

Abstract
Arylamine N-acetyltransferases (NATs) are polymorphic drug-metabolizing enzymes, acetylating arylamine carcinogens and drugs including hydralazine and sulphonamides. The slow NAT phenotype increases susceptibility to hydralazine and isoniazid toxicity and to occupational bladder cancer. The two polymorphic human NAT loci show linkage disequilibrium. All mammalian Nat genes have an intronless open reading frame and non-coding exons. The human gene products NAT1 and NAT2 have distinct substrate specificities: NAT2 acetylates hydralazine and human NAT1 acetylates p-aminosalicylate (p-AS) and the folate catabolite para-aminobenzoylglutamate (p-abaglu). Human NAT2 is mainly in liver and gut. Human NAT1 and its murine homologue are in many adult tissues and in early embryos. Human NAT1 is strongly expressed in oestrogen receptor-positive breast cancer and may contribute to folate and acetyl CoA homeostasis. NAT enzymes act through a catalytic triad of Cys, His and Asp with the architecture of the active site-modulating specificity. Polymorphisms may cause unfolded protein. The C-terminus helps bind acetyl CoA and differs among NATs including prokaryotic homologues. NAT in Salmonella typhimurium supports carcinogen activation and NAT in mycobacteria metabolizes isoniazid with polymorphism a minor factor in isoniazid resistance. Importantly, nat is in a gene cluster essential for Mycobacterium tuberculosis survival inside macrophages. NAT inhibitors are a starting point for novel anti-tuberculosis drugs. Human NAT1-specific inhibitors may act in biomarker detection in breast cancer and in cancer therapy. NAT inhibitors for co-administration with 5-aminosalicylate (5-AS) in inflammatory bowel disease has prompted ongoing investigations of azoreductases in gut bacteria which release 5-AS from prodrugs including balsalazide.
Keywords: arylamine N-acetyltransferase, catalytic triad, acetyl CoA, tuberculosis, breast cancer, hydralazine, isoniazid, pharmacogenetics
Introduction
This review describes studies of the drug-metabolizing enzyme arylamine N-aetyltransferase (NAT) over a period of some 30 years up to the present day, which was presented as the JR Vane Lecture in December 2012. The work includes early pharmacogenetic analysis, use of isoenzyme specific antibodies in Western blotting and immunohistochemistry; transgenic mice, which may have generated more questions than answers, large-scale recombinant protein expression for structural studies and development of isoenzyme specific inhibitors. Isoniazid, the anti-tubercular prodrug, is metabolized by NAT (Figure Figure 1) and prompted work on NAT in mycobacteria, stimulating investigations in drug discovery. Recent work on azoreductases, which generate arylamines from azo drug precursors, is also covered. These studies have relied on an enthusiastic band of co-investigators and collaborators including project students and graduate research students (Supporting Information Table S1). The present review adds to earlier summaries (Sim et al., 2000; 2007; 2008a–c).
Figure 1.
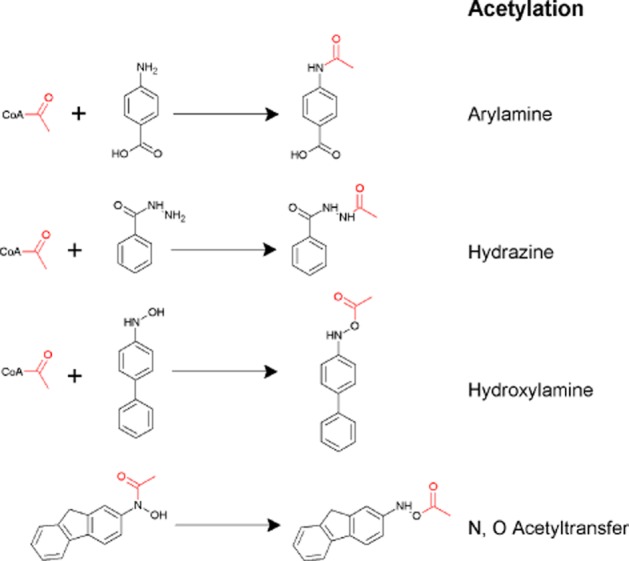
Metabolic reactions catalysed by arylamine N-acetyltransferase. Acetylation reactions from the top show N-acetylation of an arylamine, p-aminobenzoic acid; N-acetylation of isoniazid; O-acetylation of an arylhydroxylamine and the bottom reaction shows N,O acetyltransfer in a hydroxamate. The top three reactions use acetyl CoA as a cofactor while the bottom reaction does not. The bottom two reactions are associated with carcinogenesis. From Sim et al., 2010.
NATs came to prominence in identifying acetyl CoA as a key metabolic intermediate (Lippmann and Kaplan, 1946). Their role in xenobiotic metabolism has been wide-reaching for early insight into interindividual differences in the ability to metabolize drugs (Kalow, 2004; Weber and Hein, 1985 for reviews). Studies on arylamine N-acetyltransferases (NATs) were among the earliest examples of pharmacogenetic variation (Evans et al., 1960) and laid the foundation for understanding that different ethnic populations could differ considerably in drug metabolism (see Jones, 2013) with important implications for clinical trials (Tam et al., 2000). NAT polymorphism also has importance in understanding adverse side effects (see Park et al., 1992).
The corresponding author's journey in drug metabolism was triggered by attempts to understand how an environmental factor, the drug, could lead to an adverse reaction. These studies were carried out firstly in the Pharmacology Department in South Parks Road in Oxford and subsequently in the iconic building in Mansfield Road for which the funds were raised by Prof. David Smith from Squibb. The building was opened by JR Vane in the summer of 1991 (Figure 2). Since 2011, the studies have been continued at Kingston University.
Figure 2.

JR Vane at the opening of the Pharmacology Department, Oxford 1991. John Vane is on the right, David Smith, Head of Department, is in the middle and the head of research of Squibb Pharmaceuticals, who gave the endowment before the Bristol-Meyers takeover is on the left.
Adverse drug reactions
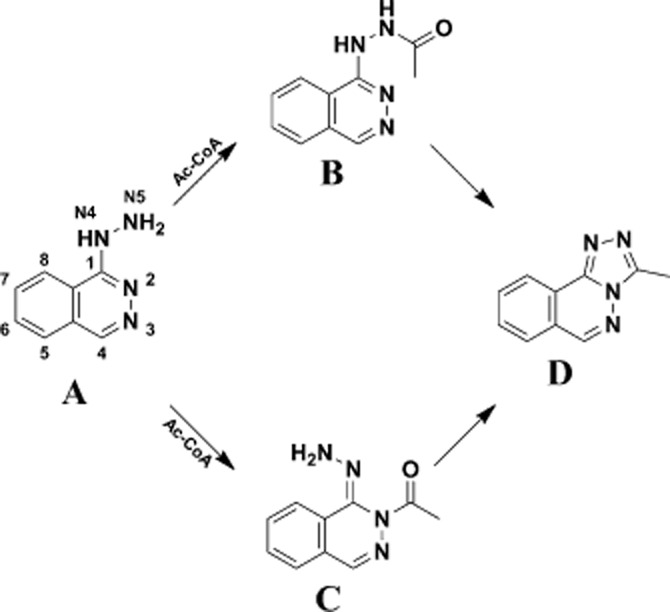
Even in the post-genomic era, identification of an environmental trigger of disease can be very difficult, and therefore, adverse drug reactions have the advantage that the drug serves as a readily identifiable environmental factor (McDowell et al., 2013). I chose to study hydralazine-induced systemic lupus erythematosus (SLE; Batchelor et al., 1980). Immune complexes become deposited in capillaries and in kidney glomeruli with inhibition of the complement cascade contributing to SLE susceptibility (Walport, 2002 for review). We showed hydralazine inhibited the solubilization of immune complexes by blocking complement component C4, which is polymorphic (Sim et al., 1984), with hydralazine inhibiting the C4A type more than the C4B type (Sim and Law, 1985). Immune system polymorphisms had a role in predisposition to hydralazine-induced SLE (Mitchell et al., 1987; 1989; Mitchell and Sim, 1989) but the most important polymorphism was the ability to metabolize hydralazine (Drayer and Reidenberg, 1977). Hydralazine is acetylated polymorphically to produce cyclized methyl triazolophthalazine (MTP; Timbrell et al., 1980). When complement component C4 is activated a thiol ester is exposed, which binds covalently to adjacent immune complex (Sim et al., 1981) leading to solubilization. Hydralazine, but not its acetylated metabolite MTP, blocked covalent binding of complement component C4 (Figure 3; Sim et al., 1984). In the process, hydralazine becomes bound to C4 (Sim and Law, 1985).
Figure 3.
Hydralazine inhibits binding of complement component C4. Radiolabelled C4 was activated by a model for immune complexes (C1s bound to Sepharose) and the inhibition of binding was compared using hydralazine or its acetylated metabolite, methyltriazolophthalazine synthesized by EW Gill. From Sim et al. (1984).
The metabolism of hydralazine has been investigated extensively and recent structural studies with recombinant NAT protein have demonstrated the mechanisms of the cyclization reaction (Figure 4; Abuhammad et al., 2010).
Figure 4.
Two possible routes of acetylation of hydralazine. From the crystal structure and the different tautommers and ionization states it is proposed that hydralazine may also be acetylated initially at the N in the heterocyclic ring. In either case, cyclization is proposed to generate methyltriazolophthalazine. From Abuhammad et al. (2010).
NAT and pharmacogenetics
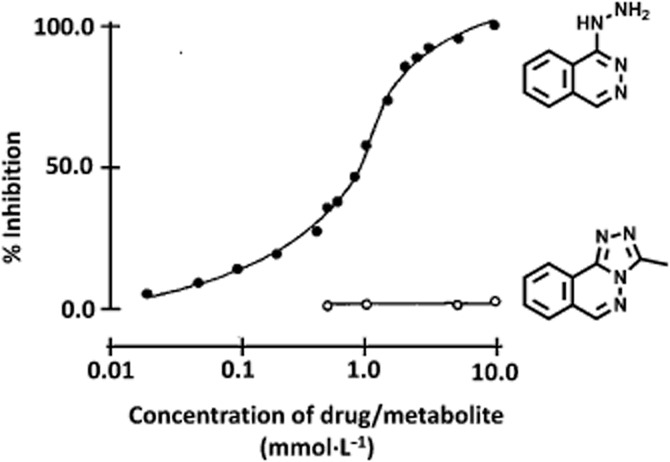
In the 1960's, it had been established that there was polymorphism in isoniazid inactivation by human NAT activity (Evans et al., 1960). This is the classic textbook pattern of distribution into fast and slow acetylators ( Figure 5). Fast and slow acetylators were classified with several drugs including isoniazid, hydralazine and sulphamethazine (see Weber and Hein, 1985). It was also identified that ethnic populations varied such that in Orientals, there were approximately 15% slow acetylators while in the Caucasian population, around 50% were slow acetylators (Harris et al., 1958; Sim and Hickman, 1991). The slow acetylator phenotype was, in early studies, found to be more prevalent in isoniazid-induced toxicity as well as in hydralazine-induced lupus (Woosley et al., 1978). More recent studies have demonstrated that isoniazid-induced hepatotoxicity have also been associated with the slow acetylator type (Gupta et al., 2013).
Figure 5.
Isoniazid pharmacokinetics in a population. After Evans et al. (1960).
In a landmark study, it was also shown that slow acetylation was a major predisposing factor in industrial bladder cancer (Cartwright et al., 1982) where bladder cancer had been known since the late 19th century to be linked to exposure to aniline dyes (Rehn, 1895; see Dietrich and Dietrich, 2001).
Some drugs were identified, which were metabolized by acetylation but there did not appear to be the same distribution among individuals into fast and slow acetylators. These drugs included para-aminosalicylate (p-AS) and the compound para-aminobenzoic acid (p-aba). These drugs were referred to as monomorphic as opposed to the drugs like procainamide, isoniazid and hydralazine, which were known as polymorphic (see Weber and Hein, 1985).
An inkling of the cause behind two different classes of substrate came from some outstanding experiments by Jenne (1965). Two enzymic activities in human liver homogenates were separated by ion exchange chromatography and the profile differed when activity was measured with p-AS and with sulphamethazine. Some 10–15 years later using molecular biology, two different human NAT isoenzymes (Blum et al., 1990; Deguchi et al., 1990) were found, now known as human NAT1 and NAT2 each catalysing the acetylation of arylamines using acetyl CoA as the acetyl donor. Only NAT2 catalyses the acetylation of hydralazine, procainamide and isoniazid while p-AS is only acetylated by human NAT1 (Figure 6). As more versatile assay methods have become available (Brooke et al., 2003a), it has been possible to establish an easy method of comparing the substrate specificities of a range of different NAT enzymes (Figure 6) and this demonstrates clearly that the human NAT enzymes have different but overlapping substrate specificity profiles (Figure 6).
Figure 6.

Specificity profile of NATs. A range of different NAT enzymes were prepared and their specificity was determined with the same panel of substrates in each case The specific activity profiles of three eukaryotic NATs and three prokaryotic NATs have been reported. Specific activities are presented as percent compared with the most active substrate for each enzyme. STNAT, NAT from S. typhimurium; PANAT, NAT from P. aeruginosa; MSNAT, NAT from M. smegmatis; SMZ, sulfamethazine; PRO, procainamide; 5-AS, 5-aminosalicylic acid; PABA, 4-aminobenzoic acid; 2-AF, 2-aminofluorene; 4-CA, 4-chloroaniline; 4-BA, 4-bromoaniline; 4-IA, 4-iodoaniline; ANS, 4-anisidine; EOA, 4-ethoxyaniline; BOA, 4-butoxyaniline; HOA, 4-hexyloxyaniline; POA, 4-phenoxyaniline; AMV, 4-aminoveratrole; INH, isoniazid; CBZ, 4-chlorobenzoic hydrazide; HDZ, hydralazine (Westwood et al., 2006).
Finding two functional human NAT proteins did not solve the basis of polymorphism in metabolism but early cloning experiments provided the clue to the polymorphism in metabolism of drugs by human NAT2. In the pre-PCR, pre-whole genomic age and specific polynucleotide probes to screen DNA libraries were identified through translation of protein sequence. We had previously chosen to purify NAT from mouse liver (Watson et al., 1990) but rabbit was a much better choice (Andres et al., 1987; 1988; Sasaki et al., 1991). Partial protein sequence from pure NAT from rabbit liver NAT was published by Andres, an inventive contributor to the NAT field (Andres et al., 1987). This paper appeared when I was on maternity leave.
Fortuitously, using the sequence information from rabbit, both a Japanese and a European group chose to clone human NAT genes (Blum et al., 1990; Ohsako and Deguchi, 1990). These studies show how the accumulation of data serves to solve apparently intractable problems. The differences, which had been observed in the distribution of fast and slow acetylation in Oriental and Caucasian populations, were reflected in the DNA clones described in these two studies. Both groups found two different genes, NAT1 and NAT2, (Blum et al., 1990; Ohsako and Deguchi, 1990). Point mutations in the human NAT2 gene leading to poor acetylation (Deguchi et al., 1990; Blum et al., 1991) were found but the point mutations in the Japanese group were different from those in Caucasians (Deguchi et al., 1990; Blum et al., 1991).
Soon afterwards, we developed a restriction fragment length polymorphism PCR genotyping method for NAT2 (Hickman and Sim, 1991; Hickman et al., 1992). Now microarray-based single nucleotide polymorphism (SNP) analyses (Hein and Doll, 2012) and cheap and comprehensive sequencing across the chromosomal region are used (Patin et al., 2006a,b; Sabbagh et al., 2011; Tilak et al., 2013). These studies have also allowed the establishment of gene frequencies in a wide range of ethnic populations and disease states (e.g. Garte et al., 2001).
