

Abstract
Contamination by present-day human and microbial DNA is one of the major hindrances for large-scale genomic studies using ancient biological material. We describe a new molecular method, U selection, which exploits one of the most distinctive features of ancient DNA—the presence of deoxyuracils—for selective enrichment of endogenous DNA against a complex background of contamination during DNA library preparation. By applying the method to Neanderthal DNA extracts that are heavily contaminated with present-day human DNA, we show that the fraction of useful sequence information increases ∼10-fold and that the resulting sequences are more efficiently depleted of human contamination than when using purely computational approaches. Furthermore, we show that U selection can lead to a four- to fivefold increase in the proportion of endogenous DNA sequences relative to those of microbial contaminants in some samples. U selection may thus help to lower the costs for ancient genome sequencing of nonhuman samples also.
High-throughput DNA sequencing and the shift to library-based sample preparation techniques have greatly facilitated genetic research on human evolution in recent years. Increasing amounts of sequence data are becoming available not only from present-day humans but also from ancient human remains, helping to uncover the evolutionary histories of present-day human populations as well as their relationship to extinct archaic groups (Stoneking and Krause 2011). In ancient DNA studies, the most accessible target is mitochondrial (mt) DNA, which is present in several hundreds of copies per cell as opposed to the diploid-only nuclear genome. Consequently, complete mtDNA genomes have been sequenced from more than a dozen remains of ancient modern humans (Ermini et al. 2008; Gilbert et al. 2008; Green et al. 2010; Fu et al. 2013b; Raghavan et al. 2014) as well as representatives of two archaic hominin groups that went extinct during the Late Pleistocene, the Neanderthals (Green et al. 2008; Briggs et al. 2009; Prüfer et al. 2014), and Denisovans (Krause et al. 2010b; Reich et al. 2010). The recent recovery of mitochondrial sequences from an ∼400,000 yr-old hominin from Sima de los Huesos in Spain indicates that more comprehensive sequence data may soon become available even from Middle Pleistocene remains (Meyer et al. 2014). Retrieving nuclear sequences from ancient human material is generally more challenging, but such data have also been generated at various scales, ranging from a few megabases of sequences to full genome sequences determined with high accuracy (see Shapiro and Hofreiter 2014 for a recent summary).
Despite these advances, sequencing ancient human DNA continues to be challenging for several reasons. First, only trace amounts of highly fragmented DNA are usually preserved in ancient bones and teeth, imposing limits on the number of sequences that can be recovered from ancient specimens. Second, DNA extracted from ancient material is in many cases dominated by microbial DNA, which often contributes to 99% or more of the sequences, making direct shotgun sequencing economically infeasible. This problem can be partially overcome by hybridization enrichment of hominin sequences, such as those of a small chromosome (Fu et al. 2013a) or, as recently proposed, of sequences from throughout the whole genome (Carpenter et al. 2013). Another approach is restriction digestion of GC-rich sequence motifs, which was performed to change the ratio between endogenous and microbial library molecules in the first study of the Neanderthal genome (Green et al. 2010). A third and particularly severe problem for working with ancient human samples is present-day human contamination, which is inevitably introduced during excavation and laboratory work. Fortunately, a solid framework for validating the authenticity of ancient human sequences can be established using the distinct pattern of substitutions caused by cytosine deamination in ancient DNA sequences (Hofreiter et al. 2001; Briggs et al. 2007). The deamination product of cytosine is uracil, which is read as thymine by most DNA polymerases. Resulting C to T substitutions (or G to A substitutions, depending on the orientation in which DNA strands are sequenced and the method used to prepare DNA libraries) are particularly frequent at the 5′ and 3′ ends of sequences due to the higher rate of cytosine deamination in single-stranded overhangs (Lindahl 1993). Importantly, the frequency of deamination-induced substitutions correlates with sample age (Sawyer et al. 2012) and is low in present-day human contamination (Krause et al. 2010a). These substitutions can thus be taken as evidence for the presence of authentic ancient human sequences. Deamination-induced substitutions have also been exploited for separating ancient sequences from present-day human contamination in silico (Skoglund et al. 2012, 2014a,b; Meyer et al. 2014; Raghavan et al. 2014). Although effective in principle, this approach is costly, because a large proportion of sequence data is excluded from downstream analysis. Furthermore, ancient DNA base damage is not determined directly and may occasionally be confounded with evolutionary sequence differences.
Here we describe a novel laboratory technique, uracil selection (“U selection”), which enables physical separation of uracil-containing DNA strands from nondeaminated strands at the stage of DNA library preparation. Our method builds on a single-stranded library preparation method, which has been shown to be particularly efficient for retrieving sequences from highly degraded ancient DNA (Meyer et al. 2012; Gansauge and Meyer 2013). We apply U selection to several Neanderthal DNA extracts and show that it is a powerful tool for enriching Neanderthal DNA sequences against a background of present-day human contamination. We also report cases where U selection drastically increases the proportion of Neanderthal DNA relative to microbial DNA.
