

Abstract
Background
Neuropathic pain (NP) is a common occurrence following spinal cord injury (SCI). Identification of specific molecular pathways that are involved in pain syndromes has become a major priority in current SCI research. We have investigated the role of a cation-dependent chloride transporter, Cl-regulatory protein Na+-K+-Cl- 1 (NKCC1), phosphorylation profile of NKCC1 and its specific involvement in neuropathic pain following contusion SCI (cSCI) using a rat model. Administration of the NKCC1 inhibitor bumetanide (BU) increases the mean hindpaw withdrawal latency time (WLT), thermal hyperalgesia (TH) following cSCI. These results demonstrate implication of NKCC1 co-transporter and BUin SCI-induced neuropathic pain. The with-no-lysine (K)–1 (WNK1) kinase has been shown to be an important regulator of NKCC1 phosphorylation in many systems, including nocioception. Mutations in a neuronal-specific exon of WNK1 (HSN2) was identified in patients that have hereditary sensory neuropathy type II (HSANII) also implicates WNK1 in nocioception, such that these patients have loss of perception to pain, touch and heat. In our ongoing research we proposed two studies utilizing our contusion SCI (cSCI) NP model of rat.
Purpose
Study 1 aimed at NKCC1 expression and activity is up-regulated following cSCI in the early edema and chronic neuropathic pain phases. Study 2 aimed at identifying the expression profile of alternatively spliced WNK1 isoforms in animals exhibiting thermal hyperalgesia (TH) following cSCI.
Methods
Adult male Sprague Dawley rats (275–300 g) following laminectomy received cSCI at T9 with the NYU impactor-device II by dropping 10 g weight from the height of 12.5 mm. Control rats obtained laminectomy but no impaction. Following injury, functional recovery was assessed by BBB locomotor scores on day 1, 7, 14, 21, 35, and 42 and development of thermal hyperalgesia on day 21, 28, 35, and 42 day of injury by monitoring hind paw withdraw latency time (WLT) in seconds compared with the baseline data before injury.
Results
Increased NKCC1 may explain observed increase in magnetic resonance imaging (MRI) T2, exhibiting NKCC1 localization in neurons. This data supports NKCC1’s role in the pathogenesis of acute and chronic phases of injury, namely spinal cord edema and chronic phase neuropathic pain. NKCC1 dependent chloride influx requires the phosphorylation at specific residues. Probing for the HSN2 exon of WNK1 reveals two key findings: i) the HSN2 exon is found in alternatively spliced neuronal isoforms found at 250 kDa and 230 kDa; ii) the 250 kDa isoform is found only in tissue that is injured.
Conclusions
This data implicates the NKCC1/WNK1/WNK1HSN2 involvement in post-injury response that contributes to the development of neuropathic pain. Targeting this system may have therapeutic benefit.
Keywords: Spinal cord injury, Neuropathic pain, Thermal hyperalgesia, NKCC1, WNK1, HSN2, Edema, MRI
Introduction
Neuropathic pain (NP) is defined as pain secondary to a primary lesion or dysfunction of the nervous system.1 More than one million suffer from spinal cord injury (SCI) in the United States alone with more than 12, 000 new cases each year. Annual global incidence 15 to 40 cases per million.2,3 NP following SCI is a significant clinical problem, affecting up to 75% of SCI patients. NP is often excruciating and can significantly impact the quality of a patient’s life.3 The mechanism underlying derangement of the GABAergic system has been implicated in spinal nocioceptive processing as a topic of research.4–6 Normal GABA function is critically dependent on the activity of the cation chloride cotransporters, Na+-K+-Cl- cotransporter 1 (NKCC1) responsible for Cl- influx, and K+-Cl- co Transporter 2 (KCC2) responsible for Cl- efflux.
We have demonstrated recently that NKCC1 activity is up-regulated for two weeks following cSCI, which precedes onset of chronic NP.4 Inhibition of NKCC1 with its potent antagonist BU significantly reduced pain behavior in rats. One current thought implicates NKCC1 and neuropathic pain to neurons. Our previous findings report that following a contusion spinal cord injury (cSCI) NKCC1 protein expression is up-regulated, and KCC2 expression is down-regulated.4 Moreover, administration of the NKCC1 inhibitor bumetanide (BU) increased the mean hindpaw withdrawal latency time (WLT). These results have implicated the NKCC1 co-transporter in observed decreased WLT following cSCI. Bumetanide-sensitive NKCC1, expressed in astrocytes and the blood brain barrier (BBB), implicates formation of cerebral edema in rodent stroke.7 Activation of NKCC1 and its regulatory pathway has been linked to altered Cl- regulation and chronic pain.8 We have reported that WNK1 is possibly involved in NKCC1 phosphorylation. These results suggest that persistent activation of NKCC1 and WNK1 may play an important role in SCI-induced neuropathic pain.5 However, it remains unknown whether other isoforms of WNK family, particularly WNK3, WNK4, also play a role in regulation of NKCC1 activity following SCI.6
WNK1 (with no lysine (K)) is a widely expressed serine/threonine protein kinase, an enzyme encoded by the WNK1 gene in humans, having mutations associated with Gordon hyperkalemia-hypertension syndrome (pseudohypoaldosteronism Type II, featuring hypertension) and congenital sensory neuropathy (HSAN Type II, featuring loss of perception to pain, touch, and heat due to a loss of peripheral sensory nerves.9 The role of this kinase was first described in kidney where it dynamically controls ion channels that regulate changes in cell volume. WNK1, through intermediates oxidative stress-responsive kinase-1 (OSR1) and STE20/SPS1-related proline/alanine-rich kinase (SPAK), phosphorylates the inwardly directed Na+-K+-Cl--cotransporter 1 (NKCC1) and the outwardly directed K+-Cl--cotransporter 2 (KCC2), activating and deactivating these channels, respectively. WNK1, NKCC1 and KCC2 are also expressed in the central nervous system (CNS). Growing evidence implicates that WNK1 plays a critical role in pathologic nervous system signaling where changes in intracellular ion concentration in response to γ-aminobutyric-acid (GABA) can activate otherwise silent pathways.10
In HSAN type II disease, a novel mutation in the WNK1/HSN2 gene corroborates the clinical consistency present in various genotypes,11 where none or declined numbers of peripheral sensory neurons result in impairment of pain, temperature, and touch sensation.12 Thus HSN2 may be implicated in the improvement and/or maintenance of peripheral sensory neurons or supportive Schwann cells. The present study was aimed at identifying the expression profile of alternatively spliced WNK1 isoforms in animals exhibiting thermal hyperalgesia following contusion SCI. Understanding the pathogenesis of spinal cord edema may help improve motor function rescue. Spinal cord edema correlates with diminished motor activity. We hypothesize that these results can be explained by NKCC1’s role in establishment and maintenance of the chloride equilibrium potential (Figure 1). In this paper we present our findings on the pathogenesis of SCI. In particular, we focused on the role of the NKCC1 in altering ionic homeostasis which results in altered osmotic and GABAergic homeostasis. Cerebral ischemia models have implicated NKCC1 in elevated T2 signal associated with post-infarct cerebral edema. We propose a similar mechanism for spinal cord injury induced edema given similar NKCC1 expression profiles as is noted post-cerebral ischemia. In the present study we planned to determine and characterize the HSN2 gene in the epicenter area of contusion spinal cord injury followed by neuropathic pain induced during the chronic phase. Understanding the upstream regulators of NKCC1 activation affords more specific therapeutic options to down-regulate the NKCC1 system. Furthermore, this work has potential to study the role of NKCC1 inhibitor, bumetanide, on edema volume and implication of neuronal WNK1 isoform (HSN2) in spinal cord induced neuropathic pain model.