In the initial cloning experiments, the Japanese used cDNA (Ohsako and Deguchi, 1990) while the Meyer group used genomic DNA (Blum et al., 1990). The latter studies identified a third pseudogene, which was not transcribed and hence was not found in the cDNA library (Blum et al., 1990). Comparison of the cDNA and genomic DNA clones showed the open reading frames of each of the NAT1 and NAT2 genes were intronless. Human NAT1 (Ebisawa and Deguchi, 1991) has a non-coding exon and we now know there are a series of upstream non-coding exons in the human NAT1 gene and that the splicing of these into the proximal region immediately in front of the coding exon may be tissue specific (Husain et al., 2004; Boukouvala and Sim, 2005; Barker et al., 2006; see Minchin et al., 2007).
Human NAT2 also has a non-coding exon spliced in frame with its single coding exon (Husain et al., 2007; see Sim et al., 2010). The extended NAT2 gene is much simpler than the multiple alternative transcripts of human NAT1. The wide spread tissue expression of human NAT1 (in most tissues including endocrine tissues, blood cells, neural tissue as well as gut and liver) compared with the more restricted expression of human NAT2 (mainly gut and liver) may underlie the more complex gene organisation (see Minchin et al., 2007). These genes as well as the human pseudogene are in close proximity in the genome in the region 8p22 (Hickman et al., 1994; Franke et al., 1994).
Although in the initial cloning experiments NAT1 was found to be invariant among individuals, we now know that human NAT1 also has different alleles (Vatsis and Weber, 1993; Grant et al., 1997; Payton and Sim, 1998). Point mutations in the open reading frame along have been described as well as alleles with no mutations in the coding exon but there were insertions and deletions at the 3′ end of the gene (Vatsis and Weber, 1993). Genotypng NAT1 using PCR-based methods were more complex than for NAT2 (Smelt et al., 1998; Pirmohamed et al., 2000; Johnson et al., 2004). Multiplex SNP analyses are still used (see Hein, 2009) but genotyping through sequencing is now the method of choice.
We know there is linkage disequilibrium across the 200kb region on chromosome 8 in which all three NAT genes are found (Cascorbi et al., 1995; 2001,; Smelt et al., 1998) and this has been exploited in molecular anthropological studies of population migration and microevolution (Patin et al., 2006a,b; Sabbagh et al., 2011). In order to understand differential expression of the two human NATs, we developed antibodies against the intact proteins, which detect both NAT1 and NAT2, as they are over 80% identical in sequence. The C-termini differ sufficiently to use the C-terminal dodecapeptide to generate a human NAT1-specific antibody, which has been widely employed (e.g. Butcher et al., 1998; Adam et al., 2003; Johansson et al., 2012). The anti-NAT2 antibody has not been so successful.
Effects of mutations in human NAT2 and human NAT1
A series of reviews of the effects of each of the mutations in human NAT1 and NAT2 has been carried out through modelling (Rodrigues-Lima and Dupret, 2002; Walraven et al., 2008; Rajasekaran et al., 2011). A full description of all NAT1 and NAT2 alleles is to be found on the web site initially curated by David Hein (http://louisville.edu/medschool/pharmacology/consensus-human-arylamine-n-acetyltransferase-gene-nomenclature/) and now hosted at http://nat.mbg.duth.gr/.
Specific highlights will be covered here.
Most alleles of human NAT1 and NAT2 are haplotypes of several point mutations with one being a signature mutation, which alone causes a change such that the resultant protein shows reduced activity. Wide phenotypic spread in the pharmacokinetics of isoniazid (Ellard and Gammon, 1976) had been identified and molecular studies confirmed the earlier twin studies of (Evans et al., 1960), namely, that slow alleles of human NAT2 are somatically inherited. While one ‘slow’ allele confers the slow acetylation phenotype, those with two ‘slow’ alleles show a more extreme phenotype (Hickman and Sim, 1991). Inherent effects of the level of ‘slowness’ conferred by different NAT2 alleles have not been fully explored (Ruiz et al., 2012).
However alleles fall into two classes, those that cause unstable protein and those which affect the activity of a folded protein.
A list of the main mutations that affect amino acids in human NAT1 and NAT2 are shown in Table 1. The premature stop codons in human NAT1 are readily understood (Hubbard et al., 1998; Hughes et al., 1998; Payton and Sim, 1998).
Table 1.
Mutations causing amino acid changes in human NAT genes
| Mutation | NAT allelic family | Amino acid | |
|---|---|---|---|
| NAT2 | C190T | 2*19 | Arg64Trp |
| G191A | 2*14 | Arg64Gln | |
| T341C | 2*5 | Ile114Thr | |
| G364A | 2*D12 | Asp122Asn | |
| A434C | 2*17 | Gln145Pro | |
| G499A | 2*10 | Glu167Lys | |
| G590A | 2*6 | Arg197Gln | |
| A803G | 2*12 | Lys268Arg | |
| G857A | 2*7 | Gly286Gln | |
| NAT1 | C97T | 1*19 | Arg33Stop |
| C190T | 1*17 | Arg64Trp | |
| C559T | 1*15 | Arg187Stop | |
| G560A | 1*14 | Arg187Gln | |
| A752T | 1*22 | Aso251Val |
Expressing variant alleles causing amino acid substitutions as recombinant enzymes has identified the effects of point mutations (Hickman et al., 1995a,b; Walraven et al., 2008 for review). One amino acid substitution with tryptophan substituted for aspartate at position 64 (W64D) occurs in both NAT1 and NAT2. Two very different elegant studies (Butcher et al., 2004; Liu et al., 2006) provided the conclusion that Trp (W) at position 64 results in an unfolded variant, which accumulates intracellularly in aggresomes for degradation via ubiquitination through the proteosomal pathway.
An example of a mutation resulting in folded but inactive protein is the change of aspartate 122 to asparagine (D122N). Asp122 is an essential part of the charge relay system in the active site (see below) and asparagine is not effective as has been confirmed by site-directed mutagenesis (Wang et al., 2004).
Apparently, healthy individuals with very little NAT1 activity have each been shown to have two defective alleles (Hughes et al., 1998; Payton and Sim, 1998). It was nevertheless shown there was an inverse correlation between NAT1 activity and folate levels in red blood cells (Ward et al., 1992), which opened up an ongoing debate on the relationship between NAT1 and folate (see Minchin et al., 2007; Butcher and Minchin, 2012).
The allele NAT1*10 has had more coverage than any other: it has no amino acid substitutions within the coding region – there are deletions and insertions at the 3′ end. In colon cancer studies, 1*10 was associated with increased activity (Bell et al., 1995), its effect has been hotly debated for other tissues where the number of copies of the 1*10 allele did not correlate with the level of NAT1 activity (Grant et al., 1997; Payton and Sim, 1998). Effects of NAT1 mutations are being revisited (Millner et al., 2012a,b) and NAT1*10 remains an interesting allele. A 1*10 homozygous individual was identified in a study of patients with Alzheimer's disease (Johnson et al., 2004) was identified and NAT1 polymorphism, including 1*10, has been implicated in epidemiological studies of neural tube defects (Lammer et al., 2004; Jensen et al., 2005; 2006).
Epigenetic control of NAT gene expression and hence enzyme activity is yet to be fully explored. CpG islands in the vicinity of the NAT genes (Matas et al., 1997) and their murine equivalents (Fakis et al., 2000) indicate methylation sites and analyses of the upstream spliced region show this is the case (Kim et al., 2008; Wakefield et al., 2010). Epigenetic control is an important area for the future.
NATs and cancer
NAT has links with cancer at many levels (Table 2).
Table 2.
Relationships between NAT and cancer
| Links between NAT and cancer |
|---|
| Carcinogenesis |
|
| Polymorphism |
|
| Gene expression |
|
| Cytogenetics |
|
Carcinogenesis
Early studies identified that many carcinogens were NAT substrates (Bartsch et al., 1973). It was debated whether the drug-metabolizing N-acetyltransferase (see Weber and Hein, 1985) was the same enzyme as the carcinogen-activating acetylating activity, which catalysed N-acetylation, O-acetylation and N,O acetyl transfer in hydroxamates (Figure 1; King, 1974). The elegant work of Hanna (Hanna, 1994) in this controversy provided balance and has further defined the mechanism of protein adduct formation with human NAT1 (Liu et al., 2009). Recently, N- and O- acetylation activity in preserved human hepatocytes has been carried out (Doll et al., 2010).
Cloning the N-hydroxylamino O-acetyltransferase from Salmonella typhimurium was a major step towards showing that this activity and NAT in humans were homologues (Watanabe et al., 1992). Strains of S. typhimurium overexpressing its own nat gene were more sensitive in the Ames test for carcinogens (Watanabe et al., 1987) and eukaryotic cells showed increased carcinogenesis when human NATs were overexpressed (Watanabe et al., 1994).
Bladder cancer
Bladder cancer is a frequently occurring occupational and smoking-related cancer (Letašiová et al., 2011 overview). In bladder cancer, there are many links with NAT. The link between bladder cancer and exposure to NAT substrates has been known for over a century (Rehn, 1895; see Dietrich and Dietrich, 2001). The phenotypic link of NAT2 slow acetylation to bladder cancer susceptibility (Cartwright et al., 1982) has been confirmed by genotyping studies (Risch et al., 1995) and has been reviewed (Vineis et al., 2001; Hein, 2002). Interestingly, NAT2 is barely expressed in bladder epithelium although NAT1 is. In some bladder tumours, NAT1 is reduced (Stanley et al., 1996) but no systematic study has been carried out.
The location of NAT genes in a hot spot on chromosome 8 (Knowles et al., 1993; Spurr et al., 1995; Hubbard et al., 1997) prompted comparison in bladder tumour and normal DNA of NAT genes in this unstable region (Stacey et al., 1996; 1997; 1999; Watters et al., 1998). Loss of heterozygosity (Thygesen et al., 1999) and gene duplication were detected but whether these changes are causative remains to be established. This region was not highlighted in a whole genome analysis (Kiemenev et al., 2008).
Colorectal cancer
The link between NAT and colorectal cancer has had a chequered history. The NAT 1*10 allele association with increased activity stimulated many studies (Bell et al., 1995). A meta-analysis of 59 studies (Liu et al., 2012) appears to confirm that there is no link between NAT1 or NAT2 polymorphism and colorectal cancer. Each of these NAT proteins is detected in the colon and the availability of specific NAT1 antibodies (Hickman et al., 1998) has clearly demonstrated staining in the crypts of the colon. This study showed the ratio of NAT1 to NAT2 among individuals varies up to 70-fold. The activity of human NAT1 in the gut has implications also for the treatment of inflammatory bowel disease (IBD) which is a prediposing factor in colon cancer. IBD is treated with 5-aminosalicylate (5-AS), which is a substrate for human NAT1 and human NAT2 and this will be considered below in relation to NAT as a target for novel drug development.
NAT and breast cancer
From proteomic and genomic studies, human NAT1 but not NAT2 was found to be overexpressed in oestrogen receptor-positive female breast cancer (Adam et al., 2003; see Sim et al., 2008a) and more recently in male breast cancer (Johansson et al., 2012). NAT2 is only marginally expressed in any breast tissue.
Understanding NAT1 overexpression in breast cancer and studies in other endocrine tumours (Butcher et al., 2007; Butcher and Minchin, 2010) has made use of cell lines (Wakefield et al., 2008a). The human breast cancer cell line ZR75 has an extremely high level of human NAT1, which is neither due to gene duplication nor to a mutant version of human NAT1.
Studies with siRNA indicate a reduction in contact-inhibited growth when NAT1 is suppressed (Tiang et al., 2011; see Butcher and Minchin, 2012). Up-regulation of NAT1 in breast cancer has been linked with improved response to tamoxifen (Bièche et al., 2004) and it has interestingly been observed that human NAT1 (Lu et al., 2001; Lee et al., 2004) and its mouse equivalent (Kawamura et al., 2008) is inhibited by tamoxifen and also bisphenol A.
There is no difference in the proportion of fast acetylating individuals for human NAT2 between a breast cancer and control group (Webster et al., 1989) and this finding illustrates further that the relationship between NAT and breast cancer involves human NAT1 only.
Human NAT1 and NAT2 in development
In adults, human NAT1 has a much wider tissue distribution than human NAT2. During development, the difference is even more marked. In placentae, human NAT1 is found throughout pregnancy (Smelt et al., 1998; 2000; Upton et al., 2000) up to term (Derewlany et al., 1994a,b). Foetal tissues have human NAT1 activity (Pacifici et al., 1986) and NAT1 transcripts were detected at the four-cell stage and in blastocysts (Smelt et al., 2000). Human NAT2 could not be detected in these studies.
Folate is essential for normal human development and a link between human NAT1 and folate was hinted at by the inverse correlation between red blood cell folate and human NAT1 activity in red cells (Ward et al., 1992). Human NAT1 was shown to acetylate the folate catabolite para-aminobenzoylglutamate (p-abaglu) in lymphocytic cell extracts (Minchin, 1995), in placenta (Upton et al., 2000) and using recombinant human NAT1 (Ward et al., 1995) but not human NAT2 (Kawamura et al., 2004).
The physiological role of human NAT1 has raised many questions (see Minchin et al., 2007) as apparently healthy adult individuals with two defective human NAT1 alleles exist (Ward et al., 1992; Hughes et al., 1998; Payton and Sim, 1998). It was necessary to establish a model system in order to study further the possible relationship between folate and NAT and to decipher the role of human NAT1 in development.
Mouse NATs
Although mouse liver had proved to be a poor choice for purification of Nat enzymes (Watson et al., 1990), mouse Nats were most suitable for recombinant protein studies and excellent for investigation of Nat in development. Two mouse genes, Nat1 and Nat2, which are over 80% identical, were initially cloned from fast and slow acetylating mouse strains with each having an intronless open reading frame (Martell et al., 1991; 1992). An upstream non-coding exon in mouse Nat2 has also been characterized (Boukouvala et al., 2003; see Sim et al., 2010).
Mouse Nat1 was identical in the fast and slow strains and showed specificity for isoniazid while mouse Nat2 catalysed p-aba acetylation and had a point mutation resulting in asparagine 99 in the fast strain and isoleucine in the slow strain resulting in a less stable enzyme (Martell et al., 1992; De Leon et al., 1995). A third functional mouse gene, Nat3 (Kelly and Sim, 1994), is highly polymorphic and less similar to the other genes (around 70% identity) but with very low activity (Fretland et al., 1997; Boukouvala et al., 2002). All three mouse genes are found in close proximity in the syngenic region to chromosome 8p22 where the human NAT genes are found (Hickman et al., 1994; Fakis et al., 2000).
The naming of mouse Nat2 is historical (Martell et al., 1991) but perversely has been retained (Hein et al., 2008). It is the functional equivalent of human NAT1; the substrate specificity profile of mouse Nat2 is like that of human NAT1 (Kawamura et al., 2008): the C-terminal sequence is the same as human NAT1 and mouse NAT2 is also recognized by human NAT1-specific antibodies raised against the C-terminus. This has allowed mouse NAT2 to be detected immunohistochemically in adult mouse tissues (Stanley et al., 1997) and traced during development (Stanley et al., 1998). Like its human counterpart, mouse NAT2 is found in early embryos in embryonic stem (ES) cells (Payton et al., 1999a). The Nat2 gene is expressed in the neonatal period unlike the other mouse Nat genes (Mitchell et al., 1999), again mirroring human NAT1. Studies of congenic mouse strains had identified the region around the Nat loci in mice as implicated in susceptibility to neural tube defects including cleft lip and palate (Karolyi et al., 1990) and intriguingly, mouse Nat2 is expressed in the developing neural tube (Stanley et al., 1998; Cornish et al., 2003; Figure 7).