Results
The U selection method
Since U selection is an extension of single-stranded library preparation, each DNA strand is converted into a library molecule and is evaluated for the presence of uracils separately. A detailed comparison of single-stranded DNA library preparation and the U selection method is provided in Figure 1. The first reaction steps are common to both protocols. Briefly, terminal phosphate groups are removed from double-stranded ancient DNA fragments, the strands are separated by heat, and biotinylated adapters are ligated to their 3′ ends. After immobilization of the ligated strands on streptavidin-coated magnetic beads, the adapter sequence is used as a priming site to initiate a polymerization reaction that copies the ancient template strands. The two protocols then diverge with regard to the scheme that is used to attach the second adapter, which is joined only to the copied strand in standard library preparation but to both strands in U selection, requiring a detour via phosphorylation of the ancient strand and nick fill-in (see Fig. 1). The separation of uracil-containing DNA strands is performed by creating incisions in the ancient template strands at uracils, which are then used to prime a nick extension reaction that displaces the biotinylated DNA strand and releases a double-stranded copy of the original template strand into solution (the “supernatant” library fraction). Library molecules without uracils remain bound to the beads and are subsequently released by heat treatment (the “bead” library fraction), analogous to standard single-stranded library preparation. It should be noted that unlike all double-stranded library preparation methods that have been used with ancient DNA, the single-stranded protocol is strictly strand-specific (Meyer et al. 2012). Thus, uracils manifest as C to T changes in the resulting sequences and no complementary G to A changes are introduced.
Figure 1.

Schematic comparison of standard single-stranded DNA library preparation and U selection. (1) Double-stranded ancient DNA molecules (gray lines) are dephosphorylated (not depicted) and heat-denatured. A deoxyuracil is denoted as a green circle. (2) Single-stranded adapter oligonucleotides carrying a biotinylated 3′ linker arm are ligated to the 3′ ends of the ancient molecules. The ligation products are then immobilized on streptavidin-coated magnetic beads (large circles). (3) An extension primer is hybridized to the adapter and Bst DNA polymerase is used to create a copy of the template strand. This reaction generates 3′ overhangs, which are subsequently removed in a blunt-end repair step using T4 DNA polymerase (data not shown). For U selection, T4 polynucleotide kinase is included in blunt-end repair to add a 5′ phosphate to the original template strand. (4a) In the standard protocol, a double-stranded adapter (blue) carrying a 5′ phosphate on one strand and a 3′ dideoxy block on the other strand is ligated to the newly synthesized strand using T4 DNA ligase. (5a) The library molecules are released from the beads by heat treatment. (4b) For U selection, a nonphosphorylated double-stranded adapter (blue) is ligated to the original template strand using T4 DNA ligase. (5b) The nick remaining in the opposite strand is removed in a fill-in reaction with the strand-displacing Bst polymerase. (6) If present, uracils are removed by uracil-DNA glycosylase, and the resulting abasic site is cut by endonuclease VIII, which leaves both 5′ and 3′ phosphates. The 3′ phosphates are removed with T4 polynucleotide kinase and the resulting 3′ hydroxyl groups are used to prime a strand-displacement polymerization reaction with Bst polymerase. (7) The supernatant now contains double-stranded library molecules originating from ancient DNA strands that carried a uracil, which (8a) are recovered in a separate tube, whereas (8b) the remaining library molecules are released by heat treatment.
Reducing present-day human contamination in Neanderthal DNA libraries
We first applied U selection to DNA extracts obtained from an infant Neanderthal individual excavated at Mezmaiskaya Cave, Russia (Golovanova et al. 1999). This individual is dated to 60,000–70,000 yr ago (Skinner et al. 2005) and both its complete mitochondrial genome (Briggs et al. 2009) as well as parts of its nuclear genome (Prüfer et al. 2014) have been sequenced in previous studies. While these studies used DNA extracts that contained around 1% present-day human contamination, several other extracts were obtained in the past from the same material that were highly contaminated with human DNA, preventing attempts to increase sequence coverage of the Mezmaiskaya genome beyond 0.5-fold. We explored two of these highly contaminated extracts further and prepared libraries using both the standard single-stranded as well as the U selection method. Although it may seem counterintuitive at first, we treated the Mezmaiskaya DNA extracts with E. coli uracil-DNA-glycosylase (UDG) prior to U selection to improve sequence data quality (Briggs et al. 2010). E. coli UDG efficiently excises uracils from the interior of molecules, but leaves most terminal uracils and those located at the 3′ penultimate position of molecules intact (Meyer et al. 2012). Uracils are thus retained where they occur most frequently and provide most power for discriminating endogenous DNA from contaminants, while C to T substitutions in the sequence data are confined to only three positions where they can easily be identified and masked out (Prüfer et al. 2014).
