

Abstract
Skull base chordomas (CD) are rare, locally aggressive, notochord-derived neoplasms for which prognostically relevant biomarkers are not well established. In a retrospective study of 28 cases of primary clival CD, we evaluated the relevance of Ki67 proliferation index (PI), p53 and EGFR expression, and 1p, 9p, 10q23, and 17p13 loss. Results were then correlated with clinical and pathologic parameters. Ki67 PI ≥5%, p53 accumulation, and EGFR expression was seen in 32%, 44%, and 8% of chordomas, respectively. 1p LOH and/or 1p36 hemizygous deletion was seen in 30% of tumors, while 9p LOH and/or 9p21 homozygous deletion was seen in 21% of cases. LOH at 10q23 and 17p13 were identified in 57% and 52% of cases, respectively. Ki67 PI ≥5% and 9p LOH were significantly associated with a shorter overall survival, while homozygous deletion at 9p21 via fluorescence in situ hybridization approached significance. No correlation with survival was found for p53 or EGFR expression, 1p36 hemizygous deletion, or LOH at 1p, 10q23, or 17p13. These results suggest that chordomas with elevated Ki67 PI or deletion at 9p21 may be at risk for a more aggressive clinical course and shorter survival, and that these biomarkers may be used to improve therapeutic stratification.
Keywords: Chordoma, Ki67, 1p36, 9p21, CDKN2A
Introduction
Chordomas are rare neoplasms that may arise from persistent notochord tissue. These tumors can arise anywhere along the axial spine, but most commonly are seen in the skull base/clivus and sacrococcygeal regions. Three variants of chordoma are recognized histologically: classic, chondroid, and dedifferentiated/sarcomatoid. Although typically slow-growing, chordomas are extremely difficult to eradicate by surgical and adjuvant means. Thus they usually recur multiple times, causing considerable mortality as evidenced by 5- and 10-year survival rates between 70–80% and 30–40%, respectively (1). Because growth rate, invasiveness, and survival after initial resection is highly variable, identification of molecular and protein prognostic markers is of interest.
In broad-based genetic studies the majority of chordomas were found to be diploid, with reduced survival in cases that had abnormal karyotypes including aneuploidy (2, 3). Additional work has identified nonrandom patters of copy number variation, including losses on 1p, 3, 9p, and 10, as well as gains on 7, with no site-specific differences (4–9). In particular, the CDKN2A locus encoding the tumor suppressor protein p16 has been found to be frequently deleted (5). Several familial chordoma cohorts have been studied extensively, with genetic mapping identifying 1p36 and 7q as key sites of loss and gain, respectively, (10–13). 1p36 loss is also seen in many sporadic chordomas (14) and was suggested by one group to be an unfavorable prognostic marker (15).
Expression of receptor tyrosine kinases (RTKs) and cell cycle proteins has also been studied in chordomas, with conflicting results. Some have shown a correlation between increased Ki67/MIB-1 proliferation index (PI) and solid pattern, recurrent tumors, shorter disease-free survival, overall survival, and/or doubling time, but not others (16–21). Aberrant overexpression of p53 correlates with increased PI and/or shorter survival in some studies, while others have not found any such link (19–23). Epidermal growth factor receptor (EGFR), an RTK well known to have oncogenic activity in a variety of neoplasms, has been consistently shown to be overexpressed and activated in chordomas, often in conjunction with other RTKs including platelet-derived growth factor receptors A and B, c-KIT, and HER2/neu (24–26). One such study found no association with RTK expression and tumor site or survival (25), but another showed that absence of c-MET correlated with an adverse outcome (27).
Thus, while chordomas are known to carry nonrandom genetic alterations and variable expression of proteins involved in cellular proliferation, a consensus regarding the prognostic utility of these biomarkers has not yet been reached. Herein we describe our retrospective study of primary clival chordoma specimens resected from 28 patients. Fluorescence in situ hybridization (FISH) on 1p36 and 9p21, and PCR-based microsatellite LOH analysis of 1p, 9p, 10q, and 17p, as well as immunohistochemical evaluation of Ki67 PI, p53, and EGFR, was performed. Data were then correlated with clinical outcomes. These results suggest that either loss at 9p21 or Ki67 PI over 5% is adverse prognostic markers. In particular, to our knowledge this study is the first to demonstrate that 9p21 deletion is an adverse prognostic biomarker. Incorporation of these markers into routine evaluation of chordomas may thus help customize postsurgical monitoring and therapeutic stratification.
Materials and methods
Cohort characteristics
The chordoma cohort used in this study was a subset of cases from a larger cohort described previously (28). Paraffin blocks and slides from 28 patients were successfully retrieved from the University of Pittsburgh Medical Center, Department of Pathology Archives (1969–2007) in accordance with Institutional Review Board guidelines. The diagnosis of chordoma and histologic subtyping on hematoxylin and eosin (H&E) stains were verified by 2 observers (GJO and RRS). Clinical follow-up data were available for all but 1 patient. The precise cause of death (i.e. death from tumor or other disease) was not available in most cases. Median follow-up interval on surviving patients was 67 months (range: <1 to 212 months). Cohort characteristics are summarized in Table 1.
Table 1.