Figure 7.
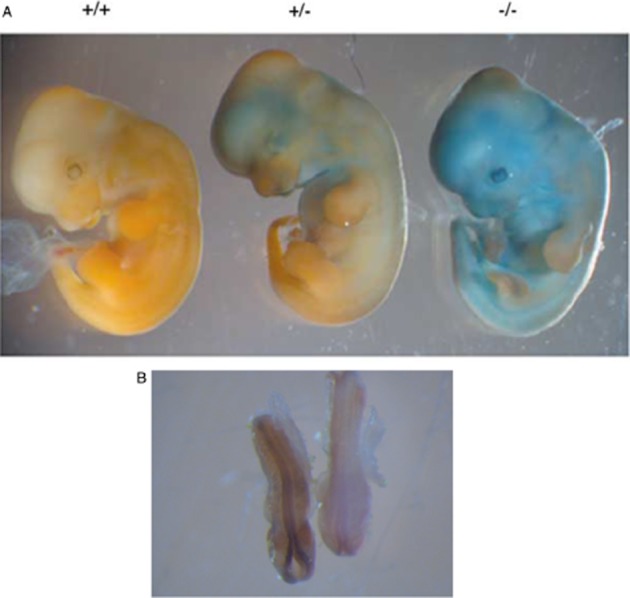
Generation of knockout mice. The Nat2 gene has been interrupted by a cassette in frame, which includes the β-galactosidase gene. Embryos can be stained with X-gal, which is converted to a blue colour in the knockout mice. Ashows 12.5 day embryos with wild type, heterozygote (Nat2+/−), a homozygote null (Nat2−/−). B shows 8.5 day whole mount embryo with wild type on the right and homozygote Nat2−/− on the left. Staining in the developing neural tube is marked. Panel A is from Cornish et al., 2003. Panel B was prepared by L. Wakefield.
Knowledge of the genetic region around the mouse Nat genes (Fakis et al., 2000) allowed creation of transgenic mice to study development. Many attempts were made to overexpress human NAT1 in mouse ES cells (Johnson, 2002; Sim et al., 2003). No normal mice were produced overexpressing human NAT1 following targeted insertion using a strong promoter, although two mice with neural tube defects were found as well as deformed embryos and evidence of resorption early in gestation (Sim et al., 2003). Random insertion of human NAT1 in mouse ES cells under the same strong promoter generated several mice overexpressing human NAT1 (Cao et al., 2005) but at a much lower level than expected. Overexpression of human NAT1 appears detrimental during development confirming a role for mouse Nat2 in teratogen-induced cleft lip and palate (Erickson et al., 2008; Erickson, 2010 for review). These studies mirror the link of human NAT1 *10 genotype to neural tube defects (Lammer et al., 2004; Jensen et al., 2005; 2006) but as discussed, the link between this allele and increased NAT1 activity, although attractive, is controversial.
It has been reasoned that excess human NAT1/mouse NAT2 is detrimental in mice during development due to increased folate catabolism driven by metabolism of the folate breakdown product p-abaglu (Wakefield et al., 2007a; Cao et al., 2010). These studies have been challenged recently but additional information linking human NAT1 and mouse NAT2 with folate has been described. These enzymes, but no other NATs tested, are specifically able to catalyse hydrolysis of acetyl CoA in the presence of folate (Rodrigues-Lima et al., 2011). Hydrolysis of acetyl CoA in all NATs tested is accelerated in the presence of an arylamine or hydrazine substrate – the basis of NAT assays (Brooke et al., 2003a). Folate itself is not a substrate for human NAT1 acetylation and these studies open up the possibility that human NAT1 and its homologue in mouse, Nat2, regulate homeostasis between acetyl CoA and folate pools. A direct link remains to be established but the observation explains why folate has been described as an inhibitor of human NAT1 (Ward et al., 1995) since it serves to deplete the cofactor acetyl CoA.
Knockout mice
Transgenic mice with mouse Nat2 deleted (Sugamori et al., 2003) or interrupted by insertion of lacZ in frame with the gene have been made (Cornish et al., 2003). The latter strain of mice allows lacZ expression under control of the mouse Nat promoter. Expression of mouse Nat2 can therefore be studied by staining whole embryos with the β-galactosidase substrate X-gal. This is illustrated dramatically in the skin of mice (Figure Figure 7) and adds further support to the similarity with human NAT1, which is also found in skin. Mouse Nat2 is expressed in embryos including in the developing heart in scattered clusters of cells including at the sino-atrial node (Wakefield et al., 2005;2008b). The detection of Nat2 expression in ES cells (Payton et al., 1999a) raises the question of whether it is cardiac stem cells, which are expressing Nat2. The expression of mouse Nat1 appears to be mainly restricted to liver and gut and is unmodified in the Nat2 −/− mice (Loehele et al., 2006).
Mice lacking Nat3 have been generated (Sugamori et al., 2007) but show no differences in drug metabolism. Mice lacking Nat1 and /or Nat2 appear healthy but show differences in drug metabolism (Sugamori et al., 2003) although there appears to be defects that emerge in long-term breeding programmes (Wakefield et al., 2007b). There appears to be a gender imbalance in the offspring on breeding heterozygotes (Cornish et al., 2003; Wakefield et al., 2008b). Gender differences in carcinogen activation have also been identified in mice with the Nat2 gene deleted (Sugamori et al., 2012).
Mouse NAT2 is found in adult mice in a similar distribution to the human NAT1 (Stanley et al., 1997) including in breast tissue (Williams et al., 2001; Wakefield et al., 2008b). The relationship between human NAT1 expression and hormonal control (Adam et al., 2003; Butcher et al., 2007) and the earlier studies on hormonal control of murine Nat (Estrada-Rodgers et al., 1998) may well be a key to the gender-linked asymmetry observed in transgenic mice and the understanding of epigenetic control of gene expression are areas of relative ignorance at present.
Structural studies
Early enzymology studies of Riddle and Jencks (1971) identified NAT activity involved formation of an acetylated enzyme intermediate in a mechanism known as ping-pong bi-bi. Cysteine was implicated in accepting the acetyl group from acetyl CoA as NAT was inhibited by classic cysteine reactive agents and by oxidative damage. One of us (ES) was particularly interested to study the NAT enzymes from experience gained with iron sulphur proteins (Sim and Vignais, 1979) although NAT turned out not to be an iron sulphur protein. Recent work has extended the oxidative sensitivity of the NAT enzymes to reactive oxygen species (see Dupret et al., 2005).
The sulphydryl reactive agents N-ethyl maleimide and bromacetanilide inhibited NAT (Andres et al., 1988; Watson et al., 1990) and it was clear from accumulated evidence (see Weber and Hein, 1985) that NAT had an active site cysteine residue. Identifying Cys69 as the active site in NAT from S. typhimurium (Watanabe et al., 1992) was followed by confirmation through site-directed mutagenesis that the equivalent residue in human NAT2 was Cys68 (Dupret and Grant, 1992).
The 3D structure of the NAT enzymes needed sufficient pure protein for crystallization and recombinant protein was the way forward. Following a well trodden path for drug-metabolizing enzymes, sufficient pure stable recombinant protein was obtained by cloning nat from S. typhimurium (Sinclair et al., 1998) after many attempts to generate sufficient human NAT as a recombinant enzyme for crystallization (Ward et al., 1995; Sinclair and Sim, 1997). While the yield of the S. typhimurium NAT was improved (Sinclair et al., 1998), its substrate specificity and characteristics were established (Figure 6). Labelling the active site cysteine with a bromacetanilide conferring selectivity included a 13C label and also 19F. The bromacetanilide binds covalently to the active site Cys (determined by mass spectrometry) and we hedged our bets with the possibility of probing the molecular environment of the active site by 13C and 19F NMR should it prove impossible to obtain crystals. It appeared we might have needed the NMR approach as on one particular rare occasion when there were diffractable crystals, the data collected at the Daresbury Synchrotron was destroyed by an electrical storm in transit to our Oxford computer. This was a particularly low point but we eventually obtained diffraction data with active S. typhimurium NAT labelled with selenomethionine to allow the structure of the unique protein fold to be solved (Sinclair et al., 2000).
When the structure was obtained, it was worth waiting for (Figure 8). The active site Cys with its specificity label was visible juxtaposed to a histidine residue and an aspartate residue – a clearly identifiable catalytic triad, which activates the Cys in order to allow the first step of catalysis in which the Cys residue becomes acetylated before being transferred to the incoming arylamine. The catalytic triad was obvious in all NATs and appears essential for activity (Sandy et al., 2005a): indeed in one of the NAT2 slow variants, the Asp of the catalytic triad has been mutated to Asn and is inactive. In all subsequent structures of NATs obtained, the catalytic triad residues are superimposable.
Figure 8.
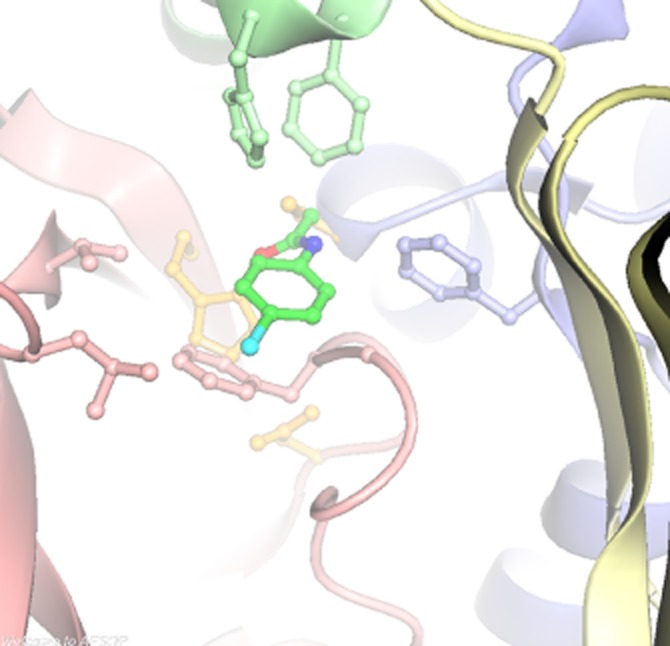
Active site of S. typhimurium NAT. The residues forming the catalytic triad are shown in pale yellow Cys69, His107 and Asp122. The active site Cys69 has been labelled with bromacetanilide (bright green). The His residue is behind the labelled Cys, and the Asp residue is in front. The pale pink, green and blue residues correspond to the three domains of the NAT structure.
Solving the protein fold allowed excellent work to be done in solving the complete structure of mammalian NAT by NMR (Zhang et al., 2006) and then the same bromacetanilide approach was used (but without the 19F and 13C labels) in studies from the Structural Genomics Consortium along with site-directed mutagenesis to improve the stability of the human NAT1 protein (Wu et al., 2007). The improvement in stability of human NAT1 was based on comparing the substrate specificity and stability of a series of chimeras of human NAT1 and human NAT2 (Goodfellow et al., 2000). The importance of residues 125–127 were identified in a landmark study.
Comparison of the crystal structures of human NAT1 and the model of human NAT2 rationalized the substrate specificity profiles of the two enzymes. Interestingly, modifying only one residue in NAT1 (phenylalanine 125 to serine; F125S) alters the specificity such that it resembles human NAT2 in which residue 125 is serine. The same is true of the mouse equivalent enzyme to human NAT1, Nat2, its specificity is altered when residue 125 is mutated from phenylalanine to serine (F125S) (Laurieri et al., 2010).
The NAT enzyme proteins, which are between 280 and 300 amino acids long (Sandy et al., 2005a; Vagena et al., 2008), consist of three almost equivalently sized domains, an α-domain; a β-domain and an α/β-C-terminal domain All active site Cys, His107 and Asp122 residues are within the first two domains and a third α-β lid folds over the active site. The length and role of the third C-terminal domain differs most among NATs. The C-terminus contributes to substrate recognition because in human NAT1, removal of the third domain results in a protein that hydrolyses acetyl CoA readily with no arylamine substrate present (Sinclair and Sim, 1997). The same is true of the enzyme from S. typhimurium (Mushtaq et al., 2002) when either the entire third domain or even the C-terminal 11 amino acids are missing. Thus, the interaction with substrate and the interaction with cofactor can be separated. Folate, which mediates hydrolysis specifically in human NAT1 and mouse NAT2, may bind to effectively unpeel the third domain of NAT from the active site. More work is needed to confirm this interaction as there is no crystal structure with folate bound. New modelling data support folate binding (Laurieri et al., 2014).
The role of the C-terminus also varies in relation to cofactor binding. The structure of human NAT 1 with CoA bound (Wu et al., 2007) and of an enzyme from Mycobacterium marinum with CoA bound (Fullam et al., 2008) and from Bacillus anthracis (Pluvinage et al., 2011) demonstrate that CoA can bind in different positions in relation to the C-terminus. Interestingly, the NAT enzyme from B. anthracis was shown as having CoA bound although the recombinant enzyme had been through several purification steps and had not been incubated with CoA (Pluvinage et al., 2011).
NATs have been crystallized from many organisms and in some cases with substrate bound including hydralazine (Abuhammad et al., 2011) or isoniazid (Sandy et al., 2005b). The ping-pong bi-bi mechanism implies that acetyl CoA binds first but there is clear evidence from crystallography and also from NMR (Delgoda et al., 2003; Kawamura et al., 2008) studies that substrate can be bound without prior binding of acetyl CoA.
NAT in bacteria
Finding NAT in bacteria prompted the question of whether there might be a NAT homologue in mycobacteria to metabolize the anti-tuberculosis agent isoniazid.
Isoniazid is a substrate of human NAT2 and isoniazid is a prodrug activated by katG in M. tuberculosis. Using gridded libraries from GlaxoSmithKline, we demonstrated that a nat gene was present and expressed in both M. smegmatis and M. tuberculosis (Payton et al., 1999b; 2001a,b). The genome of M. tuberculosis was published around the same time (Cole et al., 1998) and subsequent studies showed the gene in M. bovis BCG and M. tuberculosis were identical (see Sim et al., 2008b). The nat gene in mycobacteria is in a gene cluster, which is essential for cholesterol catabolism for intracellular fuel in these organisms (Van der Geize et al., 2007). The gene cluster, including the nat gene, is essential for intracellular survival of mycobacteria inside macrophage (Bhakta et al., 2004; Anderton et al., 2006; Yam et al., 2009).
A subgroup of M. tuberculosis strains, which had an accumulation of mutations, also had mutations in the nat gene, which contributed marginally to isoniaizid resistance in clinical isolates (Upton et al., 2001; Sholto-Douglas-Vernon et al., 2005). Overexpressing nat experimentally in mycobacteria, it was shown that NAT increases resistance to isoniazid (Payton et al., 1999a; Bhakta et al., 2004) but is not the sole factor. Making a nat gene deleted strain of M. bovis BCG, resulted in an approximately threefold increase in sensitivity to isoniazid. These gene deletion studies however showed a very exciting finding – that the nat deleted strain was defective in cell wall synthesis as well as being essential for intracellular survival of M. bovis BCG (Bhakta et al., 2004). These studies strongly suggested that the nat gene product from M. tuberculosis was a good drug target for anti-tuberculosis therapy.