Using a quantitative PCR assay that primes on the adapter sequences (Meyer et al. 2008; Gansauge and Meyer 2013), we compared the number of library molecules obtained from standard single-stranded library preparation and U selection and found that the yield of library molecules is slightly reduced with U selection, reaching 54%–62% of that achieved with standard single-stranded library preparation, presumably due to the modifications that were made to the standard method (Table 1). To determine the success of the uracil selection process, we then mapped all sequences to the human reference genome and approximated the proportion of sequences that are derived from uracil-containing molecules by dividing the number of sequences carrying a terminal C to T difference to the reference by the total number of sequences. These proportions were 57.9% and 63.2% in the supernatant library fractions obtained from the two extracts, and 0.8% in the bead library fractions (Table 1). By comparison, only 6.3% and 6.7% of the sequences generated with the standard single-stranded method from both extracts showed a terminal C to T difference from the reference, revealing an almost 10-fold enrichment of uracil-containing molecules by U selection. However, we did not achieve full separation of the two library fractions by U selection. In silico filtering for sequences with terminal C to T differences thus remains necessary to exclude sequences from nondeaminated DNA strands.
Table 1.
Comparison of standard single-stranded library preparation and U selection using two Neanderthal DNA extracts that are heavily contaminated with human DNA
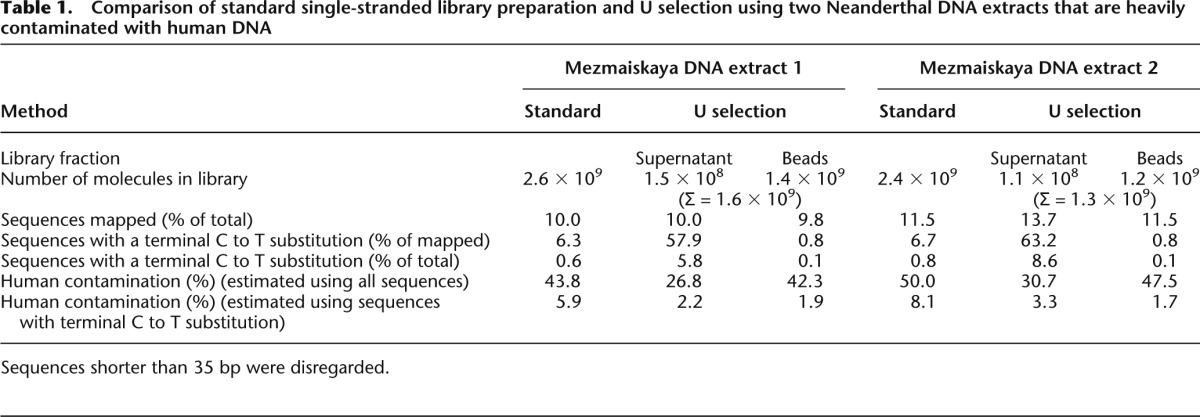
To determine whether present-day human contamination was efficiently reduced by U selection, we enriched all libraries for mtDNA and aligned the resulting sequences against the human mitochondrial reference genome. We obtained mitochondrial contamination estimates by identifying the state of sequences overlapping a set of positions where the respective Neanderthal individual differs from at least 99% of present-day humans (Green et al. 2008). Contamination estimates for the two libraries prepared with the standard single-stranded method are 43.8% and 50.0%, respectively (Table 1). By in silico filtering for sequences with terminal C to T differences to the reference genome, contamination estimates decrease to 5.9% and 8.1%, respectively. In contrast, contamination estimates obtained in the same way for the uracil-selected library fractions are 2.2% and 3.3%, respectively, and thus considerably lower.
Increasing the proportion of informative sequences
For the Mezmaiskaya libraries, U selection did not substantially change the fraction of sequences that could be mapped to the human genome (Table 1), indicating that the uracil content of the microbial contaminants is similar to that of the endogenous DNA in the Mezmaiskaya extracts, and hence that the bulk of microbial contamination present in the bone is of ancient origin. To broaden our sampling, we performed additional U-selection experiments using DNA extracts obtained from two Neanderthal bones from Vindija Cave, Croatia (Vi33.17 and Vi33.19), both undated, and an ∼39,000-yr-old Neanderthal bone from El Sidrón Cave, Spain (SD1253), which has been extensively studied before (e.g., Lalueza-Fox et al. 2005; Briggs et al. 2009; Burbano et al. 2010). We performed U selection both with and without prior excision of internal uracils and sequenced the resulting libraries directly and after mtDNA enrichment. Enrichment for uracil-containing DNA strands was even stronger than in the experiments with Mezmaiskaya DNA (Table 2), and U selection was efficient both on terminal and internal uracils (Fig. 2). For the El Sidrón Neanderthal we again detected no substantial difference in the proportion of mapped sequences when comparing the two library fractions (Table 2). However, the uracil-enriched library fractions of the Vindija samples showed a striking increase in the proportion of mapped sequences induced by U selection, from 0.6% to 3.0% for one of the extracts and from 4.1% to 15.1% for the other, indicating that the microbial contamination in the Vindija samples is largely of recent origin, a result that is congruent with previous analyses of bacterial sequences generated from these samples (Zaremba-Niedzwiedzka and Andersson 2013). Inspection of the base composition of the uracil-selected sequences reveals a mild increase in GC-content compared with the bead fraction, but overall the GC-content of the aligned sequences is close to the genome average (Supplemental Fig. 1). Analysis of the mitochondrial sequences of the Vindija and El Sidrón Neanderthals shows that the mitochondrial genome sequences of all three individuals can be completely assembled using sequences from the uracil-enriched library fraction (see Supplemental Fig. 2 for information on mitochondrial assemblies) and that present-day human contamination among the uracil-selected sequences is very low (0.1%–0.8%; see Supplemental Table 1). The two new Neanderthal mtDNA genome sequences obtained from the Vindija samples fall within the known mitochondrial variation of Neanderthals (see Supplemental Fig. 3 for a phylogenetic tree), with Vi33.19 carrying a mtDNA sequence identical to a previously published sequence of another bone (Vi33.16) from Vindija Cave (Green et al. 2008).