Fig. 1:
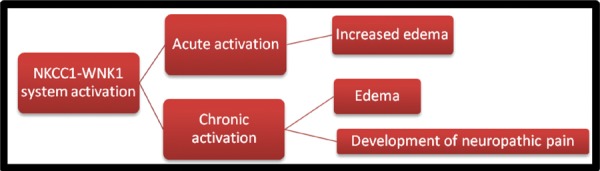
Schematic diagram of the approach to address various steps involved in mechanism of spinal cord injury induced neuropathic pain.
Methods
Sprague Dawley male rats weighing 250–270 g weight were used for different experiments. The protocol and all surgical procedures were in accordance with NIH guidelines and were approved by the Institutional Animal Care and Use Committee of the University of Wisconsin.
Spinal cord injury: Adult male Sprague Dawley rats (275–300 g) underwent contusional SCI with the NYU impactor with 12.5 g/cm injury as previously reported from our laboratory.4,13–16 Sprague Dawley rats were selected because of our experience with the assessment of TH using this strain.14,15 Briefly, following the induction of adequate inhalational anesthesia (Isoflurane, induction 5%, maintenance 2.5%, in a 50: 50 mixture of oxygen and nitrous oxide), a T9 laminectomy was performed and the rat was transferred to the impactor. A cSCI was created by dropping a 10-g weight from a height of 12.5 mm. Ten additional sham-operated controls underwent laminectomy, but were not contused. The incisions were closed and rats returned to their cages after recovering from anesthesia. Throughout the procedure, body temperature is maintained at 37°C with a heating pad. Rats underwent manual bladder expression until bladder control is re-established.
Assessment of locomotor ability and functional outcome: The functional neurological deficits due to the cSCI were assessed by behavioral analysis. Animals were observed individually in an open field testing area consisting of an unfilled plastic wading pool. Four minute observation periods were videotaped on post-impaction days 1, 7, 14, 21, 28, 35, and 42. A single blinded reviewer will score the animals’ locomotor function using the BBB locomotor rating scale.17 BBB scores were measured before injury (baseline) and on the described post injury days. These measurements are important in order to account for confounding variables between groups related to differences in neurological injury.
Assessment of Thermal Hyperalgesia (TH): TH testing developed by Hargreaves et al.18 assesses an animal’s withdrawal of a hindpaw to a thermal noxious stimulus. We have found TH to be a sensitive and reliable behavior test and used for our experiments.19–21 Briefly, an animal is placed inside the apparatus (Plantar™ Test, Stoelting, IL), and a movable focused beam of radiant heat is placed under the animal’s paw. As soon as the animal moves its paw a photocell turns off the heat, and the latency for the animal to withdraw its paw is recorded. Strength of stimulation is adjusted to produce baseline latencies of 8–10 s (typically 45–47 C). In this TH model we have found a decreased withdrawal latency time of greater than two seconds, as compared to baseline values, indicative of the development of neuropathic pain. A safety cut-off (~15 s) is used to prevent prolonged exposure to the noxious heat. Hind limb TH testing is performed the day prior to injury and then again on post injury days 21, 28, 35, and 42. We characterized the incidence of pain behavior in each cohort of animals, described the zone of allodynia, quantified its severity, and noted any changes that occur over time. Animals were sacrificed 42 days following injury in order to harvest the spinal cord for analysis.
Sample spinal cord harvesting: The epicenter and rostral tissues (7–10 mm sections) were harvested on day 1, 2, 3 (acute phase of injury) and 21, 28, 35, 42 post injury (chronic phase of injury). The sections were immediately submerged in liquid nitrogen and stored at –80°C. Spinal cord samples were harvested from animals on days 1, 3, or 7 following SCI and on days 35 and 42 following SCI, when neuropathic hyperalgesia was developed.