We had extreme difficulty in generating large quantities of the NAT from M. tuberculosis (TBNAT) as a recombinant protein (Upton et al., 2001; Abuhammad et al., 2012) as did others (Sikora et al., 2008). While we were improving the yield of the TBNAT enzyme, we used NAT from M. smegmatis (Sandy et al., 2005b) and then M. marinum (Fullam et al., 2008), which is much closer in sequence to the NAT from M. tuberculosis. M. marinum NAT is very soluble while the M. tuberculosis NAT is not (Fullam et al., 2009; Abuhammad et al., 2013 in press) and they show very different physical properties (Lack et al., 2009), including melting at different temperatures. The melting property resides in the first two domains of the protein and may reflect the ability of TBNAT to remain active in inflammatory conditions associated with intracellular infection.
We have at last, after 15 years, obtained a crystal structure of the NAT from M. tuberculosis by seeding a preparation of M. tuberculosis NAT with a crystal of NAT from M. marinum (Abuhammad et al., 2013). The substrate specificities of the M. marinum and M. tuberculosis NAT enzymes are not identical and this illustrates the importance in drug discovery of using an appropriate model or at least understanding the differences when a surrogate has to be used. While M. marinum NAT has been useful as a model, it emphasizes the need to have the target protein available for developing specific inhibitors (Fullam et al., 2009).
NAT as drug target
Development of NAT ligands
We embarked on a search for specific NAT isoenzymes in 2003 to seek possible anti-tuberculosis agents and coupled that with a search for human NAT1 inhibitors. Up-regulation of human NAT1 in breast cancer made the search for NAT inhibitors an attractive prospect, which has recently been reinforced (Butcher and Minchin, 2012). We were also interested in IBD because the presence of NATs in the gut epithelium had been established by immunohistochemistry (Hickman et al., 1998) and NAT is involved in metabolic inactivation of the drug used to treat IBD, namely 5-AS. Therefore, there might be a benefit in IBD from co-administration of 5-AS and NAT inhibitors.
Screening for NAT inhibitors
An automatable assay was established (Brooke et al., 2003a) which could be adapted to detect inhibitors (Brooke et al., 2003b). A library of some 5000 compounds was available (Russell et al., 2009) and we assembled the other tools required – sufficient of all of the enzymes we wished to use. We produced hundreds of milligrams of each of human NAT1 (Wang et al., 2005), hamster NAT2 (Kawamura et al., 2004), S. typhimurium NAT (Sinclair et al., 1998), M. smegmatis NAT (Sandy et al., 2002) and P. aeruginosa NAT (Westwood et al., 2005). The mammalian enzymes were to allow any possible toxicity to be identified early but generated very interesting results.
We compared the substrate specificity profiles of each of these enzymes (Westwood et al., 2006) (Figure Figure 6) and other NATs used in later stages of sophistication in the screen, which were available in lower quantities (Kawamura et al., 2004; 2008; Fullam et al., 2009). The screen (Westwood et al., 2011) identified compounds inhibiting either the eukaryotic enzymes or the prokaryotic enzymes specifically (Figure 7). All hits from the screen were resynthesized as in one case the isomer in the screen had been incorrectly annotated (Fullam et al., 2011).
Inhibitors and their development as potential antituberculars
Following duplicate screening, compounds inhibiting prokaryotic NAT enzymes specifically with an IC50 value of less than 10 μM were tested for inhibition of growth of mycobacteria and of Escherichia coli. Of those that initially looked interesting (Fullam et al., 2008; 2013; Westwood et al., 2010; Abuhammad et al., 2012), we have studied two of these classes in detail with structure activity analyses, identifying the mechanism of inhibition.
In the triazole class, which shows competitive inhibition with substrate, structure activity relationship (SAR) analysis of anti-mycobacterial activity and NAT inhibition are coincident and modelling of binding to the active site of TBNAT readily explains the SAR (Westwood et al., 2010). The effects of the triazole class on M. bovis BCG phenotype was very similar to that of deleting the nat gene (Bhakta et al., 2004), including inhibiting growth of M. bovis BCG inside macrophage without showing cytotoxicity for the macrophage. Based on these findings, a commercial company, now known as Summit plc, used a similar screening mechanism and generated compounds that were developed to the point of out-licencing as anti-tuberculosis leads.
The piperidinols have recently been shown to bind covalently, effectively acting in a prodrug-type manner (Figure 9). Covalent binding is a feature of excellent drugs for tuberculosis including the recently identified benzothiazines (Batt et al., 2012) as well as isoniazid, the front line prodrug. The piperidinol class was our most effective hit in inhibiting growth of M. tuberculosis and was equally effective at blocking growth of M. tuberculosis in which the nat gene is deleted (Abuhammad et al., 2012). This is now seen as a benefit as polypharmacy (as it is called) is a feature of many excellent and long-surviving drugs such as aspirin. It is good to see the demise of the dogma that a lead compound was only worth investigating if it could be demonstrated to be against only one identifiable target. A broader minded attitude is beginning to allow a flourishing pipeline for novel anti-tuberculosis agents (http://www.tballiance.org/). In view of the ever present possibility of the development of resistance, it is important that all promising targets are pursued as well as the return to the whole cell approach. Our own work has now begun to focus on another of the gene products of the ‘nat’ gene cluster in M. tuberculosis, namely, HsaD (Lack et al., 2009).
Figure 9.
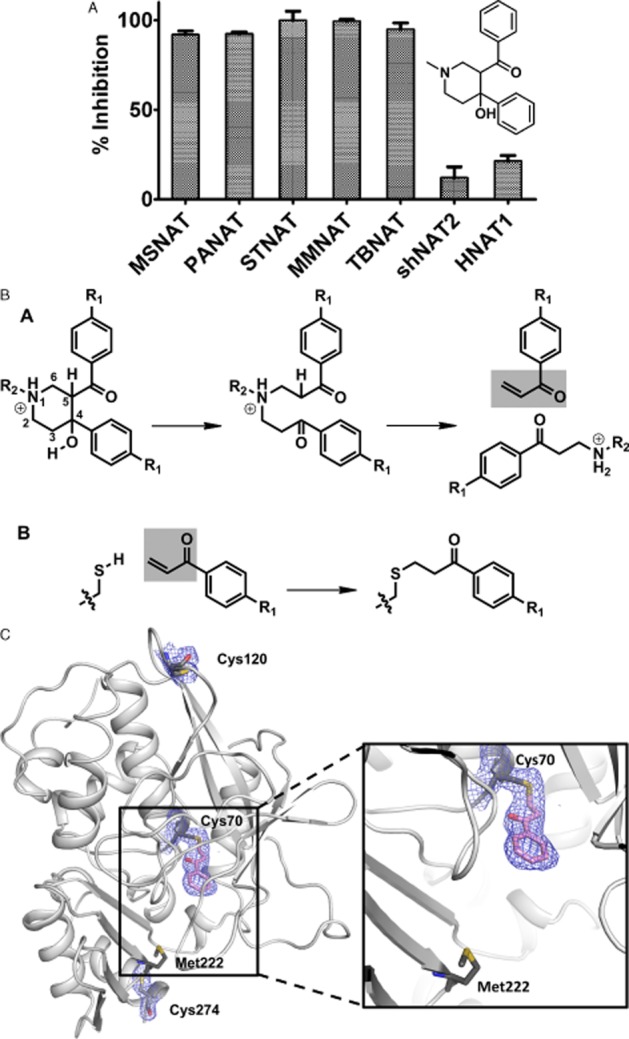
Inhibition of mycobacterial NAT by piperidinol. (A) The piperidinol inhibitor shows specificity for prokaryotic enzymes. (B) The mechanism of the reaction for the piperidinol inhibitor to NAT is through a chemical transformation to the corresponding phenyl vinyl ketone (PVK), which then binds to the active site Cys residue. C shows the inhibitor bound to the active site Cys residue. Electron density is shown around another Cys, which has no such modification.
We have also looked at the effects of plant extracts and found a terpenoid from the traditional medicine, the pepper bark tree (Warburgia salutaris), which appears to act through inhibition of NAT from mycobacteria. However, the extract was not very potent (Madikane et al., 2007).
Inhibitors and their development for use in cancer
Of the compounds identified as specific for eukaryotic NATs, we rescreened to identify those which inhibited human NAT1 and its homologues from other mammals. We pursued only two classes of compound that were effective at inhibiting human NAT1 in breast cancer cell extracts (Russell et al., 2009). One of the compounds was found to be generally cytotoxic, a rhodanine (Russell et al., 2009), but it has been exploited as a potential chemotherapeutic class (Tiang et al., 2010; see Butcher and Minchin, 2012). The Australian group are pushing forward with this approach and have also identified NAT as a breast cancer target using siRNA (Tiang et al., 2011). Using low MW inhibitors has proved difficult as a result of solubility issues.
In our own studies, a group of human NAT1-specific compounds, naphthoquinones, have proved very interesting. These compounds specifically change colour on binding to human NAT1 and to the mouse homologue mouse Nat2. The change of colour is due to a proton transfer between the protein and the compound and site-directed mutagenesis studies have allowed identification of the protein residues involved (Laurieri et al., 2010; 2013). This compound shows the same colour change with mouse NAT2 but not with any of the other NAT enzymes. To use this for detecting human NAT1 as a biomarker sensitivity will need to be improved and we are currently making fluorescent versions of the naphthoquinone (Egleton, 2012).
Inhibitors of NAT and potential in IBD
5-AS is used to treat IBD and it was reasoned that if the acetylation of 5-AS could be inhibited then this provides a method for improving the amount of p-AS, which has be generated in the treatment of IBD. 5-AS is metabolized by N-acetylation and while NATs in gut bacteria are very good at acetylating 5-AS (Delomenie et al., 2001; Westwood et al., 2006), the human enzyme human NAT1 as well as NAT2 also acetylate 5-AS.
As 5-AS is mostly given as a prodrug, balsalazide or sulphasalazine, with 5-AS released by azoreductase in the gut flora, it was necessary to understand whether potential NAT inhibitors would also inhibit the gut flora azoreductases. We commenced work on azoreductases during the sabbatical leave of the corresponding author in Dundee with Mike Coughtrie in 2005. Following bioinformatic analysis, we made recombinant azoreductases from P. aeruginosa, which are very soluble flavoproteins and crystallize readily (Wang et al., 2007). Structures of azoreductase with balasalazide bound show clearly that the relative position of the substrate in relation to the isoalloxazine ring nitrogen atoms relies on a proton relay system within the substrate resulting in the reduction of the diazo bond in balsalazide (Ryan et al., 2010a). We have also demonstrated that the antibiotic nitrofurazone is reduced by azoreducatse with the electron flow through the substrate from the flavin initially reducing the nitroso group (Ryan et al., 2011; Figure 10).
Figure 10.

Binding of substrates to azoreductase paAzoR1 from Pseudomonas aeruginosa. (A) Balsalazide (from Ryan et al., 2010a) structure was solved to 2.3 Å. Balsalazide is shown in purple and FMN cofactor is shown in yellow. (B) Nitrofurazone (Ryan et al., 2011). Structure was solved at 2.08 Å. Nitrofurazone is in brown while stably bound water molecule involved in reduction is a black ball. C shows the tautomerization and charge relay leading to reduction of the nitro group.
It was proposed (Ryan et al., 2010b) that the azoreductases would reduce quinones and we found in separate experiments that the naphthoquinone inhibitor of human NAT1 was indeed an inhibitor of azoreductase. To expand the diversity of NAT1 inhibitors, we have used the 3D shape of our best inhibitor, the naphthoquinone, to screen against shapes of all purchasable chemical space – some 35 million compounds. From a random selection of 23 compounds from the top 100 hits, almost half (10 compounds) had an IC50 of less than 10 μM (Ballester et al., 2010). Although none was better than the probe compound, the diversity of chemical classes made this a positive approach to diversification in ligand identification.
Acknowledgments
This review is dedicated to the memory of Edward W. Gill. E. S. is extremely grateful to be awarded the JR Vane medal and to the many funding bodies who have supported her work. The University of Jordan is thanked for a studentship (A. A.). E. S. thanks unreservedly all of her colleagues – technicians, project students, visitors, master's students, doctoral students and postdocs who have worked in the labs in Oxford and now in Kingston. E. S. is profoundly grateful to former mentors, especially Rodney Porter and Bill Paton as well as friends, collaborators and colleagues who have shared their ideas and acted as sounding boards for many of the more outlandish research plans. E. S. acknowledges the unstinting support and tolerance of family members both immediate (B., F., G.) and extended because without them it would have been much less fun. Who else would have succeeded in making a molecule of acetyl CoA from cupcakes!
Glossary
- 5-AS
5-aminosalicylate
- ES
embryonic stem
- IBD
inflammatory bowel disease
- MTP
methyl triazolophthalazine
- NAT
arylamine N-acetyltransferase
- p-aba
para-aminobenzoic acid
- p-abaglu
para-aminobenzoylglutamate
- p-AS
para-aminosalicylate
- SAR
structure activity relationship
- SLE
systemic lupus erythematosus
- SNP
single nucleotide polymorphism
Conflict of interest
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Collaborators and co-workers.
