

Abstract
Interleukins represent a class of immunomodulatory cytokines, small intercellular signaling proteins, that are critically involved in the regulation of immune responses. They are produced in large amounts by various cell types during inflammatory reactions, and the balance of cytokines determines the outcome of an immune response. Therefore, cytokines are regarded as interesting therapeutic targets for the treatment of patients with liver diseases. Mouse models provide a good tool for in vivo studies on cytokine function, as human and mouse cytokines share many homologies. Sophisticated mouse models either mimicking distinct pathological conditions or targeting cytokines and cytokine-signaling pathways in the liver or even in distinct cellular compartments have provided enormous insight into the different functions of interleukins during hepatic inflammation. Interleukins may have pro- as well as anti-inflammatory functions in chronic liver diseases, some interleukins even both, dependent on the inflammatory stimulus, the producing and the responding cell type. IL-17, for example, promotes hepatic fibrogenesis through activation of hepatic stellate cells and facilitates development of liver cancer through recruitment of myeloid-derived suppressor cells. IL-22, on the other hand, protects from development of fibrosis or steatohepatitis. IL-12 balances T-helper (Th)-1 and Th2 cell responses in infectious disease models. IL-13 and IL-33, two cytokines related to Th2 cells and innate lymphoid cells, promote fibrotic responses in the liver. IL-10 is the prototypic anti-inflammatory interleukin with tissue-protective functions during chronic liver injury and fibrogenesis. Despite its critical role for inducing the acute-phase response in the liver, IL-6 signaling is protective during fibrosis progression, but promotes hepatocellular carcinoma. Experimental studies in mice help to define the exact influence of a specific cytokine on the outcome of chronic liver diseases and to identify useful therapeutic targets.
Keywords: liver fibrosis, liver cirrhosis, liver cancer, interleukin, cytokine, T cell
Introduction
Cytokines are small signaling proteins that facilitate intercellular communication between various cells. They function through cell-surface receptors, and downstream signaling induces an alteration of cell function and phenotype. Interleukins are immunomodulatory cytokines that mediate the communication between leukocytes or leukocytes and their target cells.1 The term “interleukin” is used rather independently of the cytokine function for molecules that either act on leukocytes or are produced by leukocytes, mainly lymphocytes. How a specific interleukin acts on its target cells depends on the interleukin, producing and responding cell type, type of immune response, and abundance of the interleukin itself.2 Dependent on the context, interleukins can have pro- and anti-inflammatory functions.
Interleukins are produced in large amounts during all kinds of immune responses, and the outcome of an inflammatory reaction is determined by the balance of cytokines produced.1,2 Proinflammatory interleukins are induced by a variety of stimuli, including infections with pathogens, but also sterile organ damage. Their main function is to stimulate immune responses that result in the elimination of invading pathogens or damaged and dying cells. At the same time, anti-inflammatory mediators are produced to protect the host’s body from exaggerated immune responses and to limit organ damage. As soon as the pathogenic stimuli are removed, production of interleukins is no longer needed, and inflammation subsides. However, if the stimulus persists, inflammation can become chronic and induce a variety of inflammatory diseases, which often have fatal consequences for the host organism.3 Moreover, excess production of anti-inflammatory cytokines can suppress effective immune responses against malignant cells, thus promoting tumor growth.4
The role of interleukins in chronic liver diseases has been intensively investigated in the past few decades. Animal models provide a good tool for in vivo studies, due to the existence of mouse orthologues to most of the human cytokines and the availability of experimental models of human liver diseases. In this article, we focus on the major findings (summarized in Table 1) derived from experimental mouse models defining possible functional roles for interleukins in the progression of chronic liver diseases.
Table 1.
Role of interleukins in experimental mouse models of chronic liver diseases (selection)
| Interleukin | Experimental mouse model | Producing cell type | Responding cell type | Effector function | Reference(s) |
|---|---|---|---|---|---|
| Pathogenic functions of interleukins | |||||
| IL-6 | HCC | KCs | Hepatocytes | Increased IL-6 production in males leads to increased HCC development | 14,15 |
| Obesity-induced upregulation of IL-6 promotes HCC | 16 | ||||
| IL-12 | Schistosoma mansoni/Toxoplasma gondii double infection | IL-12 downregulates anti-Schistosoma Th2 response, leading to earlier death due to S. mansoni infection |
36 | ||
| Malaria | CTLs | IL-12 activates CTLs that kill infected hepatocytes | 34 | ||
| IL-13 | S. mansoni infection | Th2 | IL-13 induces production of collagen, α-SMA | 39–41 | |
| IL-17 | PBC | Th17 | High levels of IL-17 and Th17 cells increase liver inflammation | 44 | |
| NAFLD | Th17 | Hepatocytes | IL-17 increases steatosis in hepatocytes | 48 | |
| Fibrosis | Th17 | KCs, HSCs | IL-17 activates HSCs and induces collagen production | 46 | |
| HCC | γδ T cells | MDSCs | IL-17 induces recruitment of MDSCs, which inhibit CTL responses | 49 | |
| IL-22 | HCC | Infiltrating cells | IL-22-deficiency reduces HCC development | 58 | |
| IL-33 | CCl4-induced fibrosis | LSECs, HSCs | Th2, ILCs | IL-33 induces IL-13 production in Th2 and ILCs | 62,63 |
| Protective functions of interleukins | |||||
| IL-6 | CCl4-induced fibrosis | NPCs | Abrogation of IL-6 signaling in NPCs enhances fibrosis | 6 | |
| Deletion of IL-6 increases hepatocyte injury and apoptosis | 18 | ||||
| IL-10 | Fibrosis | HSCs, LSECs, KCs, lymphocytes | HSCs | IL-10 inhibits HSC activation | 26,27 |
| Trichinella spiralis infection | Th cells | IL-10 dampens the cytokine response to T. spiralis | 32 | ||
| IL-12 | S. mansoni infection | Th1 | IL-12 shifts the immune response towards Th1 (reduced granuloma formation) | 35 | |
| IL-22 | ASH, NASH | IL-22 reduces fat accumulation and steatosis | 54,55 | ||
| Fibrosis | HSCs | IL-22 induces senescence in HSCs | 56 | ||
Abbreviations: SMA, smooth-muscle actin; ASH, alcohol-induced steatohepatitis; CCl4, carbon tetrachloride; CTLs, cytotoxic T lymphocytes; HCC, hepatocellular carcinoma; HSCs, hepatic stellate cells; ILCs, innate lymphoid cells; KCs, Kupffer cells; LSECs, liver sinusoidal endothelial cells; MDSCs, myeloid-derived suppressor cells; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; NPCs, nonparenchymal cells; PBC, primary biliary cirrhosis; Th, T helper.
