

Abstract
The fiber-modified adenoviral vector Delta-24-RGD (D24RGD) offers vast therapeutic potential. Direct injection of D24RGD has been used to successfully target ovarian tumors in mice. However, systemic toxicity, especially in the liver, is a profoundly limits the efficacy of direct viral vector delivery. Mesenchymal stem cells (MSC) have the ability to function as a vector for targeted gene therapy because of their preferential engraftment into solid tumors and participation in tumor stroma formation. We show that MSC-guided delivery of D24RGD is specific and efficient and reduces the overall systemic toxicity in mice to negligible levels compared with D24RGD alone. In our model, we found efficient targeted delivery of MSC-D24RGD to both breast and ovarian cell lines. Furthermore, immunohistochemical staining for adenoviral hexon protein confirmed negligible levels of systemic toxicity in mice given MSC-D24RGD compared with those given D24RGD. These data suggest that delivery of D24RGD via MSC not only increases the targeted delivery efficiency but also reduces the systemic exposure of the virus, thereby reducing overall systemic toxicity to the host and ultimately enhancing its value as an anti-tumor therapeutic candidate.
Keywords: gene delivery, fiber-modified adenovirus, MSC, ovarian cancer
Introduction
Adenovirus, a DNA tumor virus, is able to infect and replicate in quiescent cells because viral proteins (vp) induce S-phase entry. The most efficient progression from G1 into S-phase requires binding and inactivation of the pRb family of proteins by the adenovirus E1A early vp. This interaction requires amino acids 122–129 of the E1A protein conserved region 2. Mutations within this domain result in a loss of transformation by E1A without inhibiting its transactivation function.1 For adenovirus serotype 5 (Ad5), coxsackie adenovirus receptor (CAR) expression is the major factor determining infectivity. Evidence suggests that the expression level of CAR is often low on primary ovarian cancer cells2 and on mesenchymal stem cells (MSC).3 To overcome low expression levels and thus increase infection in MSC, and subsequently in ovarian carcinoma, the fiber of Ad5–Delta-24–RGD (D24RGD) was modified by incorporating the Arginine-Glycine-Asparatate (RGD)-4C motif into the HI loop, to allow for integrin binding. This infectivity enhancement has previously been shown to dramatically increase the transduction of ovarian cancer cells.4 Moreover, because most ovarian cancer patients present with ascites, the RGD-4C modification allows partial escape from neutralizing antibodies regularly present in the ascites.4 D24RGD has been used as therapy for many types of tumors,5–7 including ovarian tumors,8 via direct injection of the virus. Direct injection of the D24 virus alone has resulted in increased survival in ovarian carcinoma mouse models, but it causes significant systemic toxicity, especially in the liver.
Numerous studies have shown the inadequacy of viral vector gene delivery is due to systemic toxicity induced in the host. Even when locally injected into the tumor site—an application that is not possible in all cancers—viral dissemination still occurs.9–11 The high toxicity levels render this therapeutic method ineffective in potential nontopical clinical applications; however, as we will show, using MSC as a delivery vehicle abrogates viral dissemination and reduces systemic toxicity to negligible levels.
MSC have vast potential for clinical use. They contribute to the maintenance and regeneration of various connective tissues and can be obtained from bone marrow, expanded in vitro, and genetically modified for therapy. Their in vivo engraftment depends on the appropriate external signals from the tissue microenvironment. It has been shown that tumors provide the local environmental signals—closely resembling wound healing and tissue repair signals—that can mediate MSC engraftment.12–15 We originally showed the preferential engraftment of MSC into solid tumors and their participation in tumor stroma development; subsequently, we and others have shown the efficacy of tumor engraftment by MSC.11, 16–18
MSC enhance the therapeutic value of viral vector–mediated gene therapy in two ways. First, they enhance the targeting specificity of the conditionally replicating D24RGD to the localized tumor environment, thereby theoretically increasing the therapeutic index by selectively replicating within tumor tissue,19 resulting in efficient tumor penetration and amplification of the anti-tumor effect. This ability has been shown by other investigators in a number of different cancer models.18 Second, the use of MSC substantially reduces the systemic toxicity and viral dissemination that occurs during standard D24RGD treatment. Adenoviral gene therapy via MSC delivery is a safe and superior therapeutic application for aggressive cancer therapies, and it need no longer be limited to localized intratumoral injections: it can be administered intraperitoneally.
The efficiency of MSC delivery of adenovirus to tumors has been investigated previously in ovarian and lung metastases animal models and glioma animal models.12,18,20,21 Contrary to Hakkarainen et al.22 who were unable to show MSC-tumor tropism, we are able to show MSC-tumor tropism with our D24RGD-loaded MSC which enhances the specificity of viral delivery and safeguards against the systemic toxicity observed in trials using viral vectors alone.
