

Abstract
The advanced stages of cutaneous T cell lymphoma (CTCL) are characterized not only by decreased levels of pro-inflammatory cytokines, resulting in high susceptibility to infections, but also by high constitutive activity of NFκB, which promotes cell survival and resistance to apoptosis. The increased expression of the proto-oncogene Bcl3 belonging to IκB family is associated with the pathogenesis of the different types of human cancer, yet, the function and regulation of Bcl3 in CTCL have not been studied. Here, we show that Bcl3 is highly expressed in CTCL Hut-78 and HH cells. The suppression of Bcl3 levels decreases the expression of the pro-survival genes cIAP1 and cIAP2, reduces cell viability, and increases CTCL apoptosis. Interestingly, Bcl3 suppression concomitantly increases expression and the release of the pro-inflammatory cytokines IL-8 and IL-17 in CTCL cells. Chromatin immunoprecipitation studies show that Bcl3 regulates cIAP1, cIAP2, IL-8 and IL-17 gene expression through direct binding to their promoters. Bcl3 expression is regulated by bortezomib (BZ)-mediated proteasome inhibition, and BZ inhibits Bcl3 recruitment to its target promoters, resulting in decreased expression of cIAP1 and cIAP2, but increased expression of IL-8 and IL-17. The Bcl3 expression is regulated through NFκB subunit exchange on Bcl3 promoter. In untreated cells, the Bcl3 promoter is occupied predominantly by p65/p50 heterodimers, inducing Bcl3 expression; however, in BZ-treated cells, the p65/50 heterodimers are replaced by p52 subunits, resulting in Bcl3 transcriptional repression. These data provide the first insights into the function and regulation of Bcl3 in CTCL, and indicate that Bcl3 has an important pro-survival and immunosuppressive role in these cells.
Keywords: Bcl3, Bortezomib, Cutaneous T-cell lymphoma, NFκB, Pro-inflammatory cytokine
1. Introduction
Cutaneous T-cell lymphoma (CTCL) encompasses a group of lymphoproliferative disorders characterized by skin invasive neoplastic T cells. Mycosis fungoides (MF) and the leukemic variant Sézary syndrome (SS) are the most common clinical forms [1–5]. NFκB activity is constitutively increased in CTCL, where it induces the expression of anti-apoptotic genes and resistance to apoptosis, and plays a central mediator between malignant cell survival and inflammatory signaling [6–11]. The advanced stages of CTCL are associated with suppressed cell-mediated immunity and decreased levels of pro-inflammatory cytokines, resulting in increased susceptibility to infection [12–17]. Despite the recent advances in elucidating the immune mechanisms responsible for the pathogenesis of CTCL, there is no effective strategy to prolong survival in the advanced stages [18–24].
Bortezomib (BZ, Velcade, PS-341) is the first FDA approved 26S proteasome inhibitor that has been widely used in the treatment of patients with multiple myeloma [25–27]. BZ has shown promising results also in patients with relapsed or refractory CTCL [28–31]. However, the precise molecular mechanisms are not fully understood. BZ has been originally developed as the inhibitor of inducible NFκB activity and NFκB-dependent transcription [25–27]. Interestingly, however, recent studies have shown that the BZ effect on NFκB-dependent transcription is gene specific; while some genes are inhibited, some are unaffected, and some genes are actually increased, depending on the subunit composition of NFκB dimers recruited to NFκB-responsive promoters [32–35].
B-cell chronic lymphatic leukemia protein 3 (Bcl3) is a member of IκB family that was first identified as a candidate proto-oncogene in some patients with chronic lymphocytic leukemia [36]. However, unlike other IκBs, Bcl3 is a predominantly nuclear protein, which contains a transactivation domain, and can be recruited to NFκB-responsive promoters, resulting in transcriptional activation or repression, depending on the subunit composition of NFκB complexes [37–43]. Elevated Bcl3 expression results in increased cell proliferation, survival and malignant potential. Studies have demonstrated increased Bcl3 expression in the different types of hematopoietic and solid cancers [44–49], yet its function and regulation in CTCL have not been investigated.
In this study, we have sought to determine the function and regulation of Bcl3 in human CTCL cells. We show that Bcl3 is highly expressed in CTCL Hut-78 and HH cells, and its suppression inhibits the expression of anti-apoptotic genes cIAP1 and cIAP2, but increases the expression of pro-inflammatory cytokines IL-8 and IL-17, indicating that Bcl3 has a pro-survival and immunosuppressive function in CTCL cells. We further demonstrate that Bcl3 expression is regulated by the BZ-mediated proteasome inhibition and by the subunit exchange of NFκB proteins. These findings provide the first insights into the function and regulation of Bcl3 in CTCL, and identify Bcl3 as a new potential target in the CTCL treatment.
2. Materials and methods
2.1. Antibodies and reagents
Purified polyclonal antibodies against human Bcl3 (sc-185), Bcl2 (sc-492), NFκB p65 (sc-372X), NFκB p50 (sc-7178X), cRel (sc-71X), RelB (sc-226X), NFκB p52 (sc-848X), and lamin B (sc-6216) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Purified polyclonal antibody against lactate dehydrogenase (LDH; 20-LG22) was from Fitzgerald Industries International (Concord, MA, USA), and actin antibody was from Sigma (St. Louis, MO, USA). Horseradish peroxidase (HRP)-conjugated anti-rabbit, anti-mouse and anti-goat secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Bortezomib was purchased from ChemieTek (Indianapolis, IN, USA). Recombinant human IL-8 and IL-17 proteins were from R&D (Minneapolis, MN, USA). All other reagents were molecular biology grade and were from Sigma (St. Louis, MO).
2.2. Cell culture
CTCL cell lines, Hut-78 (ATCC® TIB-161) and HH (ATCC® CRL-2105) cells, derived from the peripheral blood of patients with SS and non-MF/SS aggressive CTCL respectively, as well as monocytic leukemia U937 (ATCC® CRL-1593.2) and THP-1 (ATCC® TIB-202) cells were obtained from and validated by the American Type Culture Collection (ATCC; Rockville, MD, USA), and used between the passage numbers 10 and 15. Peripheral blood mononuclear cells (PBMC) from healthy human volunteers were purchased from AllCells (PB003F; Alameda, CA, USA). Cells were maintained at 37 °C in RPMI 1640 medium, supplemented with 10% heat inactivated fetal bovine serum (FBS) and 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin, in a humidified atmosphere with 5% CO2. Prior to cell treatment, cells were seeded (5 × 105 cells/ml) in 6-well plates and grown 24 h at 37 °C with 5% CO2. Bortezomib was dissolved in DMSO and stored at −80 °C. An equivalent volume of DMSO was used in all experiments as a solvent control. Cell viability was measured by using Trypan Blue exclusion.
2.3. Transfections
Prior to transfections, cells were seeded (5×105 cells/ml) into a 6-well plate and incubated in a humidified 5% CO2 atmosphere at 37 °C in antibiotic-free RPMI medium supplemented with 10% FBS for 24 h. For siRNA transfections, 50 nmol (final concentration) of control siRNA-A (sc-37007; Santa Cruz Biotechnology, CA, USA) or Bcl3 siRNA (sc-29789) was used. Cells were transfected with TransIT-siQUEST transfection reagent (Mirus Bio, Madison, WI, USA) according to the manufacturer’s instructions. For shRNA transfections, 2.5 μg of control shRNA-A (sc-108060) or Bcl3 shRNA plasmid (sc-29789-SH) was used, and cells were transiently transfected with TransIT-Jurkat transfections reagent (Mirus Bio) according to the manufacturer’s instructions. After transfection, fresh RPMI medium supplemented with FBS and antibiotics was added and cells were incubated for 48 h.