Table 2.
The effect of U selection on the proportion of sequences ≥35 bp that can be mapped to the human genome in a survey of three additional Neanderthal samples
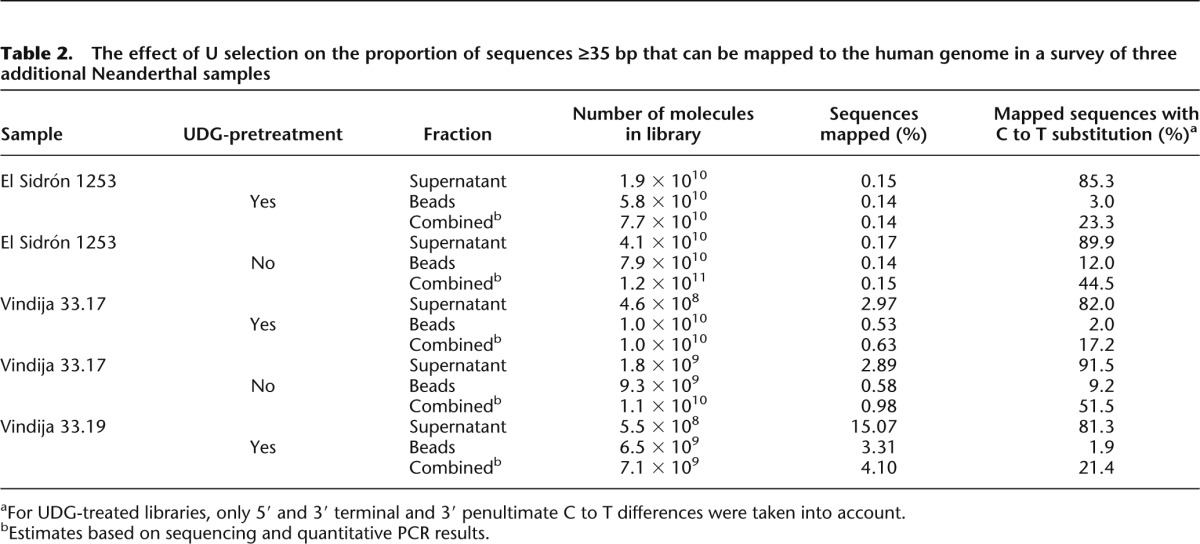
Figure 2.
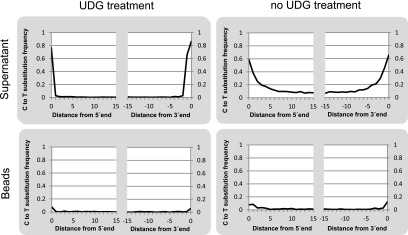
C to T substitution frequencies as functions of the alignment position in sequences obtained from a Neanderthal sample (Vi33.17) after U selection. Libraries were prepared with and without excision of internal uracils by UDG prior to U selection. Plots are shown for both library fractions (uracil-enriched supernatant and uracil-depleted bead fraction).
Based on the counts of unique library molecules obtained by quantitative PCR as well as the sequencing results, we can estimate the number of Neanderthal genomes present in the Vindija and El Sidrón libraries, which show very little present-day human contamination. This allows us to determine that between 77% and 83% of Neanderthal genomes are removed by U selection and filtering for sequences with terminal C to T substitutions (Supplemental Fig. 4). In addition, between 38% and 49% of Neanderthal sequences are lost if UDG pretreatment is performed to excise uracils from the interior of ancient DNA strands prior to U selection. Nonetheless, using this approach, 1 g of bone material from Vindija bones Vi33.17 or Vi33.19 would still yield 12- or 48-fold coverage of the nuclear genome, respectively (Supplemental Table 1).
Discussion
We show that one of the most distinct characteristics of ancient DNA, the presence of uracils, can be exploited to physically separate ancient DNA from recent contamination at the stage of library preparation. This approach represents a substantial improvement over purely computational strategies for isolating authentic ancient sequences from mixtures with present-day human contamination. First, as exemplified for the Mezmaiskaya Neanderthal DNA extracts, present-day human contamination is more effectively reduced when performing U selection because the presence of uracil is directly inferred. Second, U selection greatly reduces the costs of generating sequences from samples that are contaminated with present-day human DNA—by a factor of ∼10 in our example. It should be noted, however, that the efficiency of contamination removal is dependent on the abundance of human contamination relative to endogenous ancient DNA, the time that has passed since excavation, and possibly the conditions under which fossils are stored. This is because, first, U selection does not provide full separation between uracil-containing and uracil-free molecules, and second, uracils also start to accumulate in contaminant DNA molecules after excavation. Residual contamination in the sequence data may be further reduced by applying additional filters in silico after U selection and C to T filtering. For example, for one of the uracil-selected Mezmaiskaya sequences, residual contamination decreases from 3.3% to 2.0% when discarding sequences longer than 60 bp (Supplemental Fig. 5).