References
- Abuhammad A, Lowe E, Fullam E, Noble M, Garman E, Sim E. Probing the architecture of Mycobacterium marinum N-acetyltransferase active site. Protein Cell. 2010;1:384–392. doi: 10.1007/s13238-010-0037-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abuhammad A, Lack N, Schweichler J, Staunton D, Sim RB, Sim E. Improvement of the expression and purification of Mycobacterium tuberculosis arylamine N-acetyltransferase (TBNAT) a potential target for novel anti-tubercular agents. Protein Expr Purif. 2011;80:246–252. doi: 10.1016/j.pep.2011.06.021. [DOI] [PubMed] [Google Scholar]
- Abuhammad A, Fullam E, Lowe ED, Staunton D, Kawamura A, Sim E. Piperidinols that show anti-tubercular activity are inhibitors of arylamine N-acetyltransferase: an essential enzyme for mycobacterial survival inside macrophages. PLoS ONE. 2012;7:e52790. doi: 10.1371/journal.pone.0052790. doi: 10.1371/journal.pone.0052790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abuhammad A, Lowe ED, McDonough MA, Shaw Stewart PD, Kolek SA, Sim E, Garman EF. Structure of arylamine N-acetyltransferase from Mycobacterium tuberculosis determined by cross-seeding with the homologous protein from M. marinum: triumph over adversity. Acta Crystallogr D Biol Crystallogr. 2013;69:1433–1446. doi: 10.1107/S0907444913015126. [DOI] [PubMed] [Google Scholar]
- Adam PJ, Berry J, Loader JA, Tyson KL, Craggs G, Smith P, et al. NAT-1 is highly expressed in breast cancers and conveys enhanced growth resistance to etoposide in vitro. Mol Cancer Res. 2003;1:826–835. [PubMed] [Google Scholar]
- Anderton MC, Bhakta S, Besra GS, Jeavons P, Eltis LD, Sim E. Characterization of the putative operon containing arylamine N-acetyltransferase (nat) in Mycobacterium bovis BCG. Mol Microbiol. 2006;59:181–192. doi: 10.1111/j.1365-2958.2005.04945.x. [DOI] [PubMed] [Google Scholar]
- Andres HH, Vogel RS, Tarr GE, Johnson L, Weber WW. Purification, physicochemical, and kinetic properties of liver acetyl-CoA:arylamine N-acetyltransferase from rapid acetylator rabbits. Mol Pharmacol. 1987;31:446–456. [PubMed] [Google Scholar]
- Andres HH, Klem AJ, Schopfer LM, Harrison JK, Weber WW. On the active site of liver acetyl-CoA. Arylamine N-acetyltransferase from rapid acetylator rabbits (III/J) J Biol Chem. 1988;263:7521–7527. [PubMed] [Google Scholar]
- Ballester P, Westwood IM, Laurieri N, Sim E, Richards WG. Prospective virtual screening with Ultrafast Shape Recognition: the identification of novel inhibitors of arylamine N-acetyltransferases. J R Soc Interface. 2010;7:335–342. doi: 10.1098/rsif.2009.0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DF, Husain A, Neale JR, Martini BD, Zhang X, Doll MA, et al. Functional properties of an alternative tissue-specific promoter for human arylamine N-acetyltransferase1. Pharmacogenet Genomics. 2006;16:515–525. doi: 10.1097/01.fpc.0000215066.29342.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch H, Dworkin C, Miller EC, Miller JA. Formation of electrophilic N-acetoxyarylamines in cytosoles from rat mammary gland and other tissues by transacetylation from the carcinogen N-hydroxy-4-acetylaminobiphenyl. Biochim Biophys Acta. 1973;304:42–45. doi: 10.1016/0304-4165(73)90113-x. [DOI] [PubMed] [Google Scholar]
- Batchelor JR, Welsh KI, Mansilla Tinoco R, Dollery CT, Hughes GRV, Bernstein R. Hydralazine-induced systemic lupus erythematosus: influence of HLA-DR and sex on susceptibility. Lancet. 1980;315:1107–1109. doi: 10.1016/s0140-6736(80)91554-8. [DOI] [PubMed] [Google Scholar]
- Batt SM, Jabeen T, Bhowruth V, Quill L, Lund PA, Eggeling L, et al. Structural basis of inhibition of Mycobacterium tuberculosis DprE1 by benzothiazinone inhibitors. Proc Natl Acad Sci U S A. 2012;109:11354–11359. doi: 10.1073/pnas.1205735109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell DA, Stephens EA, Catranio T, Umbach DM, Watson M, Deakin M, et al. Polyadenylation polymorphism in the acetyltransferase 1 gene (NAT1) increases risk of colorectal cancer. Cancer Res. 1995;55:3537–3542. [PubMed] [Google Scholar]
- Bhakta S, Besra GS, Upton AM, Parish T, Sholto-Douglas-Vernon C, Gibson KJ, et al. Arylamine N-acetyltransferase is required for synthesis of mycolic acids and complex lipids in Mycobacterium bovis BCG and represents a novel drug target. J Exp Med. 2004;199:1191–1199. doi: 10.1084/jem.20031956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bièche I, Girault I, Urbain E, Tozlu S, Lidereau R. Relationship between intratumoral expression of genes coding for xenobiotic-metabolizing enzymes and benefit from adjuvant tamoxifen in estrogen receptor alpha-positive postmenopausal breast carcinoma. Breast Cancer Res. 2004;6:R252–R262. doi: 10.1186/bcr784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum M, Grant DM, McBride W, Heim M, Meyer UA. Human arylamine N-acetyltransferase genes: isolation, chromosomal localization, and functional expression. DNA Cell Biol. 1990;9:193–203. doi: 10.1089/dna.1990.9.193. [DOI] [PubMed] [Google Scholar]
- Blum M, Demierre A, Grant DM, Heim M, Meyer UA. Molecular mechanism of slow acetylation of drugs and carcinogens in humans. Proc Natl Acad Sci U S A. 1991;88:5237–5241. doi: 10.1073/pnas.88.12.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukouvala S, Sim E. Structural analysis of the genes for human arylamine N-acetyltransferases and characterisation of alternative transcripts. Basic Clin Pharmacol Toxicol. 2005;96:343–351. doi: 10.1111/j.1742-7843.2005.pto_02.x. [DOI] [PubMed] [Google Scholar]
- Boukouvala S, Price N, Sim E. Identification and functional characterization of novel polymorphisms associated with the genes for arylamine N-acetyltransferases in mice. Pharmacogenetics. 2002;12:385–394. doi: 10.1097/00008571-200207000-00006. [DOI] [PubMed] [Google Scholar]
- Boukouvala S, Price N, Plant KE, Sim E. Structure and transcriptional regulation of the Nat2 gene encoding for the drug-metabolising enzyme arylamine N-acetyltransferase type 2 in mice. Biochem J. 2003;375:593–602. doi: 10.1042/BJ20030812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooke EW, Davies SG, Mulvaney AW, Pompeo F, Sim E, Vickers RJ. An approach to identifying novel substrates of bacterial arylamine N-acetyltransferases. Bioorg Med Chem. 2003a;11:1227–1234. doi: 10.1016/s0968-0896(02)00642-9. [DOI] [PubMed] [Google Scholar]
- Brooke EW, Davies SG, Mulvaney AW, Okada M, Pompeo F, Sim E, et al. Synthesis and in vitro evaluation of novel small molecule inhibitors of bacterial arylamine N-acetyltransferases. Bioorg Med Chem Lett. 2003b;13:2527–2530. doi: 10.1016/s0960-894x(03)00484-0. [DOI] [PubMed] [Google Scholar]
- Butcher NJ, Minchin RF. Arylamine N-acetyltransferase 1 gene regulation by androgens requires a conserved heat shock element for heat shock factor-1. Carcinogenesis. 2010;31:820–826. doi: 10.1093/carcin/bgq042. [DOI] [PubMed] [Google Scholar]
- Butcher NJ, Minchin RF. Arylamine N-acetyltransferase 1: a novel drug target in cancer development. Pharmacol Rev. 2012;64:147–165. doi: 10.1124/pr.110.004275. [DOI] [PubMed] [Google Scholar]
- Butcher NJ, Ilett KF, Minchin RF. Functional polymorphism of the human arylamine N-acetyltransferase type 1 gene caused by C190T and G560A mutations. Pharmacogenetics. 1998;8:67–72. doi: 10.1097/00008571-199802000-00009. [DOI] [PubMed] [Google Scholar]
- Butcher NJ, Arulpragasam A, Minchin RF. Proteasomal degradation of N-acetyltransferase 1 is prevented by acetylation of the active site cysteine: a mechanism for the slow acetylator phenotype and substrate-dependent down-regulation. J Biol Chem. 2004;279:22131–22137. doi: 10.1074/jbc.M312858200. [DOI] [PubMed] [Google Scholar]
- Butcher NJ, Tetlow NL, Cheung C, Broadhurst GM, Minchin RF. Induction of human arylamine Nacetyltransferase type I by androgens in human prostate cancer cells. Cancer Res. 2007;67:85–92. doi: 10.1158/0008-5472.CAN-06-2635. [DOI] [PubMed] [Google Scholar]
- Cao W, Chau B, Hunter R, Strnatka D, McQueen CA, Erickson RP. Only low levels of exogenous N-acetyltransferase can be achieved in transgenic mice. Pharmacogenomics J. 2005;5:255–261. doi: 10.1038/sj.tpj.6500319. [DOI] [PubMed] [Google Scholar]
- Cao W, Strnatka D, McQueen CA, Hunter RJ, Erickson RP. N-acetyltransferase 2 activity and folate levels. Life Sci. 2010;86:103–106. doi: 10.1016/j.lfs.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright RA, Rogers HJ, Barham-Hall D, Glashan RW, Ahmad RA, Higgins E, et al. Role of N-acetyltransferasre phenotypes in bladder carcinogenesis: a pharmacogenetic epidemiological approach to bladder cancer. Lancet. 1982;320:842–846. doi: 10.1016/s0140-6736(82)90810-8. [DOI] [PubMed] [Google Scholar]
- Cascorbi I, Drakoulis N, Brockmöller J, Maurer A, Sperling K, Roots I. Arylamine N-acetyltransferase (NAT2) mutations and their allelic linkage in unrelated Caucasian individuals: correlation with phenotypic activity. Am J Hum Genet. 1995;257:581–589. [PMC free article] [PubMed] [Google Scholar]
- Cascorbi I, Roots I, Brockmöller J. Association of NAT1 and NAT2 polymorphisms to urinary bladder cancer: significantly reduced risk in subjects with NAT1*10. J Cancer Res. 2001;61:5051–5056. [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Cornish VA, Pinter K, Boukouvala S, Johnson N, Labrousse C, Payton M, et al. Generation and analysis of mice with a targeted disruption of the arylamine N-acetyltransferase Type 2 Gene. Pharmacogenomics J. 2003;3:169–177. doi: 10.1038/sj.tpj.6500170. [DOI] [PubMed] [Google Scholar]
- De Leon JH, Martell KJ, Vatsis KP, Weber WW. Slow acetylation in mice is caused by a labile and catalytically impaired mutant N-acetyltransferase (NAT2 9) Drug Metab Dispos. 1995;23:1354–1361. [PubMed] [Google Scholar]
- Deguchi T, Mashimo M, Suzuki T. Correlation between acetylator phenotypes and genotypes of polymorphic arylamine N-acetyltransferase in human liver. J Biol Chem. 1990;265:12757–12760. [PubMed] [Google Scholar]
- Delgoda R, Lian L-Y, Sandy J, Sim E. NMR investigation of the catalytic mechanism of arylamine N-acetyltransferase from Salmonella typhimurium. Biochemica et Biophysica Acta. 2003;1620:8–14. doi: 10.1016/s0304-4165(02)00500-7. [DOI] [PubMed] [Google Scholar]
- Delomenie C, Foux S, Laonguemaux S, Brahimi N, Bizet C, Picard B, et al. Identification and functional characterization of arylamine N-acetyltransferases in eubacteria: evidence for highly selective acetylation of 5-aminosalicylic acid. J Bacteriol. 2001;183:3417–3427. doi: 10.1128/JB.183.11.3417-3427.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derewlany LO, Knie B, Koren G. Human placental transfer and metabolism of p-aminobenzoic acid. J Pharmacol Exp Ther. 1994a;269:761–765. [PubMed] [Google Scholar]
- Derewlany LO, Knie B, Koren G. Arylamine N-acetyltransferase activity of the human placenta. J Pharmacol Exp Ther. 1994b;269:756–760. [PubMed] [Google Scholar]
- Dietrich H, Dietrich B. Ludwig Rehn (1849–1930) – pioneering findings on the aetiology of bladder tumours. World J Urol. 2001;19:151–153. doi: 10.1007/s003450100211. [DOI] [PubMed] [Google Scholar]
- Doll MA, Zang Y, Moeller T, Hein DW. Codominant expression of N-acetylation and O-acetylation activities catalyzed by N-acetyltransferase 2 in human hepatocytes. J Pharmacol Exp Ther. 2010;334:540–544. doi: 10.1124/jpet.110.168567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drayer DE, Reidenberg MM. Clinical consequences of polymorphic acetylation of basic drugs. Clin Pharmacol Ther. 1977;22:251–258. doi: 10.1002/cpt1977223251. [DOI] [PubMed] [Google Scholar]
- Dupret JM, Grant DM. Site-directed mutagenesis of recombinant human arylamine N-acetyltransferase expressed in Escherichia coli. Evidence for direct involvement of Cys68 in the catalytic mechanism of polymorphic human NAT2. J Biol Chem. 1992;267:7381–7385. [PubMed] [Google Scholar]
- Dupret JM, Dairou J, Atmane N, Rodrigues-Lima F. Inactivation of human arylamine N-acetyltransferase 1 by hydrogen peroxide and peroxynitrite. Methods Enzymol. 2005;400:215–229. doi: 10.1016/S0076-6879(05)00012-1. [DOI] [PubMed] [Google Scholar]
- Ebisawa T, Deguchi T. Structure and restriction fragment length polymorphism of genes for human liver arylamine N-acetyltransferases. Biochem Biophys Res Commun. 1991;177:1252–1257. doi: 10.1016/0006-291x(91)90676-x. [DOI] [PubMed] [Google Scholar]
- Egleton J. 2012. Ligands of arylamine N-acetyltransferase. Part II. Thesis Chemistry Oxford.