2.4. Preparation of whole cell extracts, cytoplasmic and nuclear extracts, and western blotting
Whole cell extracts, and nuclear (NE) and cytoplasmic extracts (CE) were prepared as described previously [33–35], and separated on 12% SDS gels. To determine equal protein loading, membranes were stripped and re-probed with anti-actin antibody as described [34,35]. The contamination of nuclear and cytoplasmic fractions by cytoplasmic and nuclear proteins, respectively, was determined by western analysis using lactate dehydrogenase (LDH) and lamin B as specific markers as described [34,35].
2.5. Real time RT-PCR
Total RNA was isolated by using RNeasy mini-kit (Qiagen, Valencia, CA, USA). The iScript one-step RT-PCR kit with SYBR Green (BioRad, Hercules, CA, USA) was used as a supermix and 20 ng of RNA was used as template on a Bio-Rad MyIQ Single Color Real-Time PCR Detection System (BioRad). The primers used for the quantification of Bcl3, cIAP1, cIAP2, Bcl2, IL-8, IL-17, and actin mRNA were purchased from SA Biosciences (Frederick, MD, USA).
2.6. Chromatin immunoprecipitation (ChIP)
ChIP analysis was performed as described previously [34,35]. Briefly, proteins and DNA were cross-linked by formaldehyde, cells were washed and sonicated. The lysates were centrifuged (15,000 g, 10 min, 4 °C), and the supernatant extracts were diluted with ChIP dilution buffer and pre-cleared with Protein A/G Agarose (Santa Cruz, CA) for 2 h at 4 °C. Immunoprecipitation was performed overnight at 4 °C, with Bcl3, p65, p50, cRel, RelB or p52 antibodies. Following immunoprecipitation, the samples were incubated with Protein A/G Agarose (1 h, 4 °C), and the immune complexes were collected by centrifugation (150 g, 5 min, 4 °C), washed, and extracted with 1% SDS–0.1 M NaHCO3. After reversing the cross-linking, proteins were digested with proteinase K, and the samples were extracted with phenol/chloroform, followed by precipitation with ethanol. The pellets were resuspended in nuclease-free water and subjected to real time PCR. Immunoprecipitated DNA was analyzed by real-time PCR (25 μl reaction mixture) using the iQ SYBR Green Supermix and the Bio-Rad MyIQ Single Color Real-Time PCR Detection System (Bio-Rad). Each immunoprecipitation was performed four times using different chromatin samples, and the occupancy was calculated by using the ChIP-qPCR Human IGX1A Negative Control Assay (SA Biosciences, Frederick, MD) as a negative control and corrected for the efficiency of the primers, which detect specific genomic DNA sequences within ORF-free intergenic regions or “promoter deserts” lacking any known or predicted structural genes. The ChIP primers for IL-8 and IL-17 were purchased from Qiagen (Valencia, CA, USA). The ChIP primers for Bcl3, cIAP1, cIAP2 and Bcl2 were the following: Bcl3: forward, 5′-TTGCGGAGAGAAACACCTACT-3′ and reverse, 5′-CGCTCTCTCTGCCTCTGTT-3′; cIAP1: forward, 5′-TGACTGGCAGGCAGAAATGA-3′ and reverse, 5′-TTTGCCCGTTGAATCCGAT-3′; cIAP2: forward, 5′-TTCAGTAAATGCCGCGAAGAT-3′ and reverse, 5′-TGGTTTGCATGTGCACTGGT-3′; and Bcl2: forward, 5′-TGCATCTCAT GCCAAGGG-3′ and reverse, 5′-CCCCAGAGAAAGAAGAGGAGTT-3.
2.7. Apoptosis assay
Apoptosis was evaluated with a cell death detection ELISA kit that quantifies the release of nucleosomes into the cytoplasm (Cell Death Detection ELISAPLUS, Roche, Indianapolis, IN, USA) as described [32]. The assay was performed at the indicated time points as per the manufacturer’s instructions.
2.8. ELISA
Cytokine release was measured in cell culture supernatants by commercially available ELISA kits (R&D, Minneapolis, MN, USA) as previously described [34,35].
2.9. Statistical analysis
The results represent at least three independent experiments. Numerical results are presented as means ± SE. Data were analyzed by using an InStat software package (GraphPAD, San Diego, CA, USA). Statistical significance was evaluated by using Mann–Whitney U test with Bonferroni correction for multiple comparisons, and p < 0.05 was considered significant.
3. Results
3.1. Bcl3 is highly expressed in CTCL cells, and its expression is inhibited by BZ
To determine whether Bcl3 is expressed in CTCL cells and whether its expression is regulated by proteasome, we have analyzed the Bcl3 protein levels in whole cell extracts prepared from CTCL Hut-78 and HH cells incubated 24 h with increasing BZ concentrations. As shown in Fig. 1, Bcl3 is expressed in Hut-78 (Fig. 1A) and HH (Fig. 1B) CTCL cells, and proteasome inhibition by BZ decreases its protein levels in both cell lines. BZ also significantly suppressed Bcl3 mRNA levels in CTCL cells. Compared to untreated cells, 100 nM BZ that approximately corresponds to the clinically used BZ concentrations [50], inhibited more than 90% of Bcl3 mRNA expression in Hut-78 cells (Fig. 1C). The inhibition of Bcl3 mRNA expression by BZ was time dependent (Fig. 1D).
Fig. 1.

Bcl3 is highly expressed in CTCL cells, and its expression is inhibited by BZ. Western blotting of whole cell extracts prepared from CTCL Hut-78 (A) and HH cells (B) treated with increasing concentrations of BZ for 24 h, and analyzed by using Bcl3 antibody. To confirm equal protein loading, the membranes were stripped and re-probed with actin antibody. Each lane corresponds to approximately 5 × 104 cells. (C) Real time RT-PCR analysis of Bcl3 mRNA levels in Hut-78 cells treated 24 h with increasing BZ concentrations. (D) Real time RT-PCR analysis of Bcl3 mRNA levels in Hut-78 cells treated 0, 6, 24 and 48 h with 10 nM BZ. The values represent the mean ± SE of four experiments. Asterisks denote a statistically significant (p < 0.05) inhibition compared to control untreated (UT) cells. (E) Western blotting of whole cell extracts prepared from Hut-78, HH, U937, THP1 and PBMC cells analyzed by Bcl3 and control actin antibodies; each lane corresponds to approximately 5 × 104 cells.
To compare the Bcl3 protein levels in CTCL cells to other leukocytes, we have analyzed the Bcl3 expression in CTCL Hut-78 and HH cells, in monocytic leukemia cell lines U937 and THP1, and in normal human peripheral blood mononuclear cells (PBMC). As shown in Fig. 1E, compared to the monocytic U937 and THP1 cells and normal human PBMC, the CTCL Hut-78 and HH cell lines express considerably more Bcl3.
3.2. Suppression of Bcl3 regulates survival in CTCL cells
To obtain a first insight into the Bcl3 function in CTCL, we have analyzed cell viability and cytoplasmic nucleosome enrichment in Hut-78 cells transfected with Bcl3 siRNA, as well as with control non-silencing siRNA. Transfection with Bcl3 siRNA resulted in approximately 70% reduction in total cellular Bcl3 protein levels compared to cells transfected with control non-silencing siRNA (Fig. 2A, B). The suppression of Bcl3 resulted in approximately 40% decreased Hut-78 cell viability measured by Trypan Blue staining (Fig. 2C), and 60% increased nucleosome enrichment in the cytoplasm, indicating apoptosis (Fig. 2D). These results have suggested that Bcl3 is involved in the regulation of cell survival in CTCL cells.
Fig. 2.

Bcl3 suppression induces apoptosis in CTCL cells. (A) Western blotting of whole cell extracts prepared from Hut-78 cells transfected with control non-silencing and Bcl3 specific siRNA, and analyzed by using Bcl3 and actin specific antibodies. (B) Densitometric evaluation of Bcl3 densities showed in panel A. The Bcl3/actin value in cells transfected with control siRNA was arbitrarily set to 100%, and the Bcl3/actin value in cells transfected with Bcl3 siRNA is presented relative to this value. The data represent the mean of four experiments ± SE, and the asterisk denotes a statistically significant (p < 0.05) change compared to cells transfected with control siRNA. (C) Cell viability and (D) the cytoplasmic nucleosome enrichment assay in Hut-78 cells transfected with control and Bcl3 siRNA. The values represent the mean ± SE of four experiments; the asterisk denotes a statistically significant (p < 0.05) change compared to cells transfected with control siRNA.