We also show that U selection can, at least in some instances, reduce the proportion of sequences derived from microbial DNA. Among the five Neanderthal samples studied here, we identified two, both from Vindija Cave, in which uracil selection substantially increases the proportion of mapped sequences, in one of the samples from 4.1% to 15.1%. This greatly improves the suitability of this sample for deeper shotgun sequencing. However, this effect is not seen in all Neanderthal samples studied here and future work will show how frequently U selection can be used as a means to enrich endogenous DNA over microbial contamination, also in nonhuman samples. It may be particularly interesting, for example, to apply U selection to samples from permafrost, where substantial bacterial growth may occur after excavation. Indeed, since U selection allows for directly assessing the frequency of deamination-induced substitutions in microbial sequences, the method opens the prospect of studying in greater resolution the progression in which microorganisms enter fossils and how they contribute to bone diagenesis.
An obvious drawback of U selection is that not all ancient molecules carry uracils. The exact proportion of uracil-containing molecules varies among samples and cannot be accurately predicted from the age of a sample alone. For example, only 3.5% of DNA sequences obtained from Ötzi, a 5300-yr-old mummy found in glacier ice in the Alps, show deamination-induced C to T substitutions at their 5′ end (Keller et al. 2012), while these frequencies are as high as 16% in sequences from a 7000-yr-old human tooth excavated from La Braña-Arintero, a cave site in Northwestern Spain (Olalde et al. 2014), and around 40% in the Neanderthals from Vindija Cave (Green et al. 2010), which were also studied here. As shown above, ∼20% of the endogenous molecules in the Vindija samples carry a terminal uracil. Extrapolating from this number we would thus expect only 8% of molecules to be recovered by U selection from the La Braña individual and 2% from Ötzi, respectively. It should be noted, however, that the uracil-depleted library fractions are not lost and remain available for further experiments, for instance, using hybridization capture approaches.
Since uracil originates from cytosine, U selection slightly increases the sequence representation of GC-rich regions of the genome. However, since single-stranded library preparation leads to an overrepresentation of AT-rich sequences, the overall GC-content of uracil-selected sequences moves closer to the genome average. More data will be needed to evaluate in detail how U selection affects coverage in genomic regions of different GC-content or the ability to detect methylation marks in ancient DNA (Pedersen et al. 2014), but as shown above, base composition biases do not prevent the assembly of complete mitochondrial genomes at moderate sequencing depth.
In summary, we have shown that U selection enables cost-effective shotgun sequencing of ancient hominin samples that are contaminated with present-day human DNA. Such samples are becoming increasingly relevant in evolutionary research as ancient DNA studies expand to very poorly preserved samples containing only trace amounts of endogenous DNA, such as the hominin remains of Sima de los Huesos (Meyer et al. 2013). We also report a striking case, that of the Vindija Neanderthals, where U selection greatly increases the fraction of endogenous DNA relative to microbial contamination, putting another high-coverage Neanderthal genome into reach. Given the small number of samples analyzed here, it seems very likely that more such cases will be identified in the future.
Methods
DNA extraction and library preparation
Two DNA extracts were generated using 107 and 90 mg of bone material of the Mezmaiskaya Neanderthal infant following the methodology described by Rohland and Hofreiter (2007). DNA was extracted from two Vindija Neanderthal individuals as well as the El Sidrón individual using the method described in Dabney et al. (2013). DNA libraries were prepared using the single-stranded library preparation method detailed in Gansauge and Meyer (2013), with or without uracil excision by E. coli UDG, or following the U selection protocol described below. Libraries were prepared using 5% or 20% of the extract volume as input (see Supplemental Table 1 for an overview of all libraries prepared for this study). Extraction and library preparation blank controls, using water instead of bone or DNA extract, were performed in parallel to the experiments with sample DNA. In addition, two positive controls were included in each experiment, using 1 μL of a 0.1-μM dilution of an oligonucleotide carrying a deoxyuracil (CL103: 5′-Pho-TCGTCGTUTGGTATGGCTTCATTCAGCTCCGGTTCCCAACGATCAAGGCGAGTTACATGA-Pho-3′) and no deoxyuracil (CL104: 5′-Pho-TCGTCGTTTGGTATGGCTTCATTCAGCTCCGGTTCCCAACGATCAAGGCGAGTTACATGA-Pho-3′).