Sample preparation and western blotting
Spinal cord segments were homogenized in anti-phosphatase buffer (pH 7.4, mmol/L: 145 NaCl, 1.8 NaH2PO4, 8.6 Na2HPO4, 100 NaF, 10 Na4P2O7, 2 Na3VO4, 2 EDTA) containing protease inhibitors.6 Briefly, the homogenate was centrifuged at 7000 rpm for 15 min at 4°C. The supernatant was retained and protein content of the supernatant was determined by the BCA protein assay (Pierce; Rockford, Ill). Protein samples (60 μg/lane) and pre-stained molecular mass markers (Bio-Rad; Hercules, CA) were denatured in SDS sample buffer. The samples were then electrophoretically separated on 8% SDS gels, and the resolved proteins were electrophoretically transferred to a nitrocelluose membrane. The blots were incubated in 7.5% nonfat dry milk in Tris-buffered saline (TBS), and then incubated overnight with a primary antibody at 4ºC. The blots were rinsed with TBST and incubated with horseradish peroxidase-conjugated secondary IgG for 1 h. Bound antibody was visualized using the enhanced chemiluminescence assay (ECL, Amersham Corp; Piscataway, NJ). Monocolonal antibody against non-phosphorylated NKCC1 [T4, 1:3000; Developmental Studies Hybridoma Bank; Iowa City, IA), and monoclonal anti-phosphorylated-NKCC1 (BL74611:1000; gifts from Dr. Kahle of Harvard University) were used for detection of total NKCC1 (t-NKCC1) and phosphorylated NKCC1 (p-NKCC1), respectively. Anti-βIII-tubulin monoclonal antibody was used as a loading control (Promega; Madison, WI). Densitometric measurement of each protein band was performed with Image J22 and average pixel intensity was recorded.
MRI Imaging
T2 image sequence using a 4.7T Varian system: A Variant 4.7 Tesla/40 cm horizontal bore MRI System, with a 400 mT/m, 120 mm inner diameter gradient insert were used with a laboratory-built 3 cm diameter coil for transmitting and receiving the MRI signal at the WIMR small animal imaging facility at the University of Wisconsin School of Medicine and Public Health. Rats were placed in prone position (ventral recumbence) on a laboratory-built Plexiglas holder that also incorporates the matching and tuning electronic circuitry of the radiofrequency (RF) coil. Animals were placed in the MRI bore tunnel. Anesthesia was maintained in all animals with inhaled Isoflurane at 1–2%. Respiratory rate was monitored continuously using the monitoring & gating system for small animals (Model 1024; S.A. Instruments Inc., Stony Brook, NY). Animals were anesthetized during imaging. During prolonged imaging (>45 minutes) animals were kept warm with MRI compatible heating pads. Consecutive series of either axial or sagittal T2 weighted images were acquired each hour for 2 hours while the animal were under general anesthesia.
Results
We have shown in our SCI-induced NP model that NKCC1 is up-regulated during the first two weeks of injury. Changes in expression of p-WNK1 and t-WNK1 proteins in spinal cord epicenter region following acute phase of contusion spinal cord injury on 1, 3, and 7 days are presented in Figure 2. Densitometricanalysis of the ratio of p-WNK1/t-WNK1 intensity shows increased p-WNK1 expression in injured spinal cord tissues.
Fig. 2:
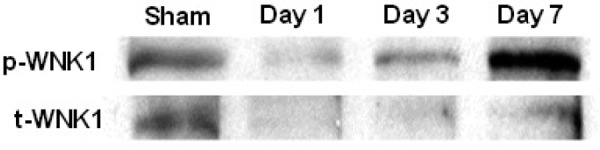
Changes in expression of p-WNK1 and t-WNK1 proteins in spinal cord epicenter following acute phase of contusion injury. A WNK1 protein expression in epicenter spinal cord tissues of rats following SCI at day 1, 3, and 7. Sham (S) samples were obtained from animals subject to laminectomy without subsequent spinal cord contusion. The blot was probed with anti-p-WNK1 (R&D Systems, 1:2000) and anti-t-WNK1 (R&D Systems, 1:1000).
Changes in expression of p-NKCC1 and t-NKCC1 proteins in epicenter spinal cord following chronic phase of SCI on day 35 and 42 in Figure 3. We observed changes in NKCC1 phosphorylation levels following injury in the chronic phase of NP as the ultimate source of GABAergic signal derangement. Since WNK1 is a well-established stimulator of NKCC1, we also examined how NKCC1 phosphorylation levels change relative to expression changes in both WNK1 isoforms identified in the spinal cord.
Fig. 3:
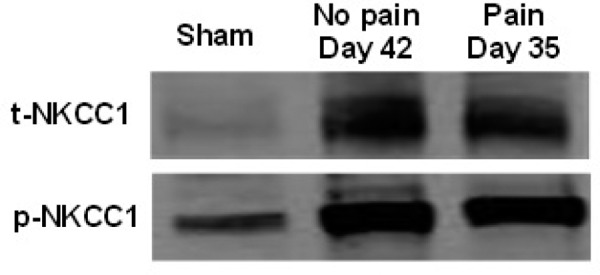
Changes in expression of p-NKCC1 and t-NKCC1 proteins in epicenter during chronic phase of contusion SCI. A. NKCC1 protein expression in epicenter spinal cord tissues of rats (NP = No-Pain and P = pain) following SCI at day 42 (NP) and day 35 (P). Sham (S) samples were obtained from animals subject to laminectomy without subsequent spinal cord contusion. The blot was probed with anti-p-NKCC1 (BL7461 antibody, 1:1000; secondary anti-rabbit 1:2000) and anti-t-NKCC1 (T4 monoclonal antibody, 1:3000; goat anti-mouse 1:3000).
WNK1/HSN2 expression in different tissues including spinal cord, brain, kidney, liver of spinal cord injured rat was studied to determine tissue specific expression of WNK1/HSN2. Only epicenter tissue of spinal cord exhibited WNK1/HSN2 expression during chronic phase of injury on day 35 post SCI (Figure 4). No expression of WNK1/HSN2 was observed in brain, kidney, liver of spinal cord injured rats.
Fig. 4:
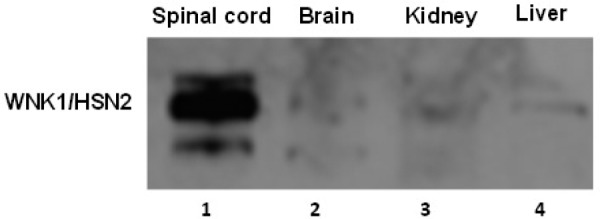
WNK1/HSN2 expression in different tissues including spinal cord epicenter, brain, kidney, liverof spinal cord injured rat. Tissue samples: 1 = Spinal cord epicenter; 2 = Brain; 3 = Kidney; 4 = Liver were acquired from animalson day 35 following laminectomyand spinal cord contusion injury. The blot was probed with primary WNK1/HSN2 antibody, 1:800; secondary anti-rabbit 1:2000). Western blots exposed for 3 minutes.