Interleukin 6
IL-6 has long been recognized as an important proinflammatory cytokine whose expression is associated with many inflammatory disorders. Serum levels of IL-6 increase rapidly after infection or organ inflammation, and are therefore used in clinical practice as a diagnostic marker to detect inflammatory conditions, especially sepsis.5 Serum and intrahepatic levels of IL-6 are also strongly elevated in patients with acute and chronic liver diseases.6 IL-6 belongs to a family of cytokines comprising IL-6, IL-11, LIF, OSM, CNTF, NNT-1/BAFF-3, and CT-1 (Figure 1A).7 IL-6 binds directly to hepatocytes by interacting with an 80 kD membrane glycoprotein (gp80) that complexes with a signal-transducing transmembrane molecule named gp130 (Figure 1B).8 Binding of gp130 leads to dimerization of the intracellular domains of two gp130 molecules, which promotes association with receptor associated Janus kinases (JAKs) JAK1 and JAK2 and tyrosine kinase, and phosphorylation of different tyrosine residues on the gp130 molecule. Depending on the location of the phosphorylated tyrosines, STAT proteins (mainly STAT3) and also the Ras/MAPK become activated and trigger numerous downstream effects mediated by the signaling of IL-6 and related cytokines (Figure 1B).7 An important negative regulator of IL-6 signaling is SOCS3.9
Figure 1.
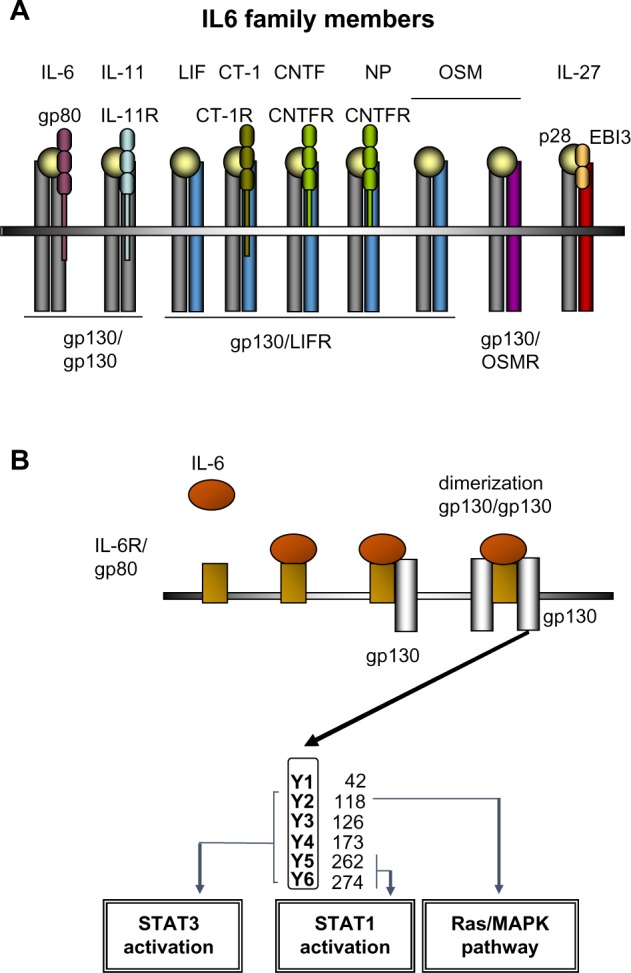
The IL-6 cytokine family and IL6 signaling in the liver.
Notes: (A) Selected members of the IL-6 cytokine family and their receptors (schematic). (B) IL-6 binds to IL-6R/gp80, eg, on hepatocytes. IL-6-gp80 then complexes with the signal-transducing molecule gp130. The complex of IL-6, gp80 (IL-6R), and two gp130 molecules mediates IL-6 signaling via phosphorylation of tyrosine (Y) residues of the intracellular gp130 molecule. Depending on the location of the phosphorylated tyrosines, STAT proteins (mainly STAT3), and also the Ras/MAPK pathway become activated and trigger the downstream effects.
Abbreviation: R, receptor.
IL-6-dependent signaling in the liver is critical for the induction of the acute-phase response.10 In experimental models of liver injury, mice deficient for the gp130 receptor in hepatocytes showed an abolished acute-phase response and an increased susceptibility to lipopolysaccharide-induced liver failure or to bacterial infections.11,12 In a model of ConA-induced hepatitis, pretreatment with IL-6 can protect mice from liver injury. This protection from ConA-induced liver damage requires gp130 signaling in hepatocytes and is mediated via the gp130/STAT3 signaling cascade, resulting in the upregulation of other cytokines, such as the IL-8 orthologue KC (CXCL1) and SAA2.13
In the context of chronic liver diseases, IL-6 has been identified as a major factor associated with the sex disparity observed in liver cancer. Men are much more prone to develop hepatocellular carcinoma (HCC) than women, and the same is true for male and female mice.14,15 Experimental studies on chemically induced HCC in mice have shown a correlation between IL-6 levels and hepatocarcinogenesis. Naugler et al observed that treatment with diethylnitrosamine (DEN) or carbon tetrachloride (CCl4) induced higher IL-6 levels in male than in female mice and that administration of estradiol reduced IL-6 levels.15 Accordingly, administration of estradiol before HCC induction reduced HCC development in male mice, and treatment with exogenous IL-6 enhanced HCC development in female mice. These results were further supported by a study by Yeh et al showing that DEN treatment induced the release of IL-1α from hepatocytes, which in turn activated Kupffer cells to produce IL-6.14 IL-6 then regulated NF-κB expression in hepatocytes, which led to HCC development. Again, IL-6 production was downregulated by estrogens.