3.3. Suppression of Bcl3 inhibits expression of anti-apoptotic genes, but increases expression of pro-inflammatory genes in CTCL cells
To determine whether Bcl3 regulates pro-survival genes in CTCL cells, we have analyzed the expression of NFκB-dependent anti-apoptotic genes cIAP1, cIAP2 and Bcl2 in Hut-78 cells transfected with Bcl3 specific siRNA or shRNA, or corresponding non-silencing controls. Bcl3 suppression by both siRNA and shRNA significantly decreased the mRNA (Fig. 3A) and protein levels (Fig. 3B, C) of cIAP1 and cIAP2, while Bcl2 levels were not affected, suggesting that Bcl3 increases the survival of CTCL cells by inducing the transcription of cIAP1 and cIAP2.
Fig. 3.

Suppression of Bcl3 inhibits expression of anti-apoptotic genes, but induces expression of pro-inflammatory genes in CTCL cells. (A) Real time RT-PCR analysis of cIAP1, cIAP2, Bcl2, IL-8 and IL-17 mRNA levels in Hut-78 cells transfected with control non-silencing siRNA or shRNA (full columns), and Bcl3 specific siRNA (empty columns) or shRNA (gray columns). The values represent the mean ± SE of four experiments. Asterisks denote a statistically significant (p < 0.05) change compared to cells transfected with the corresponding control RNA. (B) Western analysis of total protein levels of Bcl3, cIAP1, cIAP2, Bcl2, and control actin, in Hut-78 cells transfected with control non-silencing and Bcl3 specific siRNA or shRNA. (C) Densitometric evaluation of cIAP1, cIAP2 and Bcl2 densities shown in panel B. The cIAP1, cIAP2 and Bcl2 protein/actin values in cells transfected with control RNA were arbitrarily set to 100%, and the protein/actin values in cells transfected with Bcl3 siRNA or shRNA are presented relative to those values. The data represent the mean of four experiments ± SE, and the asterisks denote a statistically significant (p < 0.05) change compared to cells transfected with the corresponding controls. (D) IL-8 and IL-17 release measured by ELISA in cell culture supernatants of Hut-78 cells transfected with control non-silencing siRNA or shRNA (full columns) and Bcl3 specific siRNA (empty columns) or shRNA (gray columns). The values represent the mean ± SE of four experiments. Asterisks denote a statistically significant (p < 0.05) change compared to cells transfected with the corresponding controls.
In addition to the anti-apoptotic genes, we have investigated the effect of Bcl3 suppression on the NFκB-dependent pro-inflammatory cytokines IL-8 and IL-17, since both cytokines were implicated in the pathogenesis of CTCL [51–54]. Interestingly, in contrast to the anti-apoptotic genes cIAP1 and cIAP2 that were inhibited by Bcl3 suppression, Bcl3 suppression significantly increased both mRNA levels (Fig. 3A) and cytokine release (Fig. 3D) of IL-8 and IL-17 in Hut-78 cells. Compared to cells transfected with control siRNA and shRNA, cells transfected with Bcl3 siRNA and shRNA exhibited approximately 2.5 and 3.8 fold higher IL-8 mRNA levels, respectively (Fig. 3A). IL-17 gene expression was increased approximately 3.5 and 4.5 folds in cells transfected with siRNA and shRNA, respectively, compared to cells transfected with corresponding controls (Fig. 3A). At the protein level, the IL-8 and IL-17 release was barely detectable in CTCL Hut-78 cells transfected with control siRNA or shRNA (Fig. 3D). However, the suppression of Bcl3 with siRNA or shRNA increased the IL-8 release to about 50 and 40 pg/ml, respectively. The IL-17 release was increased in cells transfected with Bcl3 specific siRNA or shRNA to approximately 10 and 25 pg/ml, respectively (Fig. 3D). The IL-8 and IL-17 release levels in Bcl3-suppressed cells are comparable to the IL-8 and IL-17 serum levels in healthy adults [55–57].
3.4. Bcl3 is recruited to cIAP1, cIAP2, IL-8 and IL-17 promoters in CTCL cells, and BZ inhibits the Bcl3 recruitment
Bcl3 can be recruited to gene promoters, resulting in transcriptional repression or activation, depending on the transcription factors or co-regulators present in the transcriptional complex [58–60]. Since the mRNA expression of cIAP1, cIAP2, IL-8 and IL-17 was dependent on Bcl3 (Fig. 3A), we have investigated the possibility that Bcl3 is recruited to cIAP1, cIAP2, IL-8 and IL-17 promoters in CTCL cells. Hut-78 cells were incubated 24 h with 0 and 10 nM BZ, cross-linked, lysed, and chromatin was sheared by sonication. Bcl3 recruitment to cIAP1, cIAP2, Bcl2, IL-8 and IL-17 promoters was analyzed by chromatin immunoprecipitation (ChIP) and quantified by real time PCR. The NFκB binding sites of the above genes are shown in Fig. 4A.
Fig. 4.
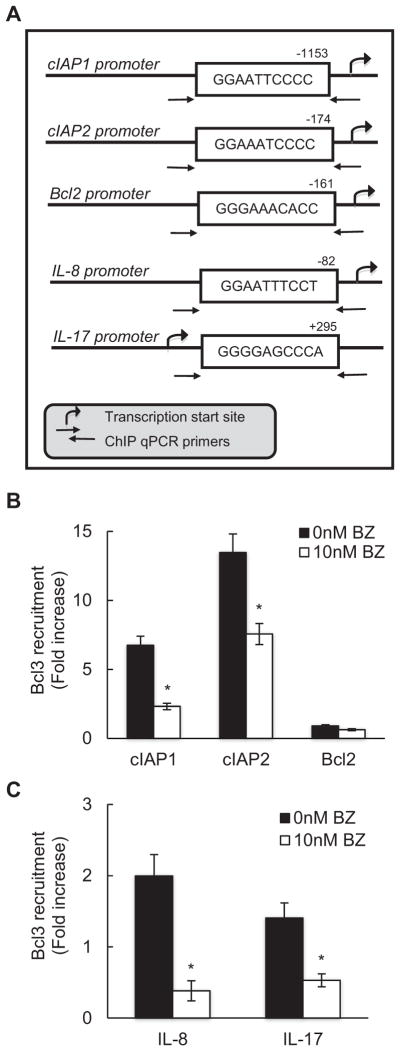
BZ inhibits Bcl3 recruitment to cIAP1, cIAP2, IL-8 and IL-17 promoters in CTCL cells. (A) Schematic illustration of NFκB binding sites in human cIAP1, cIAP2, Bcl2, IL-8 and IL-17 promoters, and the ChIP primers used in the ChIP assay. Bcl3 recruitment to the anti-apoptotic gene promoters cIAP1, cIAP2 and Bcl2 (B), and the pro-inflammatory gene promoters IL-8 and IL-17 (C) was analyzed by ChIP in untreated Hut-78 cells and cells treated 24 h with 10 nM BZ. The data are presented as the change in occupancy over the human IGX1A (SA Biosciences) sequence control and represent the mean ± SE of four experiments. Asterisks denote a statistically significant (p < 0.05) change compared to untreated cells.