In an effort to overcome the systemic toxicity initiated by high-titer adenoviral treatments, mammalian cells have been explored as delivery vehicles. Gene delivery vectors must possess several characteristics to be considered clinically applicable. To date, several cells have been examined as potential vehicles, including hematopoietic stem cells,32 fibroblasts,33 keritinoctes,34 and myoblasts,35 in an attempt to overcome the immunological barriers to direct administration of viral vectors, including rapid degradation (immune clearance) and high toxicity.18,36 MSC are an attractive tool for the delivery of anti-cancer agents to tumors because of their selective engraftment in injured or regenerating tissues, such as tumors. 15
In our in vivo mouse model, we show the delivery of D24RGD via MSC increases the targeted delivery efficiency and reduces the overall systemic toxicity to the host by in vivo imaging, histology, and immunohistochemical staining.
Materials and methods
Cell isolation and culture
Human MSC were isolated as previously reported.12 Human ovarian cancer SKOV-3, OVCAR-3, and HEY cells were cultured as previously described,23 as were human breast cancer MDA231 and melanoma A375SM cell lines.17 Human kidney fibroblast 293s cells were a gift from Dr. Richard Cristiano (Department of Genitourinary Oncology, M. D. Anderson Cancer Center, Houston, TX). The cells were maintained in Dulbecco’s modified Eagle medium (MEM) supplemented with 10% fetal bovine serum, L-glutamine, and penicillin-streptomycin mixture (Gibco/Invitrogen, Carlsbad, CA).
Adenoviruses
Green fluorescent protein (GFP)–tagged adenovirus was created in our laboratory as reported previously.24 The replication-competent Ad5-D24RGD adenovirus was provided by Juan Fueyo (The University of Texas M. D. Anderson Cancer Center, Houston, TX). This virus contains a 24-nucleotide deletion from Ad5 bp 923 to 946 (both included) that corresponds to the amino acid sequence L122TCHEAGF129 of the E1A protein and is known to be necessary for Rb protein binding.23 Details of the tumor-specific replication of this virus have been presented elsewhere,25
D24RGD also contains recombinant RGD fiber. Briefly, an E1 fragment containing the 24-bp Ad5 deletion was isolated from the plasmid pXC1-Δ24 (originally used to construct D24)25 and cloned by homologous recombination into the ClaI-digested plasmid pVK503 containing the RGD fiber. The genome of the new virus was released from the plasmid backbone by digestion with PacI, and the resulting fragment was used to transfect 293 cells to rescue Ad5-D24RGD. Details of this process have been published elsewhere.26
The control virus used throughout this experiment was UV-inactivated D24RGD (UVD24RDG). It was prepared as follows: D24RGD was diluted 1:1000 in serum-free alpha MEM, irradiated on ice eight times with 125mL, and then used immediately.
MSC replication
MSC were plated at 20,000 cells per well in a six-well plate (Becton Dickinson, Franklin Lakes, NJ). Every 24 hours, one well was washed with PBS, and cells from that well were lifted with Trypsin-EDTA (Gibco, Carlsbad, CA), counted ten times on a hemacytometer, and the average number determined.
D24RGD replication in MSC
MSC were plated at 100,000 cells per well in a six-well plate (Becton Dickinson). Then, 24 hours later, MSC were infected for 2 hours in serum-free medium (alpha MEM) at 37ºC in 5% CO2 with increasing concentrations of D24RGD. Forty-eight hours later, cells were stained with crystal violet (0.2% in 10% phosphate-buffered formalin) for 1 hour. The wells were then washed with H2O and allowed to dry, and photographs were taken at 4× magnification. Imaging was performed with a Zeiss Axioplan2 microscope (Carl Zeiss Inc., Thornwood, NY) equipped with a charge coupled device (CCD) camera (Hamamatsu Corp., Bridgewater, NJ) and Adobe Photoshop software (Adobe Systems Inc., San Jose, CA). D24RGD replication in MSC was confirmed by the detection of hexon protein levels (IDEIA kit, dakocytomation). Burst size data, measuring the viral replication in one round of MSC replication was measured after 24 hours of MSC replication. Briefly, 24 hours after MSC were plated at 250,000 cells per 10cm dish, MSC were infected for 2 hours with D24RGD at increasing MOI. 24 hours later, the MSC were trypsinized, spun down and resuspended in TE buffer (10uM) before being freeze-thawed 3 times. DNA was extracted by phenol:chloroform:isoamyl alchoholwashed in 100% ethanol and resuspended in water before being analyzed by real time PCR (ABI-7900) using Sybr-green super mix (Bio-Rad, Hercules, CA) and E1a primers. E1a expressed was standardized against diluted concentrations of pure virus titer at known concentrations and expressed as viral particles per microliter.