IL-6 was also found to account for increased HCC development in obese mice. Mice fed a high-fat diet (HFD) expressed higher levels of IL-6 as well as TNFα, and developed more HCC after DEN treatment than mice fed a normal diet.16 Consistently, IL-6-deficient or TNF receptor-deficient mice displayed reduced hepatocarcinogenesis after DEN treatment and an HFD, which was also accompanied by reduced expression of STAT3. Furthermore, disruption of IL-6 signaling reduced the obesity-induced levels of TNFα and vice versa, confirming the important role of IL-6 and TNFα for promoting fat-induced liver inflammation and carcinogenesis. Additionally, deletion of SOCS3 has been shown to promote HCC development. Due to the loss of negative regulation, SOCS3−/− mice display increased STAT3 activation in the liver, which is associated with reduced apoptosis and accelerated proliferation of hepatocytes, leading to enhanced HCC development.17
In patients with fulminant hepatic failure or chronic liver diseases, IL-6 expression in serum and liver tissue correlates with disease progression.6,11 In contrast to its pathogenic role in liver cancer, IL-6 has been associated with protective functions during hepatic fibrogenesis (Figure 2). Kovalovich et al showed that IL-6-deficient mice display increased hepatocyte injury and apoptosis after just a single CCl4 injection, which resulted in higher liver fibrosis upon chronic treatment.18 Similar results were obtained in a different study, where deletion of gp130 in nonparenchymal liver cells also resulted in increased hepatic fibrogenesis.6 In contrast, gp130 deletion in hepatocytes did not influence fibrosis development. These results suggest a protective function for IL-6/gp130 signaling in nonparenchymal cells, possibly through downregulation of hepatic stellate cells (HSCs), the major extracellular matrix-producing cell type in liver fibrosis. Furthermore, a more recent study by Nasir et al showed that systemic injection of IL-6 followed by intrahepatic transplantation of mesenchymal stem cells was also able to reduce hepatocyte apoptosis and liver fibrogenesis after CCl4 treatment.19 The authors concluded that mesenchymal stem cell transplantation might be considered as a therapeutic option for patients with liver fibrosis. Other possible therapeutic approaches interfering with IL-6 and its signaling include the humanized anti-IL-6 receptor (IL-6R) antibody tocilizumab, soluble IL-6Rs, inhibitors of SOCS3 or signaling molecules, IL-6R-binding aptamers, or an agonistic designer cytokine called “hyper-IL-6”, which is a fusion protein of IL-6 and the soluble IL-6R, with considerably higher efficacy than the combination of the natural proteins.20–22
Figure 2.

Role of interleukins in development of liver fibrosis.
Notes: Interleukins are important immunologic mediators that critically regulate immune responses. During liver fibrogenesis, these cytokines can have profibrotic as well as antifibrotic functions. Typical profibrotic cytokines include IL-13 and IL-17, which directly mediate activation of hepatic stellate cells (HSCs), but also IL-33 and IL-5, which induce IL-13 production in lymphocytes and eosinophilic granulocytes, respectively. IL-10 and IL-22 have been associated with protection from hepatofibrogenesis, as they can prevent activation of HSCs and production of collagen. Furthermore, IL-12 also reduces fibrogenesis through downregulation of profibrotic Th-2 responses.
Abbreviations: Eo, eosinophilic granulocyte; hepa, hepatocyte; ILC, innate lymphoid cell; KC, Kupffer cell; LSEC, liver sinusoidal endothelial cell; MFB, myofibroblast; TC, T cell; Th, T helper.
Interleukin 10
IL-10 is one of the major anti-inflammatory cytokines, with important roles in counterbalancing hyperactive immune responses to protect the body from excessive cell and organ damage. Moreover, IL-10 is critically involved in tolerance against potential allergens, and impairment in IL-10 signaling may result in autoimmune reactions like asthma.23 IL-10 can be produced by regulatory T cells but also monocytes and B cells.24 In the liver, IL-10 can be produced by a variety of cell types, including hepatocytes, Kupffer cells, sinusoidal endothelial cells, HSCs, and lymphocytes.25 It is upregulated upon liver inflammation under various conditions, and is clearly associated with protective functions during chronic liver disease (Figure 2). Upon fibrosis induction through CCl4 treatment, IL-10-deficient mice show higher liver fibrosis with larger inflammatory infiltrates compared to wild-type (WT) mice.26,27 Independent studies demonstrated that HSCs express the IL-10R and also produce IL-10 when in a quiescent stage.28,29 It seems likely that this pathway suppresses HSCs’ profibrogenic function and keeps them in the quiescent status. Furthermore, IL-10 has been shown to directly affect collagen production and TGFβ secretion, thereby further limiting experimental fibrosis in mice.30 Additionally, in thioacetamide-induced liver fibrosis, IL-10 gene therapy reverts hepatic fibrosis and prevents cell apoptosis after fibrosis has already been established, suggesting a therapeutic potential for treatment with IL-10.31
In parasitic infection of the liver, IL-10 controls the host’s immune response to the parasite and therefore prevents exaggerated necrotic liver damage. IL-10-deficient mice infected with Trichinella spiralis show increased necrotic lesions along with eosinophilic infiltrates and higher production of proinflammatory cytokines.32 Bliss et al showed that IL-10 had no influence on the establishment or survival of the parasite but T cell-derived IL-10 was able to reduce the cytokine response induced by T. spiralis. Thereby, IL-10 dampens the inflammatory response and limits organ damage.32
Interleukin 12
IL-12 is a proinflammatory cytokine that plays an important role in the stimulation of T cells and other lymphocytes. Its major function is the induction of a T-helper (Th)-1 phenotype in Th cells and inhibition of the Th2 phenotype.33 Th1 immune responses are crucial for protective immunity against infections with (mainly intracellular) bacteria and fungi, and thus defective IL-12 expression or signaling may result in uncontrolled infections. On the other hand, overproduction of IL-12 can lead to severe autoimmune disorders due to the constant activation of Th1 cells.