- Ellard GA, Gammon PT. Pharmacokinetics of isoniazid metabolism in man. J Pharmacokinet Biopharm. 1976;4:83–113. doi: 10.1007/BF01086149. [DOI] [PubMed] [Google Scholar]
- Erickson RP, Cao W, Acuna DK, Strnatka DW, Hunter RJ, Chau BT, et al. Confirmation of the role of N-acetyltransferase 2 in teratogen-induced cleft palate using transgenics and knockouts. Mol Reprod Dev. 2008;75:1071–1076. doi: 10.1002/mrd.20852. [DOI] [PubMed] [Google Scholar]
- Erickson RPJ. Genes, environment, and orofacial clefting: N-acetyltransferase and folic acid. Craniofac Surg. 2010;21:1384–1387. doi: 10.1097/SCS.0b013e3181ec6992. [DOI] [PubMed] [Google Scholar]
- Estrada-Rodgers L, Levy GN, Weber WW. Characterization of a hormone response element in the mouse N-acetyltransferase 2 (Nat2*) promoter. Gene Expr. 1998;7:13–24. [PMC free article] [PubMed] [Google Scholar]
- Evans DA, Manley KA, McKusch VA. Genetic control of isoniazid metabolism in man. Br Med J. 1960;2:485–491. doi: 10.1136/bmj.2.5197.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakis G, Boukouvala S, Buckle V, Payton M, Denning C, Sim E. Chromosome mapping of the genes for murine arylamine N-acetyltransferases (NATs). Enzymes involved in the metabolism of carcinogens: identification of a novel upstream noncoding exon for murine Nat2. Cytogenet Cell Genet. 2000;90:134–138. doi: 10.1159/000015648. [DOI] [PubMed] [Google Scholar]
- Franke S, Klawitz I, Schnakenberg E, Rommel B, Van de Ven W, Bullerdiek J, et al. Isolation and mapping of a cosmid clone containing the human NAT2 gene. Biochem Biophys Res Commun. 1994;199:52–55. doi: 10.1006/bbrc.1994.1192. [DOI] [PubMed] [Google Scholar]
- Fretland AJ, Doll MA, Gray K, Feng Y, Hein DW. Cloning, sequencing and recombinant expression of NAT1, NAT2 and NAT3 derived from the C3H/HeJ (rapid) and A/HeJ (slow) acetylator inbred mouse: functional characterization of the activation and detoxification of aromatic amine carcinogens. Toxicol Appl Pharmacol. 1997;1432:360–366. doi: 10.1006/taap.1996.8036. [DOI] [PubMed] [Google Scholar]
- Fullam E, Westwood IM, Anderton MC, Lowe ED, Sim E, Noble ME. Divergence of co-factor recognition across evolution: coenzyme A binding in a prokaryotic Arylamine N-acetyltransferase. J Mol Biol. 2008;375:178–191. doi: 10.1016/j.jmb.2007.10.019. [DOI] [PubMed] [Google Scholar]
- Fullam E, Kawamura A, Wilkinson H, Abu-Hammad A, Westwood I, Sim E. Comparison of the arylamine N-acetyltransferase from Mycobacterium marinum and Mycobacterium tuberculosis. Protein J. 2009;28:281–295. doi: 10.1007/s10930-009-9193-0. [DOI] [PubMed] [Google Scholar]
- Fullam E, Abuhammad A, Wilson D, Anderton M, Davies SG, Russell A, Sim E. Analysis of beta-amino alcohols as inhibitors of the potential anti-tubercular target N-acetyltransferase. Bioorg Med Chem Lett. 2011;15:1185–1190. doi: 10.1016/j.bmcl.2010.12.099. [DOI] [PubMed] [Google Scholar]
- Fullam E, Talbot J, Abuhammed A, Westwood IM, Davies SG, Russell AJ, et al. Design, synthesis and structure-activity relationships of 3,5-diaryl-1H-pyrazoles as inhibitors of arylamine N-acetyltransferase. Bioorg Med Chem Lett. 2013;23:2759–2764. doi: 10.1016/j.bmcl.2013.02.052. [DOI] [PubMed] [Google Scholar]
- Garte S, Carcorbi L, Alexandrie A-K, Ambrosone C, Autrup H, Autrup J-L, et al. Metabolic gene polymorphism frequencies in control populations. Cancer Epidemiol Biomarkers Prev. 2001;10:1239–1248. [PubMed] [Google Scholar]
- Goodfellow GH, Dupret JM, Grant DM. Identification of amino acids imparting acceptor substrate selectivity to human arylamine acetyltransferases NAT1 and NAT2. Biochem J. 2000;348:159–166. [PMC free article] [PubMed] [Google Scholar]
- Grant DM, Hughes NC, Janezic SA, Goodfellow GH, Chen HJ, Gaedigk A, Yu VL, Grewal R. Human acetyltransferase polymorphisms. Mutat Res. 1997;376:61–70. doi: 10.1016/s0027-5107(97)00026-2. [DOI] [PubMed] [Google Scholar]
- Gupta VH, Amarapurkar DN, Singh M, Sasi P, Joshi JM, Baijal R, et al. Association of N-acetyltransferase 2 and cytochrome P450 2E1 gene polymorphisms with antituberculosis drug-induced hepatotoxicity in Western India. J Gastroenterol Hepatol. 2013;28:1368–1374. doi: 10.1111/jgh.12194. [DOI] [PubMed] [Google Scholar]
- Hanna PE. N-acetyltransferases, O-acetyltransferases, and N,O-acetyltransferases: enzymology and bioactivation. Adv Pharmacol. 1994;27:401–430. doi: 10.1016/s1054-3589(08)61041-8. [DOI] [PubMed] [Google Scholar]
- Harris HW, Knight RA, Selin MJ. Comparison of isoniazid concentrations in the blood of people of Japanese and European descent; therapeutic and genetic implications. Am Rev Tuberc. 1958;78:944–948. doi: 10.1164/artpd.1958.78.6.944. [DOI] [PubMed] [Google Scholar]
- Hein D. Molecular genetics and function of NAT1 and NAT2: role in aromatic amine metabolism and carcinogenesis. Mutat Res. 2002;506–507:65–77. doi: 10.1016/s0027-5107(02)00153-7. [DOI] [PubMed] [Google Scholar]
- Hein DW. N-acetyltransferase SNPs: emerging concepts serve as a paradigm for understanding complexities of personalized medicine. Expert Opin Drug Metab Toxicol. 2009;5:353–366. doi: 10.1517/17425250902877698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein DW, Doll MA. Accuracy of various human NAT2 SNP genotyping panels to infer rapid, intermediate and slow acetylator phenotypes. Pharmacogenomics. 2012;13:31–41. doi: 10.2217/pgs.11.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein DW, Boukouvala S, Grant DM, Minchin RF, Sim E. Changes in consensus arylamine N-acetyltransferase gene nomenclature. Pharmacogenet Genomics. 2008;18:367–368. doi: 10.1097/FPC.0b013e3282f60db0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman D, Sim E. N-acetyltransferase polymorphism. Comparison of phenotype and genotype in humans. Biochem Pharm. 1991;42:1007–1014. doi: 10.1016/0006-2952(91)90282-a. [DOI] [PubMed] [Google Scholar]
- Hickman D, Risch A, Camilleri R, Sim E. Allele-specific amplification of human polymorphic arylamine N-acetyltransferase: identification of new alleles. Pharmacogenetics. 1992;2:217–226. doi: 10.1097/00008571-199210000-00004. [DOI] [PubMed] [Google Scholar]
- Hickman D, Risch A, Buckle V, Spurr NK, McCarthy A, Sim E. Chromosomal localization of human genes for Arylamine N-acetyltransferase. Biochem J. 1994;297:441–445. doi: 10.1042/bj2970441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman D, Palamanda JR, Unadkat JD, Sim E. Enzyme kinetic properties of human recombinant arylamine N-acetyltransferase 2 allotypic variants expressed in Escherichia coli. Biochem Pharmacol. 1995a;50:697–703. doi: 10.1016/0006-2952(95)00182-y. [DOI] [PubMed] [Google Scholar]
- Hickman D, Palamanda J, Unadkat J, Sim E. Expression and activities of the common alleles of human arylamine N-acetyltransferase type 2. Biochem Pharmacol. 1995b;48:276–283. doi: 10.1016/0006-2952(95)00182-y. [DOI] [PubMed] [Google Scholar]
- Hickman D, Pope J, Patil SD, Fakis G, Smelt V, Stanley LA, et al. Expression of arylamine N-acetyltransferase in human intestine. Gut. 1998;42:402–409. doi: 10.1136/gut.42.3.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard AL, Harrison DJ, Moyes C, Wyllie AH, Cunningham C, Mannion A, et al. N-acetyltransferase 2 genotype in colorectal cancer and selective gene retention in cancers with chromosome 8p deletions. Gut. 1997;41:229–234. doi: 10.1136/gut.41.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard AL, Moyes C, Wyllie AH, Smith CA, Harrison DJ. N-acetyl transferase 1: two polymorphisms in coding sequence identified in colorectal cancer patients. Br J Cancer. 1998;9:913–916. doi: 10.1038/bjc.1998.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes NC, Janezic SA, McQueen KL, Jewett MA, Castranio T, Bell DA, et al. Identification and characterization of variant alleles of human acetyltransferase NAT1 with defective function using p-aminosalicylate as an in-vivo and in-vitro probe. Pharmacogenetics. 1998;8:55–66. doi: 10.1097/00008571-199802000-00008. [DOI] [PubMed] [Google Scholar]
- Husain A, Barker DF, States JC, Doll MA, Hein DW. Identification of the major promoter and non-coding exons of the human arylamine N-acetyltransferase 1 gene (NAT1) Pharmacogenetics. 2004;14:397–406. doi: 10.1097/01.fpc.0000114755.08559.6e. [DOI] [PubMed] [Google Scholar]
- Husain A, Zhang X, Doll MA, States JC, Barker DF, Hein DW. Identification of N-acetyltransferase 2 (NAT2) transcription start sites and quantitation of NAT2-specific mRNA in human tissues. Drug Metab Dispos. 2007;35:721–727. doi: 10.1124/dmd.106.014621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenne JW. Partial purification and properties of the isoniazid transacetylase in human liver. Its relationship to the acetylation of p-aminosalicylic acid. J Clin Invest. 1965;44:1992–2002. doi: 10.1172/JCI105306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LE, Hoess K, Whitehead AS, Mitchell LE. The NAT1 C1095A polymorphism, maternal multivitamin use and smoking, and the risk of spina bifida. Birth Defects Res A Clin Mol Teratol. 2005;73:512–516. doi: 10.1002/bdra.20143. [DOI] [PubMed] [Google Scholar]
- Jensen LE, Hoess K, Mitchell LE, Whitehead AS. Loss of function polymorphisms in NAT1 protect against spina bifida. Hum Genet. 2006;120:52–57. doi: 10.1007/s00439-006-0181-6. [DOI] [PubMed] [Google Scholar]
- Johansson I, Nilsson C, Berglund P, Lauss M, Ringnér M, Olsson H, et al. Gene expression profiling of primary male breast cancers reveals two unique subgroups and identifies N-acetyltransferase-1 (NAT1) as a novel prognostic biomarker. Breast Cancer Res. 2012;14:R31. doi: 10.1186/bcr3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson N. 2002. Mammalian arylamine N-acetyltransferases. DPhil Thesis. University of Oxford.
- Johnson A, Bell P, Jonovska V, Budge M, Sim E. NAT gene polymorphisms and susceptibility to Alzheimer's disease: identification of a novel NAT1 allelic variant. BMC Med Genet. 2004;17:6–13. doi: 10.1186/1471-2350-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DS. How personalized medicine became genetic, and racial: Werner Kalow and the formations of pharmacogenetics. J Hist Med Allied Sci. 2013;68:1–48. doi: 10.1093/jhmas/jrr046. [DOI] [PubMed] [Google Scholar]
- Kalow W. Human pharmacogenomics: the development of a science. Hum Genomics. 2004;1:375–380. doi: 10.1186/1479-7364-1-5-375. doi: 10.1186/1479-7364-1-5-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolyi J, Erickson RP, Liu S, Killewald L. Major effects on teratogen-induced facial clefting in mice determined by a single genetic region. Genetics. 1990;126:201–205. doi: 10.1093/genetics/126.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura A, Graham J, Mushtaq A, Tsiftsoglou SA, Vath GM, Hanna PE, et al. Eukaryotic arylamine N-acetyltransferase: investigation of substrate specificity by high-throughput screening. Biochem Pharmacol. 2004;69:347–359. doi: 10.1016/j.bcp.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Kawamura A, Westwood I, Wakefield L, Long H, Zhang N, Walters K, et al. Mouse N-acetyltransferase type 2, the homologue of human N-acetyltransferase type 1. Biochem Pharmacol. 2008;75:1550–1560. doi: 10.1016/j.bcp.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly SL, Sim E. Arylamine N-acetyltransferase in Balb/c mice: identification of a novel mouse isoenzyme by cloning and expression in vitro. Biochem J. 1994;302:347–353. doi: 10.1042/bj3020347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiemenev LA, Thorlacius S, Sulem P, Geller F, Aben KKH, Stacey SN. Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nat Genet. 2008;40:1307–1312. doi: 10.1038/ng.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Kang HS, Chang HL, Jung YC, Sim HB, Lee KS, et al. Promoter hypomethylation of the N-acetyltransferase 1 gene in breast cancer. Oncol Rep. 2008;19:663–668. [PubMed] [Google Scholar]
- King CM. Mechanism of reaction, tissue distribution, and inhibition of arylhydroxamic acid acyltransferase. Cancer Res. 1974;34:1503–1515. [PubMed] [Google Scholar]
- Knowles MA, Shaw ME, Proctor AJ. Deletion mapping of chromosome 8 in cancers of the urinary bladder using restriction fragment length polymorphisms and microsatellite polymorphisms. Oncogene. 1993;8:1357–1364. [PubMed] [Google Scholar]
- Lack N, Kawamura A, Fullam E, Laurieri N, Beard S, Russell AJ, et al. Temperature stability of proteins essential for the intracellular survival of Mycobacterium tuberculosis. Biochemical J. 2009;418:369–378. doi: 10.1042/BJ20082011. [DOI] [PubMed] [Google Scholar]
- Lammer EJ, Shaw GM, Iovannisci DM, Finnell RH. Periconceptional multivitamin intake during early pregnancy, genetic variation of acetyl-N-transferase 1 (NAT1), and risk for orofacial clefts. Birth Defects Res A Clin Mol Teratol. 2004;70:846–852. doi: 10.1002/bdra.20081. [DOI] [PubMed] [Google Scholar]
- Laurieri N, Crawford M, Kawamura A, Westwood I, Robinson J, Matsuno A, et al. Small molecule colorimetric probes for specific detention of human arylamine N-acetyltransferase 1, a potential breast cancer biomarker. J Am Chem Soc. 2010;132:3238–3239. doi: 10.1021/ja909165u. [DOI] [PubMed] [Google Scholar]
- Laurieri N, Egleton J, Varney A, Dairou J, Rodrigues-Lima F, Sim E, et al. A novel color change mechanism for breast cancer biomarker detection: naphthoquinones as specific ligands of human arylamine N-acetyltransferase 1 under consideration. PLoS ONE. 2013;8:e70600. doi: 10.1371/journal.pone.0070600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurieri N, Dairou J, Egleton J, Stanley LA, Russell A, Dupret J-M, et al. From arylamine N-acetyltransferase to folate-dependent acetyl CoA hydrolase: impact of folic acid on the activity of (human) NAT1 and its homologue (mouse) NAT2. PLoS ONE. 2014 doi: 10.1371/journal.pone.0096370. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Lu HF, Wang DY, Chen DR, Su CC, Chen YS. Effects of tamoxifen on DNA adduct formation and arylamines N-acetyltransferase activity in human breast cancer cells. Res Commun Mol Pathol Pharmacol. 2004;115–116:217–233. [PubMed] [Google Scholar]
- Letašiová S, Medveďová A, Šovčíková M, Dušinská M, Volkovová K, Mosoiu K, et al. Bladder cancer, a review of the environmental risk factors. Environ Health. 2011;11(Suppl. 1):S1–S11. doi: 10.1186/1476-069X-11-S1-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann F, Kaplan NO. A common factor in the enzymatic acetylation of sulfanilamide and of choline. J Biol Chem. 1946;162:743–744. [Google Scholar]
- Liu F, Zhang N, Zhou X, Hanna PE, Wagner CR, Koop P, et al. Arylamine N-acetyltransferase aggregation and constitutive ubiquitylation. J Mol Biol. 2006;361:482–492. doi: 10.1016/j.jmb.2006.06.029. [DOI] [PubMed] [Google Scholar]