Following anti-WNK1/HSN2 antibody staining WNK1 exhibited increased expression and is up-regulated throughout the first two weeks of injury as well. In present findings HSN2 has been shown to be strongly up-regulated in epicenter region of the injured spinal cord. WNK1 presence in two isoforms in injured tissue at 250 and 230 kDa is of interest (Figure 5). Two bands are seen at 230 kDa (*) and 250 kDa (**) only at the site of injury in epicenter (E) region of the spinal cord, however rostral (R) region to the injury site did not express the 250 kDa band. The intensity of the 250 kDa band seems to correspond with animal’s change in hindpaw withdrawal latency time (WLT) tested for thermal hyperalgesia (TH). Furthermore, the intensity of the 250 kDa band seems to correspond with animal’s change in hindpaw withdrawal latency time (WLT) tested for (TH). Regression analysis of data showing correlation of 250 kDa band intensity with WLT suggesting that elevated 250 kDa WNK1 isoform expression may play a role in TH.
Fig. 5:
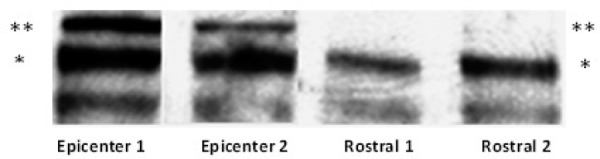
Immunoblotting with anti-WNK1/HSN2 antibody. Two bands are seen at 230 kDa (*) and 250 kDa (**). Only at the site of injury epicenter (E) are both bands expressed, however rostral (R) areas to the injury site do not express the 250 kDa band.
Immunobloting of WNK1 expression was confirmed by immunohistochemistry employed on epicenter and rostral regions following on day of contusion injury. pWNK1 and tWNK1 staining results are shown in the Figure 6.
Fig. 6:
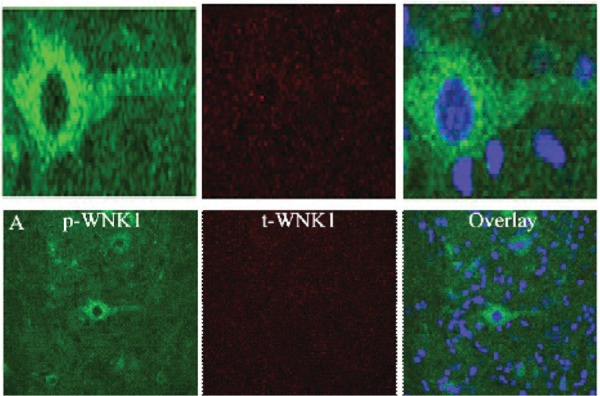
WNK1 expression at epicenter and rostral site following SCI at Day 1. Epicenter. pWNK1 staining (R&D Systems, 1:100, Left), tWNK1 staining (R&D Systems, 1:100, Middle), and overlay with pWNK1, tWNK1, and ToPro3 nuclei staining (Right).
T2 magnetic resonance imaging of contused spinal cord following day 1, 7, 14 of injury exhibited hyperintense regions of T2 signal are shown in Figure 7.
Fig. 7:
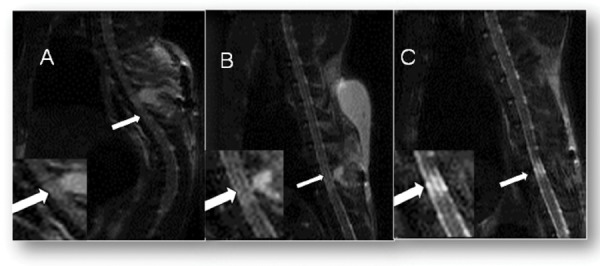
T2 Magnetic Resonance Imaging of contused spinal cord 24 hours after injury (A), 7 days after injury (B) and 14 days after injury (C). Arrows indicate hyperintense regions of T2 signal. Panel A and C are from different animals, although both animals very low Basso, Beattie, and Bresnahan (BBB) scores.
Discussion
GABA receptors are found trans-synaptically in areas of the spinal cord thought to be involved with sensory and pain perception, namely primary afferent terminals, interneurons in laminae I-IV in the dorsal horn.23 Subarachnoid transplantation of GABAergic neurons into the spinal cord following excitotoxic injury attenuates chronic allodynia and hyperalgesia.24 Elevation of NKCC1 expression increases intracellular Cl-, which causes GABA to act primarily as an excitatory neurotransmitter in the brain during early development.25 Unfortunately, effective analgesic therapies are not available for chronic neuropathic pain (CNP). The majority of patients with SCI suffer from CNP; however, current treatment strategies are inadequate and refractive because the cellular mechanisms that provide the substrate for CNP are poorly understood.26–29 Use of GABA antagonist, gabapentin in postherpetic neuralgia,30 SCI induced chronic pain,31 muscle spasticity, induced as well as spontaneous autonomic dysreflexia following complete SCI32 studies demonstrated considerable promise.
Increased NKCC1 expression following spinal cord injury presents a mechanism that explains the pathogenesis of spinal cord edema and NP. Our preliminary MRI T2 images are confirmed by previous work that there is extensive spinal cord edema following injury. The underlying pathophysiology of this edema is probably rooted in both destruction of the blood spinal-cord barrier (BSCB) from trauma in the first days following injury. Disrupted BSCB would cause vasogenic edema. However, by the second week following injury, it has been noted that the BSCB is restored. Persistence of edema may therefore be due to ionic flux. Cerebral ischemia models have implicated NKCC1 in elevated T2 signal associated with post-infarct cerebral edema. We propose a similar mechanism for spinal cord injury induced edema given similar NKCC1 expression profiles as is noted post-cerebral ischemia.