MSC-D24RGD in ovarian cell lines
MSC were plated at 100,000 cells per well in a six-well plate (Becton Dickinson). Then, 24 hours later, MSC were infected for 2 hours in serum-free medium (alpha MEM) at 37ºC in 5% CO2 with increasing concentrations of D24RGD. Wells were then washed three times with PBS, and medium (alpha MEM and 20% fetal calf serum [FCS]) was replaced. Supernatants were collected after 48 hours and applied to OVCAR-3 and SKOV-3 cells (initially plated at 50,000 cells/well in a 12-well plate 24 hours earlier). Results are shown as viable cells (percentage of control) after 72 hours and were determined using trypan blue cell counts.
MSC-D24RGD in Multiple Cell Lines
MSC were plated at 100,000 cells per well in a six-well plate (Becton Dickinson). Then, 24 hours later, MSC were infected for 2 hours in serum-free medium (alpha MEM) at 37ºC in 5% CO2 with 50 vp/cell of D24RGD. Wells were then washed three times with PBS, and medium (alpha MEM and 20% FCS) was replaced. Supernatants were applied 72 hours later to OVCAR-3, SKOV-3, HEY, A375sm (melanoma), MDA231 (breast cancer), or 293s cells (initially plated at 300,000 cells/well 24 hours earlier). Cells were photographed 24 and 48 hours after the application of the MSC- D24RGD supernatants. After 48 hours, cells were stained with crystal violet (0.2% in 10% phosphate-buffered formalin) for 1 hour. The wells were then washed with DiH2O and allowed to dry, and photographs were taken at 4× magnification.
Mice, cell administration, tumors, and survival analysis
Female CB-17 SCID mice were purchased from Harlan (Indianapolis, IN). Mice were used in accordance with institutional guidelines under approved protocols. Cells were administered as a suspension in 1 ml of PBS. Mice were injected intraperitoneally with 4 x 106 SKOV-3 cells (day 0). Then, 12 days later, treatments began that consisted of 4 weekly injections of either 1 x 106 MSC-D24RGD (50 vp/cell D24RGD, n=21), MSC-GFP (3000 vp/cell, n=10), MSC alone (n=5), or MSC-UVD24RGD (50 vp/cell, n=5). One control group received no treatments (n=4). Survival was measured as the day of tumor cell injection until the day of death. Differences in survival were determined using a two-tailed log-rank test. Statistical analysis was performed using Statistica software (StatSoft Inc., Tulsa, OK).
Tissue processing and imaging studies
Tumors and other organs were embedded in OTC compound (Miles, Inc., Elkhart, IN) and then snap-frozen in liquid nitrogen and stored at –80ºC. Frozen tissue was sectioned (6–8 μm) and processed for hematoxylin-eosin (HE) or hexon immunohistochemical staining. The appropriate slides were also checked for the presence of GFP.
Immunohistochemical analysis
Adenoviral hexon protein was detected in tumor xenografts using frozen sections of mouse tumors. Sections were fixed for 5 minutes in neutral buffered formalin, then endogenous peroxidase activity was quenched by incubating the sections in 0.3% hydrogen peroxide in methanol for 30 minutes. The sections were treated with goat anti-hexon antibody (diluted 1:1000; Chemicon, Temecula, CA). Peroxidase substrate (Vector Laboratories, Burlingame, CA) was developed using an 3-amino-9-ethylcarbazole substrate kit (Vector Laboratories). Slides were then counterstained with hematoxylin QS (Vector Laboratories), and aqueously mounted with aqueous mounting medium (low viscosity) (Scytek Laboratories, Logan, UT).
Results
Replication of D24RGD in MSC
The D24RGD replication pattern closely resembles that of the MSC growth curve (Figure 1). D24RGD replication in MSC was confirmed by the detection of hexon protein levels (Figure 2) in the MSC supernatant over a six-day period. The increase in hexon protein correlates with replication of the virus in MSC. MSC-D24RGD showed evidence of a cytopathic effect (CPE) within 48 hours compared with UV-irritated controls after infection with 5, 50, 500, 1000, or 2500 vp/cell (Figure 2). D24RGD replication in MSC was confirmed by the detection of E1a expression levels in the MSC following one round of replication. The E1a expression was quantified by real-time PCR using known, sequentially diluted concentrations of pure virus as standard curve. E1a expression in infected MSC was graphed as viable vp produced per cell, or burst size (Figure 2d).
Figure 1. MSC replication.

The pattern of MSC growth is a sigmoidal curve. MSC were initially plated at 20,000 cells per well in a six-well plate. After 2 days, MSC exit the lag phase and begin to divide rapidly, about once every 24 hours. This continues through day 7, when the cells are near confluence.
Figure 2. D24RGD efficiently replicates in MSC.