Bcl3 was recruited to cIAP1 and cIAP2 (Fig. 4B), and IL-8 and IL-17 (Fig. 4C) promoters in untreated cells, indicating that Bcl3 regulates the transcription of these genes in CTCL cells. Proteasome inhibition by 10 nM BZ, which greatly reduced the cellular Bcl3 protein levels (Fig. 1A), also significantly reduced the Bcl3 occupancy at these promoters (Fig. 4B, C). In contrast, Bcl3 was not recruited to Bcl2 promoter (Fig. 4B), which is consistent with the data demonstrating that Bcl3 does not regulate the Bcl2 expression in CTCL cells (Fig. 3A).
3.5. BZ-mediated proteasome inhibition induces IL-8 and IL-17 expression in CTCL cells
Since BZ decreased the Bcl3 recruitment to IL-8 and IL-17 promoters (Fig. 4C), and Bcl3 suppression increased the IL-8 and IL-17 expression (Fig. 3A, D), we hypothesized that proteasome inhibition by BZ might increase the IL-8 and IL-17 expression in CTCL cells. To test this possibility, we have analyzed IL-8 and IL-17 mRNA levels and cytokine release in Hut-78 and HH cells treated 24 h with increasing BZ concentrations. Indeed, BZ significantly increased IL-8 and IL-17 mRNA levels in both CTCL cell types (Fig. 5A, B). Furthermore, BZ also increased the IL-18 and IL-17 release from CTCL cells (Fig. 5C, D). In untreated Hut-78 and HH cells, the IL-8 and IL-17 release levels were barely detectable, which is in an agreement with the immunosuppressive nature of these cells [61]. However, 100 nM BZ increased the IL-8 and IL-17 release levels in both CTCL cells to about 60 and 15 pg/ml, respectively (Fig. 5C, D).
Fig. 5.

BZ induces IL-8 and IL-17 expression in CTCL cells. Real time RT-PCR analysis of IL-8 (A) and IL-17 (B) mRNA levels in Hut-78 (full columns) and HH (empty columns) cells treated 24 h with increasing BZ concentrations. ELISA of released IL-8 (C) and IL-17 (D) levels measured in cell culture supernatants of Hut-78 and HH cells treated 24 h with increasing BZ concentrations. Cell viability of Hut-78 cells incubated 24 h with increasing concentrations of human recombinant IL-8 (E) and IL-17 (F) proteins, measured by Trypan Blue exclusion. The values represent the mean ± SE of four experiments. Asterisks denote a statistically significant (p < 0.05) change compared to untreated cells.
To investigate whether the BZ-increased IL-8 and IL-17 release might directly affect the CTCL cell viability, we have measured the effect of recombinant human IL-8 and IL-17 proteins on the viability of Hut-78 cells. As shown in Fig. 5E and F, 50 and 15 pg/ml concentrations of IL-8 and IL-17, which are approximately induced by 100 nM BZ in Hut-78 cells (Fig. 5C, D), decrease the CTCL cell viability by about 10%, suggesting that the BZ-induced IL-8 and IL-17 might contribute to the previously observed BZ-mediated inhibition of CTCL cell viability [30].
3.6. Bcl3 mediates the BZ-induced IL-8 and IL-17 expression, and cIAP1 and cIAP2 inhibition
Our results have suggested that the BZ-induced expression of IL-8 and IL-17, and the inhibition of cIAP1 and cIAP2 are mediated, at least partly, by Bcl3. To test this hypothesis, we have transfected Hut-78 cells with Bcl3 (or control) siRNA, and then incubated 24 h with 0, 10 and 100 nM BZ. The suppression of Bcl3 expression further decreased Hut-78 cell viability in BZ-treated cells (Fig. 6A). In addition, the suppression of Bcl3 significantly decreased cIAP1 and cIAP2 mRNA (Fig. 6B), and increased IL-8 and IL-17 mRNA levels (Fig. 6C) in BZ-treated cells. In contrast, Bcl3 suppression did not have any significant effect on the Bcl2 mRNA levels in BZ-treated Hut-78 cells (Fig. 6B); this is in a good agreement with the data demonstrating that Bcl3 is not recruited to Bcl2 promoter (Fig. 4B) and Bcl2 expression is not regulated by BZ [33]. These results indicate that the BZ-induced IL-8 and IL-17 expression, and cIAP1 and cIAP2 inhibition in Hut-78 cells, are mediated by Bcl3.
Fig. 6.

Bcl3 mediates the BZ-induced IL-8 and IL-17 expression, and cIAP1 and cIAP2 inhibition in Hut-78 cells. (A) Suppression of Bcl3 mRNA (top left panel) and protein (lower left panel) levels by Bcl3 siRNA decreases viability in BZ-treated Hut-78 cells, measured by Trypan Blue exclusion (right panel). (B) Real time RT-PCR analysis of cIAP1, cIAP2 and Bcl2 mRNA levels in Hut-78 cells transfected with control siRNA (full columns) or Bcl3 specific siRNA (empty columns) and treated 24 h with 0, 10 and 100 nM BZ. (C) Real time RT-PCR analysis of IL-8 and IL-17 mRNA levels in Hut-78 cells transfected with control siRNA (full columns) or Bcl3 specific siRNA (empty columns) and treated 24 h with 0, 10 and 100 nM BZ. The values represent the mean ± SE of four experiments. Asterisks denote a statistically significant (p < 0.05) change compared to cells transfected with the corresponding control siRNA.
3.7. The BZ-mediated inhibition of Bcl3 expression is associated with decreased recruitment of p65 and p50, but increased recruitment of p52, to Bcl3 promoter in CTCL cells
Since previous studies have suggested that Bcl3 expression is regulated by NFκB [62,63], we wanted to determine which NFκB subunits regulate the Bcl3 transcription in CTCL cells. To this end, we have first analyzed which NFκB subunits are localized in the nucleus in Hut-78 cells. As shown in Fig. 7A, p65, p50 and RelB were both in the cytoplasm and in the nucleus in untreated cells, but 10 and 100 nM BZ reduced the RelB nuclear levels. In contrast, while cRel and p52 were localized mainly in the cytoplasm in untreated cells, 100 nM BZ increased their nuclear accumulation, especially that of p52 (Fig. 7A).
Fig. 7.

Inhibition of Bcl3 expression by BZ is associated with decreased p65 and p50 recruitment, but increased p52 recruitment, to Bcl3 promoter in CTCL cells. (A) Immunoblotting of cytoplasmic (CE) and nuclear (NE) extracts prepared from Hut-78 cells treated 24 h with increasing BZ concentrations, and analyzed by using p65, p50, RelB, cRel and p52 NFκB antibodies. The presence of cytoplasmic proteins in nuclear fraction was evaluated by re-probing the membrane with lactate dehydrogenase (LDH) antibody. Nuclear contamination in the cytoplasmic fraction was assessed by using lamin B specific antibody. To confirm equal protein loading, the membranes were stripped and re-probed with actin antibody. Each lane corresponds to approximately 5 × 104 cells. (B) Schematic illustration of the NFκB binding site in human Bcl3 promoter and primers used in the ChIP assay. (C) Recruitment of NFκB p65, p50, cRel, RelB and p52 subunits to Bcl3 promoter in Hut-78 cells treated 24 h with 0, 10 and 100 nM BZ was analyzed by ChIP and quantified by real time PCR. The data are presented as the change in occupancy over the human IGX1A (SA Biosciences) sequence control and represent the mean ± SE of four experiments. Asterisks denote a statistically significant (p < 0.05) change compared to control untreated cells. (D) Model of the regulation of Bcl3 transcription by the exchange of NFκB subunits in CTCL cells.