Consistently, current knowledge about the role of IL-12 in chronic liver diseases has been mainly derived from animal models of bacterial or parasitic infection. Depending on the kind of pathogen, IL-12 can have protective as well as pathogenic functions. In malaria infection, a classic intracellular pathogen inducing Th1 responses, IL-12 expression is associated with increased liver injury.34 IL-12 levels increase during malaria infection, and IL-12-deficient mice show less liver injury compared to WT mice. However, inflammatory infiltrates in the liver did not differ between the strains. Adachi et al were able to show that the induction of IL-12 occurred via Toll-like receptor-mediated pathways, as liver injury and IL-12 expression were dependent on the signaling adaptor MyD88.34 IL-12 production then led to activation of cytotoxic lymphocytes that were able to kill infected hepatocytes. Therefore, liver pathogenesis was mediated by IL-12 production.
In contrast, during infection with Schistosoma mansoni, IL-12 was able to reduce liver pathology. S. mansoni usually induces a Th2-type immune response, which leads to the formation of large granulomas around parasite eggs in the liver. Sensitization of mice with S. mansoni eggs together with IL-12 before establishment of the parasite shifted the immune reaction toward a Th1 response with high expression of IL-12, IFNγ, and TNFα.35 Granuloma formation was also reduced in these mice. Neutralization of these Th1-associated cytokines with specific antibodies restored the Th2-type response and increased granuloma formation. The authors concluded that the conversion of the immune response from Th2 to Th1 phenotype by IL-12 is protective in this setting (Figure 2).
A more complex study using mice double-infected with S. mansoni, a Th2-inducing extracellular parasite, and Toxoplasma gondii, a Th1-inducing intracellular protozoan, further underlines the importance of IL-12 for the balance between Th1 and Th2 responses.36 Double-infected WT mice show increased liver damage and mortality compared to monoinfected ones, whereas double-infected IL-12-deficient mice display reduced liver damage and longer survival. This is associated with a reduction in Th1-associated cytokines (IFNγ, TNFα) and elevation of Th2-associated cytokines (IL-5). Accordingly, due to the loss of IL-12 and Th1 responses, these mice show uncontrolled replication of T. gondii. The authors concluded that IL-12 induced by T. gondii suppresses the Th2 response needed for protection against S. mansoni, leading to increased liver pathology and mortality in double-infected WT mice.36 At the same time, S. mansoni infection also renders mice more susceptible to T. gondii infection, because Th2 responses also downregulate IL-12 expression.
Interleukin 13 and interleukin 5
IL-13 is a Th2 effector cytokine that is produced mainly by activated Th2 cells, and is therefore involved in protective immunity against extracellular parasites like helminths but also allergic disorders.33 In the liver, IL-13 is associated with progression of fibrosis, as it can induce the production of collagen, TGFβ, and other fibrosis-associated genes in HSCs.37,38 Its role in chronic liver diseases has been most extensively studied in mice with S. mansoni infection leading to granuloma formation and subsequent fibrosis development in the liver. Several studies showed that IL-13 is the predominant profibrotic Th2 cytokine in S. mansoni infection, and that blockade of IL-13 prevents liver fibrogenesis.39–41 IL-13-deficient mice or mice treated with an IL-13 inhibitor developed significantly less liver fibrosis than mice with intact IL-13 signaling. By using different WT strains with differential susceptibility to fibrosis development, Chiaramonte et al showed that IL-13 levels were similar in all strains, and that the ratio of IL-13 to Th1 cytokines correlated with the degree of fibrosis.40 Furthermore, blockade of IL-13 was also helpful after liver fibrosis was already established, suggesting a therapeutic potential of IL-13 neutralization for patients with liver fibrosis. Interestingly, IL-13 played no role in the establishment of the parasite, nor did IL-13 blockade change the expression of other cytokines. The predominance of IL-13 over other Th2 cytokines was demonstrated by the fact that IL-4-deficient mice did not show such a profound reduction in collagen deposition and fibrosis development compared to IL-13-deficient mice.39,41
Reiman et al showed that IL-13 production during S. mansoni production is also regulated by IL-5, another Th2-associated cytokine.42 IL-5-deficient mice display formation of fewer and smaller granulomas in the liver, accompanied by reduced fibrosis and lower numbers of infiltrating eosinophilic granulocytes. Establishment or morbidity of the infection was not influenced by IL-5 deletion. The authors concluded that IL-5 mediates the recruitment of eosinophils to the liver, which are a source of IL-13 during S. mansoni infection (Figure 2). Therefore, IL-5 blockade might also be helpful for the treatment of fibrosis.
Interleukin 17
IL-17 is the prototypic and most widely studied member of the IL-17 family of cytokines. IL-17 is mainly produced by Th17 cells, but also other lymphocytes and neutrophils. Th17 cells, the third major proinflammatory subset of Th cells, are involved in host defense against extracellular pathogens, but also in a variety of autoimmune disorders, including asthma, multiple sclerosis, inflammatory bowel disease, and others.43
Evidence exists that Th17 cells are preferentially induced in the liver, and they have been associated with pathogenic functions in chronic liver inflammation. IL-2Rα−/− mice that spontaneously develop liver pathology resembling human primary biliary cirrhosis show elevated serum levels of IL-17 and IL-17-positive infiltrates in the liver.44 This is consistent with the observation that expression of IL-2 usually negatively regulates Th17 cell differentiation.45 Lan et al showed that the number of Th17 cells in the liver is higher than that observed in the spleen in both WT and IL-2Rα−/− mice, and that hepatic Th17 cells produce higher levels of IL-17 than splenic Th17 cells.44 Furthermore, coculture of CD4+ T cells with nonparenchymal liver cells induced IL-17 production in the T cells, suggesting that the intrahepatic milieu preferentially induces differentiation of Th17 cells.