Chordoma cohort demographics and histological features. This cohort reflected the age and gender distribution of skull base chordomas. Most cases showed at least partial chondroid differentiation, while 32% showed a significant solid component.
| Demographics | Histology |
|---|---|
| Median age: 39 years (12–77) 61% male 39% female Median followup time: 67 months 36% dead |
Conventional: 39% (11/28) |
| Chondroid: 61% (17/28) | |
| >5% solid: 32% (9/28) |
Tissue Microarray (TMA) Construction and Immunohistochemistry
The method for TMA construction was described previously (28). Briefly, a manual tissue arrayer (MTA-1, Beecher Instruments, (Sun Prairie, WI, USA) was used to select 0.6 mm cores from each paraffin block to a blank recipient paraffin block and arrayed in triplicate. Use of decalcified blocks could not be avoided in 50% (13/26) cases. Paraffin sections from the constructed TMA block were cut and incubated with antibodies to p53 (1:100, DO7, Dako, Carpinteria, CA, USA), Ki67 (1:25, ki-55, Dako, Carpinteria, CA, USA), and EGFR (1:500, H11, Dako, Carpinteria, CA, USA). Staining was visualized using the ImmPRESS™ (Vector Labs, Burlingame, CA, USA) detection system with 2-diaminobenzidine as the substrate chromogen. Each antibody was scored manually between two observers (GJO and RRS). Staining intensity of p53 was graded on a scale of 0 (negative) to 3 (strongly positive) and multiplied by the percent of cells with nuclear staining to produce a p53 score. Any case with a p53 score greater than or equal to 25 was considered positive. Ki67 PI was determined for each core and assigned a score of 0 if less than 5% or 1 if greater than or equal to 5%. Discrepancies between observers were resolved by simultaneous review.
Fluorescence in situ hybridization (FISH)
FISH protocols were similar to that described previously (29). Formalin-fixed paraffin-embedded TMA sections were mounted and serially sectioned at 5-μm intervals. H&E sections were used by one of the authors (CH) to determine the area of the tissue to be targeted for analysis. FISH slides were deparaffinized in xylene twice for 10 minutes, dehydrated twice with 100% ethanol, and then pretreated using the Vysis Paraffin Pretreatment Kit. Slides were digested for 18 minutes in protease solution (0.5 mg/ml) at 37 C. For 1p36, hybridization was performed using a spectrum orange labeled probe for 1p36 and spectrum green labeled control probe for 1q25 (Vysis dual-color probe set, LSI 1p36/LSI 1p25, Downers Grove, IL, USA). For 9p21, hybridization was performed using a spectrum orange labeled probe for CDKN2A (p16) and a spectrum green labeled chromosome 9 centromeric probe (CEP 9) (Vysis, Downers Grove, IL, USA). The target slide and probe were co-denatured at 95 C for 8 minutes and incubated overnight at 37 C in a humidified chamber. Post-hybridization washes were performed using 2XSSC/0.3% Igepal at 72 C for 2 minutes. Slides were air-dried in the dark and counterstained with DAPI (Vysis, Downers Grove, IL, USA). Analysis was performed using a Nikon Optiphot-2 (Nikon, Inc., Melville, NY, USA) and Quips Genetic Workstation equipped with Chroma Technology filter set with single band excitors for SpectrumOrange, FITC, DAPI (uv 360 nm) (Vysis, Downers Grove, IL, USA). Only individual and well delineated cells were scored. Overlapping cells were excluded from the analysis. At least 60 nuclei were counted in each case. Cases were considered positive for 1p36 deletion if ≥ 20% of nuclei showed deletion. For 9p21, only homozygous deletion was counted and was defined by loss of both 9p21 signals in ≥ 20% of nuclei with at least one CEP 9 signal. These cutoff points were derived using nonneoplastic autopsy brain tissue as controls.
PCR-based microsatellite LOH analysis
Manual microdissection of the tissue sample was performed to include tumor tissue. Matched nonneoplastic tissue was not available. Specimens with the minimum of 50% of tumor cells in a microdissection target were accepted for the analysis. DNA was isolated using standard laboratory procedures. Optical density readings were obtained. The assay utilized 7 microsatellite markers on chromosome 1p22-36 (D1S171, D1S162, D1S199, D1S1172, D1S1161, D1S407 and D1S226), 3 on 9p21-22 (CDKN2A gene), 3 on 17p13 (TP53 gene), and 2 on 10q23 (PTEN gene). PCR was performed and the PCR products were analyzed using capillary gel electrophoresis on GeneMapper ABI 3730 (Foster City, CA). Relative fluorescence was determined for individual alleles and the ratio of peaks was calculated. Neoplastic tissue was then analyzed to detect loss of heterozygosity. As normal tissue was not available, peak height ratios falling outside of 2 standard deviations beyond the mean of previously validated normal values for each polymorphic allele paring were assessed as showing loss of heterozygosity. Only LOH at 2 or more informative loci on 1p and 9p, and LOH at 1 or more informative loci on 10q or 17p, were scored as loss.
Data Analysis
Survival analysis was performed using the Kaplan Meier method with SPSS software, version 17.0 (SPSS, Chicago, IL, USA). Group-wise comparisons were made using the log-rank test.