The first steps of U selection were performed exactly following steps 1–15 of the protocol described in Gansauge and Meyer (2013) with two modifications: (1) In step 1, endonuclease VIII and Afu UDG were replaced by 0.5 μL of 1 U/μL USER enzyme mix (New England Biolabs) where applicable. (2) In step 8, the volume of streptavidine beads was increased to 100 μL per reaction. U selection was then continued as follows: A blunt-end reaction mix was prepared by combining 83.1 μL water, 10 μL 10× Tango buffer (Thermo Scientific), 2.5 μL 1% Tween 20, 0.4 μL 25 mM dNTP, 1 μL 100 mM ATP, 1 μL 5 U/μL T4 DNA polymerase and 2 μL 10 U/μL T4 polynucleotide kinase (both Thermo Scientific). The beads were pelleted in a magnetic rack and the supernatant was discarded. The beads were then resuspended in the blunt-end reaction mix, incubated for 15 min at 25°C, and washed exactly as described in step 19 of Gansauge and Meyer (2013). A ligation reaction mix was prepared by combining 34.75 μL water, 5 μL 10× T4 DNA ligase buffer (Thermo Scientific), 5 μL 50% PEG-4000 solution, 1.25 μL 1% Tween-20, 2 μL 100 μM double-stranded adapter and 2 μL 30 U/μL (HC) T4 DNA ligase (Thermo Scientific). The adapter oligonucleotides CL53 and CL73 of Gansauge and Meyer (2013) were replaced by CL49 (5′-ACACTCTTTCCCTACACGACGCTCTTCC-3′) and CL50 (5′-GGAAGAGCGTCG-3′). After discarding the wash buffer, the beads were resuspended in the ligation reaction mix and incubated for 1 h at 22°C. The beads were again washed as described in step 19 and the wash buffer was discarded. A fill-in reaction mix was prepared by combining 42.5 μL water, 5 μL 10× Isothermal amplification buffer (New England Biolabs), 0.5 μL 25 mM dNTP, and 2 μL 8 U/μL Bst DNA polymerase 2.0 (New England Biolabs). The wash buffer was discarded and the beads were resuspended in the fill-in reaction mix and incubated for 15 min at 37°C. The beads were then pelleted and the reaction mix discarded. After washing the beads with 200 μL wash buffer A and 200 μL wash buffer B (see Gansauge and Meyer 2013 for wash buffer recipes), they were resuspended in an excision reaction mix comprised of the following components: 34.25 μL water, 5 μL 10× GeneAmp PCR buffer II (Life Technologies), 4 μL 25 mM MgCl2, 1.25 μL 1% Tween-20, 0.5 μL 25 mM dNTP, 0.75 μL 10 U/μL endonuclease VIII, and 0.25 μL 1 U/μL E. coli UDG (both New England Biolabs). The reaction mix was incubated at 37°C. After 5 min, 2 μL 10 U/μL T4 polynucleotide kinase (Thermo Scientific) was added and the reaction mix was incubated again at 37°C. After another 10 min, 2 μL of 8 U/μL Bst DNA polymerase 2.0 (New England Biolabs) was added and the reaction was completed by incubation for 15 min at 37°C. The beads were then pelleted and the supernatant (corresponding to the ‘supernatant library fraction’) was transferred to another tube and incubated for 1 min at 95°C to inactivate the enzymes. To recover the residual library molecules (corresponding to the ‘bead library fraction’), the beads were washed with 200 μL wash buffer A and 200 μL wash buffer B and resuspended in 50 μL EBT. The library molecules were then released into the supernatant by incubation at 95°C for 1 min.
Following library preparation, the number of molecules in each library was determined using quantitative PCR assays as described in Gansauge and Meyer (2013). Libraries were then split into aliquots of 25 μL, each of which was amplified in a 100-μL PCR reaction using AccuPrime Pfx DNA polymerase (Life Technologies) as described elsewhere (Dabney and Meyer 2012). Amplification was carried out using a double-indexing scheme (Kircher et al. 2012), which introduces library-specific barcodes onto both adapter sequences. PCR products from the same libraries were then pooled and purified using the MinElute PCR purification kit (Qiagen).
Enrichment of mitochondrial DNA, sequencing, and sequence analysis
For shotgun sequencing, two library pools were generated, one from the Mezmaiskaya libraries and another from the Vindija and El Sidrón libraries. Each pool was sequenced on one run of Illumina’s MiSeq platform using a recipe for paired-end sequencing (2 × 76 cycles) with two index reads (Kircher et al. 2012). In addition, all libraries were enriched for mtDNA using human mtDNA probes and following the protocol detailed in Fu et al. (2013a). The enriched libraries were pooled and sequenced on another MiSeq run as described above.
After discarding sequences that did not perfectly match one of the expected index combinations, overlapping paired-end reads were merged into single sequences to reconstruct full-length molecule sequences (Meyer et al. 2012). Merged sequences were then aligned against the human reference genome (GRCh37/1000 Genomes Project release) or against the revised Cambridge mtDNA reference sequence using BWA (Li and Durbin 2009) with parameters adjusted for the mapping of ancient sequences (Meyer et al. 2012). Sequences shorter than 35 bp were discarded and duplicates were removed by calling a consensus from sequences with identical alignment start and end coordinates. Summary statistics and further analyses were performed using perl scripts. Mitochondrial contamination estimates were obtained by analyzing diagnostic positions in the mtDNA genome where the mtDNA genome sequence of the respective Neanderthal individual (Green et al. 2008; Briggs et al. 2009; Prüfer et al. 2014) differs from those of a panel of 311 present-day humans (Green et al. 2008).