Our previous studies have demonstrated significant changes in increased expression of NKCC1 in epicenter spinal cord tissues on day 2, 7, and 14 following contusion SCI of Sprague Dawley rats.4 In addition to increased NKCC1 activity, WNK1 activity is increased. Understanding the upstream regulators of NKCC1 activation affords more specific therapeutic options to down-regulate the NKCC1 system. NKCC1 has been characterized to play a role in many physiological and pathological systems in organs ranging from brain to kidney. Previously, we have shown that NKCC1 plays a critical role in the development of neuropathic pain development. The later condition is reversed using an NKCC1 specific inhibitor bumetanide (BU). Specifically, BU administration increased withdrawal latency time (WLT).4,5 We hypothesize that these results can be explained by NKCC1’s role in maintenance and establishment of the chloride equilibrium potential. This determines the GABAergic system’s ability to be excitatory or inhibitory. The GABAergic system has been implicated in spinal nocioceptive processing. Normal GABA function is critically dependent on the activity of the cation chloride cotransporters, Na+-K+-Cl- cotransporter 1 (NKCC1) responsible for Cl- influx and K+-Cl- cotransporter 2 (KCC2) responsible for Cl- efflux. An important consequence of NKCC1 activation is increased intracellular chloride.
NKCC1 has been implicated in cerebral edema following cerebral ischemia as well; however, these studies examined NKCC1 activity within 24 hours following injury.7 Brain sustains homeostasis by regulating the flow of solute and water across its major cellular barriers, failure to do so can result in cerebral edemafollowing stroke and ischemic brain injury. Resulting cerebral edema is an outcome of weakened regulation of astrocytic cell volume, permeability alterations and dependent on activity of specific ion channels and transporters. Furthermore, pharmacological inhibition or genetic deficiency of NKCC1 decreases ischemia-induced cell swelling, BBB breakdown, cerebral edema, and neurotoxicity. A therapeutic approach utilizing combination pharmacological strategies has been suggested to inhibit action of NKCC1 expression, thus precisely regulating the neuronal homeostasis of Cl- ions. We present data that NKCC1 expression and activity is up-regulated following SCI in the early and chronic phases. Increased NKCC1 may explain observed increase in magnetic resonance imaging (MRI) T2. Additionally, NKCC1 is localized to neurons. These data support NKCC1’s role in the pathogenesis of acute and chronic phases of injury, specifically spinal cord edema and chronic phase NP.
Understanding the pathogenesis of spinal cord edema may help improve motor functional return. Spinal cord edema correlates with diminished motor activity. In the brain, it is has been shown to play an important role in post-cerebral ischemia edema formation.7 NKCC1 has also been implicated in the pathogenesis of cerebral edema following middle cerebral artery occlusion and diabetic ketogenic acidosis. The pathological accumulation of fluid in the brain’s intracellular and extracellular spacesis a major cause of morbidity and mortality following stroke and other forms of ischemic brain injury. Molecular pathogenesis results from impaired astrocytic cell volume regulation and permeability alterations in the cerebral microvasculature, both of which arise from pathological changes in the activities of specific ion channels and transporters.7 Furthermore, the implication of bumetanide-sensitive NKCC1, an electroneutral cotransporter expressed in astrocytes and the BBB, in cerebral edema formation in several different rodent models of stroke has been demonstrated.7 This pharmacological strategyusing NKCC1 inhibitor, bumetanide, in spinal cord injury induced neuropathic pain already proved beneficial for the treatment of ischemic and potentially other types ofcerebral edema.
HSAN are clinically and genetically heterogeneous disorders characterized by axonal atrophy and degeneration, exclusively or predominantly affecting the sensory and autonomic neurons.33 So far, disease-associated mutations have been identified in seven genes: two genes for autosomal dominant (SPTLC1 and RAB7) and five genes for autosomal recessive forms of HSAN (WNK1/HSN2, NTRK1, NGFB, CCT5 and IKBKAP). Overall disease-associated mutations were found in 19% of the studied patient group, suggesting that additional genes are associated with HSAN. Their genotype-phenotype correlation study broadens the spectrum of HSAN and provides additional insights for molecular and clinical diagnosis. Probing the HSN2 exon of WNK1 reveals three key findings: i) the HSN2 exon is found in alternatively spliced neuronal isoforms found at 250 kDa and 230 kDa; ii) the 250 kDa is found only in tissue that is injured; iii) the intensity of the 250 kDa band appears to be proportional to the degree of thermal hyperalgesia that animal exhibits. The presence of an injury specific 250 kDa WNK1 isoform with the HSN2 exon can be explained through analysis of the alternatively splicing sites within the WNK1 gene. Previous work has cited exon 11 and exon 12 to be crucial exons in the regulation of WNK1 function and there splicing to be tightly regulated.34 Shekarabi et al.9 probedneuronal specific HSN2 exon with the “anti-WNK1/HSN2” antibody. One possible explanation why Shekarabi et al.9 did not observe an HSN2 detected WNK1 isoform above 230 kDa in spinal cord is that this novel 250 kDa WNK1 isoform is only induced in injury or stressed situations (e.g. following spinal cord injury). An important series of experiments will be needed to confirm the exact sequences of the 250 kDa and 230 kDa bands.
Preliminary data of regression analysis correlates 250 kDa band intensity with the WLT suggests that elevated 250 kDa WNK1 isoform expression may play a role in thermal hyperalgesia. Increasing sample size is the first step to elucidate this relationship. Furthermore, we characterized this relationship at early post-injury time points (day 1, 3, 7) and late post injury (day 35, and 42). We believe that changes in NKCC1 phosphorylation levels following injury in the chronic phase of neuropathic pain is the ultimate source of GABAergic signal derangement.6 Since WNK1 is a well-established stimulator of NKCC1 we will also examine how NKCC1 phosphorylation levels change relative to expression changes in both WNK1 isoforms identified in the spinal cord. Investigating WNK1 expression in the CNS after SCI, where altered neuronal signaling could underlie pathological states such as NP has future perspective.10 Recently a novel homozygous mutation in SCN9A with autosomal recessive HSAN (HSAN type IID) has been identified causinginstabilities in the sensory, olfactory, and autonomic nervous systemscoupled with additional symptomsas hyposmia, hypogeusia, hearing loss, bone dysplasia.35
Following a spinal cord injury (SCI), evidence has shown the presence of a threonine protein kinase, WNK1, being widely expressed in the central nervous system has been reported in a mini review from our laboratory.10 This kinase could potentially be a cause of neuropathic pain (NP) following the injury. WNK1 activates and deactivates the Na+-K+-Cl- cotransporter 1 (NKCC1) and K+-Cl- cotransporter 2 (KCC2) respectively; WNK1 achieves this through intermediates oxidative stress-responsive kinase 1 (OSR1) and STE20/SP21-related proline/alanine-rich kinase (SPAK), which phosphorylates the channels. It is suggested that WNK1 plays a role in this nervous system situation as its response to γ-aminobutyric-acid (GABA) leads to changes in the intracellularion concentration, leading to the activation of pathways that would have otherwise not been able to function.