During liver fibrogenesis induced by CCl4 treatment or bile-duct ligation, IL-17 is strongly upregulated in the liver, as well as in the circulation. In the liver, it is mainly produced by T cells, but also Kupffer cells.46 Meng et al demonstrated that abrogation of IL-17 signaling by deletion of IL-17RA protected mice from hepatofibrogenesis. Furthermore, the authors showed that the major IL-17-responsive cell types in the liver are Kupffer cells and HSCs, which contribute to fibrosis development upon activation (Figure 2).46 IL-17 directly induced production of collagen and α-smooth-muscle actin in HSCs through activation of STAT3, as STAT3−/− HSCs failed to produce collagen upon IL-17 treatment and STAT3-deficient mice displayed less fibrosis development compared to WT mice. A different study by Zheng et al showed also that blockade of IL-17 itself reduced liver fibrosis, whereas treatment with recombinant IL-17 resulted in increased fibrosis development.47 Furthermore, the transfer of bone marrow-derived stem cells decreased IL-17 levels in recipient mice and thus ameliorated hepatic fibrosis.
Induction of Th17 cells has also been associated with nonalcoholic fatty liver disease. Mice fed an HFD display higher levels of IL-17 and greater numbers of Th17 cells in the liver than mice fed normal chow.48 The liver damage observed after lipopolysaccharide injection into HFD-fed mice was ameliorated through treatment with an IL-17-blocking antibody, which resulted in reduced serum transaminase levels and fewer infiltrates in the liver. Furthermore, Tang et al showed that addition of IL-17 increased hepatocyte steatosis in an in vitro culture system. This suggests a proinflammatory role for Th17 cells also in nonalcoholic fatty liver disease.48
A very recent study on hepatocarcinogenesis in mice induced by DEN or transplantation of Hepa1–6 cells identified a pathogenic role for IL-17 in this setting as well. Ma et al demonstrated that IL-17-deficient mice display reduced tumor growth with increased numbers of cytotoxic T lymphocytes (CTLs) and decreased numbers of myeloid derived suppressor cells (MDSCs) in the liver.49 Administration of recombinant IL-17 enhanced tumor growth, increased MDSC numbers, and decreased CTL numbers in these mice. In contrast to the previously described studies on chronic liver disease models where IL-17 was predominantly produced by Th17 cells, γδ T cells were the major IL-17 producers in this tumor model. Consistently, depletion of γδ T cells reduced tumor growth in WT mice. Furthermore, the authors showed that IL-17 induced MDSC recruitment to the liver and that MDSCs in turn promoted IL-17 production by γδ T cells, thus acting as a positive feedback loop on each other.49 MDSCs then suppress antitumor CTL responses and thus promote tumor growth. Interestingly, γδ T cells are also a major source of IL-17 during experimental fibrosis induction in mice, but the antifibrogenic functions exerted by γδ T cells in hepatofibrogenesis are independent from their IL-17 synthesis.50,51
Interleukin 22
IL-22 belongs to the IL-10 family of cytokines, and plays important roles in the modulation of tissue immune responses to inflammation. IL-22 can be produced by adaptive CD4+ Th cells, including Th17 cells, but also lymphocytes of the innate immune system, like natural killer cells or γδ T cells.52 IL-22 is expressed in various chronic inflammatory conditions, and may fulfill pro- as well as anti-inflammatory functions. IL-22R is mainly expressed by epithelial cells, including hepatocytes, and IL-22 has therefore been described to exert tissue-protective functions at epithelial surfaces.52 Several studies have shown that IL-22 can reduce inflammation-induced damage of hepatocytes in vitro and in vivo by promoting their survival and inhibiting apoptosis.53
In alcoholic as well as nonalcoholic steatohepatitis, IL-22 reduced liver-fat content and subsequent liver injury.54,55 In mice fed with ethanol or an HFD, IL-22 was strongly upregulated in the liver, and treatment with exogenous IL-22 reduced liver injury and lipogenesis in both models. Moreover, Ki et al showed that this protective function is dependent on STAT3 signaling, as STAT3-deficient mice were not protected when treated with IL-22.54 In CCl4-induced liver fibrogenesis, IL-22 is protective through induction of senescence in HSCs, the major fibrogenic cell type in the liver (Figure 2). A study by Kong et al showed that HSCs express IL-22R and that IL-22 prevents HSC apoptosis in vitro and in vivo without affecting their proliferation.56 Interestingly, overexpression of IL-22 or treatment with recombinant IL-22 also reduced fibrosis in vivo, although HSC survival was increased. The authors showed that this was the result of HSC senescence induced by IL-22. Induction of senescence was dependent on STAT3 signaling and further promoted by activation of SOCS3.56
Furthermore, IL-22 is also involved in the restoration of liver mass after organ damage. Liver progenitor cells have been shown to express IL-22R, and IL-22 derived from inflammatory cells induces liver progenitor cell proliferation. Concordantly, mice treated with IL-22 show increased ductular reactions after a 3,5-diethoxycarbonyl-1,4-dihydrocollidine diet, suggesting that IL-22 helps in restoring functional liver mass after hepatocyte loss.57
However, IL-22 can also have pathogenic functions in chronic liver disease, as demonstrated by a study on DEN-induced carcinogenesis in mice. IL-22 is upregulated in the liver upon DEN treatment, and liver tumors express even higher levels of IL-22 than normal liver parenchyma.58 Furthermore, IL-22+ cells accumulate around hepatic tumor nodules and IL-22-deficient mice develop less HCC. This suggests a tumor-promoting role for IL-22 in HCC development.