MSC were infected with 5–2500 vp/cell of (a) D24RGD or (b) UV D24RGD. A CPE was seen within 48 hours in cells infected with D24RGD but not those infected with UVD24RGD. (c) These data were confirmed by detection of hexon levels in the supernatants of MSC infected with D24RGD or UVD24RGD (5 or 2500 vp/cell). MSC that were infected with D24RGD showed an increase in hexon levels, consistent with amplification of the virus. (d) Burst size data based on E1a expression shows that an MSC infected with an MOI of 50, after 24 hours, was able to produce over 1500 vp per cell.
MSC-D24RGD effects on multiple cell lines
The 72-hour supernatant from D24RGD-infected MSC (50 vp/cell) was applied to six different cell lines: A375sm, MDA231, HEY, SKOV3, OVCAR-3, and 293s. Within 24 hours, a CPE was evident in all cell lines, and progressive amplification continued through 48 hours. The demonstrated CPE is consistent with efficient replication. These data confirm that not only does D24RGD replicate in MSC, but the viable virus can infect, replicate in, and lyse target cells in vitro (Figure 3).
Figure 3. MSC-D24RGD induces CPE in multiple cell lines.

Seventy-two-hour supernatants from MSCs infected with (a) D24RGD or (b) UVD24RGD were applied to multiple cell lines. Evidence of CPE is seen in panel a, confirming that D24RGD can replicate in MSC and produce virus that is able to infect, replicate in, and lyse target cells in vitro.
We also counted cell death by measuring cell viability of two human ovarian carcinoma lines, OVCAR-3 and SKOV-3, after 72 hours of incubation with supernatants from MSC-D24RGD that had been conditioned for 48 hours previously. (Figure 4) The decrease in cell number is inversely proportional to the titer of virus used to infect the MSCs. Because D24RGD replicates in these tumor cells, as the length of the assay increased, number of viable cells decreased (data not shown).
Figure 4. Viability of tumor cell lines treated with D24RGD-infected MSC supernatant.

Supernatants of MSCs conditioned for 48 hours with D24RGD or UVD24RGD were applied to human ovarian carcinoma cell lines OVCAR-3 or SKOV-3. Then, 72 hours later, viable cells were determined via trypan blue counts and displayed as percentage of control counts. The decrease in cell number was inversely proportional to the increase in virus titers used to infect MSC.
MSC-D24RGD as treatment for ovarian carcinoma in vivo
MSC-D24RGD (P<0.001) and D24RGD alone (P<0.001) nearly doubled the survival of mice, compared with survival of untreated control mice. The average increase in survival was 55 days (Figure 5a). The other control groups—those receiving MSC-GFP (P=.151), MSC (P=.024), and MSC-UVD24RGD (P=.096)—had no significant increase or decrease in survival time.
Figure 5. Intraperitoneal administration of MSC-D24RGD increases survival of SCID mice.

Anti-cancer effect of targeted D24RGD to established Skov-3 ovarian carcinoma via intraperitoneal administration of with and without the MSC vehicle. (a) MSC-D24RGD (4 injections, 1×/week) show a survival benefit (P≤.0001) over that of MSC delivery the UVD24RGD control virus. Survival of SCID mice treated with MSC-D24RGD or D24RGD increased by 55 days on average, compared with survival of control mice. (b) Hexon staining of tumor sections from mice representative of the group receiving D24RGD alone, MSC-D24RGD, and MSC alone treated mice.
Although the increase survival due to tumor burden using MSC-D24RGD was similar to that of using D24RGD alone, we hypothesized that using MSC to deliver the virus would allow for a targeted, local delivery of D24RGD to the tumor. This was determined via immunohistochemical analysis. The presence of the hexon staining within tumor sections is more evident in the MSC-D24RGD as opposed to the D24RGD where the hexon staining is evident only around the outer edge of the tumor (Figure 5b).
Systemic biodistribution
To determine the systemic dispersion of virus, SKOV-3 SCID mice were injected intraperitoneally with either MSC-D24RGD (50 vp/cell, n=3) or D24RGD (5 x 107 vp/mouse, n=3). Immunohistochemical analysis of tissues from mice after 1 or 3 days showed significant differences in the diffusion of hexon (Figure 6). Intraperitoneal injections of the virus alone resulted in a more systemic distribution of virus. One day after injection of virus, both the muscle behind the tumor and muscle in the opposite limb had hexon-positive spots, as did the spleen, kidney, and liver. It should be noted that the liver had many more positive spots. In addition, inclusion bodies can be found in HE-stained sections of the livers of mice treated with D24RGD alone (data not shown). Hexon protein positivity is evidence of viral infection and leads to systemic toxicity that can be deadly. We did not observe high levels of hexon staining in any tissues, and mild staining was seen in the MSC-D24RGD–treated group. Tissues examined 1 day after MSC-D24RGD injection were negative, except for a few small spots in the liver (Figure 6a) compared with tissues from D24RGD-treated mice (Figure 6b). By day 3, the D24RGD-treated groups displayed decreased hexon staining (Figure 6d), and MSC- D24RGD treated tissues remained negative for hexon, with the exception of the liver, which displayed a decrease in staining from the day 1 sample (Figure 6c). Foci quantitation of the hexon-stained tissue highlights the minor presence of the virus in the liver (indicator of systemic toxicity) of MSC-D24RGD–treated mice (Figure 7). These data demonstrate the ability of MSC to locally deliver D24RGD to the tumor microenvironment with a dramatic decrease in systemic spread compared with the use of D24RGD alone.