Results
Cohort demographics and histology
Our retrospective cohort consisted of 28 clival chordomas resected from 11 female and 17 male patients, with a median age of 39 years (range 12–77 years) (Table 1). All tumors were initial resections; recurrences and previously treated tumors were excluded. At the end of the study 10 patients had died and 1 was lost to follow-up with a (Kaplan-Meier) median survival of 169 months. The (arithmetic) median follow-up on surviving patients was 67 months. 39% (11/28) of the chordomas were conventional (Figure 1A–B), 61% (17/28) were chondroid (Figure 1C), and none were dedifferentiated. 32% (9/28) had a solid component greater than 5% (Figure 1B).
Figure 1.
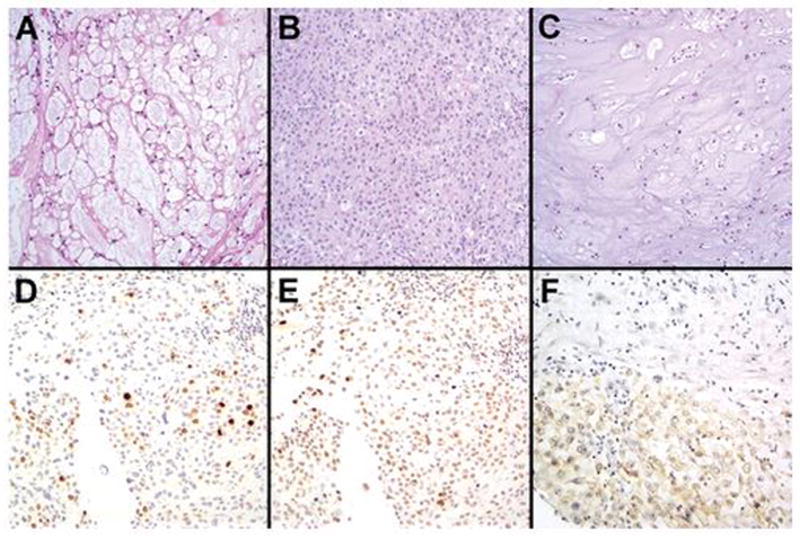
Histologic and immunohistochemical characterization of skull base chordomas. H&E stains showed both conventional (A, B) and chondroid (C) histologic subtypes of chordomas in the cohort. 32% had a prominent solid component (B), 32% showed elevated Ki67 PI (D), and 44% showed increased p53 accumulation (E). EGFR expression, in contrast, was seen in only 8% of cases (F).
Ki67, p53, and EGFR Immunohistochemistry
Immunohistochemical analysis of Ki67 showed that 32% (8/25) tumors had nuclear Ki67 expression in 5% or more of tumor cells (Table 2, PFigure 1D). Such cases had a 56% reduction in mean survival time compared to patients whose chordomas had a PI less than 5% (69.3 versus 159.3 months, = 0.005) (Figure 3A).
Table 2.
Correlation of immunohistochemical and molecular biomarkers with survival in skull base chordomas. Of the biomarkers studied, Ki67 PI 5% and 9p LOH were significantly associated with shorter overall survival (OS), while chordomas with 9p21 homozygous deletion trended toward shortened OS. P53 accumulation, EGFR expression, and deletions on 1p, 10q, and 17p were not significantly associated with survival.
| Biomarker | Result | Frequency (%) | Mean OS (mos) | 95% CI (mos) | P |
|---|---|---|---|---|---|
| Ki67 PI | <5% | 17/25 (68) | 159.3 | 118.9 – 199.7 | 0.005 |
| ≥5% | 8/25 (32) | 69.3 | 28.7 – 110.0 | ||
| P53 expression score | <25 | 14/25 (56) | 145.1 | 102.3 – 187.9 | 0.282 |
| ≥25 | 11/25 (44) | 96.6 | 65.4 – 127.8 | ||
| EGFR expression | negative | 22/24 (92) | 137.6 | 99.1 – 176.1 | 0.270 |
| positive | 2/24 (8) | 57.7 | 16.4 – 98.9 | ||
| 1p36 FISH | intact | 19/23 (83) | 134.5 | 97.2 – 171.7 | 0.628 |
| hemizygous deletion | 4/23 (17) | 122.7 | 91.7 – 153.6 | ||
| 1p LOH | negative | 15/24 (62) | 142.0 | 91.0 – 192.9 | 0.820 |
| positive | 9/24 (38) | 120.4 | 87.5 – 153.2 | ||
| 9p21 FISH | intact | 18/23 (78) | 151.4 | 115.1 – 187.7 | 0.083 |
| homozygous deletion | 5/23 (22) | 73.8 | 47.9 – 99.7 | ||
| 9p LOH | negative | 22/25 (88) | 146.2 | 109.8 – 182.5 | 0.033 |
| positive | 3/25 (12) | 71.7 | 42.5 – 100.9 | ||
| 10q LOH | negative | 9/21 (43) | 130.7 | 75.8 – 185.6 | 0.855 |
| positive | 12/21 (57) | 117.1 | 83.3 – 150.9 | ||
| 17p LOH | negative | 11/23 (48) | 120.4 | 69.2 – 171.6 | 0.565 |
| positive | 12/23 (52) | 125.1 | 92.9 – 157.3 |
CI = confidence interval.