Interleukin 33
IL-33 is a quite recently identified cytokine of the IL-1 family, and is mainly expressed by stromal cells.59 It has been shown to strongly induce the Th2 phenotype in Th cells, and therefore promotes progression of Th2-related diseases like asthma. However, IL-33 also exerts protective functions in cardiovascular diseases, including obesity and atherosclerosis.60,61
IL-33 has recently gained attention in the context of liver fibrogenesis, as Th2 cells are strongly associated with fibrosis progression. Consistently, IL-33 has been shown to promote hepatic fibrosis through the induction of Th2 cells. Marvie et al showed that IL-33 is upregulated in human and murine fibrosis, and is mainly produced by sinusoidal endothelial cells and activated HSCs.62 Further studies in mice demonstrated that the IL-33 receptor is expressed on intrahepatic T cells and that IL-33 induces expression of the Th2-related cytokines IL-4, IL-5, and IL-13 in these cells (Figure 2). Accordingly, IL-33 levels also correlated with collagen levels in fibrotic livers.
A recent study by McHedlidze et al identified a second mechanism of IL-33-mediated fibrogenesis. IL-33 was upregulated in CCl4- or thioacetamide-treated mice and facilitated the accumulation of innate lymphoid cells in fibrotic livers.63 Innate lymphoid cells are potent producers of IL-13 and promote activation of HSCs and therefore fibrosis (Figure 2). Interestingly, vector-encoded overexpression of IL-33 specifically in the liver was sufficient to induce fibrosis without administration of chemicals, demonstrating the powerful profibrotic role of IL-33-mediated IL-13 induction.
Conclusion and outlook
Taken together, sophisticated mouse models either mimicking distinct pathological conditions by induction of liver injury or targeting cytokines and cytokine-signaling pathways in the liver or even in distinct cellular compartments have provided enormous insight into the different functions of interleukins during chronic liver diseases. Interleukins critically regulate the immune responses, leading to the development of chronic diseases. The exact effect of a cytokine depends on different factors, including the producing cell type, the responding cell type, and the disease entity. These effects can be protective, pathogenic, or both, depending on the disease model studied (summarized in Table 1). This makes blockade of proinflammatory or induction of anti-inflammatory interleukins attractive strategies for the treatment of liver diseases. For example, inhibition of IL-17 or IL-33 might prevent the development of fibrosis, and blockade of IL-13 might even revert liver pathology when fibrosis is already established. Likewise, IL-10 gene therapy has been proven to induce resolution of fibrosis in a mouse model and might also be considered for patients with liver fibrosis. However, some cytokines can have opposing functions, depending on the liver disease studied, and might thus not be optimal therapeutic targets. IL-6 or IL-22, for example, prevent fibrogenesis, but promote hepatocellular carcinoma. Therefore, induction of IL-6 or IL-22 could be helpful for the treatment of patients with liver fibrosis, but would also potentially increase the risk of tumor development in the liver. Mouse models provide a unique opportunity to clarify the contribution of a specific cytokine to liver pathology, and to optimize treatment strategies with respect to interleukin targeting or cell-specific delivery.
Acknowledgments
The authors thank all members of the Tacke lab and the Medical Clinic III for helpful discussions. This work was supported by the German Research Foundation (DFG Ta434/2-1, DFG SFB/TRR57) and by the Interdisciplinary Center for Clinical Research (IZKF) Aachen.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Brocker C, Thompson D, Matsumoto A, Nebert DW, Vasiliou V. Evolutionary divergence and functions of the human interleukin (IL) gene family. Hum Genomics. 2010;5:30–55. doi: 10.1186/1479-7364-5-1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Commins SP, Borish L, Steinke JW. Immunologic messenger molecules: cytokines, interferons, and chemokines. J Allergy Clin Immunol. 2010;125:S53–S72. doi: 10.1016/j.jaci.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 3.Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–835. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 4.Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity. 2013;39:61–73. doi: 10.1016/j.immuni.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jekarl DW, Lee SY, Lee J, et al. Procalcitonin as a diagnostic marker and IL-6 as a prognostic marker for sepsis. Diagn Microbiol Infect Dis. 2013;75:342–347. doi: 10.1016/j.diagmicrobio.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 6.Streetz KL, Tacke F, Leifeld L, et al. Interleukin 6/gp130-dependent pathways are protective during chronic liver diseases. Hepatology. 2003;38:218–229. doi: 10.1053/jhep.2003.50268. [DOI] [PubMed] [Google Scholar]
- 7.Tacke F, Luedde T, Trautwein C. Inflammatory pathways in liver homeostasis and liver injury. Clin Rev Allergy Immunol. 2009;36:4–12. doi: 10.1007/s12016-008-8091-0. [DOI] [PubMed] [Google Scholar]
- 8.Taga T, Kishimoto T. Gp130 and the interleukin-6 family of cytokines. Annu Rev Immunol. 1997;15:797–819. doi: 10.1146/annurev.immunol.15.1.797. [DOI] [PubMed] [Google Scholar]
- 9.Bode JG, Nimmesgern A, Schmitz J, et al. LPS and TNFα induce SOCS3 mRNA and inhibit IL-6-induced activation of STAT3 in macrophages. FEBS Lett. 1999;463:365–370. doi: 10.1016/s0014-5793(99)01662-2. [DOI] [PubMed] [Google Scholar]
