

Abstract
Traditional dynamic hydrogels have been designed to respond to changes in physicochemical inputs, such as pH and temperature, for a wide range of biomedical applications. An emerging strategy that may allow for more specific “bio-responsiveness” in synthetic hydrogels involves mimicking or exploiting nature’s dynamic proteins. Hundreds of proteins are known to undergo pronounced conformational changes in response to specific biochemical triggers, and these responses represent a potentially attractive toolkit for design of dynamic materials. This “emerging area” review focuses on the use of protein motions as a new paradigm for design of dynamic hydrogels. In particular, the review emphasizes early examples of dynamic hydrogels that harness well-known protein motions. These examples then serve as templates to discuss challenges and suggest emerging directions in the field. Successful early examples of this approach, coupled with the fundamental properties of nature’s protein motions, suggest that protein-based materials may ultimately achieve specific, multiplexed responses to a range of biochemical triggers. Applications of this new class of materials include drug delivery, biosensing, bioactuation, and tissue engineering.
1. Introduction
Traditional dynamic hydrogels have been designed to respond to a range of physicochemical inputs, most notably temperature, pH, and ionic strength. For example, hydrogels composed of poly(N-isopropyl acrylamide), poly(acrylic acid), and poly-(methacrylic acid) can undergo substantial changes in volume, shape, mesh size, mechanical stiffness, and optical transparency in response to physicochemical inputs.1–3 These pronounced changes in soft material properties can be useful in sensors, soft actuators, drug delivery systems, and cell culture substrates. Indeed, dynamic hydrogels serve as enabling tools throughout soft materials science, particularly in applications that require environmental-responsiveness. Despite the importance of dynamic hydrogels, their inability to respond to specific biomolecular inputs presents an important challenge. In particular, a number of potential biological applications of dynamic hydrogels may not be sufficiently robust to accommodate changes in environmental pH, temperature, or ionic strength. Biological systems typically exist and function under regulated homeostatic conditions, which may not tolerate significant changes in physicochemical parameters. For example, a drug delivery strategy may benefit from triggered release of a drug in response to a local increase in the temperature, but that temperature increase could lead to a concomitant, and unintended, change in the behavior of cells in the local environment. The challenges associated with traditional dynamic hydrogels call for new approaches, in which soft materials dynamically vary their properties in response to specific biochemical triggers.
An emerging strategy that may allow for more specific “bio-responsiveness” involves using nature’s dynamic molecules as an inspiration. Nature’s soft materials are often capable of undergoing substantial changes in their physical, chemical, and biological properties in response to highly specific biochemical ligands. Remarkably, the pronounced changes in nature’s macroscopic material properties can often be traced to dynamic interactions at the molecular scale. Several natural examples help to illustrate this phenomenon. Muscle contraction relies on the molecular “lever arm” motion of myosin along networks of actin filaments.4 Dynamic tissue remodeling often requires allosteric conformational shifts in proteases,5 which in turn degrade proteins in the extracellular matrix. Virus assembly into nano-metre-scale “shells” involves dynamic protein–protein interactions that can be driven by protein motion (e.g. the 20° “hinge motion” of the tomato bushy stunt virus).6 In each case, a dynamic motion of protein molecules translates into a pronounced change in properties of soft biological materials.
The existence proof provided by nature’s dynamic materials suggests an opportunity to generate new classes of synthetic soft materials that change their structure and function in useful ways. In essence, the dynamic molecular motions prevalent in nature represent a unique toolkit for dynamic materials design. Protein motions may be particularly advantageous due to their prevalence, adaptability, specificity, and controllable responsiveness. There are hundreds of protein conformational changes that have been well-characterized,7,8 and recent computational models of protein function suggest that protein motion may be a ubiquitous regulator of protein–protein interactions.9 Dynamic protein domains can also be modified and exploited in a variety of ways using routine recombinant DNA technologies, thereby expanding on an already large library of protein motions. Furthermore, our understanding of the fundamentals of protein motion is perhaps progressing toward a paradigm in which de novo design or directed evolution of dynamic protein units may become realistic. Therefore, protein motions represent a large and rapidly expanding toolkit that is primed for use in dynamic materials. Finally, the characteristics of natural protein motions suggest that they may enable new levels of bio-specificity and multiplexed responsiveness when compared to traditional synthetic polymer units. That is, many protein conformational changes occur in response to binding of specific biochemical triggers, and there is the potential to mix multiple responsive proteins in a single material.