Figure 3.
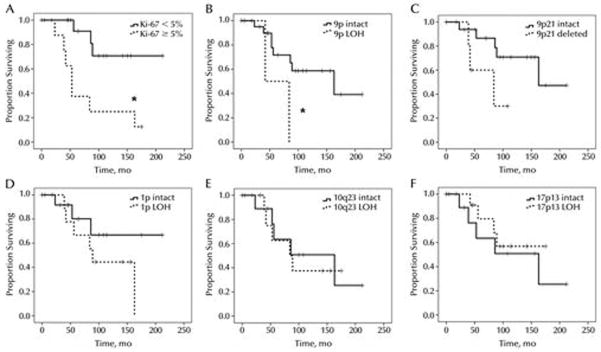
Overall survival of skull base chordomas with proliferative and molecular alterations. Chordomas with elevated Ki67 PI showed a significant reduction in overall survival (A, *P = 0.005), as did cases with large LOH on 9p (B, *P = 0.03). Homozygous deletion of 9p21 trended toward correlation with shorter overall survival (C). LOH on 1p (D), 10q23 (E), or 17p13 (F), on the other hand, showed no differences in survival; neither did 1p36 hemizygous deletion via FISH, chromosome 9 monosomy, p53 protein accumulation, EGFR expression, presence of a solid tumor component, or histologic subtype (not shown).
P53 accumulation was seen in 44% of tumors (11/25) (Figure 1E). Survival in patients with tumors expressing low levels of p53 (145.1 months, CI = 102.3—187.9) was not significantly different from those with increased p53 expression (96.6 months, CI = 65.4—127.8, P = 0.282).
EGFR expression was uncommon in chordomas, with only 8% (2/24) cases showing any positivity (Figure 1F). While survival in these 2 patients was shorter than those with EGFR-negative tumors, the difference was not statistically significant (57.7 months, CI = 16.4—98.9 versus 137.6 months, 99.1—176.1, P = 0.27).
FISH and PCR-based LOH on 1p, 9p, 10q, and 17p
Large LOH of 1p was seen in 38% of tumors (9/24) (Figure 2C), while 17% of chordomas had hemizygous loss of the 1p36 locus (4/23, Figure 2G, Table 2). LOH on 1p (120.4 months, CI = 87.5—153.2) was not associated with a difference in survival compared to 1p intact tumors (142.0 months, CI = 91.0—192.9, P = 0.820) (Figure 3D). Likewise, overall survival was similar in 1p36 deleted tumors (122.7 months, CI = 91.7—153.6) versus 1p36 intact tumors (134.5 months, CI = 97.2—171.7, P = 0.628).
Figure 2.
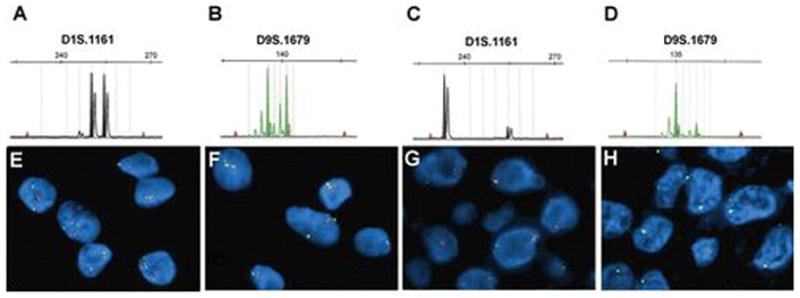
Frequency of 1p and 9p deletions in skull base chordomas. 58% of tumors showed no microsatellite LOH on 1p (A) nor 9p (B), 38% had 1p LOH (C), and 12% had LOH on 9p (D). 70% of chordomas had neither 1p36 (E) nor 9p21 (F) deletions via FISH. 17% of cases showed heterozygous deletion of 1p36 (G), while homozygous deletion of CDKN2A on 9p21 was identified in 22% of tumors (H). Orange probes identify 1p36 (E, G) and 9p21 (F, H), while green probes correspond to 1q25 (E, G) and CEP9 (F, H).
Microsatellite-based LOH analysis identified 9p loss in 12% of tumors (Figure 2D), while FISH identified homozygous loss of the 9p21 locus in 22% (5/23) of cases (Figure 2H, Table 2). 9p loss via LOH analysis was associated with a 51% decrease in mean survival time (71.7 months, CI 42.5—100.9) compared to 9p intact cases (146.2 months, CI 109.8—182.5, P = 0.033) (Figure 3B). Survival in tumors with 9p21 homozygous deletion showed a similar trend toward reduced survival compared to 9p21 intact cases (73.8 months, CI 47.9 – 99.7 versus 151.4 months, CI 115.1—187.7, P = 0.083) (Figure 3C). 35% (9/26) tumors had chromosome 9 monosomy in at least some cells, but did not correlate with survival (not shown).