To determine whether the nuclear NFκB subunits are recruited to the Bcl3 promoter, we have analyzed the human Bcl3 promoter using TFSEARCH program for searching transcription factor binding sites [64], and we have identified a new potential NFκB binding site in the Bcl3 promoter (Fig. 7B). This site (5′-GGGACACCCC-3′) shows a high homology to the consensus NFκB site 5′-GGGRNWYYCC-3′, where R is purine, N is any nucleotide, W is adenine or thymine, and Y is pyrimidine. Using ChIP, we have analyzed the recruitment of p65, p50, cRel, RelB and p52 to the Bcl3 promoter in Hut-78 cells incubated 24 h with 0, 10 and 100 nM BZ. As shown in Fig. 7C, both p65 and p50 NFκB were heavily recruited to the Bcl3 promoter in untreated cells, indicating that the Bcl3 promoter site is occupied by NFκB p65/p50 heterodimers, resulting in the high Bcl3 expression in untreated CTCL cells. Proteasome inhibition by BZ almost completely inhibited the recruitment of p65 and p50 (Fig. 7C), despite their high levels in the nucleus (Fig. 7A). These findings suggest that the high expression of Bcl3 in CTCL cells is mediated by p65/p50 heterodimers, and that proteasome inhibition suppresses the Bcl3 transcription by inhibiting the p65/p50 recruitment.
Unlike p65 and p50, the recruitment of cRel and RelB was low, and was not affected by BZ, indicating that the Bcl3 transcription in CTCL cells is not regulated by cRel or RelB. Interestingly however, even though the recruitment of p52 to Bcl3 promoter was low in untreated cells, it was significantly increased in 100 nM BZ-treated cells (Fig. 7C), suggesting that proteasome inhibition replaces p65/p50 heterodimers with p52 subunits, resulting in the transcriptional repression of Bcl3 (Fig. 7D).
4. Discussion
The advanced stages of CTCL are characterized not only by the increased survival of malignant T cells and their resistance to apoptosis, but also by severe immunodeficiency that is associated with the decreased production of pro-inflammatory cytokines [1–4]. Here, we have analyzed the function and regulation of the proto-oncogene and transcriptional regulator Bcl3, which has been associated with the pathogenesis of the different types of human cancer, but its regulation and function in CTCL have never been investigated. We show that compared to other leukocytes including normal human PBMC, Bcl3 is highly expressed in CTCL Hut-78 and HH cells (Fig. 1). The Bcl3 expression in CTCL is further supported by previous studies indicating increased Bcl3 gene expression in about 50% of CTCL patients [44], and increased Bcl3 protein levels in about 25% of patients with MF [65]. Our results indicate that Bcl3 induces the expression of the pro-survival genes cIAP1 and cIAP2, while it concomitantly inhibits the expression of the pro-inflammatory cytokines IL-8 and IL-17 in CTCL cells (Fig. 8).
Fig. 8.

Proposed model of Bcl3 function and regulation in CTCL. In CTCL cells, Bcl3 induces expression of anti-apoptotic genes cIAP1 and cIAP2, but inhibits expression of pro-inflammatory genes IL-8 and IL-17, thus contributing to the high survival and immunosuppressive nature of these cells. Proteasome inhibition by BZ decreases the Bcl3 expression, resulting in the increased apoptosis and restoration of the “physiological” levels of IL-8 and IL-17.
Unlike other members of IκB family, Bcl3 contains a transactivation domain, and can be recruited to promoters, resulting in transcriptional activation or repression depending on the transcriptional complex [58–60]. We have found that Bcl3 is recruited not only to the NFκB binding sites of the pro-survival genes cIAP1 and cIAP2, but also to the pro-inflammatory cytokines IL-8 and IL-17 in CTCL cells, and that pro-teasome inhibition by BZ inhibits this recruitment (Fig. 4). Interestingly however, while proteasome inhibition and the resulting decreased Bcl3 promoter occupancy are associated with the decreased expression of cIAP1 and cIAP2, they concurrently increase the expression of IL-8 and IL-17. Even though the release of IL-8 and IL-17 from untreated CTCL Hut-78 and HH cells is barely detectable, which is a hallmark of the advanced stages of CTCL, the suppression of Bcl3 by siRNA (Fig. 3) or BZ (Fig. 5) increases their release to about 60 and 15 pg/ml, respectively. While these IL-8 and IL-17 levels are lower compared to the release from stimulated inflammatory cells or during chronic inflammatory disorders, they correspond to the physiological levels in healthy donors [55–57]. Our results show that IL-8 and IL-17, at the BZ-induced concentrations, inhibit the viability of Hut-78 cells (Fig. 5E, F), indicating that the BZ-induced IL-8 and IL-17 might contribute to the previously observed BZ-mediated inhibition of CTCL cell viability [30]. However, it seems likely, that the BZ-induced IL-8 and IL-17 might have also a paracrine effect, and regulate the viability and function of neutrophils, PBMC, and other leukocytes.
Bortezomib is the first clinically approved proteasome inhibitor that has been very effective in the treatment of multiple myeloma, and has shown promising results in other hematological malignancies as well, including CTCL [25–31]. One of the main mechanisms of BZ function is the inhibition of inducible NFκB activity and the expression of NFκB-dependent genes [25–27]. Interestingly however, recent studies have demonstrated that the proteasome inhibition has a differential effect on the expression of NFκB-dependent genes. While some NFκB-regulated genes are inhibited, some are unaffected, and some genes are in fact increased by the proteasome inhibition [31–35]. We have previously shown that while the BZ-mediated proteasome inhibition does not affect the expression of Bcl2 that is regulated predominantly by p50/50 homodimers, it decreases the expression of cIAP1 and cIAP2 regulated by p65/p50 heterodimers, resulting in increased CTCL apoptosis [33]. Our present data show that in addition to the inhibition of cIAP1 and cIAP2 expression, BZ induces the expression of IL-8 and IL-17 in CTCL cells, and increases their cellular levels to those seen in healthy adults (Fig. 5). Furthermore, our results indicate that the BZ effect on the expression of these NFκB-dependent genes is mediated through Bcl3 (Fig. 6).
Previous studies have suggested that Bcl3 forms transcriptional complexes mainly with NFκB p50 and p52 subunits [58–60]. However, our data indicate that Bcl3 does not regulate the transcription of Bcl2 (Fig. 3), which is regulated predominantly by p50/50 homodimers [33]. Furthermore, our results indicate that Bcl3 regulates the transcription of cIAP1 and cIAP2 genes (Fig. 3), which are regulated by p65/p50 heterodimers in CTCL cells [33]. These data suggest that the regulation of NFκB-dependent genes by Bcl3 is more complex, and may include additional transcription factors and co-regulators. This is further supported by the finding that Bcl3 promoter binding concomitantly increases (cIAP1, cIAP2) and decreases (IL-8, IL-17) the expression of NFκB-responsive genes (Fig. 3, 4). In this context, Bcl3 was shown to interact also with other transcriptional regulators, including the AP-1 transcription factors c-Jun and c-fos, STAT1, histone deacetylase 1 (HDAC1), and peroxisome proliferator-activated receptor-γ [66–69]. In lung cells infected with respiratory virus, Bcl3 recruits HDAC1 to the IL-8 promoter, resulting in the transcriptional repression of IL-8 [67]. Thus, it seems likely that Bcl3 mediates the recruitment of additional transcriptional regulators to the promoter regions of cIAP1, cIAP2, IL-8 and IL-17 genes in CTCL cells, resulting in their activation or repression depending on the recruited factor.
In this study, we have identified a new NFκB binding site (GGGACA CCCC) in human Bcl3 promoter, located from −298 to −289 (Fig. 7B). This site has a high homology to the consensus NFκB binding site, and ChIP data demonstrate that NFκB p65 and p50 subunits are highly recruited to this site in CTCL cells. Interestingly, however, in BZ-treated CTCL cells, the p65 and p50 subunits are replaced by p52 NFκB (Fig. 7C), indicating that the Bcl3 expression in CTCL cells is regulated through NFκB subunit exchange on Bcl3 promoter. The increased p52 recruitment to Bcl3 promoter correlates with the increased nuclear levels of p52 in BZ-treated cells (Fig. 7A). However, since p65 and p50 subunits are also present at high levels in the nucleus of BZ-treated CTCL cells, these data suggest that the p52 subunits might have higher affinity for Bcl3 promoter than the p65/p50 subunits. Our data support a model where in untreated cells, the Bcl3 promoter is occupied predominantly by p65/p50 heterodimers, resulting in the high Bcl3 expression; however, in BZ-treated cells, the p65/50 heterodimers are replaced by p52 subunits, resulting in Bcl3 transcriptional repression (Fig. 7D).