Data access
The mtDNA sequence of Vi33.17 has been submitted to the NCBI GenBank (http://www.ncbi.nlm.nih.gov/genbank/) under accession number KJ533544. Sequence data generated for this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number PRJEB6014.
Acknowledgments
We thank Qiaomei Fu for constituting the phylogenetic tree, Isabelle Glocke for help in the laboratory, Jesse Dabney, Alexander Hübner, Petra Korlević, Svante Pääbo, Kay Prüfer, and Viviane Slon for helpful discussions and comments on the manuscript, Barbara Höber and Antje Weihmann for performing the sequencing runs, Gabriel Renaud and Udo Stenzel for processing the raw sequence data, and Dejana Brajkovic, Vladimir B. Doronichev, Javier Fortea, Liubov V. Golovanov, Ivan Gušic, Željko Kucan, Carles Lalueza-Fox, Marco de la Rasilla, Antonio Rosas, and Pavao Rudan for providing samples. This work was funded by the Max Planck Society.
Author contributions: M.T.G. and M.M. conceived the method, M.T.G. performed the experiments, M.M. analyzed the data, and M.T.G. and M.M. wrote the paper.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.174201.114.
References
- Briggs AW, Stenzel U, Johnson PL, Green RE, Kelso J, Prüfer K, Meyer M, Krause J, Ronan MT, Lachmann M, et al. 2007. Patterns of damage in genomic DNA sequences from a Neandertal. Proc Natl Acad Sci 104: 14616–14621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs AW, Good JM, Green RE, Krause J, Maricic T, Stenzel U, Lalueza-Fox C, Rudan P, Brajkovic D, Kucan Z, et al. 2009. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science 325: 318–321 [DOI] [PubMed] [Google Scholar]
- Briggs AW, Stenzel U, Meyer M, Krause J, Kircher M, Pääbo S. 2010. Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Res 38: e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbano HA, Hodges E, Green RE, Briggs AW, Krause J, Meyer M, Good JM, Maricic T, Johnson PL, Xuan Z. 2010. Targeted investigation of the Neandertal genome by array-based sequence capture. Science 328: 723–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter ML, Buenrostro JD, Valdiosera C, Schroeder H, Allentoft ME, Sikora M, Rasmussen M, Gravel S, Guillen S, Nekhrizov G, et al. 2013. Pulling out the 1%: whole-genome capture for the targeted enrichment of ancient DNA sequencing libraries. Am J Hum Genet 93: 852–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabney J, Meyer M. 2012. Length and GC-biases during sequencing library amplification: a comparison of various polymerase-buffer systems with ancient and modern DNA sequencing libraries. Biotechniques 52: 87–94 [DOI] [PubMed] [Google Scholar]
- Dabney J, Knapp M, Glocke I, Gansauge MT, Weihmann A, Nickel B, Valdiosera C, Garcia N, Pääbo S, Arsuaga JL, et al. 2013. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc Natl Acad Sci 110: 15758–15763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermini L, Olivieri C, Rizzi E, Corti G, Bonnal R, Soares P, Luciani S, Marota I, De Bellis G, Richards MB, et al. 2008. Complete mitochondrial genome sequence of the Tyrolean Iceman. Curr Biol 18: 1687–1693 [DOI] [PubMed] [Google Scholar]
- Fu Q, Meyer M, Gao X, Stenzel U, Burbano HA, Kelso J, Pääbo S. 2013a. DNA analysis of an early modern human from Tianyuan Cave, China. Proc Natl Acad Sci 110: 2223–2227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, Mittnik A, Johnson PL, Bos K, Lari M, Bollongino R, Sun C, Giemsch L, Schmitz R, Burger J, et al. 2013b. A revised timescale for human evolution based on ancient mitochondrial genomes. Curr Biol 23: 553–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gansauge MT, Meyer M. 2013. Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nat Protoc 8: 737–748 [DOI] [PubMed] [Google Scholar]
- Gilbert MT, Kivisild T, Gronnow B, Andersen PK, Metspalu E, Reidla M, Tamm E, Axelsson E, Gotherstrom A, Campos PF, et al. 2008. Paleo-Eskimo mtDNA genome reveals matrilineal discontinuity in Greenland. Science 320: 1787–1789 [DOI] [PubMed] [Google Scholar]
- Golovanova LV, Hoffecker JF, Kharitonov VM, Romanova GP. 1999. Mezmaiskaya Cave: a Neanderthal occupation in the Northern Caucasus. Curr Anthropol 40: 77–86 [Google Scholar]
- Green RE, Malaspinas AS, Krause J, Briggs AW, Johnson PL, Uhler C, Meyer M, Good JM, Maricic T, Stenzel U, et al. 2008. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell 134: 416–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH, et al. 2010. A draft sequence of the Neandertal genome. Science 328: 710–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofreiter M, Jaenicke V, Serre D, von Haeseler A, Pääbo S. 2001. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res 29: 4793–4799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Graefen A, Ball M, Matzas M, Boisguerin V, Maixner F, Leidinger P, Backes C, Khairat R, Forster M, et al. 2012. New insights into the Tyrolean Iceman’s origin and phenotype as inferred by whole-genome sequencing. Nat Commun 3: 698. [DOI] [PubMed] [Google Scholar]