Of note, correlation between FISH and PCR-based LOH results on 1p and 9p was not perfect. 2 of 4 cases with 1p36 loss on FISH showed LOH of 1p, whereas 6 of 8 cases with 1p LOH did not show any deletion of 1p36 (LOH succeeded but FISH failed on the ninth case). Likewise, 3 of 5 cases with homozygous 9p21 deletion via FISH did not show LOH on 9p, while 1 of 3 tumors had 9p LOH but not 9p21 homozygous deletion.
LOH of 10q23 and 17p13 showed no correlation with overall survival (Table 2, Figure 3E & F), nor did 17p LOH correlate with increased p53 staining (not shown).
Discussion
The primary therapy in chordoma management is surgical resection, although the recurrence rate is so high that it is generally regarded as incurable even with radical resection (30). High-dose radiotherapy has demonstrated some success in delaying recurrence and progression, but because the tumor is usually located in a delicate area and shows a wide range of invasiveness and tumor doubling time, oncologists have difficulty deciding whether postsurgical radiation is always appropriate on a case-by-case basis, or how often to monitor patients for recurrence. Thus, effective biomarkers that predict behavior would be most useful in the management of these tumors. Our results in a retrospective cohort of clival chordomas suggest that loss of 9p21 or Ki67 PI over 5% are adverse prognostic markers associated with reduced survival. In particular, detection of LOH in at least 2 microsatellite loci on 9p21 was significantly associated with shorter survival, while homozygous deletion of 9p21 approached significance. P53 and EGFR expression and loss of 1p36, 10q, or 17p, on the other hand, did not correlate with survival in this analysis.
Ki67 is a protein expressed in cells that are not in the G0 phase of the cell cycle, and appears to play a role in chromatin structuring during replication (31). It has been used for decades as a marker of cellular proliferation in both neoplastic and nonneoplastic conditions, and often correlates with biological behavior in a variety of tumors. In other studies on chordomas the average PI was usually between 2–5%, in keeping with its overall slow growth rate. Our results are consistent with most prior work wherein chordomas with a PI over 5% had a shorter doubling time and/or shorter overall survival (16, 18–20), but not all studies have found such a link (17, 21).
Loss of 1p and/or 1p36 was identified by both FISH and LOH in nearly 40% of chordomas. These results are consistent with prior work showing that such losses are quite common in chordomas, with reported frequencies varying between 30–85% (4, 14, 15). 1p LOH or 1p36 deletion did not correlate with survival in this cohort, in contrast to the only other study wherein 1p36 status and outcome was analyzed (15). An isochromosome 1q has been reported in a subset of recurrent chordomas (6), and familial kindreds of clival chordomas often feature deletion of 1p36 as a key molecular defect (10). Together these findings reinforce the idea that key tumor suppressor genes may be located on 1p, in particular 1p36, but the precise genes involved are unclear. While this locus is likely critical in oncogenesis, its utility as a prognostic biomarker is questionable.
In this study, 9p losses were less common than 1p losses; nearly 25% of all chordomas had 9p loss detectable by either FISH or LOH assays. Specific deletions of CDKN2A on 9p21 have been identified in up to 70% of all chordomas, and 9p21 has been shown to be lost in unbalanced translocations with 1p36 in familial tumors (5, 7, 10). The current study, however, is the first to correlate 9p21 loss with worse outcome. That such a deletion could promote aggressive behavior is readily explained by the presence of the aforementioned CDKN2A gene on 9p21, which encodes the critical cell cycle checkpoint protein p16. Homozygous loss of this gene and/or loss of p16 expression are well-known markers of high grade behavior in a variety of neoplasms, including gliomas (32). Other genes on 9p21 could be involved, but as is the case with the 1p36 locus such candidates are speculative as of yet.
The relatively low correlation between FISH and LOH analyses of 1p and 9p loci is interesting, and such discrepancies have been documented previously (33). There are several possible reasons for this. One, the FISH probes for 1p36 and 9p21 are commercial, proprietary probes for which the exact gene sequence is not publicly available. So other than knowing that the probes are designed to target 1p36 and include the CDKN2A region of 9p21, respectively, exact correlation with specific microsatellite loci is not possible. Small losses could thus be detected by one assay and not the other if the FISH probes do not include the microsatellite sequences. In particular, the commercially available 9p21 LSI probe is rather large (190kb) and also covers p14, p15 and a portion of the MTAP gene which is frequently co-deleted with CDKN2Ap16 gene. In our study we did not investigate status of these genes by LOH analysis, but we believe that deletion of these genes might be an underlying reason for deletions detected by FISH but not PCR-based LOH analysis. Another possibility is that since matched nonneoplastic DNA was not available, the increased frequency of noninformative loci may have obscured true LOH in some cases, particularly those that had homozygous deletion at similar loci by FISH. Third, this cohort is relatively small due to the rarity of chordomas. In a larger cohort, stronger correlation between FISH and LOH might be found.
Loss on 10q23 has drawn interest in oncology research because a key gene, PTEN, encoding the phosphatase and tensin homolog protein, is located on 10q23 and is mutated or deleted in many neoplasms. Loss of PTEN functioning results in unregulated PI3K/Akt/mTOR pathway activation, promoting cellular proliferation, migration, and radioresistance (34). It may also act to promote overall genomic stability. Loss of 10q has been identified as either a high grade marker and/or an adverse prognostic indicator in many tumors including gliomas. In chordomas, either part of the long arm or all of chromosome 10 has been shown to be deleted in up to 70% of cases, including 1 kindred of familial tumors (5, 7, 9, 10). In this series 10q23 LOH was seen 57% of all tumors with informative loci, but did not correlate with a higher PI or shorter survival. Nevertheless, the frequency of 10q23 deletion suggests that this region contains key genes in tumorigenesis, including PTEN.