5. Conclusions
In conclusion, our study shows that Bcl3 is highly expressed in Hut-78 and HH cells, and suggests that Bcl3 has a dual role in these cells. On the one hand, Bcl3 is highly recruited to cIAP1 and cIAP2 promoters, resulting in their high transcription, and likely contributing to the high survival of CTCL cells. On the other hand, Bcl3 is recruited to the IL-8 and IL-17 promoters, resulting in the IL-8 and IL-17 suppression, which may contribute to the immunosuppressive nature of CTCL cells (Fig. 8). Together, our data indicate that Bcl3 has a pro-survival and immunosuppressive role in CTCL cells, and identify Bcl3 as a new potential target in the treatment of CTCL.
Acknowledgments
This work was supported by NIH grants AI085497 and CA173452 to I. Vancurova.
Abbreviations
- Bcl3
B-cell chronic lymphatic leukemia protein 3
- BZ
bortezomib
- ChIP
chromatin immunoprecipitation
- CTCL
cutaneous T cell lymphoma
- MF
mycosis fungoides
- SS
Sézary syndrome
References
- 1.Siegel RS, Kuzel TM. Cutaneous T-cell lymphoma/leukemia. Curr Treat Options Oncol. 2000;1:43–50. doi: 10.1007/s11864-000-0014-0. [DOI] [PubMed] [Google Scholar]
- 2.Berger CL, Tigelaar R, Cohen J, Mariwalla K, Trinh J, Wang N, Edelson RL. Cutaneous T-cell lymphoma: malignant proliferation of T-regulatory cells. Blood. 2005;105:1640–1647. doi: 10.1182/blood-2004-06-2181. [DOI] [PubMed] [Google Scholar]
- 3.Kim EJ, Hess S, Richardson SK, Newton S, Showe LC, Benoit BM, Vittorio CC, Junkins-Hopkins JM, Wysocka M, Rook AH. Immuno-pathogenesis and therapy of cutaneous T cell lymphoma. J Clin Invest. 2005;115:798–812. doi: 10.1172/JCI24826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krejsgaard T, Odum N, Geisler C, Wasik MA, Woetmann A. Regulatory T cells and immunodeficiency in mycosis fungoides and Sezary syndrome. Leukemia. 2012;26:424–432. doi: 10.1038/leu.2011.237. [DOI] [PubMed] [Google Scholar]
- 5.Wilcox RA. Cutaneous T cell lymphoma: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol. 2011;86:928–948. doi: 10.1002/ajh.22139. [DOI] [PubMed] [Google Scholar]
- 6.Chang TP, Vancurova I. NFκB function and regulation in cutaneous T-cell lymphoma. Am J Cancer Res. 2013;3:433–445. [PMC free article] [PubMed] [Google Scholar]
- 7.Qin JZ, Nestle FO, Häffner A, Dummer R, Burg G, Döbbeling U. Cutaneous T cell lymphoma cells contain constitutive NFκB complexes. J Invest Derm. 1997:108–225. [Google Scholar]
- 8.Izban KF, Ergin M, Qin JZ, Martinez RL, Pooley RJ, Saeed S, Alkan S. Constitutive expression of NFκB is a characteristic feature of mycosis fungoides: implications for apoptosis resistance and pathogenesis. Hum Pathol. 2000;31:1482–1490. doi: 10.1053/hupa.2000.20370. [DOI] [PubMed] [Google Scholar]
- 9.Sors A, Jean-Louis F, Pellet C, Laroche L, Dubertret L, Courtois G, Bachelez H, Michel L. Down-regulating constitutive activation of the NFκB canonical pathway overcomes the resistance of cutaneous T-cell lymphoma to apoptosis. Blood. 2006;107:2354–2363. doi: 10.1182/blood-2005-06-2536. [DOI] [PubMed] [Google Scholar]
- 10.Sors A, Jean-Louis F, Bégué F, Parmentier L, Dubertret L, Dreano M, Courtois G, Bachelez H, Michel L. Inhibition of IκB kinase subunit 2 in cutaneous T-cell lymphoma down-regulates NFκB constitutive activation, induces cell death, and potentiates the apoptotic response to antineoplastic chemotherapeutic agents. Clin Cancer Res. 2008;14:901–911. doi: 10.1158/1078-0432.CCR-07-1419. [DOI] [PubMed] [Google Scholar]
- 11.Kiessling MK, Klemke CD, Kaminski MM, Galani IE, Krammer PH, Gülow K. Inhibition of constitutively activated NFκB induces reactive oxygen species- and iron-dependent cell death in cutaneous T-cell lymphoma. Cancer Res. 2009;69:2365–2374. doi: 10.1158/0008-5472.CAN-08-3221. [DOI] [PubMed] [Google Scholar]
- 12.Rook RH, Kubin M, Cassin M, Vonderheid EC, Vowels BR, Wolfe JT, Wolf SF, Singh A, Trinchieri G, Lessin SR. IL-12 reverses cytokine and immune abnormalities in Sezary syndrome. J Immunol. 1995;154:1491–1498. [PubMed] [Google Scholar]
- 13.Yoo EK, Cassin M, Lessin SR, Rook AH. Complete molecular remission during biologic response modifier therapy for Sézary syndrome is associated with enhanced helper T type 1 cytokine production and natural killer cell activity. J Am Acad Dermatol. 2001;45:208–216. doi: 10.1067/mjd.2001.116345. [DOI] [PubMed] [Google Scholar]
- 14.Chong BF, Wilson AJ, Gibson HM, Hafner MS, Luo Y, Hedgcock CJ, Wong HK. Immune function abnormalities in peripheral blood mononuclear cell cytokine expression differentiates stages of cutaneous T-cell lymphoma/mycosis fungoides. Clin Cancer Res. 2008;14:646–653. doi: 10.1158/1078-0432.CCR-07-0610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abraham RM, Zhang Q, Odum N, Wasik MA. The role of cytokine signaling in the pathogenesis of cutaneous T-cell lymphoma. Cancer Biol Ther. 2011;12:1019–1022. doi: 10.4161/cbt.12.12.18144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guenova E, Watanabe R, Teague JE, Desimone JA, Jiang Y, Dowlatshahi M, Schlapbach C, Schaekel K, Rook AH, Tawa M, Fisher DC, Kupper TS, Clark RA. TH2 cytokines from malignant cells suppress TH1 responses and enforce a global TH2 bias in leukemic cutaneous T-cell lymphoma. Clin Cancer Res. 2013;19:3755–3763. doi: 10.1158/1078-0432.CCR-12-3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Q, Wang HY, Wei F, Liu X, Paterson JC, Roy D, Mihova D, Woetmann A, Ptasznik A, Odum N, Schuster SJ, Marafioti T, Riley JL, Wasik MA. Cutaneous T Cell Lymphoma Expresses Immunosuppressive CD80 (B7-1) Cell Surface Protein in a STAT5-Dependent Manner. J Immunol. 2014;192:2913–2919. doi: 10.4049/jimmunol.1302951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Querfeld C, Rosen ST, Guitart J, Kuzel TM. The spectrum of cutaneous T-cell lymphomas: new insights into biology and therapy. Curr Opin Hematol. 2005;12:273–278. doi: 10.1097/01.moh.0000166498.64515.03. [DOI] [PubMed] [Google Scholar]
- 19.Duvic M, Foss FM. Mycosis fungoides: pathophysiology and emerging therapies. Semin Oncol. 2007;34:S21–S28. doi: 10.1053/j.seminoncol.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 20.Gardner JM, Evans KG, Musiek A, Rook AH, Kim EJ. Update on treatment of cutaneous T-cell lymphoma. Curr Opin Oncol. 2009;21:131–137. doi: 10.1097/CCO.0b013e3283253190. [DOI] [PubMed] [Google Scholar]