- Kircher M, Sawyer S, Meyer M. 2012. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res 40: e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause J, Briggs AW, Kircher M, Maricic T, Zwyns N, Derevianko A, Pääbo S. 2010a. A complete mtDNA genome of an early modern human from Kostenki, Russia. Curr Biol 20: 231–236 [DOI] [PubMed] [Google Scholar]
- Krause J, Fu Q, Good JM, Viola B, Shunkov MV, Derevianko AP, Pääbo S. 2010b. The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature 464: 894–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalueza-Fox C, Sampietro ML, Caramelli D, Puder Y, Lari M, Calafell F, Martinez-Maza C, Bastir M, Fortea J, de la Rasilla M, et al. 2005. Neandertal evolutionary genetics: mitochondrial DNA data from the Iberian peninsula. Mol Biol Evol 22: 1077–1081 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T 1993. Instability and decay of the primary structure of DNA. Nature 362: 709–715 [DOI] [PubMed] [Google Scholar]
- Meyer M, Briggs AW, Maricic T, Hober B, Hoffner B, Krause J, Weihmann A, Pääbo S, Hofreiter M. 2008. From micrograms to picograms: quantitative PCR reduces the material demands of high-throughput sequencing. Nucleic Acids Res 36: e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Kircher M, Gansauge MT, Li H, Racimo F, Mallick S, Schraiber JG, Jay F, Prüfer K, de Filippo C, et al. 2012. A high-coverage genome sequence from an archaic Denisovan individual. Science 338: 222–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Fu Q, Aximu-Petri A, Glocke I, Nickel B, Arsuaga JL, Martinez I, Gracia A, de Castro JM, Carbonell E, et al. 2014. A mitochondrial genome sequence of a hominin from Sima de los Huesos. Nature 505: 403–406 [DOI] [PubMed] [Google Scholar]
- Olalde I, Allentoft ME, Sanchez-Quinto F, Santpere G, Chiang CW, DeGiorgio M, Prado-Martinez J, Rodriguez JA, Rasmussen S, Quilez J, et al. 2014. Derived immune and ancestral pigmentation alleles in a 7,000-year-old Mesolithic European. Nature 507: 225–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen JS, Valen E, Velazquez AM, Parker BJ, Rasmussen M, Lindgreen S, Lilje B, Tobin DJ, Kelly TK, Vang S, et al. 2014. Genome-wide nucleosome map and cytosine methylation levels of an ancient human genome. Genome Res 24: 454–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prüfer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, Heinze A, Renaud G, Sudmant PH, de Filippo C, et al. 2014. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan M, Skoglund P, Graf KE, Metspalu M, Albrechtsen A, Moltke I, Rasmussen S, Stafford TW Jr, Orlando L, Metspalu E, et al. 2014. Upper Palaeolithic Siberian genome reveals dual ancestry of Native Americans. Nature 505: 87–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Green RE, Kircher M, Krause J, Patterson N, Durand EY, Viola B, Briggs AW, Stenzel U, Johnson PL, et al. 2010. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 468: 1053–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohland N, Hofreiter M. 2007. Ancient DNA extraction from bones and teeth. Nat Protoc 2: 1756–1762 [DOI] [PubMed] [Google Scholar]
- Sawyer S, Krause J, Guschanski K, Savolainen V, Pääbo S. 2012. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS ONE 7: e34131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro B, Hofreiter M. 2014. A paleogenomic perspective on evolution and gene function: new insights from ancient DNA. Science 343: 1236573. [DOI] [PubMed] [Google Scholar]
- Skinner AR, Blackwell BAB, Martin S, Ortega A, Blickstein JIB, Golovanov LV, Doronichev VB. 2005. ESR dating at Mezmaiskaya Cave, Russia. Appl Radiat Isot 62: 219–224 [DOI] [PubMed] [Google Scholar]
- Skoglund P, Malmstrom H, Raghavan M, Stora J, Hall P, Willerslev E, Gilbert MT, Gotherstrom A, Jakobsson M. 2012. Origins and genetic legacy of Neolithic farmers and hunter-gatherers in Europe. Science 336: 466–469 [DOI] [PubMed] [Google Scholar]
- Skoglund P, Northoff BH, Shunkov MV, Derevianko AP, Pääbo S, Krause J, Jakobsson M. 2014a. Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proc Natl Acad Sci 111: 2229–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoglund P, Storåb J, Götherström A, Jakobsson M. 2014b. Accurate sex identification of ancient human remains using DNA shotgun sequencing. J Archaeol Sci 40: 4477–4482 [Google Scholar]
- Stoneking M, Krause J. 2011. Learning about human population history from ancient and modern genomes. Nat Rev Genet 12: 603–614 [DOI] [PubMed] [Google Scholar]
- Zaremba-Niedzwiedzka K, Andersson SGE. 2013. No ancient DNA damage in actinobacteria from the Neanderthal bone. PLoS ONE 8: e62799. [DOI] [PMC free article] [PubMed] [Google Scholar]