P53 is a well-studied tumor suppressor gene located on 17p13. It is such a critical regulator of the cell cycle that over half of all human neoplasias contain some alteration of the protein. A commonly used pair of surrogate markers for identifying a p53-driven tumor when a full mutation analysis is not possible is 17p13 LOH and accumulation of p53 via immunohistochemistry. It should be noted, though, that mechanisms of p53 inactivation and mutations are complex and incompletely understood, and there are most likely other important tumor suppressors on 17p13. P53 mutations have yet to be detected in chordomas by sequencing (23), but accumulation of p53 has been described in multiple studies with about 30% of cases showing at least some immunopositivity, a proportion similar to this cohort, in agreement with some studies (17, 20) but not others (19, 23).
Prior work has identified EGFR and other RTK activity in some chordomas (26). Only one study has identified a link between RTK expression and survival in chordomas, wherein presence of c-MET expression was associated with favorable outcome (27). In our cohort, all cases showed strong c-MET expression (data not shown), rendering evaluation of its prognostic utility impossible. Nevertheless, the suggestion that chordomas do express RTKs raises the possibility of targeted therapy with a small molecule inhibitor like erlotinib or imatinib (35). It will be of interest to determine whether RTK status can predict response to such inhibitors, as is routinely done with trastuzumab in breast carcinomas.
In summary, our findings suggest that biomarkers enhancing prognostic accuracy in chordomas exist. 9p21/CDKN2A deletion is associated with shorter survival, a novel finding in chordomas. This study also confirms prior results suggesting that Ki67 PI is a simple, cost-effective method to identify tumors at increased risk of aggressive behavior. Chordomas may therefore soon join the growing list of tumors for which prognostic and predictive tests routinely enhance quality of care.
Acknowledgments
The authors thank Kimberly Fuhrer and Cary Sipos for their histological and immunohistochemical expertise, as well as John Salvatore and Carol Sherer for their work in fluorescence in situ hybridization. The authors would also like to thank Jennifer Ridge-Hetrick and Kim Adams for their administrative support.
Support: CH was supported by a Callie Rohr/American Brain Tumor Association Fellowship. This work was also supported by a pilot from the Head and Neck SPORE NIH 1P50 CA097190 (awarded to RRS). The study was also supported by the Stout Family Fund for Head and Neck Cancer Research at the Eye & Ear Foundation of Pittsburgh and the Head and Neck Oncology Registry.
References
- 1.Yoneoka Y, Tsumanuma I, Fukuda M, Tamura T, Morii K, Tanaka R, Fujii Y. Cranial base chordoma--long term outcome and review of the literature. Acta Neurochir (Wien) 2008;150:773–778. doi: 10.1007/s00701-008-1600-3. discussion 778. [DOI] [PubMed] [Google Scholar]
- 2.Schoedel KE, Martinez AJ, Mahoney TM, Contis L, Becich MJ. Chordomas: pathological features; ploidy and silver nucleolar organizing region analysis. A study of 36 cases. Acta Neuropathol. 1995;89:139–143. doi: 10.1007/BF00296357. [DOI] [PubMed] [Google Scholar]
- 3.Almefty KK, Pravdenkova S, Sawyer JR, Al-Mefty O. Impact of cytogenetic abnormalities on the management of skull base chordomas. J Neurosurg. 2009 doi: 10.3171/2008.9.JNS08285. [DOI] [PubMed] [Google Scholar]
- 4.Bayrakli F, Guney I, Kilic T, Ozek M, Pamir MN. New candidate chromosomal regions for chordoma development. Surg Neurol. 2007;68:425–430. doi: 10.1016/j.surneu.2006.11.046. discussion 430. [DOI] [PubMed] [Google Scholar]
- 5.Hallor KH, Staaf J, Jonsson G, Heidenblad M, Vult von Steyern F, Bauer HC, Ijszenga M, Hogendoorn PC, Mandahl N, Szuhai K, Mertens F. Frequent deletion of the CDKN2A locus in chordoma: analysis of chromosomal imbalances using array comparative genomic hybridisation. Br J Cancer. 2008;98:434–442. doi: 10.1038/sj.bjc.6604130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sawyer JR, Husain M, Al-Mefty O. Identification of isochromosome 1q as a recurring chromosome aberration in skull base chordomas: a new marker for aggressive tumors? Neurosurg Focus. 2001;10:E6. doi: 10.3171/foc.2001.10.3.7. [DOI] [PubMed] [Google Scholar]