An effect of an autosomal recessive mutation in the exon “HSN2” on the WNK1 serine-threonine kinase will lead to a rare early onset of severe sensory loss in distal limbs, called hereditary sensory and autonomic neuropathy type 2 (HSNAII).36 While the function of WNK1 in the nervous system is not well known, studies suggest that there is an interaction between KCC2 and WNK1/HSN2 that leads to transcription independent of KCC2’s activation. The mutation of WNK1 would therefore induce the KCC2 expression, and HSNAII symptoms would arise from the hindered proper peripheral sensory nerve development that stemmed from the KCC2 overexpression.
A confined alternatively spliced WNK1 is an autosomal recessive mutation can lead to hereditary sensory and autonomic neuropathy type 2 (HSANII), which is the early loss of sensory perception.36 While looking at the relationship between WNK1 and KCC2, WNK1 has been known to phosphorylate KCC2 which in turn regulates its functioning levels. In regards to HSANII, the interactions between WNK1 and KCC2 have led to KCC2 to be a theory of regulating neurogenesis. If phosphorylation occurs, the mutation in WNK1/HSN2 leads to a decreased functioning in nervous system development linked with HSANII. There will be an imbalance in the level of KCC2, lowering the amount of transcription and development of a proper peripheral nervous system.
Fig. 8:
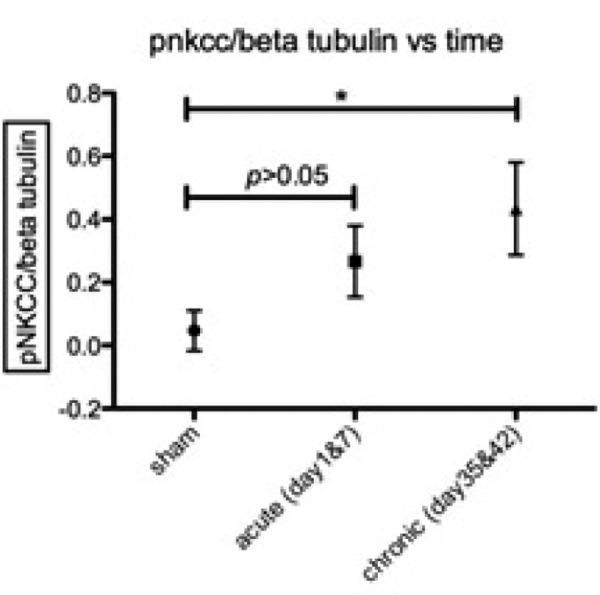
Time course of NKCC1 co-transporter phosphorylation following cSCI. p-NKCC was analyzed on western blots using the anti-p-NKCC antibody R5 and normalized to beta-tubulin. This value reflects the normalized amount of NKCC1 that is phosphorylated. Data are means ± s. e. m., n = 3 at sham, acute and chronic phases. One-way ANOVA determined a significant increase in the chronic phase (p<0.05). Asterisks indicate a significant difference (p<0.05) from sham.
Fig. 9:
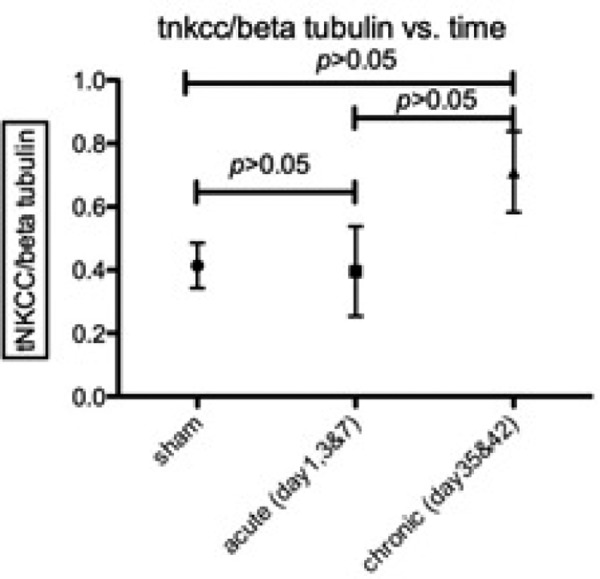
Time course of NKCC1 co-transporter expression following cSCI. t-NKCC1 was analyzed on western blots using the anti-total-NKCC antibody and normalized to beta-tubulin. Data are means ± s.e.m., n = 4 at sham, acute and chronic phases. One way ANOVA did not show significance, but is most likely due to low sample number.
Fig. 10:
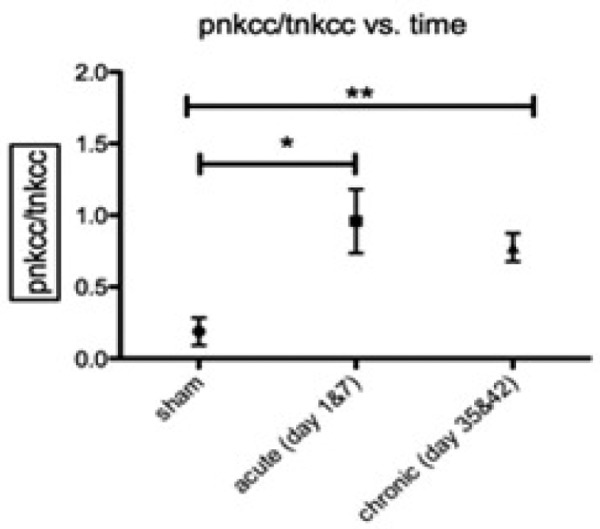
Time course of NKCC1 co-transporter phosphorylation following cSCI. p-NKCC was analyzed on western blots using the anti-p-NKCC antibody R5 and normalized total NKCC1. This value reflects the percentage of NKCC1 phosphorylated. Data are means ± s.e.m., n = 5–6 at sham, acute and chronic phases. One-way ANOVA determined a significant increase in acute (P<0.05) and chronic (P<0.01). Asterisks indicate a significant difference (P<0.05) sham, **= p<0.01, *= p<0.05).
