regulation of early growth response gene 1 (EGR-1) via ERK1/2 signals attenuates sulindac sulfide-mediated cytotoxicity in the human intestinal epithelial cells. Toxicol Appl Pharmacol 223: 155–163 [DOI] [PubMed] [Google Scholar]
- Mukherji A, Kobiita A, Ye T, Chambon P. 2013. Homeostasis in intestinal epithelium is orchestrated by the circadian clock and microbiota cues transduced by TLRs. Cell 153: 812–827 [DOI] [PubMed] [Google Scholar]
- Pott J, Hornef M. 2012. Innate immune signalling at the intestinal epithelium in homeostasis and disease. EMBO Rep 13: 684–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pott J, Stockinger S, Torow N, Smoczek A, Lindner C, McInerney G, Bäckhed F, Baumann U, Pabst O, Bleich A, et al. . 2012. Age-dependent TLR3 expression of the intestinal epithelium contributes to rotavirus susceptibility. PLoS Pathog 8: e1002670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawls JF, Mahowald MA, Ley RE, Gordon JI. 2006. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell 127: 423–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, Neph S, Sabo P, Kim JM, Liao W, et al. . 2012. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 151: 153–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall H-U, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F. 2013. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-β-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17: 225–235 [DOI] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. . 2012. A map of the cis-regulatory sequences in the mouse genome. Nature 488: 116–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulzhenko N, Morgun A, Hsiao W, Battle M, Yao M, Gavrilova O, Orandle M, Mayer L, Macpherson AJ, McCoy KD, et al. . 2011. Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nat Med 17: 1585–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer F, Bäckhed F. 2013. The gut microbiota—masters of host development and physiology. Nat Rev Microbiol 11: 227–238 [DOI] [PubMed] [Google Scholar]
- Song L, Crawford GE. 2010. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harbor Protocols 2010 doi:10.1101/pdb.prot5384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CE. 1977. Comparative physiology of the digestive system. In Duke’s physiology of domestic animals, 9th ed. (ed. Swenson MJ), pp. 216–232. Cornell University Press, Ithaca, NY. [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. . 2012. The accessible chromatin landscape of the human genome. Nature 489: 75–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, Ley R, Wakeland EK, Hooper LV. 2011. The antibacterial lectin RegIIIγ promotes the spatial segregation of microbiota and host in the intestine. Science 334: 255–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier LG, Clevers H. 2009. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol 71: 241–260 [DOI] [PubMed] [Google Scholar]
- von Furstenberg RJ, Gulati AS, Baxi A, Doherty JM, Stappenbeck TS, Gracz AD, Magness ST, Henning SJ. 2011. Sorting mouse jejunal epithelial cells with CD24 yields a population with characteristics of intestinal stem cells. Am J Physiol Gastrointest Liver Physiol 300: G409–G417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhou Y, Jackson LN, Johnson SM, Chow C-W, Evers BM. 2011. Nuclear factor of activated T cells (NFAT) signaling regulates PTEN expression and intestinal cell differentiation. Mol Biol Cell 22: 412–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhou Y, Rychahou P, Liu C, Weiss HL, Evers BM. 2013. NFAT5 represses canonical Wnt signaling via inhibition of β-catenin acetylation and participates in regulating intestinal cell differentiation. Cell Death Dis 4: e671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Song Y, Zhao D, Verkman AS. 2005. Phenotype analysis of aquaporin-8 null mice. Am J Physiol Cell Physiol 288: C1161–C1170 [DOI] [PubMed] [Google Scholar]