- 10.Zhang D, Sun M, Samols D, Kushner I. STAT3 participates in transcriptional activation of the C-reactive protein gene by interleukin-6. J Biol Chem. 1996;271:9503–9509. doi: 10.1074/jbc.271.16.9503. [DOI] [PubMed] [Google Scholar]
- 11.Streetz KL, Wüstefeld T, Klein C, et al. Lack of gp130 expression in hepatocytes promotes liver injury. Gastroenterology. 2003;125:532–543. doi: 10.1016/s0016-5085(03)00901-6. [DOI] [PubMed] [Google Scholar]
- 12.Wuestefeld T, Klein C, Streetz KL, et al. Lack of gp130 expression results in more bacterial infection and higher mortality during chronic cholestasis in mice. Hepatology. 2005;42:1082–1090. doi: 10.1002/hep.20912. [DOI] [PubMed] [Google Scholar]
- 13.Klein C, Wüstefeld T, Assmus U, et al. The IL-6-gp130-STAT3 pathway in hepatocytes triggers liver protection in T cell-mediated liver injury. J Clin Invest. 2005;115:860–869. doi: 10.1172/JCI200523640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeh SH, Chen PJ. Gender disparity of hepatocellular carcinoma: the roles of sex hormones. Oncology. 2010;78(Suppl 1):172–179. doi: 10.1159/000315247. [DOI] [PubMed] [Google Scholar]
- 15.Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 16.Park EJ, Lee JH, Yu GY, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogata H, Kobayashi T, Chinen T, et al. Deletion of the SOCS3 gene in liver parenchymal cells promotes hepatitis-induced hepatocarcinogenesis. Gastroenterology. 2006;131:179–193. doi: 10.1053/j.gastro.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 18.Kovalovich K, DeAngelis RA, Li W, Furth EE, Ciliberto G, Taub R. Increased toxin-induced liver injury and fibrosis in interleukin-6-deficient mice. Hepatology. 2000;31:149–159. doi: 10.1002/hep.510310123. [DOI] [PubMed] [Google Scholar]
- 19.Nasir GA, Mohsin S, Khan M, et al. Mesenchymal stem cells and interleukin-6 attenuate liver fibrosis in mice. J Transl Med. 2013;11:78. doi: 10.1186/1479-5876-11-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest. 2011;121:3375–3383. doi: 10.1172/JCI57158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rose-John S. The biology of interleukin-6 in the 21st century. Semin Immunol. 2014;26:1. doi: 10.1016/j.smim.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 22.Galun E, Rose-John S. The regenerative activity of interleukin-6. Methods Mol Biol. 2013;982:59–77. doi: 10.1007/978-1-62703-308-4_4. [DOI] [PubMed] [Google Scholar]
- 23.Borish L, Aarons A, Rumbyrt J, Cvietusa P, Negri J, Wenzel S. Interleukin-10 regulation in normal subjects and patients with asthma. J Allergy Clin Immunol. 1996;97:1288–1296. doi: 10.1016/s0091-6749(96)70197-5. [DOI] [PubMed] [Google Scholar]
- 24.Del Prete G, De Carli M, Almerigogna F, Giudizi MG, Biagiotti R, Romagnani S. Human IL-10 is produced by both type 1 helper (Th1) and type 2 helper (Th2) T cell clones and inhibits their antigen-specific proliferation and cytokine production. J Immunol. 1993;150:353–360. [PubMed] [Google Scholar]
- 25.Wan S, LeClerc JL, Schmartz D, et al. Hepatic release of interleukin-10 during cardiopulmonary bypass in steroid-pretreated patients. Am Heart J. 1997;133:335–339. doi: 10.1016/s0002-8703(97)70229-1. [DOI] [PubMed] [Google Scholar]
- 26.Thompson K, Maltby J, Fallowfield J, McAulay M, Millward-Sadler H, Sheron N. Interleukin-10 expression and function in experimental murine liver inflammation and fibrosis. Hepatology. 1998;28:1597–1606. doi: 10.1002/hep.510280620. [DOI] [PubMed] [Google Scholar]
- 27.Louis H, Van Laethem JL, Wu W, et al. Interleukin-10 controls neutrophilic infiltration, hepatocyte proliferation, and liver fibrosis induced by carbon tetrachloride in mice. Hepatology. 1998;28:1607–1615. doi: 10.1002/hep.510280621. [DOI] [PubMed] [Google Scholar]
- 28.Wang SC, Ohata M, Schrum L, Rippe RA, Tsukamoto H. Expression of interleukin-10 by in vitro and in vivo activated hepatic stellate cells. J Biol Chem. 1998;273:302–308. doi: 10.1074/jbc.273.1.302. [DOI] [PubMed] [Google Scholar]
- 29.Mathurin P, Xiong S, Kharbanda KK, et al. IL-10 receptor and coreceptor expression in quiescent and activated hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2002;282:G981–90. doi: 10.1152/ajpgi.00293.2001. [DOI] [PubMed] [Google Scholar]
- 30.Reitamo S, Remitz A, Tamai K, Uitto J. Interleukin-10 modulates type I collagen and matrix metalloprotease gene expression in cultured human skin fibroblasts. J Clin Invest. 1994;94:2489–2492. doi: 10.1172/JCI117618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hung KS, Lee TH, Chou WY, et al. Interleukin-10 gene therapy reverses thioacetamide-induced liver fibrosis in mice. Biochem Biophys Res Commun. 2005;336:324–331. doi: 10.1016/j.bbrc.2005.08.085. [DOI] [PubMed] [Google Scholar]
- 32.Bliss SK, Alcaraz A, Appleton JA. IL-10 prevents liver necrosis during murine infection with Trichinella spiralis. J Immunol. 2003;171:3142–3147. doi: 10.4049/jimmunol.171.6.3142. [DOI] [PubMed] [Google Scholar]
- 33.O’Garra A, Arai N. The molecular basis of T helper 1 and T helper 2 cell differentiation. Trends Cell Biol. 2000;10:542–550. doi: 10.1016/s0962-8924(00)01856-0. [DOI] [PubMed] [Google Scholar]
- 34.Adachi K, Tsutsui H, Kashiwamura S, et al. Plasmodium berghei infection in mice induces liver injury by an IL-12- and Toll-like receptor/myeloid differentiation factor 88-dependent mechanism. J Immunol. 2001;167:5928–5934. doi: 10.4049/jimmunol.167.10.5928. [DOI] [PubMed] [Google Scholar]
- 35.Hoffmann KF, Caspar P, Cheever AW, Wynn TA. IFN-gamma, IL-12, and TNF-alpha are required to maintain reduced liver pathology in mice vaccinated with Schistosoma mansoni eggs and IL-12. J Immunol. 1998;161:4201–4210. [PubMed] [Google Scholar]