Conclusion and future direction
In our present study, MRI T2 images show spinal cord edema following injury, correlating with our previous findings where we have reported an increase in NKCC1 expression in the lesion epicenter during day 2–14 post-SCI and peaked on day 14 post-SCI. This up-regulated NKCC1 expression following SCI presents a mechanism that may explain the pathogenesis of spinal cord edema followed by neuropathic pain. The basic pathophysiology of this may be rooted in damage of the blood spinal-cord barrier following the first days ofinjury. Targeting the NKCC1/WNK1/WNK1HSN2 system that contributes towards the development of spinal cord injury-induced neuropathic pain may have therapeutic implications. Furthermore, in future studieswe will focus on assessing whether i) NKCC1 inhibitor, bumetanide, silences the WNK1/HSN2 gene after spinal cord injury; ii) silencing the WNK1/HSN2 gene by bumetanide decreases neuropathic pain induced by SCI. The knowhow of suchunidentified mechanisms may be supportive while exploringnovel therapeutic agents for treating SCI-induced neuropathic pain.
Acknowledgements
Support for this study was provided by Herman and Gwendolyn Shapiro Foundation (MMA), American Academy of Neurology Medical Student Fellowship (MMA), Department of Neurological Surgery, University of Wisconsin School of Medicine and Public Health, Congress of Neurological Surgeons Synthes Spine Fellowship Award. Thanks to Dr. Dandan Sun for supervision of Chloe Lee research in the department of Neurological Surgery at the University of Wisconsin, Madison.
Footnotes
The article complies with International Committee of Medical Journal editor’s uniform requirements for manuscript.
Conflict of Interests: None, Source of funding: None
References
- 1.Ibrahim M. M., Deng H, Zvonok A et al. Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: pain inhibition by receptors not present in the CNS. Proc. Natl. Acad. Sci. U.S.A. 2003;100:10529–33. doi: 10.1073/pnas.1834309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.National Spinal Cord Injury Statistical Center.2010. Annual Report for the Spinal Cord Injury Model Systems, National Spinal Cord Injury Statistical Center, Birmingham, Alabama, which is funded by grant number H133A060039 from the National Institute on Disability and Rehabilitation Research, Office of Special Education and Rehabilitative Services, U.S. Department of Education. [Google Scholar]
- 3.Siddall PJ, McClelland JM, Rutkowski SB et al. A longitudinal study of the prevalence and characteristics of pain in the first 5 years following spinal cord injury. Pain. 2003;103:249–257. doi: 10.1016/S0304-3959(02)00452-9. [DOI] [PubMed] [Google Scholar]
- 4.Cramer SW, Baggott C, Cain J et al. The role of cation-dependent chloride transporters in neuropathic pain following spinal cord injury. Mol Pain. 2008;4:36. doi: 10.1186/1744-8069-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hasbargen T, Ahmed MM, Miranpuri G, Li L et al. Role of NKCC1 and KCC2 in the development of chronic neuropathic pain following spinal cord injury. Ann N Y Acad Sci. 2010;1198:168–72. doi: 10.1111/j.1749-6632.2010.05462.x. [DOI] [PubMed] [Google Scholar]
- 6.Lee HK, Ahmed MM, King KC et al. Persistent phosphorylation of NKCC1 and WNK1 in the epicenter of the spinal cord following contusion injury. Spine J. 2014;14:777–81. doi: 10.1016/j.spinee.2013.06.100. [DOI] [PubMed] [Google Scholar]
- 7.Kahle KT, Simard JM, Staley KJ et al. Molecular mechanisms of ischemic cerebral edema: role of electroneutral ion transport. Physiology (Bethesda) 2009;24:257–65. doi: 10.1152/physiol.00015.2009. [DOI] [PubMed] [Google Scholar]
- 8.Galan A, Cervero F. Painful stimuli induce in vivo phosphorylation and membrane mobilization of mouse spinal cord NKCC1 co-transporter. Neuroscience. 2005;133:245–252. doi: 10.1016/j.neuroscience.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 9.Shekarabi M, Girard N, Rivière JB et al. Mutations in the nervous system–specific HSN2 exon of WNK1 cause hereditary sensory neuropathy type II. J Clin Invest. 2008;118:2496–505. doi: 10.1172/JCI34088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krueger EM, Miranpuri GS, Resnick DK. Emerging role of WNK1 in pathologic central nervous system signaling. Annals of Neurosciences. 2011;18:2. doi: 10.5214/ans.0972.7531.1118212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pacheco-Cuellar G, González-Huerta LM, Valdés-Miranda JM et al. Hereditary sensory and autonomic neuropathy II due to novel mutation in the HSN2 gene in Mexican families. J Neurol. 2011;258:1890–1892. doi: 10.1007/s00415-011-6025-x. [DOI] [PubMed] [Google Scholar]
- 12.Lafreniere RG, MacDonald ML, Dube MP et al. Study of Canadian Genetic Isolates. Identification of a novel gene (HSN2) causing hereditary sensory and autonomic neuropathy type II through the Study of Canadian Genetic Isolates. Am J Hum Genet. 2004;74:1064–73. doi: 10.1086/420795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Resnick DK, Schmitt C, Miranpuri GS et al. Molecular evidence of repair and plasticity following spinal cord injury. Neuroreport. 2004;15:837–9. doi: 10.1097/00001756-200404090-00020. [DOI] [PubMed] [Google Scholar]
- 14.DomBourian MG, Turner NA, Gerovac TA et al. B1 and TRPV-1 receptor genes and their relationship to hyperalgesia following spinal cord injury. Spine (Phila Pa 1976) 2006;31:2778–82. doi: 10.1097/01.brs.0000245865.97424.b4. [DOI] [PubMed] [Google Scholar]