- 21.Wong HK, Mishra A, Hake T, Porcu P. Evolving insights in the pathogenesis and therapy of cutaneous T-cell lymphoma (mycosis fungoides and Sezary syndrome) Br J Haematol. 2011;155:150–166. doi: 10.1111/j.1365-2141.2011.08852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roncolato F, Gazzola A, Zinzani PL, Pileri SA, Piccaluga PP. Targeted molecular therapy in peripheral T-cell lymphomas. Expert Rev Hematol. 2011;4:551–562. doi: 10.1586/ehm.11.55. [DOI] [PubMed] [Google Scholar]
- 23.Li JY, Horwitz S, Moskowitz A, Myskowski PL, Pulitzer M, Querfeld C. Management of cutaneous T cell lymphoma: new and emerging targets and treatment options. Cancer Manag Res. 2012;4:75–89. doi: 10.2147/CMAR.S9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jain S, Zain J, O’Connor O. Novel therapeutic agents for cutaneous T-cell lymphoma. J Hematol Oncol. 2012;5:24. doi: 10.1186/1756-8722-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adams J, Kauffman M. Development of the proteasome inhibitor Velcade (Bortezomib) Cancer Investig. 2004;22:304–311. doi: 10.1081/cnv-120030218. [DOI] [PubMed] [Google Scholar]
- 26.Shah JJ, Orlowski RZ. Proteasome inhibitors in the treatment of multiple myeloma. Leukemia. 2009;23:1964–1979. doi: 10.1038/leu.2009.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuhn DJ, Orlowski RZ. The immunoproteasome as a target in hematologic malignancies. Semin Hematol. 2012;49:258–262. doi: 10.1053/j.seminhematol.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zinzani PL, Musuraca G, Tani M, Stefoni V, Marchi E, Fina M, Pellegrini C, Alinari L, Derenzini E, De Vivo A, Sabattini E, Pileri S, Baccarani M. Phase II trial of proteasome inhibitor bortezomib in patients with relapsed or refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:4293–4297. doi: 10.1200/JCO.2007.11.4207. [DOI] [PubMed] [Google Scholar]
- 29.Horwitz SM. Novel therapies for cutaneous T-cell lymphomas. Clin Lymphoma Myeloma. 2008;(Suppl 5):S187–S192. doi: 10.3816/CLM.2008.s.015. [DOI] [PubMed] [Google Scholar]
- 30.Heider U, Rademacher J, Lamottke B, Mieth M, Moebs M, von Metzler I, Assaf C, Sezer O. Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in cutaneous T cell lymphoma. Eur J Haematol. 2009;82:440–449. doi: 10.1111/j.1600-0609.2009.01239.x. [DOI] [PubMed] [Google Scholar]
- 31.Biskup E, Kamstrup MR, Manfé V, Gniadecki R. Proteasome inhibition as a novel mechanism of the proapoptotic activity of γ-secretase inhibitor I in cutaneous T-cell lymphoma. Br J Dermatol. 2013;168:504–512. doi: 10.1111/bjd.12071. [DOI] [PubMed] [Google Scholar]
- 32.Vu HY, Juvekar A, Ghosh C, Ramaswami S, Le DH, Vancurova I. Proteasome inhibitors induce apoptosis of prostate cancer cells by inducing nuclear translocation of IκBα. Arch Biochem Biophys. 2008;475:156–163. doi: 10.1016/j.abb.2008.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Juvekar A, Manna S, Ramaswami S, Chang TP, Vu HY, Ghosh CC, Celiker MY, Vancurova I. Bortezomib induces nuclear translocation of IκBα resulting in gene-specific suppression of NFκB-dependent transcription and induction of apoptosis in CTCL. Mol Cancer Res. 2011;9:183–194. doi: 10.1158/1541-7786.MCR-10-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manna S, Singha B, Phyo SA, Gatla HR, Chang TP, Sanacora S, Ramaswami S, Vancurova I. Proteasome inhibition by bortezomib increases IL-8 expression in androgen-independent prostate cancer cells: the role of IKKα. J Immunol. 2013;191:2837–2846. doi: 10.4049/jimmunol.1300895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singha B, Gatla HR, Manna S, Chang TP, Sanacora S, Poltoratsky V, Vancura A, Vancurova I. Proteasome inhibition increases recruitment of IκB kinase β (IKKβ), S536P-p65, and transcription factor EGR1 to interleukin-8 (IL-8) promoter, resulting in increased IL-8 production in ovarian cancer cells. J Biol Chem. 2014;289:2687–2700. doi: 10.1074/jbc.M113.502641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohno H, Takimoto G, McKeithan TW. The candidate proto-oncogene bcl-3 is related to genes implicated in cell lineage determination and cell cycle control. Cell. 1990;60:991–997. doi: 10.1016/0092-8674(90)90347-h. [DOI] [PubMed] [Google Scholar]
- 37.Kerr LD, Duckett CS, Wamsley P, Zhang Q, Chiao P, Nabel G, McKeithan TW, Baeuerle PA, Verma IM. The proto-oncogene bcl-3 encodes an IκB protein. Genes Dev. 1992;6:2352–2363. doi: 10.1101/gad.6.12a.2352. [DOI] [PubMed] [Google Scholar]
- 38.Wulczyn FG, Naumann M, Scheidereit C. Candidate proto-oncogene bcl-3 encodes a subunit-specific inhibitor of transcription factor NFκB. Nature. 1992;358:597–599. doi: 10.1038/358597a0. [DOI] [PubMed] [Google Scholar]
- 39.Bours V, Franzoso G, Azarenko V, Park S, Kanno T, Brown K, Siebenlist U. The oncoprotein Bcl-3 directly transactivates through kappa B motifs via association with DNA-binding p50 homodimers. Cell. 1993;72:729–739. doi: 10.1016/0092-8674(93)90401-b. [DOI] [PubMed] [Google Scholar]
- 40.Nolan GP, Fujita T, Bhatia K, Huppi C, Liou HC, Scott ML, Baltimore D. The bcl-3 proto-oncogene encodes a nuclear IκB-like molecule that preferentially interacts with NFκB p50 and p52 in a phosphorylation-dependent manner. Mol Cell Biol. 1993;13:3557–3566. doi: 10.1128/mcb.13.6.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Q, Didonato JA, Karin M, McKeithan TW. BCL3 encodes a nuclear protein which can alter the subcellular location of NFκB proteins. Mol Cell Biol. 1994;14:3915–3926. doi: 10.1128/mcb.14.6.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McKeithan TW, Takimoto GS, Ohno H, Bjorling VS, Morgan R, Hecht BK, Dubé I, Sandberg AA, Rowley JD. BCL3 rearrangements and t(14;19) in chronic lymphocytic leukemia and other B-cell malignancies: a molecular and cytogenetic study. Genes Chromosomes Cancer. 1997;20:64–72. [PubMed] [Google Scholar]
- 43.Ge B, Li O, Wilder P, Rizzino A, McKeithan TW. NFκB regulates BCL3 transcription in T lymphocytes through an intronic enhancer. J Immunol. 2003;171:4210–4218. doi: 10.4049/jimmunol.171.8.4210. [DOI] [PubMed] [Google Scholar]
- 44.Mathas S, Johrens K, Joos S, Lietz A, Hummel F, Jundt F, Anagnostopoulos I, Bommert K, Lichter P, Stein H, Scheidereit C, Dörken B. Elevated NFκB p50 complex formation and Bcl-3 expression in classical Hodgkin, anaplastic large-cell, and other peripheral T-cell lymphomas. Blood. 2005;106:4287–4293. doi: 10.1182/blood-2004-09-3620. [DOI] [PubMed] [Google Scholar]
- 45.Martin-Subero JI, Wlodarska I, Bastard C, Picquenot JM, Höppner J, Giefing M, Klapper W, Siebert R. Chromosomal rearrangements involving the BCL3 locus are recurrent in classical Hodgkin and peripheral T-cell lymphoma. Blood. 2006;108:401–402. doi: 10.1182/blood-2005-09-3843. [DOI] [PubMed] [Google Scholar]