- 36.Araujo MI, Bliss SK, Suzuki Y, Alcaraz A, Denkers EY, Pearce EJ. Interleukin-12 promotes pathologic liver changes and death in mice coinfected with Schistosoma mansoni and Toxoplasma gondii. Infect Immun. 2001;69:1454–1462. doi: 10.1128/IAI.69.3.1454-1462.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sugimoto R, Enjoji M, Nakamuta M, et al. Effect of IL-4 and IL-13 on collagen production in cultured LI90 human hepatic stellate cells. Liver Int. 2005;25:420–428. doi: 10.1111/j.1478-3231.2005.01087.x. [DOI] [PubMed] [Google Scholar]
- 38.Shimamura T, Fujisawa T, Husain SR, Kioi M, Nakajima A, Puri RK. Novel role of IL-13 in fibrosis induced by nonalcoholic steatohepatitis and its amelioration by IL-13R-directed cytotoxin in a rat model. J Immunol. 2008;181:4656–4565. doi: 10.4049/jimmunol.181.7.4656. [DOI] [PubMed] [Google Scholar]
- 39.Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J Clin Invest. 1999;104:777–785. doi: 10.1172/JCI7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chiaramonte MG, Cheever AW, Malley JD, Donaldson DD, Wynn TA. Studies of murine schistosomiasis reveal interleukin-13 blockade as a treatment for established and progressive liver fibrosis. Hepatology. 2001;34:273–282. doi: 10.1053/jhep.2001.26376. [DOI] [PubMed] [Google Scholar]
- 41.Fallon PG, Richardson EJ, McKenzie GJ, McKenzie AN. Schistosome infection of transgenic mice defines distinct and contrasting pathogenic roles for IL-4 and IL-13: IL-13 is a profibrotic agent. J Immunol. 2000;164:2585–2591. doi: 10.4049/jimmunol.164.5.2585. [DOI] [PubMed] [Google Scholar]
- 42.Reiman RM, Thompson RW, Feng CG, et al. Interleukin-5 (IL-5) augments the progression of liver fibrosis by regulating IL-13 activity. Infect Immun. 2006;74:1471–1479. doi: 10.1128/IAI.74.3.1471-1479.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hammerich L, Heymann F, Tacke F. Role of IL-17 and Th17 cells in liver diseases. Clin Dev Immunol. 2011;2011:345803. doi: 10.1155/2011/345803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lan RY, Salunga TL, Tsuneyama K, et al. Hepatic IL-17 responses in human and murine primary biliary cirrhosis. J Autoimmun. 2009;32:43–51. doi: 10.1016/j.jaut.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laurence A, Tato CM, Davidson TS, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 46.Meng F, Wang K, Aoyama T, et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology. 2012;143:765. e1–776. e3. doi: 10.1053/j.gastro.2012.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng L, Chu J, Shi Y, et al. Bone marrow-derived stem cells ameliorate hepatic fibrosis by down-regulating interleukin-17. Cell Biosci. 2013;3:46. doi: 10.1186/2045-3701-3-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tang Y, Bian Z, Zhao L, et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin Exp Immunol. 2011;166:281–290. doi: 10.1111/j.1365-2249.2011.04471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma S, Cheng Q, Cai Y, et al. IL-17A produced by γδ T cells promotes tumor growth in hepatocellular carcinoma. Cancer Res. 2014;74:1969–1982. doi: 10.1158/0008-5472.CAN-13-2534. [DOI] [PubMed] [Google Scholar]
- 50.Hammerich L, Bangen JM, Govaere O, et al. Chemokine receptor CCR6-dependent accumulation of γδ T cells in injured liver restricts hepatic inflammation and fibrosis. Hepatology. 2014;59:630–642. doi: 10.1002/hep.26697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hammerich L, Tacke F. Role of gamma-delta T cells in liver inflammation and fibrosis. World J Gastrointest Pathophysiol. 2014;5:107–113. doi: 10.4291/wjgp.v5.i2.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004;21:241–254. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 53.Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology. 2004;39:1332–1342. doi: 10.1002/hep.20184. [DOI] [PubMed] [Google Scholar]
- 54.Ki SH, Park O, Zheng M, et al. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatology. 2010;52:1291–1300. doi: 10.1002/hep.23837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang L, Zhang Y, Wang L, et al. Amelioration of high fat diet induced liver lipogenesis and hepatic steatosis by interleukin-22. J Hepatol. 2010;53:339–347. doi: 10.1016/j.jhep.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 56.Kong X, Feng D, Wang H, et al. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology. 2012;56:1150–1159. doi: 10.1002/hep.25744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feng D, Kong X, Weng H, et al. Interleukin-22 promotes proliferation of liver stem/progenitor cells in mice and patients with chronic hepatitis B virus infection. Gastroenterology. 2012;143:188–198. e7. doi: 10.1053/j.gastro.2012.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang R, Tan Z, Deng L, et al. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology. 2011;54:900–909. doi: 10.1002/hep.24486. [DOI] [PubMed] [Google Scholar]
- 59.Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 60.Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie AN, Lee RT. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J Clin Invest. 2007;117:1538–1549. doi: 10.1172/JCI30634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller AM, Xu D, Asquith DL, et al. IL-33 reduces the development of atherosclerosis. J Exp Med. 2008;205:339–346. doi: 10.1084/jem.20071868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marvie P, Lisbonne M, L’Helgoualc’h A, et al. Interleukin-33 overexpression is associated with liver fibrosis in mice and humans. J Cell Mol Med. 2010;14:1726–1739. doi: 10.1111/j.1582-4934.2009.00801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McHedlidze T, Waldner M, Zopf S, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity. 2013;39:357–371. doi: 10.1016/j.immuni.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]