In view of the potential to mimic and exploit protein motions, this “emerging area” review focuses on protein motions as a new paradigm for design of dynamic materials. Protein motions are first introduced in general, followed by early examples of dynamic materials that harness protein motions. These examples are used as templates to suggest emerging directions in the field. Traditional dynamic hydrogels based on synthetic polymers are reviewed elsewhere1–3 and will not be detailed here. In addition, other types of dynamic molecular motions, including those that result from reversible intermolecular interactions and DNA folding, have been reviewed elsewhere10–12 and will not be included here.
2. The nature of protein motions
Proteins undergo a wide range of molecular motions, often termed “conformational changes”, in response to physical and biochemical stimuli. Commonly cited examples include the light-induced conformational changes in the light harvesting complex of plants, and the motions of motor proteins such as myosin and kinesin in the presence of adenosine triphosphate (ATP). However, recent studies indicate that protein motions may play a key role in a much broader array of biological processes. Computer algorithms have enabled prediction and classification of protein domain structures and flexible linker regions,8,13–15 and these algorithms suggest that large scale structural flexibility of proteins is the norm rather than the exception.9
Two descriptive terms have emerged in the structural biology literature9,16,17 to describe protein motions: hinge motion and shear motion. These terms describe relative motions of discrete, linked units in a protein, and the motions are typically associated with a ligand binding event or a protein–protein interaction. The following sections briefly introduce and summarize types of protein motions. We use the simple hinge and shear descriptions as a lexicon for simplicity, though it is noteworthy that many protein motions include components of both hinge and shear in distinct portions of a protein. The reader is referred to excellent reviews for a more comprehensive treatment of protein motions.9,16,17 Although there is clear significance to subtle movements of individual epitopes in proteins, the focus here is on global conformational changes that may be used as building blocks in dynamic materials. In addition, for practical relevance to dynamic material applications, we also focus on motions of single protein domains or domain fragments, and only briefly mention larger scale motions (e.g. F1 ATPase) that involve cooperative movements of high molecular weight protein subunits and complex quaternary structure changes. These more complex motions, though biologically important, are more difficult to envision as functional components of synthetic materials in emerging near-term approaches.
Hinge motion
Proteins that undergo hinge motion typically include two distinct domains connected by a flexible and metastable linker region (see Fig. 1–3 for examples). The hinge exists within the linker region, and the axis of rotation passes through the flexible linker.18 Thus, these proteins can often exist in either an “open” conformation, in which the linker is extended, or a “closed” conformation, in which the linker is bent or collapsed. A variety of studies have demonstrated that there is a low energy barrier between open and closed states of hinge motion proteins,9 which suggests that these proteins exist in metastable states. In addition, the properties of the flexible linker region have been shown to be important in defining the stability of the open state of the hinge,15 and the resulting ability to respond to a ligand binding event.
Fig. 1.
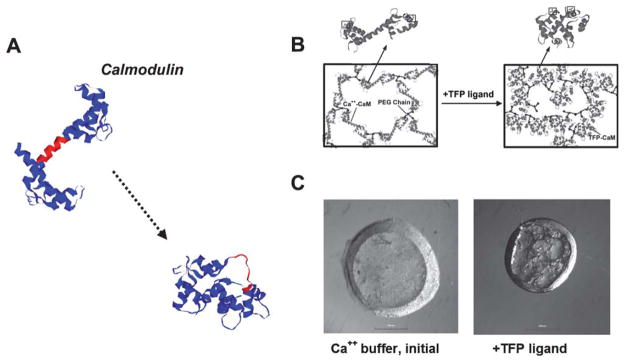
Calmodulin’s “hinge motion” translates into dramatic changes in hydrogel properties. (A) Calmodulin crystal structures before and after binding of a trifluoperazine (TFP) ligand (PDB: 1cll → 1lin). A central region is shown in red as a point of reference to demonstrate the hinge motion. (B) Schematic representing design of poly(ethylene glycol) (PEG) hydrogels that include calmodulin (CaM). (C) Top view of a cylindrical hydrogel before and after TFP binding, demonstrating calmodulin’s dynamic function within a hydrogel (scale bar =1 mm) (B and C from Sui et al.38, copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission).
Fig. 3.