- 7.Kuzniacka A, Mertens F, Strombeck B, Wiegant J, Mandahl N. Combined binary ratio labeling fluorescence in situ hybridization analysis of chordoma. Cancer Genet Cytogenet. 2004;151:178–181. doi: 10.1016/j.cancergencyto.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 8.Scheil S, Bruderlein S, Liehr T, Starke H, Herms J, Schulte M, Moller P. Genome-wide analysis of sixteen chordomas by comparative genomic hybridization and cytogenetics of the first human chordoma cell line, U-CH1. Genes Chromosomes Cancer. 2001;32:203–211. doi: 10.1002/gcc.1184. [DOI] [PubMed] [Google Scholar]
- 9.Tallini G, Dorfman H, Brys P, Dal Cin P, De Wever I, Fletcher CD, Jonson K, Mandahl N, Mertens F, Mitelman F, Rosai J, Rydholm A, Samson I, Sciot R, Van den Berghe H, Vanni R, Willen H. Correlation between clinicopathological features and karyotype in 100 cartilaginous and chordoid tumours. A report from the Chromosomes and Morphology (CHAMP) Collaborative Study Group. J Pathol. 2002;196:194–203. doi: 10.1002/path.1023. [DOI] [PubMed] [Google Scholar]
- 10.Dalpra L, Malgara R, Miozzo M, Riva P, Volonte M, Larizza L, Fuhrman Conti AM. First cytogenetic study of a recurrent familial chordoma of the clivus. Int J Cancer. 1999;81:24–30. doi: 10.1002/(sici)1097-0215(19990331)81:1<24::aid-ijc5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 11.Kelley MJ, Korczak JF, Sheridan E, Yang X, Goldstein AM, Parry DM. Familial chordoma, a tumor of notochordal remnants, is linked to chromosome 7q33. Am J Hum Genet. 2001;69:454–460. doi: 10.1086/321982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miozzo M, Dalpra L, Riva P, Volonta M, Macciardi F, Pericotti S, Tibiletti MG, Cerati M, Rohde K, Larizza L, Fuhrman Conti AM. A tumor suppressor locus in familial and sporadic chordoma maps to 1p36. Int J Cancer. 2000;87:68–72. [PubMed] [Google Scholar]
- 13.Yang XR, Beerman M, Bergen AW, Parry DM, Sheridan E, Liebsch NJ, Kelley MJ, Chanock S, Goldstein AM. Corroboration of a familial chordoma locus on chromosome 7q and evidence of genetic heterogeneity using single nucleotide polymorphisms (SNPs) Int J Cancer. 2005;116:487–491. doi: 10.1002/ijc.21006. [DOI] [PubMed] [Google Scholar]
- 14.Riva P, Crosti F, Orzan F, Dalpra L, Mortini P, Parafioriti A, Pollo B, Fuhrman Conti AM, Miozzo M, Larizza L. Mapping of candidate region for chordoma development to 1p36.13 by LOH analysis. Int J Cancer. 2003;107:493–497. doi: 10.1002/ijc.11421. [DOI] [PubMed] [Google Scholar]
- 15.Longoni M, Orzan F, Stroppi M, Boari N, Mortini P, Riva P. Evaluation of 1p36 markers and clinical outcome in a skull base chordoma study. Neuro Oncol. 2008;10:52–60. doi: 10.1215/15228517-2007-048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holton JL, Steel T, Luxsuwong M, Crockard HA, Revesz T. Skull base chordomas: correlation of tumour doubling time with age, mitosis and Ki67 proliferation index. Neuropathol Appl Neurobiol. 2000;26:497–503. doi: 10.1046/j.1365-2990.2000.00280.x. [DOI] [PubMed] [Google Scholar]
- 17.Kilgore S, Prayson RA. Apoptotic and proliferative markers in chordomas: a study of 26 tumors. Ann Diagn Pathol. 2002;6:222–228. doi: 10.1053/adpa.2002.35397. [DOI] [PubMed] [Google Scholar]
- 18.Naka T, Boltze C, Samii A, Herold C, Ostertag H, Iwamoto Y, Oda Y, Tsuneyoshi M, Kuester D, Roessner A. Skull base and nonskull base chordomas: clinicopathologic and immunohistochemical study with special reference to nuclear pleomorphism and proliferative ability. Cancer. 2003;98:1934–1941. doi: 10.1002/cncr.11756. [DOI] [PubMed] [Google Scholar]
- 19.Pallini R, Maira G, Pierconti F, Falchetti ML, Alvino E, Cimino-Reale G, Fernandez E, D’Ambrosio E, Larocca LM. Chordoma of the skull base: predictors of tumor recurrence. J Neurosurg. 2003;98:812–822. doi: 10.3171/jns.2003.98.4.0812. [DOI] [PubMed] [Google Scholar]
- 20.Sakai K, Hongo K, Tanaka Y, Nakayama J. Analysis of immunohistochemical expression of p53 and the proliferation marker Ki-67 antigen in skull base chordomas: relationships between their expression and prognosis. Brain Tumor Pathol. 2007;24:57–62. doi: 10.1007/s10014-007-0222-4. [DOI] [PubMed] [Google Scholar]