Figure 6. Immunohistochemical analysis of mice after D24RGD treatment.

Analysis of tissues from SKOV-3 SCID mice 1 or 3 days after intraperitoneal injection of either MSC-D24RGD or D24RGD showed significant differences in the dispersion of hexon. (a) Day 1 after injection of MSC-D24RGD. All tissues except for a small spot in the liver are negative. (b) Day 1 after injection of D24RGD. All tissues except for the lung are positive. The liver is bright with hexon-positive staining. (c) Day 3 after injection of MSC-D24RGD. All tissues again are negative, with the exception of a small spot in the liver. (d) Day 3 after injection of D24RGD. Virus has cleared from muscle tissues, and there is a decrease in liver concentration.
Figure 7. Relative intensity of hexon staining in tissue samples from in vivo analysis.
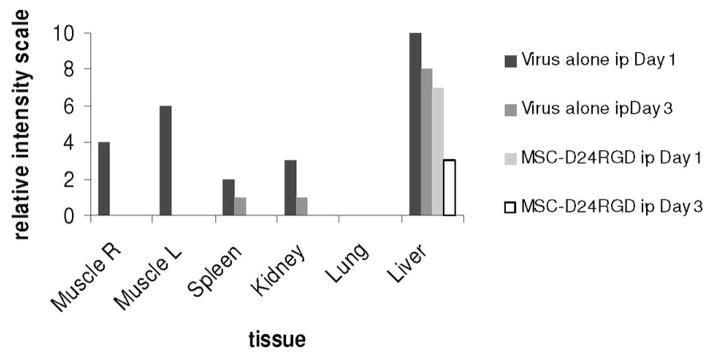
Hexon staining was measured as relative intensity among tissue samples. MSC- D24RGD displayed the least systemic toxicity, which was limited to the liver.
Discussion
Ovarian cancer usually presents at an advanced stage, typically with peritoneal metastasis, which has the worst prognosis among ovarian cancer metasases. Gene therapy trials published thus far have used intraperitoneal administration for treatment. This can be conveniently achieved with a catheter, and the peritoneal cavity allows a degree of compartmentalization. However, we show that the systemic spread that occurs after intraperitoneal injection of D24RGD used alone is highly toxic. We have shown toxicity to be the limiting factor in the value of D24RGD for treatment of peritoneally disseminated ovarian cancer when used it is without a delivery vehicle. In a recent study, adenovirus alone, even when directly injected into the tumor site was ineffective, even though it had an anti-tumor effect, because of the shortened survival attributed to liver toxicity caused by viral dissemination.11
We show the use of MSC as delivery vehicles for anti-cancer therapy in an attempt to overcome the immunological barriers to direct administration of viral vectors, including rapid degradation (immune clearance) and high systemic and organ toxicity.18,36 MSC are also easily transduced with viral vectors (e.g., adenovirus, retrovirus, and adeno-associated virus) to express high levels of transgene product that can be maintained over several passages and through differentiation.39 D24RGD is cell cycle–dependent (like other conditionally replicative adenovirus [CRAds]), and can therefore replicate when the cell is actively dividing (the S-phase). Although it has been shown that replication of CRAds is greater in proliferating normal cells than in matched, nonproliferating normal cells, it is important to note that the replication in the proliferating normal cells is still several orders of magnitude less than that seen in cancer cells.1 MSC growth closely resembles the viral replication curve (Figure 1) implying that the MSC are adequate replication vehicles. In addition, the self-renewable MSC can be extensively expanded in culture and are able to undergo site-specific differentiation by taking signals from their immediate environment, without any evidence of ectopic tissue formation.37,38
We have shown that MSC support D24RGD replication in vitro, and 3-day supernatants of MSC-D24RGD (50 vp/cell) were able to infect and lyse OVCAR-3, SKOV-3, and a number of other tumor cell lines. Our in vivo studies were conducted to target established ovarian carcinoma tumors via intraperitoneal administration of MSC-D24RGD (4 injections, 1×/week), and we found a survival benefit (P≤.0001) over that of MSC delivery the UVD24RGD control virus.