- Liu J, Ding D, Wang X, Chen Y, Li R, Zhang Y, et al. N-acetyltransferase polymorphism and risk of colorectal adenoma and cancer: a pooled analysis of variations from 59 studies. PLoS ONE. 2012;7:e42797. doi: 10.1371/journal.pone.0042797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wagner CR, Hanna PE. Isoform-selective inactivation of human arylamine N-acetyltransferases by reactive metabolites of carcinogenic arylamines. Chem Res Toxicol. 2009;22:1962–1974. doi: 10.1021/tx9002676. [DOI] [PubMed] [Google Scholar]
- Loehele JA, Cornish V, Wakefield L, Doll MA, Neale JA, Zang Y, et al. N-acetlytransferase (Nat) 1 and 2 expression in Nat2 knockout mice. J Pharmacol Exp Ther. 2006;310:724–728. doi: 10.1124/jpet.106.108662. [DOI] [PubMed] [Google Scholar]
- Lu KH, Lin KL, Hsia TC, Hung CF, Chou MC, Hsiao YM. Tamoxifen inhibits arylamine N-acetyltransferase activity and DNA-2-aminofluorene adduct in human leukaemic HL-60 cells. Res Comm Mol Pathol. 2001;109:319–331. [PubMed] [Google Scholar]
- Madikane E, Bhakta S, Russell A, Campbell W, Claridge T, Elisha G, et al. Inhibition of mycobacterial arylamine N-acetyltransferase contributes to anti-mycobacterial activity of Warburgia salutaris. Bioorg Med Chem. 2007;15:3579–3586. doi: 10.1016/j.bmc.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Martell KJ, Vatsis KP, Weber WW. Molecular genetic basis of rapid and slow acetylation in mice. Mol Pharmacol. 1991;40:218–227. [PubMed] [Google Scholar]
- Martell KJ, Levy GN, Weber WW. Cloned mouse N-acetyltransferases: enzymatic properties of expressed Nat-1 and Nat-2 gene products. Mol Pharmacol. 1992;42:265–272. [PubMed] [Google Scholar]
- Matas N, Thygesen P, Stacey M, Risch A, Sim E. Mapping AAC1, AAC2 and AACP, the genes for arylamine N-acetyltransferases, carcinogen metabolizing enzymes on human chromosome 8p22, a region frequently deleted in tumours. Cytogenet Cell Genet. 1997;77:290–295. doi: 10.1159/000134601. [DOI] [PubMed] [Google Scholar]
- McDowell SE, Thomas SK, Coleman JJ, Aronson JK, Ferner RF. A practical guide to monitoring for adverse drug reactions during antihypertensive drug therapy. J R Soc Med. 2013;106:87–95. doi: 10.1258/jrsm.2012.120137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millner LM, Doll MA, Cai J, States C, Hein DW. Phenotype of the most common ‘Slow Acetylator’ arylamine N-acetyltransferase 1 genetic variant (NAT1*14B) is substrate-dependent. Drug Metab Dispos. 2012a;40:198–204. doi: 10.1124/dmd.111.041855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millner LM, Doll MA, Stepp MW, States JC, Hein DW. Functional analysis of arylamine N-acetyltransferase 1 (NAT1NAT1*10 haplotypes in a complete NATb mRNA construct. Carcinogenesis. 2012b;33:348–355. doi: 10.1093/carcin/bgr273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minchin RF. Acetylation of p-aminobenzoylglutamate, a folic acid catabolite, by recombinant human arylamine N-acetyltransferase and U937 cells. Biochem J. 1995;307:1–3. doi: 10.1042/bj3070001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minchin RF, Hanna PE, Dupret J-M, Wagner CR, Rodrigues-Lima F, Butcher NJ. Arylamine N-acetyltransferase type 1. Int J Biochem Cell Biol. 2007;39:1999–2005. doi: 10.1016/j.biocel.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Mitchell JA, Sim E. CR1 size polymorphism in hydralazine-induced SLE. Complement Inflamm. 1989;6:88–93. doi: 10.1159/000463079. [DOI] [PubMed] [Google Scholar]
- Mitchell JA, Batchelor JR, Chapel HM. Spiers CN, Sim E. Erythrocyte complement receptor type I (CR1) expression and circulating immune complexes (CIC) levels in hydralazine-SLE. Clin Exp Immunol. 1987;68:446–456. [PMC free article] [PubMed] [Google Scholar]
- Mitchell JA, Sim RB, Sim E. CR1 polymorphism in hydralazine-induced systemic lupus erythematosus: DNA restriction fragment length polymorphism. Clin Exp Immunol. 1989;78:354–358. [PMC free article] [PubMed] [Google Scholar]
- Mitchell KM, Futscher BW, McQueen CA. Developmental expression of N-acetyltransferase in C57Bl/6 mice. Drug Metab Dispos. 1999;27:261–264. [PubMed] [Google Scholar]
- Mushtaq A, Payton M, Sim E. The COOH terminus of arylamine N-acetyltransferase from Salmonella typhimurium controls enzymic activity. J Biol Chem. 2002;277:12175–12181. doi: 10.1074/jbc.M104365200. [DOI] [PubMed] [Google Scholar]
- Ohsako S, Deguchi T. Cloning and expression of cDNAs for polymorphic and monomorphic arylamine N-acetyltransferases from human liver. J Biol Chem. 1990;265:4630–4634. [PubMed] [Google Scholar]
- Pacifici GM, Bencini C, Rane A. Acetyltransferase in humans: development and tissue distribution. Pharmacology. 1986;32:283–291. doi: 10.1159/000138181. [DOI] [PubMed] [Google Scholar]
- Park BK, Pirmohamed M, Kitteringham NK. Idiosyncratic drug reactions: a mechanistic evaluation of risk factors. Br J Clin Pharmacol. 1992;34:377–395. doi: 10.1111/j.1365-2125.1992.tb05647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patin E, Barreiro LB, Sabeti PC, Austerlitz F, Luca F, Sajantila A, et al. Deciphering the ancient and complex evolutionary history of human arylamine N-acetyltransferase genes. Am J Hum Genet. 2006a;78:423–436. doi: 10.1086/500614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patin E, Harmant C, Kidd KK, Kidd J, Froment A, Mehdi SQ, et al. Sub-Saharan African coding sequence variation and haplotype diversity at the NAT2 gene. Hum Mutat. 2006b;27:720–725. doi: 10.1002/humu.9438. [DOI] [PubMed] [Google Scholar]
- Payton M, Sim E. A simple method for genotyping arylamine N-acetyltransferase type1: identification of a new mutation associated with low NAT1 activity. Biochem Pharmacol. 1998;55:361–366. doi: 10.1016/s0006-2952(97)00478-4. [DOI] [PubMed] [Google Scholar]
- Payton M, Smelt V, Upton A, Sim E. A method for genotyping murine arylamine N-acetyltransferase type 2 (NAT2): a gene expressed in preimplantation embryonic stem cells encoding an enzyme acetylating the folate catabolite p-aminobenzoylglutamate. Biochem Pharmacol. 1999a;58:779–785. doi: 10.1016/s0006-2952(99)00171-9. [DOI] [PubMed] [Google Scholar]
- Payton M, Auty R, Delgoda R, Everett M, Sim E. Cloning and characterisation of arylamine N-acetyltransferase genes from Mycobacterium smegmatis and M. tuberculosis: increased expression results in isoniazid resistance. J Bacteriol. 1999b;181:1343–1347. doi: 10.1128/jb.181.4.1343-1347.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payton M, Gifford C, Schartau P, Hagemeier C, Mushtaq A, Lucas S, et al. Evidence towards the role of arylamine N-acetyltransferase in Mycobacterium smegmatis and development of a specific antiserum against the homologous enzyme of Mycobacterium tuberculosis. Microbiology. 2001a;147:3295–3302. doi: 10.1099/00221287-147-12-3295. [DOI] [PubMed] [Google Scholar]
- Payton M, Mushtaq A, Yu T, Wu L, Sinclair J, Sim E. Eubacterial arylamine N-acetyltransferases – identification and comparison of 18 members of the protein family with conserved active site cysteine, histidine and aspartate residues. Microbiology. 2001b;147:1137–1147. doi: 10.1099/00221287-147-5-1137. [DOI] [PubMed] [Google Scholar]
- Pirmohamed M, Alfirevic A, Vilar J, Stalford A, Wilkins EG, Sim E, et al. Association analysis of drug metabolizing enzyme gene polymorphisms in HIV-positive patients with co-trimoxazole hypersensitivity. Pharmacogenetics. 2000;10:705–713. doi: 10.1097/00008571-200011000-00005. [DOI] [PubMed] [Google Scholar]
- Pluvinage B, Li de la Sierra-Gallay I, Kubiak X, Xu X, Dairou J, Dupret JM, et al. The Bacillus anthracis arylamine N-acetyltransferase ((BACAN)NAT1) that inactivates sulfamethoxazole, reveals unusual structural features compared with the other NAT isoenzymes. FEBS Lett. 2011;585:3947–3952. doi: 10.1016/j.febslet.2011.10.041. [DOI] [PubMed] [Google Scholar]
- Rajasekaran M, Abirami S, Chen C. Effects of single nucleotide polymorphisms on human N-acetyltransferase 2 structure and dynamics by molecular dynamics simulation. PLoS ONE. 2011;6:e25801. doi: 10.1371/journal.pone.0025801. doi: 10.1371/journal.pone.0025801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehn L. Blasengeschulste bei fuchsin-arbeitern. Arch Klin Chir. 1895;50:588–600. [Google Scholar]
- Riddle B, Jencks WP. Acetyl-coenzyme A: arylamine N-acetyltransferase. Role of the acetyl-enzyme intermediate and the effects of substituents on the rate. J Biol Chem. 1971;246:3250–3258. [PubMed] [Google Scholar]
- Risch A, Wallace DMA, Bathers S, Sim E. Slow N-acetylation genotype is a susceptibility factor in occupational and smoking related bladder cancer. Hum Mol Genetics. 1995;4:231–236. doi: 10.1093/hmg/4.2.231. [DOI] [PubMed] [Google Scholar]
- Rodrigues-Lima F, Dupret JM. 3D model of human arylamine N-acetyltransferase 2: structural basis of the slow acetylator phenotype of the R64Q variant and analysis of the active-site loop. Biochem Biophys Res Commun. 2002;291:116–123. doi: 10.1006/bbrc.2002.6414. [DOI] [PubMed] [Google Scholar]
- Rodrigues-Lima F, Dairou J, Laurieri N, Busi F, Dupret JM. Pharmacogenomics, biochemistry, toxicology, microbiology and cancer research in one go. Pharmacogenomics. 2011;12:1091–1093. doi: 10.2217/pgs.11.59. [DOI] [PubMed] [Google Scholar]
- Ruiz JD, Martínez C, Anderson K, Gross M, Lang NP, Garcia-Martin E, et al. The differential effect of NAT2 variant alleles permits refinement in phenotype inference and identifies a very slow acetylation genotype. PLoS ONE. 2012;7:e44629. doi: 10.1371/journal.pone.0044629. doi: 10.1371/journal.pone.0044629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell AJ, Westwood IM, Crawford MHJ, Robinson J, Kawamura A, Redfield C. Selective small molecule inhibitors of the potential breast cancer marker, human arylamine N-acetyltransferase 1, and its murine homologue, mouse arylamine N-acetyltransferase 2. Bioorg Med Chem. 2009;17:905–918. doi: 10.1016/j.bmc.2008.11.032. [DOI] [PubMed] [Google Scholar]
- Ryan A, Laurieri N, Westwood I, Wang CJ, Lowe E, Sim E. A novel mechanism for azoreduction. J Mol Biol. 2010a;400:24–37. doi: 10.1016/j.jmb.2010.04.023. [DOI] [PubMed] [Google Scholar]
- Ryan A, Wang C-J, Laurieri N, Westwood I, Sim E. Reaction mechanism of azoreductases suggests convergent evolution with quinone oxidoreductases. Protein Cell. 2010b;1:780–790. doi: 10.1007/s13238-010-0090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan A, Kaplan E, Laurieri N, Lowe E, Sim E. Activation of nitrofurazone by azoreductases: multiple activities in one enzyme. Sci Rep NPG. 2011;1:Article number: 63. doi: 10.1038/srep00063. doi: 10.1038/srep00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbagh A, Darlu P, Crouau-Roy B, Poloni ES. Arylamine N-acetyltransferase 2 (NAT2) genetic diversity and traditional subsistence: a worldwide population survey. PLoS ONE. 2011;6:e18507. doi: 10.1371/journal.pone.0018507. doi: 10.1371/journal.pone.0018507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandy J, Mushtaq A, Kawamura A, Sinclair J, Sim E, Noble M. The structure of Arylamine N-acetyltransferase from Mycobacteria smegmatis – an enzyme which inactivates the anti-tubercular drug, isoniazid. J Mol Biol. 2002;318:1071–1083. doi: 10.1016/S0022-2836(02)00141-9. [DOI] [PubMed] [Google Scholar]
- Sandy J, Mushtaq A, Holton SJ, Schartau P, Noble MEM, Sim E. Investigation of the catalytic triad of arylamine N-acetyltransferases: essential residues required for acetyl transfer to arylamines. Biochem J. 2005a;390:115–123. doi: 10.1042/BJ20050277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandy J, Holton S, Fullam E, Sim E, Noble M. Binding of the anti-tubercular drug isoniazid to the arylamine N-acetyltransferase protein from Mycobacterium smegmatis. Protein Sci. 2005b;14:775–782. doi: 10.1110/ps.041163505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Ohsako S, Deguchi T. Molecular and genetic analyses of arylamine N-acetyltransferase polymorphism of rabbit liver. J Biol Chem. 1991;266:13243–13250. [PubMed] [Google Scholar]
- Sholto-Douglas-Vernon C, Sandy J, Victor TC, Sim E, van Helden PD. Mutational and expression analysis of tbnat and its response to isoniazid. J Med Microbiol. 2005;54:1189–1197. doi: 10.1099/jmm.0.46153-0. [DOI] [PubMed] [Google Scholar]
- Sikora AL, Frankel BA, Blanchard JS. Kinetic and chemical mechanism of arylamine N-acetyltransferase from Mycobacterium tuberculosis. Biochemistry. 2008;47:10781–10789. doi: 10.1021/bi800398c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim E, Hickman D. Polymorphism in human N-acetyltransferase – the case of the missing allele. Trends Pharm Sci. 1991;12:211–213. doi: 10.1016/0165-6147(91)90553-5. [DOI] [PubMed] [Google Scholar]
- Sim E, Law SKA. Hydralazine binds to complement component C4. Differential reactivity of C4A and C4B. FEBS Letts. 1985;184:323–327. doi: 10.1016/0014-5793(85)80631-1. [DOI] [PubMed] [Google Scholar]
- Sim E, Vignais PM. Comparison of the membrane-bound and detergent-solubilised hydrogenase from Paracoccus denitrificans. Isolation of the hydrogenase. BBA Enzymol. 1979;570:43–55. doi: 10.1016/0005-2744(79)90199-2. [DOI] [PubMed] [Google Scholar]
- Sim E, Gill EW, Sim RB. Drugs that induce systemic lupus erythematosus inhibit complement component C4. Lancet. 1984;ii:422–424. doi: 10.1016/s0140-6736(84)92905-2. [DOI] [PubMed] [Google Scholar]
- Sim E, Payton M, Noble MEM, Minchin R. An update on genetic, structural and functional studies of arylamine N-acetyltransferases in eucaryotes and procayotes. Hum Mol Genet. 2000;9:2435–2441. doi: 10.1093/hmg/9.16.2435. [DOI] [PubMed] [Google Scholar]
- Sim E, Pinter K, Mushtaq A, Upton A, Sandy J, Bhakta S, et al. Arylamine N-acetyltransferases: a pharmacogenetic approach to drug metabolism and endogenous function. Biochem Soc Trans. 2003;31:615–619. doi: 10.1042/bst0310615. [DOI] [PubMed] [Google Scholar]
- Sim E, Westwood I, Fullam E. Arylamine N-acetyltransferases. Expert Opin Drug Metab Toxicol. 2007;3:169–184. doi: 10.1517/17425255.3.2.169. [DOI] [PubMed] [Google Scholar]
- Sim E, Walters K, Boukouvala S. Arylamine N-acetyltransferases: from structure to function. Drug Metab Rev. 2008a;40:479–510. doi: 10.1080/03602530802186603. [DOI] [PubMed] [Google Scholar]
- Sim E, Sandy J, Evangelopoulos D, Fullam E, Bhakta S, Westwood I, et al. Arylamine N-acetyltrasferases in mycobacteria. Curr Drug Metab. 2008b;9:510–519. doi: 10.2174/138920008784892100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim E, Lack N, Wang CJ, Long H, Westwood I, Fullam E, et al. Arylamine N-acetyltrasferase: structural and functional implications of polymorphisms. Toxicology. 2008c;254:170–183. doi: 10.1016/j.tox.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Sim E, Fullam E, Wakefield L. Arylamine N-acetyltransferases. Compr Toxicol. 2010;4:385–412. [Google Scholar]