- 21.Naka T, Fukuda T, Chuman H, Iwamoto Y, Sugioka Y, Fukui M, Tsuneyoshi M. Proliferative activities in conventional chordoma: a clinicopathologic, DNA flow cytometric, and immunohistochemical analysis of 17 specimens with special reference to anaplastic chordoma showing a diffuse proliferation and nuclear atypia. Hum Pathol. 1996;27:381–388. doi: 10.1016/s0046-8177(96)90112-4. [DOI] [PubMed] [Google Scholar]
- 22.Matsuno A, Sasaki T, Nagashima T, Matsuura R, Tanaka H, Hirakawa M, Murakami M, Kirino T. Immunohistochemical examination of proliferative potentials and the expression of cell cycle-related proteins of intracranial chordomas. Hum Pathol. 1997;28:714–719. doi: 10.1016/s0046-8177(97)90181-7. [DOI] [PubMed] [Google Scholar]
- 23.Naka T, Boltze C, Kuester D, Schulz TO, Schneider-Stock R, Kellner A, Samii A, Herold C, Ostertag H, Roessner A. Alterations of G1-S checkpoint in chordoma: the prognostic impact of p53 overexpression. Cancer. 2005;104:1255–1263. doi: 10.1002/cncr.21296. [DOI] [PubMed] [Google Scholar]
- 24.Dobashi Y, Suzuki S, Sugawara H, Ooi A. Involvement of epidermal growth factor receptor and downstream molecules in bone and soft tissue tumors. Hum Pathol. 2007;38:914–925. doi: 10.1016/j.humpath.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 25.Fasig JH, Dupont WD, LaFleur BJ, Olson SJ, Cates JM. Immunohistochemical analysis of receptor tyrosine kinase signal transduction activity in chordoma. Neuropathol Appl Neurobiol. 2008;34:95–104. doi: 10.1111/j.1365-2990.2007.00873.x. [DOI] [PubMed] [Google Scholar]
- 26.Tamborini E, Miselli F, Negri T, Lagonigro MS, Staurengo S, Dagrada GP, Stacchiotti S, Pastore E, Gronchi A, Perrone F, Carbone A, Pierotti MA, Casali PG, Pilotti S. Molecular and biochemical analyses of platelet-derived growth factor receptor (PDGFR) B, PDGFRA, and KIT receptors in chordomas. Clin Cancer Res. 2006;12:6920–6928. doi: 10.1158/1078-0432.CCR-06-1584. [DOI] [PubMed] [Google Scholar]
- 27.Naka T, Kuester D, Boltze C, Scheil-Bertram S, Samii A, Herold C, Ostertag H, Krueger S, Roessner A. Expression of hepatocyte growth factor and c-MET in skull base chordoma. Cancer. 2008;112:104–110. doi: 10.1002/cncr.23141. [DOI] [PubMed] [Google Scholar]
- 28.Oakley GJ, Fuhrer K, Seethala RR. Brachyury, SOX-9, and podoplanin, new markers in the skull base chordoma vs chondrosarcoma differential: a tissue microarray-based comparative analysis. Mod Pathol. 2008;21:1461–1469. doi: 10.1038/modpathol.2008.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horbinski C, Dacic S, McLendon RE, Cieply K, Datto M, Brat DJ, Chu CT. Chordoid Glioma: A Case Report and Molecular Characterization of Five Cases. Brain Pathol. 2008 doi: 10.1111/j.1750-3639.2008.00196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Samii A, Gerganov VM, Herold C, Hayashi N, Naka T, Mirzayan MJ, Ostertag H, Samii M. Chordomas of the skull base: surgical management and outcome. J Neurosurg. 2007;107:319–324. doi: 10.3171/JNS-07/08/0319. [DOI] [PubMed] [Google Scholar]
- 31.Scholzen T, Endl E, Wohlenberg C, van der Sar S, Cowell IG, Gerdes J, Singh PB. The Ki-67 protein interacts with members of the heterochromatin protein 1 (HP1) family: a potential role in the regulation of higher-order chromatin structure. J Pathol. 2002;196:135–144. doi: 10.1002/path.1016. [DOI] [PubMed] [Google Scholar]
- 32.Solomon DA, Kim JS, Jean W, Waldman T. Conspirators in a capital crime: co-deletion of p18INK4c and p16INK4a/p14ARF/p15INK4b in glioblastoma multiforme. Cancer Res. 2008;68:8657–8660. doi: 10.1158/0008-5472.CAN-08-2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Broholm H, Born PW, Guterbaum D, Dyrbye H, Laursen H. Detecting chromosomal alterations at 1p and 19q by FISH and DNA fragment analysis--a comparative study in human gliomas. Clin Neuropathol. 2008;27:378–387. doi: 10.5414/npp27378. [DOI] [PubMed] [Google Scholar]
- 34.Endersby R, Baker SJ. PTEN signaling in brain: neuropathology and tumorigenesis. Oncogene. 2008;27:5416–5430. doi: 10.1038/onc.2008.239. [DOI] [PubMed] [Google Scholar]
- 35.Casali PG, Messina A, Stacchiotti S, Tamborini E, Crippa F, Gronchi A, Orlandi R, Ripamonti C, Spreafico C, Bertieri R, Bertulli R, Colecchia M, Fumagalli E, Greco A, Grosso F, Olmi P, Pierotti MA, Pilotti S. Imatinib mesylate in chordoma. Cancer. 2004;101:2086–2097. doi: 10.1002/cncr.20618. [DOI] [PubMed] [Google Scholar]