Replication and subsequent oncolysis were measured via hexon (replication) levels and viable cell counts, respectively (Figure 2), and we found that the virus was able to replicate within cells. In vivo, cell turnover and viral dispersion dynamics are likely to be different because of the natural milieu of the host, including the three-dimensional nature of solid tumors. However, trends similar to those in our in vitro studies were seen in mice given intraperitoneal injections of MSC-D24RGD (Figure 5). Importantly, we found that using MSC to deliver D24RGD not only successfully achieved a targeted gene delivery that increased the infection of tumor cells over cells infected with D24RGD alone but also limited the systemic spread of the virus. The decreased tumor burden in the mice receiving D24RGD or MSC-D24RGD were both significant compared to tumor alone. However, the decrease in tumor burden was comparable between the mice given D24RGD and those given MSC-D24RGD (Figure 5), but the overall systemic toxicity was significantly decreased in mice receiving the MSC-D24RGD injections (Figure 6). While it is understood that the human adenovirus does not replicate as efficiently in mouse tissues as it does in human, there are still significant amounts of hexon staining in the liver, kidney, spleen and even muscle that is not observed in the MSC-delivered D24RGD (Figure 6). With this in mind, the systemic toxicity of the adenovirus is assumably higher, and thus the effectiveness of the MSC delivery system may be even greater.
Similar to previous studies40, we have achieved a targeted delivery system using MSC as a vector to selectively distribute D24RGD to the tumor microenvironment and significantly increase survival in SCID mice with SKOV-3 ovarian carcinoma. More importantly, we were able to overcome a major hurdle in the viral gene therapy field: systemic toxicity. Using MSC as a “Trojan horse,” we were able to limit the systemic distribution of the virus and evade the host innate immune response. Evasion of viral dissemination and overall systemic toxicity of the adenovirus through the use of MSC as a delivery vehicle is a significant finding in the advancement of cancer gene therapy and facilitates the progression of clinical trials that have been thwarted by the toxic side effects of viral vector therapeutic agents.
References
- 1.Heise C, Hermiston T, Johnson L, Brooks G, Sampson-Johannes A, Williams A, et al. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat Med. 2000;6:1134–1139. doi: 10.1038/80474. [DOI] [PubMed] [Google Scholar]
- 2.Kelly FJ, Miller CR, Buchsbaum DJ, Gomez-Navarro J, Barnes MN, Alvarez RD, et al. Selectivity of tag-72-targeted adenovirus gene transfer to primary ovarian carcinoma cells versus autologous mesothelial cells in vitro. Clin Cancer Res. 2000;6:4323–4333. [PubMed] [Google Scholar]
- 3.Tsuda H, Wada T, Ito Y, Uchida H, Dehari H, Nakamura K, et al. Efficient BMP2 gene transfer and bone formation of mesenchymal stem cells by a fiber-mutant adenoviral vector. Mol Ther. 2003;7:354–365. doi: 10.1016/s1525-0016(02)00062-x. [DOI] [PubMed] [Google Scholar]
- 4.Hemminki A, Belousova N, Zinn KR, Liu B, Wang M, Chaudhuri TR, et al. An adenovirus with enhanced infectivity mediates molecular chemotherapy of ovarian cancer cells and allows imaging of gene expression. Mol Ther. 2001;4:223–231. doi: 10.1006/mthe.2001.0446. [DOI] [PubMed] [Google Scholar]
- 5.Bauerschmitz GJ, Kanerva A, Wang M, Herrmann I, Shaw DR, Strong TV, et al. Evaluation of a selectively oncolytic adenovirus for local and systemic treatment of cervical cancer. Int J Cancer. 2004;111:303–309. doi: 10.1002/ijc.20217. [DOI] [PubMed] [Google Scholar]
- 6.Fueyo J, Alemany R, Gomez-Manzano C, Fuller GN, Khan A, Conrad CA, et al. Preclinical characterization of the antiglioma activity of a tropism-enhanced adenovirus targeted to the retinoblastoma pathway. J Natl Cancer Inst. 2003;95:652–660. doi: 10.1093/jnci/95.9.652. [DOI] [PubMed] [Google Scholar]