- 46.Brenne AT, Fagerli UM, JD, Shaughnessy TK, Våtsveen TB, Rø H, Hella F, Zhan B, Bariogie A, Børset Sundan M, Waage A. High expression of BCL3 in human myeloma cells is associated with increased proliferation and inferior prognosis. Eur J Haematol. 2009;82:354–363. doi: 10.1111/j.1600-0609.2009.01225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Courtois G, Gilmore TD. Mutations in the NFκB signaling pathway: implications for human disease. Oncogene. 2006;25:6831–6843. doi: 10.1038/sj.onc.1209939. [DOI] [PubMed] [Google Scholar]
- 48.Maldonado V, Melendez-Zajgla J. Role of Bcl-3 in solid tumors. Mol Cancer. 2011;10:152. doi: 10.1186/1476-4598-10-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wakefield A, Soukupova J, Montagne A, Ranger J, French R, Muller WJ, Clarkson RW. Bcl3 selectively promotes metastasis of ERBB2-driven mammary tumors. Cancer Res. 2013;73:745–755. doi: 10.1158/0008-5472.CAN-12-1321. [DOI] [PubMed] [Google Scholar]
- 50.Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res. 1999;59:2615–2622. [PubMed] [Google Scholar]
- 51.Hansen ER, Vejlsgaard GL, Lisby S, Heidenheim M, Baadsgaard O. Epidermal interleukin 1 alpha functional activity and interleukin 8 immunoreactivity are increased in patients with cutaneous T-cell lymphoma. J Investig Dermatol. 1991;97:818–823. doi: 10.1111/1523-1747.ep12489011. [DOI] [PubMed] [Google Scholar]
- 52.Wismer JM, McKenzie RC, Sauder DN. IL-8 immunoreactivity in epidermis of cutaneous T-cell lymphoma patients. Lymphokine Cytokine Res. 1994;13:21–27. [PubMed] [Google Scholar]
- 53.Krejsgaard T, Ralfkiaer U, Clasen-Linde E, Eriksen KW, Kopp KL, Bonefeld CM, Geisler C, Dabelsteen S, Wasik MA, Ralfkiaer E, Woetmann A, Odum N. Malignant cutaneous T-cell lymphoma cells express IL-17 utilizing the Jak3/Stat3 signaling pathway. J Investig Dermatol. 2011;131:1331–1338. doi: 10.1038/jid.2011.27. [DOI] [PubMed] [Google Scholar]
- 54.Krejsgaard T, Litvinov IV, Wang Y, Xia L, Willerslev-Olsen A, Koralov SB, Kopp KL, Bonefeld CM, Wasik MA, Geisler C, Woetmann A, Zhou Y, Sasseville D, Odum N. Elucidating the role of IL-17 F in cutaneous T-cell lymphoma. Blood. 2013;122:943–950. doi: 10.1182/blood-2013-01-480889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Polyak SJ, Khabar KS, Rezeiq M, Gretch DR. Elevated levels of IL-8 in serum are associated with hepatitis C virus infection and resistance to interferon therapy. J Virol. 2001;75:6209–6211. doi: 10.1128/JVI.75.13.6209-6211.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arican O, Aral M, Sasmaz S, Ciragil P. Serum levels of TNF-α, IFN-γ, IL-6, IL-8, IL-12, IL-17, and IL-18 in patients with active psoriasis and correlation with disease severity. Mediat Inflamm. 2005;2005:273–279. doi: 10.1155/MI.2005.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caproni M, Antiga E, Melani L, Volpi W, Del Bianco E, Fabbri P. Serum levels of IL-17 and IL-22 are reduced by etanercept, but not by acitretin, in patients with psoriasis: a randomized-controlled trial. J Clin Immunol. 2009;29:210–214. doi: 10.1007/s10875-008-9233-0. [DOI] [PubMed] [Google Scholar]
- 58.Wang VY, Huang W, Asagiri M, Spann N, Hoffmann A, Glass C, Ghosh G. The transcriptional specificity of NF-κB dimers is coded within the κB DNA response elements. Cell Rep. 2012;2:824–839. doi: 10.1016/j.celrep.2012.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carmody RJ, Ruan Q, Palmer S, Hilliard B, Chen YH. Negative regulation of toll-like receptor signaling by NF-κB p50 ubiquitination blockade. Science. 2007;317:675–678. doi: 10.1126/science.1142953. [DOI] [PubMed] [Google Scholar]
- 60.Collins PE, Kiely PA, Carmody RJ. Inhibition of transcription by B cell Leukaemia 3 (Bcl-3) requires interaction with nuclear factor (NF)-κB p50. J Biol Chem. 2014;289:7059–7067. doi: 10.1074/jbc.M114.551986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miyagaki T, Sugaya M. Immunological milieu in mycosis fungoides and Sézary syndrome. J Dermatol. 2014;41:11–18. doi: 10.1111/1346-8138.12305. [DOI] [PubMed] [Google Scholar]
- 62.Brasier AR, Lu M, Hai T, Lu Y, Boldogh I. NFκB-inducible BCL-3 expression is an autoregulatory loop controlling nuclear p50/NFκB1 residence. J Biol Chem. 2001;276:32080–32093. doi: 10.1074/jbc.M102949200. [DOI] [PubMed] [Google Scholar]
- 63.Kim YM, Sharma N, Nyborg JK. The proto-oncogene Bcl3, induced by Tax, represses Tax-mediated transcription via p300 displacement from the human T-cell leukemia virus type 1 promoter. J Virol. 2008;82:11939–11947. doi: 10.1128/JVI.01356-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV, Ignatieva EV, Ananko EA, Podkolodnaya OA, Kolpakov FA, Podkolodny NL, Kolchanov NA. Databases on transcriptional regulation: TRANSFAC, TRRD, and COMPEL. Nucleic Acids Res. 1998;26:364–370. doi: 10.1093/nar/26.1.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Canoz O, Rassidakis GZ, Admirand JH, Medeiros LJ. Immunohistochemical detection of BCL-3 in lymphoid neoplasms: a survey of 353 cases. Mod Pathol. 2004;17:911–917. doi: 10.1038/modpathol.3800140. [DOI] [PubMed] [Google Scholar]
- 66.Na SY, Choi JE, Kim HJ, Jhun BH, Lee YC, Lee JW. Bcl3, an IκB protein, stimulates activating protein-1 transactivation and cellular proliferation. J Biol Chem. 1999;274:28491–28496. doi: 10.1074/jbc.274.40.28491. [DOI] [PubMed] [Google Scholar]
- 67.Jamaluddin M, Choudhary S, Wang S, Casola A, Huda R, Garofalo RP, Ray S, Brasier AR. Respiratory syncytial virus-inducible BCL-3 expression antagonizes the STAT/IRF and NFκB signaling pathways by inducing histone deacetylase 1 recruitment to the interleukin-8 promoter. J Virol. 2005;79:15302–15313. doi: 10.1128/JVI.79.24.15302-15313.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang J, Williams RS, Kelly DP. Bcl3 interacts cooperatively with peroxisome proliferator-activated receptor gamma (PPARγ) coactivator 1α to coactivate nuclear receptors estrogen-related receptor α and PPARα. Mol Cell Biol. 2009;29:4091–4102. doi: 10.1128/MCB.01669-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kreisel D, Sugimoto S, Tietjens J, Zhu J, Yamamoto S, Krupnick AS, Carmody RJ, Gelman AE. Bcl3 prevents acute inflammatory lung injury in mice by restraining emergency granulopoiesis. J Clin Invest. 2011;121:265–276. doi: 10.1172/JCI42596. [DOI] [PMC free article] [PubMed] [Google Scholar]