- Sim RB, Twose TM, Paterson DS, Sim E. The covalent binding reaction of complement component C3. Biochem J. 1981;193:115–127. doi: 10.1042/bj1930115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair J, Sim E. A fragment consisting of the first 204 amino terminal amino acids of arylamine N-acetyltransferase is active in the first transacetylation step of catalysis. Biochem Pharmacol. 1997;53:11–16. doi: 10.1016/s0006-2952(96)00592-8. [DOI] [PubMed] [Google Scholar]
- Sinclair JC, Delgoda R, Noble MEM, Jarmin S, Goh NK, Sim E. Purification, characterization, and crystallization of an hydroxyarylamine O-acetyltransferase from Salmonella typhimurium. Protein Expr Purif. 1998;12:371–380. doi: 10.1006/prep.1997.0856. [DOI] [PubMed] [Google Scholar]
- Sinclair JC, Sandy J, Delgoda R, Sim E, Noble MEM. Structure of arylamine N-acetyltransferase reveals a catalytic triad. Nat (Struct Biol) 2000;7:560–564. doi: 10.1038/76783. [DOI] [PubMed] [Google Scholar]
- Smelt VA, Mardon HJ, Sim E. Placental expression of arylamine N-acetyltransferases: evidence for linkage disequilibrium between NAT1*10 and NAT2*4 alleles of the two human arylamine N-acetyltransferase loci NAT1 and NAT2. Pharmacol Toxicol. 1998;83:149–157. doi: 10.1111/j.1600-0773.1998.tb01461.x. [DOI] [PubMed] [Google Scholar]
- Smelt VA, Upton A, Adjaye J, Payton MA, Boukouvala S, Johnson N, et al. Expression of arylamine N-acetyltransferases in pre-term placentas and in human pre-implantation embryos. Hum Mol Genet. 2000;9:1101–1107. doi: 10.1093/hmg/9.7.1101. [DOI] [PubMed] [Google Scholar]
- Spurr NK, Blanton S, Bookstein R, Clarke R, Cottingham R, Daiger S, et al. Report of the second international workshop on human chromosome 8 mapping 1994. Cytogen Cell Genet. 1995;68:147–164. doi: 10.1159/000133908. [DOI] [PubMed] [Google Scholar]
- Stacey M, Thygesen P, Stanley L, Matas N, Risch A, Sim E. Arylamine N-acetyltransferase as a potential biomarker in bladder cancer: fluorescent in situ hybridization and immunohistochemistry studies. Biomarkers. 1996;1:55–61. doi: 10.3109/13547509609079347. [DOI] [PubMed] [Google Scholar]
- Stacey M, Payton M, Fakis G, Pope J, Sim E. Characterization of arylamine N-acetyltransferase in the human urothelial cell line RT112. Pathogenesis. 1997;12:99–105. [Google Scholar]
- Stacey M, Matas N, Drake M, Payton M, Fakis G, Greenland J, et al. Arylamine N-acetyltransferases type 2 (NAT2), Chromosome 8 aneuploidy and identification of novel nat1 cosmid clone: an investigation of bladder cancer by interphase FISH. Genes Chromosomes Cancer. 1999;25:378–383. doi: 10.1002/(sici)1098-2264(199908)25:4<376::aid-gcc10>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Stanley LA, Coroneos E, Cuff R, Hickman D, Ward A, Sim E. Immunochemical detection of arylamine N-acetyltransferase in normal and neoplastic bladder. J Histochem Cytochem. 1996;44:1059–1067. doi: 10.1177/44.9.8773572. [DOI] [PubMed] [Google Scholar]
- Stanley LA, Mills IG, Sim E. Localization of polymorphic N-acetyltransferase (NAT2) in tissues of inbred mice. Pharmacogenetics. 1997;7:121–130. doi: 10.1097/00008571-199704000-00005. [DOI] [PubMed] [Google Scholar]
- Stanley LA, Copp AJ, Pope J, Rolls S, Smelt V, Perry V, et al. Immunochemical detection of arylamine N-acetyltransferase during mouse embryonic development and in adult mouse brain. Teratology. 1998;58:174–182. doi: 10.1002/(SICI)1096-9926(199811)58:5<174::AID-TERA3>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Sugamori KS, Wong S, Gaedigk A, Yu V, Abramovici H, Rozmahel R, et al. Generation and functional characterization of arylamine N-acetyltransferase Nat1/Nat2 double-knockout mice. Mol Pharmacol. 2003;64:170–179. doi: 10.1124/mol.64.1.170. [DOI] [PubMed] [Google Scholar]
- Sugamori KS, Brenneman D, Wong S, Gaedigk A, Yu V, Abramovici H, et al. Effect of arylamine acetyltransferase Nat3 gene knockout on N-acetylation in the mouse. Drug Metab Dispos. 2007;35:1064–1070. doi: 10.1124/dmd.107.015396. [DOI] [PubMed] [Google Scholar]
- Sugamori KS, Brenneman D, Sanchez O, Doll MA, Hein DW, Pierce WM, Jr, et al. Reduced 4-aminobiphenyl-induced liver tumorigenicity but not DNA damage in arylamine N-acetyltransferase null mice. Cancer Lett. 2012;318:206–213. doi: 10.1016/j.canlet.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam CM, Chan SL, Kam KM, Sim E, Staples D, Sole KM, et al. Rifapentine and isoniazid in the continuation phase of a 6-month regimen. Interim report: no activity of isoniazid in the continuation phase. Int J Tuber Lung Dis. 2000;4:262–267. [PubMed] [Google Scholar]
- Thygesen P, Risch A, Takle LA, Knowles MA, Sim E. Genes for human arylamine N-acetyltransferases in relation to loss of the short arm of chromosome 8 in bladder cancer. Pharmacogenetics. 1999;9:1–8. doi: 10.1097/00008571-199902000-00001. [DOI] [PubMed] [Google Scholar]
- Tiang JM, Butcher NJ, Minchin RF. Small molecule inhibition of arylamine N-acetyltransferase Type I inhibits proliferation and invasiveness of MDA-MB-231 breast cancer cells. Biochem Biophys Res Commun. 2010;393:95–101. doi: 10.1016/j.bbrc.2010.01.087. [DOI] [PubMed] [Google Scholar]
- Tiang JM, Butcher NJ, Cullinane C, Humbert PO, Minchin RF. RNAi-mediated knock-down of Arylamine N-acetyltransferase-1 expression induces E-cadherin up-regulation and cell-cell contact growth inhibition. PLoS ONE. 2011;6:1–10. doi: 10.1371/journal.pone.0017031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilak AV, Iyer SN, Mukherjee MS, Singhal RS, Lele SS. Full-gene-sequencing analysis of N-acetyltransferase-2 in an adult Indian population. Genet Test Mol Biomarkers. 2013;17:188–194. doi: 10.1089/gtmb.2012.0258. [DOI] [PubMed] [Google Scholar]
- Timbrell JA, Harland SJ, Facchini V. Polymorphic acetylation of hydralazine. Clin Pharmacol Ther. 1980;28:350–355. doi: 10.1038/clpt.1980.173. [DOI] [PubMed] [Google Scholar]
- Upton A, Smelt V, Mushtaq A, Aplin R, Johnson N, Mardon H, et al. Placental arylamine N-acetyltransferase type 2: potential contributory source of urinary folate catabolite p-acetamidobenzoylglutamate during pregnancy. Biochim Biophys Acta. 2000;1524:143–148. doi: 10.1016/s0304-4165(00)00149-5. [DOI] [PubMed] [Google Scholar]
- Upton A, Everett M, Mushtaq A, van Helden P, Victor T, Wagner R, et al. Arylamine N-acetyltransferase of Mycobacterium tuberculosis is a polymorphic enzyme and a site of isoniazid metabolism. Mol Microbiol. 2001;42:309–319. doi: 10.1046/j.1365-2958.2001.02648.x. [DOI] [PubMed] [Google Scholar]
- Vagena E, Fakis G, Boukouvala S. Arylamine N-acetyltransferases in prokaryotic and eukaryotic genomes: a survey of public databases. Curr Drug Metab. 2008;9:628–660. doi: 10.2174/138920008785821729. [DOI] [PubMed] [Google Scholar]
- Van der Geize R, Heuser T, Yam K, Wilbrink M, Hara H, Anderton M. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci U S A. 2007;104:1947–1952. doi: 10.1073/pnas.0605728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatsis KP, Weber WW. Structural heterogeneity of Caucasian N-acetyltransferase at the NAT1 gene locus. Arch Biochem Biophys. 1993;301:71–76. doi: 10.1006/abbi.1993.1116. [DOI] [PubMed] [Google Scholar]
- Vineis P, Marinelli D, Autrup H, Brockmoller J, Cascorbi I, Daly AK, et al. Current smoking, occupation, N-acetyltransferase −2 and bladder cancer: a pooled analysis of genotype-based studies. Cancer Epidemiol Biomarkers Prev. 2001;10:1249–1252. [PubMed] [Google Scholar]
- Wakefield L, Cornish V, Broackes-Carter F, Sim E. Arylamine N-acetyltransferase 2 expression in the developing heart. J Histochem Cytochem. 2005;53:583–592. doi: 10.1369/jhc.4A6496.2005. [DOI] [PubMed] [Google Scholar]
- Wakefield L, Cornish V, Long H, Griffiths WJ, Sim E. Deletion of a xenobiotic metabolizing gene in mice affects folate metabolism. Biochem Biophys Rea Commun. 2007a;364:556–560. doi: 10.1016/j.bbrc.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield L, Long H, Lack N, Sim E. Ocular defects associated with a null mutation in the mouse 3 arylamine N-acetyltransferase 2 gene. Mamm Genome. 2007b;18:270–276. doi: 10.1007/s00335-007-9010-z. [DOI] [PubMed] [Google Scholar]
- Wakefield L, Cornish V, Long H, Kawamura A, Zhang XY, Hein DW, et al. Mouse arylamine N-acetyltransferase 2 (Nat2) expression in the developing neuroendocrine system. Biomarkers. 2008a;13:106–118. doi: 10.1080/13547500701673529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield L, Robinson J, Long H, Ibbitt JC, Cooke S, Hurst H, et al. Arylamine N-acetyltransferase 1 expression in breast cancer cell lines: a potential marker in oestrogen receptor positive tumours. Genes Chromosomes Cancer. 2008b;47:118–126. doi: 10.1002/gcc.20512. [DOI] [PubMed] [Google Scholar]
- Wakefield L, Boukouvala S, Sim E. Characterisation of CpG methylation in the upstream control region of mouse Nat2: evidence for a gene-environment interaction in a polymorphic gene implicated in folate metabolism. Gene. 2010;452:16–21. doi: 10.1016/j.gene.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Walport M. Complement and systemic lupus erythematosus. Arthritis Res. 2002;4:S279–S293. doi: 10.1186/ar586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walraven JM, Trent JO, Hein DW. Structure-function analyses of single nucleotide polymorphisms in human N-acetyltransferase 1. Drug Metab Rev. 2008;40:169–184. doi: 10.1080/03602530701852917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Hagemeier C, Rahman N, Lowe E, Noble M, Coughtrie M, et al. Molecular cloning, characterisation and ligand-bound structure of an azoreductase from Pseudomonas aeruginosa. J Mol Biol. 2007;373:1213–1228. doi: 10.1016/j.jmb.2007.08.048. [DOI] [PubMed] [Google Scholar]
- Wang H, Vath GM, Gleason KJ, Hanna PE, Wagner CR. Probing the mechanism of hamster arylamine N-acetyltransferase 2 acetylation by active site modification, site-directed mutagenesis, and pre-steady state and steady state kinetic studies. Biochemistry. 2004;43:8234–8246. doi: 10.1021/bi0497244. [DOI] [PubMed] [Google Scholar]
- Wang H, Vath GM, Kawamura A, Bates CA, Sim E, Hanna PE, et al. Overexpression, purification, and characterisation of recombinant human arylamine N-acetyltransferase 1. Protein J. 2005;24:65–77. doi: 10.1007/s10930-004-1513-9. [DOI] [PubMed] [Google Scholar]
- Ward A, Hickman D, Sim E. N-acetyltransferase in human red blood cells. Biochem Pharmacol. 1992;44:1099–1104. doi: 10.1016/0006-2952(92)90373-q. [DOI] [PubMed] [Google Scholar]
- Ward A, Summers ME, Sim E. Purification of recombinant human N-acetyltransferase type 1 expressed in E. coli and characterization of its potential role in folate metabolism. Biochem Pharmacol. 1995;49:1759–1767. doi: 10.1016/0006-2952(95)00087-g. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Nohmi T, Oshidate M., Jr New tester strains of Salmonella typhimurium highly sensitive to mutagenic nitroarenes. Biochem Biophys Res Commun. 1987;147:974–979. doi: 10.1016/s0006-291x(87)80165-1. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Sofuni T, Nohmi T. Involvement of Cys69 residue in the catalytic mechanism of N-hydroxyarylamine O-acetyltransferase of Salmonella typhimurium. Sequence similarity at the amino acid level suggests a common catalytic mechanism of acetyltransferase for S. typhimurium and higher organisms. J Biol Chem. 1992;267:8429–8436. [PubMed] [Google Scholar]
- Watanabe M, Matsuoka A, Yamazaki N, Hayashi M, Deguchi T, Nohmi T, et al. New sublines of Chinese hamster CHL stably expressing human NAT1 or NAT2 N-acetyltransferases or Salmonella typhimurium O-acetyltransferase: comparison of the sensitivities to nitroarenes and aromatic amines using the in vitro micronucleus test. Cancer Res. 1994;54:1672–1677. [PubMed] [Google Scholar]
- Watson AP, Wang P, Sim E. Arylamine N-acetyltransferase of fast (C57BL6) and slow (A/J) N-acetylating strains of mice. Biochem Pharmacol. 1990;39:647–654. doi: 10.1016/0006-2952(90)90141-7. [DOI] [PubMed] [Google Scholar]
- Watters AD, Stacey MW, Going JJ, Grigor KM, Cooke TG, Sim E, et al. Genetic alterations of N-acetyl transferase in transitional cell carcinoma of the bladder. Br J Cancer. 1998;78:154–162. [Google Scholar]
- Weber WW, Hein DW. N-acetylation pharmacogenetics. Pharmacol Rev. 1985;37:25–79. [PubMed] [Google Scholar]
- Webster DJ, Flook D, Jenkins J, Hutchings A, Routledge PA. Drug acetylation in breast cancer. Br J Cancer. 1989;60:236–237. doi: 10.1038/bjc.1989.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwood I, Bhakta S, Russell AJ, Fullam E, Anderton MC, Kawamura A, et al. Identification of arylamine N-acetyltransferase inhibitors as an approach towards novel anti-tuberculars. Protein Cell. 2010;1:82–96. doi: 10.1007/s13238-010-0006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwood IM, Holton SJ, Rodrigues-Lima F, Dupret J-M, Noble MEM, Bhakta S, et al. Expression, purification, characterisation and structure of Pseudomonas aeruginosa arylamine N-acetyltransferase. Biochem J. 2005;385:605–612. doi: 10.1042/BJ20041330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwood IM, Kawamura A, Fullam E, Russell AJ, Davies SG, Sim E. Structure and mechanism of arylamine N-acetyltransferases. Curr Top Med Chem. 2006;6:1641–1654. doi: 10.2174/156802606778108979. [DOI] [PubMed] [Google Scholar]
- Westwood IM, Kawamura A, Russell AJ, Sandy J, Davies SG, Sim E. Novel small molecule inhibitors of arylamine N-acetyltransferases: drug discovery by high throughput screening. Comb Chem High Throughput Screen. 2011;14:117–124. doi: 10.2174/138620711794474051. [DOI] [PubMed] [Google Scholar]
- Williams JA, Stone EM, Fakis G, Johnson N, Cordell JA, Meinl W, et al. N-acetyltransferases, sulfotransferases and heterocyclic amine activation in the breast. Pharmacogenetics. 2001;11:373–388. doi: 10.1097/00008571-200107000-00002. [DOI] [PubMed] [Google Scholar]
- Woosley RL, Drayer DE, Reidenberg MM, Nies AS, Carr K, Oates JA. Effect of acetylator phenotype on the rate at which procainamide induces antinuclear antibodies and the lupus syndrome. New Engl J Med. 1978;298:1157–1159. doi: 10.1056/NEJM197805252982101. [DOI] [PubMed] [Google Scholar]
- Wu H, Dombrovsky L, Tempel W, Martin F, Loppnau P, Goodfellow GH, et al. Structural basis of substrate-binding specificity of human arylamine N-acetyltransferases. J Biol Chem. 2007;282:30189–30197. doi: 10.1074/jbc.M704138200. [DOI] [PubMed] [Google Scholar]
- Yam KC, D'Angelo I, Zhu H, Wang J-X, Snieckus V, Ly LH, et al. Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PloS Pathog. 2009;5:e1000344. doi: 10.1371/journal.ppat.1000344. doi: 10.1371/journal.ppat.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Liu L, Liu F, Wagner CR, Hanna PE, Walters KJ. NMR-based model reveals the structural determinants of mammalian arylamine N-acetyltransferase substrate specificity. J Mol Biol. 2006;363:188–200. doi: 10.1016/j.jmb.2006.08.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Collaborators and co-workers.