- 7.Stolarek R, Gomez-Manzano C, Jiang H, Suttle G, Lemoine MG, Fueyo J. Robust infectivity and replication of Delta-24 adenovirus induce cell death in human medulloblastoma. Cancer Gene Ther. 2004;11:713–720. doi: 10.1038/sj.cgt.7700731. [DOI] [PubMed] [Google Scholar]
- 8.Bauerschmitz GJ, Lam JT, Kanerva A, Suzuki K, Nettelbeck DM, Dmitriev I, et al. Treatment of ovarian cancer with a tropism modified oncolytic adenovirus. Cancer Res. 2002;62:1266–1270. [PubMed] [Google Scholar]
- 9.Wang Y, Hu JK, Krol A, Li YP, Li CY, Yuan F. Systemic dissemination of viral vectors during intratumoral injection. Mol Cancer Ther. 2003;2:1233–1242. [PubMed] [Google Scholar]
- 10.Yu P, Wang X, Fu Y. Enhanced local delivery with reduced systemic toxicity: Delivery, delivery, and delivery. Gene Ther. 2006;13:1131–1132. doi: 10.1038/sj.gt.3302760. [DOI] [PubMed] [Google Scholar]
- 11.Kanehira M, Xin H, Hoshino K, Maemondo M, Mizuguchi H, Hayakawa T, et al. Targeted delivery of NK4 to multiple lung tumors by bone marrow-derived mesenchymal stem cells. Cancer Gene Ther. 2007;14:894–903. doi: 10.1038/sj.cgt.7701079. [DOI] [PubMed] [Google Scholar]
- 12.Studeny M, Marini FC, Dembinski JL, Zompetta C, Cabreira-Hansen M, Bekele BN, et al. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. JNatlCancer Inst. 2004;96:1593–1603. doi: 10.1093/jnci/djh299. [DOI] [PubMed] [Google Scholar]
- 13.Hall B, Dembinski J, Sasser AK, Studeny M, Andreeff M, Marini F. Mesenchymal stem cells in cancer: tumor-associated fibroblasts and cell-based delivery vehicles. IntJHematol. 2007;86:8–16. doi: 10.1532/IJH97.06230. [DOI] [PubMed] [Google Scholar]
- 14.Hall B, Andreeff M, Marini F. The participation of mesenchymal stem cells in tumor stroma formation and their application as targeted-gene delivery vehicles. HandbExpPharmacol. 2007;(180):263–283. doi: 10.1007/978-3-540-68976-8_12. [DOI] [PubMed] [Google Scholar]
- 15.Spaeth E, Klopp A, Dembinski J, Andreeff M, Marini F. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008 doi: 10.1038/gt.2008.39. [DOI] [PubMed] [Google Scholar]
- 16.Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002;62:3603–3608. [PubMed] [Google Scholar]
- 17.Studeny M, Marini FC, Dembinski JL, Zompetta C, Cabreira-Hansen M, Bekele BN, et al. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. JNatlCancer Inst. 2004;96:1593–1603. doi: 10.1093/jnci/djh299. [DOI] [PubMed] [Google Scholar]
- 18.Komarova S, Kawakami Y, Stoff-Khalili MA, Curiel DT, Pereboeva L. Mesenchymal progenitor cells as cellular vehicles for delivery of oncolytic adenoviruses. Mol Cancer Ther. 2006;5:755–766. doi: 10.1158/1535-7163.MCT-05-0334. [DOI] [PubMed] [Google Scholar]
- 19.Kirn D. Replication-selective oncolytic adenoviruses: Virotherapy aimed at genetic targets in cancer. Oncogene. 2000;19:6660–6669. doi: 10.1038/sj.onc.1204094. [DOI] [PubMed] [Google Scholar]
- 20.Stoff-Khalili MA, Rivera AA, Le LP, Stoff A, Everts M, Contreras JL, et al. Employment of liver tissue slice analysis to assay hepatotoxicity linked to replicative and nonreplicative adenoviral agents. Cancer Gene Ther. 2006;13:606–618. doi: 10.1038/sj.cgt.7700934. [DOI] [PubMed] [Google Scholar]
- 21.Nakamizo A, Marini F, Amano T, Khan A, Studeny M, Gumin J, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65:3307–3318. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]
- 22.Hakkarainen T, Sarkioja M, Lehenkari P, Miettinen S, Ylikomi T, Suuronen R, et al. Human mesenchymal stem cells lack tumor tropism but enhance the antitumor activity of oncolytic adenoviruses in orthotopic lung and breast tumors. Hum Gene Ther. 2007;18:627–641. doi: 10.1089/hum.2007.034. [DOI] [PubMed] [Google Scholar]
- 23.Egan C, Jelsma TN, Howe JA, Bayley ST, Ferguson B, Branton PE. Mapping of cellular protein-binding sites on the products of early-region 1A of human adenovirus type 5. MOL CELL BIOL. 1988;8:3955–3959. doi: 10.1128/mcb.8.9.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yotnda P, Zompeta C, Heslop HE, Andreeff M, Brenner MK, Marini F. Comparison of the efficiency of transduction of leukemic cells by fiber-modified adenoviruses. HumGene Ther. 2004;15:1229–1242. doi: 10.1089/hum.2004.15.1229. [DOI] [PubMed] [Google Scholar]