- 15.Rajpal S, Gerovac TA, Turner NA et al. Antihyperalgesic effects of vanilloid-1 and bradykinin-1 receptor antagonists following spinal cord injury in rats. J Neurosurg Spine. 2007;6:420–4. doi: 10.3171/spi.2007.6.5.420. [DOI] [PubMed] [Google Scholar]
- 16.Ahmed MM, Rajpal S, Sweeney C et al. Cannabinoid subtype-2 receptors modulate the antihyperalgesic effect of WIN 55, 212-2 in rats with neuropathic spinal cord injury pain. Spine J. 2010;10:1049–54. doi: 10.1016/j.spinee.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 17.Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- 18.Hargreaves K, Dubner R, Brown F et al. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 19.Miletic G, Miletic V. Long-term changes in sciatic-evoked A-fiber dorsal horn field potentials accompany loose ligation of the sciatic nerve in rats. Pain. 2000;84:353–359. doi: 10.1016/s0304-3959(99)00227-4. [DOI] [PubMed] [Google Scholar]
- 20.Miletic G, Miletic V. Increases in the concentration of brain derived neurotrophic factor in the lumbar spinal dorsal horn are associated with pain behavior following chronic constriction injury in rats. NeurosciLett. 2002;319:137–140. doi: 10.1016/s0304-3940(01)02576-9. [DOI] [PubMed] [Google Scholar]
- 21.Kim HT, Kim T, Novotny B et al. Thermal hyperalgesia assessment for rats after spinal cord injury: developing a valid and useful pain index. Spine J. 2014 Jun 1;14:984–9. doi: 10.1016/j.spinee.2013.09.051. [DOI] [PubMed] [Google Scholar]
- 22.Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- 23.Bowery NG, Hudson AL, Price GW. GABAA and GABAB receptor site distribution in the rat central nervous system. Neuroscience. 1987;20:365–383. doi: 10.1016/0306-4522(87)90098-4. [DOI] [PubMed] [Google Scholar]
- 24.Eaton MJ, Wolfe SQ, Martinez M et al. Subarachnoid transplant of a human neuronal cell line attenuates chronic allodynia and hyperalgesia after excitotoxic spinal cord injury in the rat. J Pain. 2007;8:33–50. doi: 10.1016/j.jpain.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 25.Ben Ari Y. Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–39. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- 26.Hulsebosch CE, Xu GY, Perez-Polo JR, Westlund KN, Taylor CP, McAdoo DJ. Rodent model of chronic central pain after spinal cord contusion injury and effects of gabapentin. J. Neurotrauma. 2000;17:1207–1217. doi: 10.1089/neu.2000.17.1205. [DOI] [PubMed] [Google Scholar]
- 27.Hulsebosch CE. From discovery to clinical trials: treatment strategies for central neuropathic pain after spinal cord injury. Curr. Pharm. Des. 2005;11:1411–1420. doi: 10.2174/1381612053507864. [DOI] [PubMed] [Google Scholar]
- 28.Hulsebosch CE. Gliopathy ensures persistent inflammation and chronic pain after spinal cord injury. Exp. Neurol. 2008;214:6–9. doi: 10.1016/j.expneurol.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hulsebosch CE, Hains BC, Crown ED et al. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res. Rev. 2009;60:202–213. doi: 10.1016/j.brainresrev.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rice ASC, Malton S. Gabapentin in postherpetic neuralgia: a randomised, double blind, placebo controlled study. Pain. 2001;2001;94:215–24. doi: 10.1016/S0304-3959(01)00407-9. [DOI] [PubMed] [Google Scholar]
- 31.Amr YM. Multi-day low dose ketamine infusion as adjuvant to oral gabapentin in spinal cord injury related chronic pain: a prospective, randomized, double blind trial. Pain Physician. 2010;13:245–9. [PubMed] [Google Scholar]
- 32.Rabchevsky AG, Patel SP, Lyttle TS et al. Effects of gabapentin on muscle spasticity and both induced as well as spontaneous autonomic dysreflexia after complete spinal cord injury. Front Physiol. 2012;3:329. doi: 10.3389/fphys.2012.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rotthier A, Baets J, Vriendt ED et al. Genes for hereditary sensory and autonomic neuropathies: a genotype-phenotype correlation. Brain. 2009;132:2699–711. doi: 10.1093/brain/awp198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delaloy C, Lu J, Houot AM, Disse-Nicodeme S et al. Multiple promoters in the WNK1 gene: one controls expression of a kidney-specific kinase-defective isoform. Mol Cell Biol. 2003;23:9208–21. doi: 10.1128/MCB.23.24.9208-9221.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan J, Matsuura E, Higuchi Y et al. Hereditary sensory and autonomic neuropathy type IID caused by an SCN9A mutation. Online before print April 17, 2013. doi: 10.1212/WNL.0b013e3182904fdd. doi: 10.1212/WNL.0b013e3182904fdd Neurology 10.1212/WNL.0b013e 3182904fdd. [DOI] [PubMed] [Google Scholar]
- 36.Bercier V, Brustein E, Liao M et al. WNK1/HSN2 Mutation in Human Peripheral Neuropathy Deregulates KCC2 Expression and Posterior Lateral Line Development in Zebrafish (Daniorerio). PLoS Genet. 2013 Jan;9(1): doi: 10.1371/journal.pgen.1003124. Published online Jan 3, 2013. doi: 10.1371/journal. pgen.1003124PMCID: PMC3536653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan Y, Dempsey RJ, Flemmer A et al. Inhibition of Na(+)-K(++)-Cl(-) cotransporter during focal cerebral ischemia decreases edema and neuronal damage. Brain Res. 2003;961:22–31. doi: 10.1016/s0006-8993(02)03832-5. [DOI] [PubMed] [Google Scholar]