- 25.Fueyo J, Gomez-Manzano C, Alemany R, Lee PSY, McDonnell TJ, Mitlianga P, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19:2–12. doi: 10.1038/sj.onc.1203251. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki K, Fueyo J, Krasnykh V, Reynolds PN, Curiel DT, Alemany R. A conditionally replicative adenovirus with enhanced infectivity shows improved oncolytic potency. Clin Cancer Res. 2001;7:120–126. [PubMed] [Google Scholar]
- 27.Stone D, Liu Y, Li Z, Tuve S, Strauss R, Lieber A. Comparison of Adenoviruses From Species B, C, E, and F After Intravenous Delivery. MolTher. 2007;15:2146–2153. doi: 10.1038/sj.mt.6300319. [DOI] [PubMed] [Google Scholar]
- 28.Liu Q, Muruve DA. Molecular basis of the inflammatory response to adenovirus vectors. Gene Ther. 2003;10:935–940. doi: 10.1038/sj.gt.3302036. [DOI] [PubMed] [Google Scholar]
- 29.Bauerschmitz GJ, Barker SD, Hemminki A. Adenoviral gene therapy for cancer: from vectors to targeted and replication competent agents (review) Int J Oncol. 2002;21:1161–1174. [PubMed] [Google Scholar]
- 30.Chu RL, Post DE, Khuri FR, Van Meir EG. Use of replicating oncolytic adenoviruses in combination therapy for cancer. Clin Cancer Res. 2004;10:5299–5312. doi: 10.1158/1078-0432.CCR-0349-03. [DOI] [PubMed] [Google Scholar]
- 31.Rein DT, Breidenbach M, Hille S, Curiel DT. Current developments in adenovirus-based cancer gene therapy. Future Oncol. 2006;2:137–144. doi: 10.2217/14796694.2.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larochelle A, Dunbar CE. Genetic manipulation of hematopoietic stem cells. Semin Hematol. 2004;41:257–271. doi: 10.1053/j.seminhematol.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 33.Roni V, Habeler W, Parenti A, Indraccolo S, Gola E, Tosello V, et al. Recruitment of human umbilical vein endothelial cells and human primary fibroblasts into experimental tumors growing in SCID mice. Exp Cell Res. 2003;287:28–38. doi: 10.1016/s0014-4827(03)00133-2. [DOI] [PubMed] [Google Scholar]
- 34.Del Rio M, Gache Y, Jorcano JL, Meneguzzi G, Larcher F. Current approaches and perspectives in human keratinocyte-based gene therapies. Gene Ther. 2004:11. doi: 10.1038/sj.gt.3302370. [DOI] [PubMed] [Google Scholar]
- 35.Li S, Kimura E, Fall BM, Reyes M, Angello JC, Welikson R, et al. Stable transduction of myogenic cells with lentiviral vectors expressing a minidystrophin. Gene Ther. 2005;12:1099–1108. doi: 10.1038/sj.gt.3302505. [DOI] [PubMed] [Google Scholar]
- 36.Pereboeva L, Komarova S, Mikheeva G, Krasnykh V, Curiel DT. Approaches to utilize mesenchymal progenitor cells as cellular vehicles. Stem Cells. 2003;21:389–404. doi: 10.1634/stemcells.21-4-389. [DOI] [PubMed] [Google Scholar]
- 37.Liechty KW, Mackenzie TC, Shaaban AF, Radu A, Moseley AB, Deans R, et al. Human mesenchymal stem cells engraft and demonstrate site-specific differentiation after in utero transplantation in sheep. Nat Med. 2000;6:1282–1286. doi: 10.1038/81395. [DOI] [PubMed] [Google Scholar]
- 38.Devine SM, Bartholomew AM, Mahmud N, Nelson M, Patil S, Hardy W, et al. Mesenchymal stem cells are capable of homing to the bone marrow of non-human primates following systemic infusion. Exp Hematol. 2001;29:244–255. doi: 10.1016/s0301-472x(00)00635-4. [DOI] [PubMed] [Google Scholar]
- 39.Lee K, Majumdar MK, Buyaner D, Hendricks JK, Pittenger MF, Mosca JD. Human mesenchymal stem cells maintain transgene expression during expansion and differentiation. Mol Ther. 2001;3:857–866. doi: 10.1006/mthe.2001.0327. [DOI] [PubMed] [Google Scholar]
- 40.Sonabend AM, Ulasov, Tyler MA, Rivera AA, Mathis JM, Lesniak MS. Mesenchymal stem cells effectively deliver an oncolytic adenovirus to intracranial glioma. Stem Cells. 2008;26:831–841. doi: 10.1634/stemcells.2007-0758. [DOI] [PubMed] [Google Scholar]