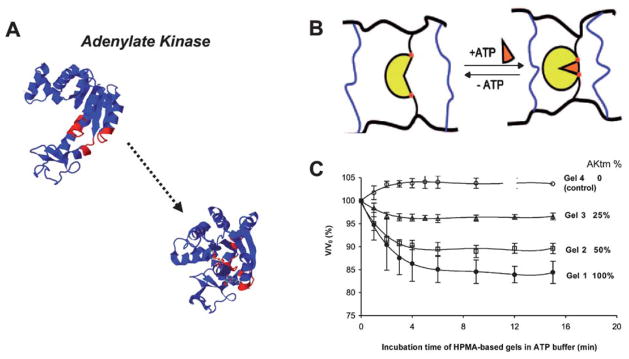
(A) Adenylate kinase crystal structures before and after binding to ATP (PDB: 1ake → 4ake). A central region is shown in red as a point of reference to demonstrate the hinge motion. (B) Schematic representing design of N-(2-hydroxypropyl)methacrylamide (HPMA) hydrogels that include adenylate kinase (yellow). (C) Dynamic relative volume decreases of hydrogels containing varying amounts of adenylate kinase, in the presence of ATP (B and C reprinted with permission from Yuan et al.40, copyright 2008 American Chemical Society).
Hinge motion is perhaps the type of natural protein motion that is most clearly relevant to dynamic materials studies, as one can envision directly translating nanometre-scale hinge bending into macroscopic volume decreases. Indeed, the conformational changes in proteins undergoing hinge motion can be considered somewhat analogous to the conformational changes in synthetic polymer chains commonly used in dynamic materials, such as poly(NiPAAm) and poly(acrylic acid).19 However, a unique advantage of protein hinges when compared to synthetic polymer chains is their ability to respond to highly specific biochemical ligands (e.g. glucose, ATP) as opposed to more general physicochemical inputs (e.g. pH, temperature).
A variety of natural biological functions are associated with hinge motion, and each is of potential relevance to dynamic material applications. For example, hinge motion can provide a mechanism for protein assembly into tightly packed structures, as demonstrated by the viral coat formed upon hinge motion of the tomato bushy stunt virus.6 Alternatively, the closing of a hinge can result in ligand recognition and sequestering, as in the case of iron sequestering by lactoferrin.20 The hinge movement in response to ligand binding can also result in varied protein–protein interactions. A notable example is calmodulin’s ability to interact with certain binding partners only in the presence of calcium.21,22 Finally, hinge motion can be associated with enzyme catalysis, exemplified by adenylate kinase’s transfer of a phosphate group from ATP to a substrate.23 In this case the hinge closes upon substrate binding, and re-opens after the catalytic event. Whether hinge motion is associated with binding, assembly, or catalysis, it provides an attractive potential mechanism for bio-responsiveness.
Shear motion
Shear motion is characterized by lateral movement of two distinct protein domains that retain a common interface during the motion. This type of motion often involves cooperative shear movement of multiple domains, analogous to tectonic plates sliding laterally. The biological consequence of shear motion often involves closing of enzyme domains around a substrate (e.g. citrate synthase, aspartate amino transferase, and glyceraldehyde 3-phosphate dehydrogenase) or revealing the ligand-binding site in a protein (e.g. trp repressor).17 These biological functions suggest the potential to use shear motion to achieve dynamic epitope presentation to cells, a concept that has been achieved in previous studies using dynamic synthetic polymers (reviewed in ref. 24). Further, one can envision how this type of motion could be used to laterally modulate the structure of a material in response to the presence of a substrate, or as a molecular “switch” to move material components into and out of orientation with one another.
Motor proteins
Motor proteins are able to convert chemical energy (e.g. ATP hydrolysis) into mechanical work. The movement of motor proteins can include hinge and/or shear motions, but they are somewhat distinct from other protein motions in their ability to move along a “track”, and thus perform mechanical work. Motor proteins can often move along the surface of a protein assembly,25 exemplified by myosin movement on actin filaments and kinesin movement along microtubules. They can also move along the surface of DNA assemblies, exemplified by type I restriction enzyme translocation along wound DNA strands in the cell.26 In this way, motor protein interactions with protein or DNA assemblies provide a useful template for design of some types of synthetic, dynamic materials.
An illustrative example is the monomeric kinesin motor KIF1A, which transitions from a tight microtubule-binding state to a weak binding, diffusive state upon ATP hydrolysis.27 The variable strength of microtubule binding allows KIF1A to move along tracks of microtubules in the cell. Recent studies have demonstrated that the transport properties of kinesin along immobilized microtubules,28 as well as the ability of immobilized kinesin motors to actively transport microtubules,29 can be used for a variety of nanotechnology applications. Examples include directional transport of molecules,28,30,31 active transport of microscale materials,29,32 and biosensing.33 Analogous approaches have used immobilized myosin motors for actin transport, including applications in directional transport,34 transport of nanoscale cargo,35 and biosensing (reviewed in ref. 36).
3. Synthetic, dynamic biomaterials that harness protein motions: early studies
Recent studies have begun to demonstrate that protein motions can be translated into macroscopic changes in material properties. An initial demonstration of this approach came in 2007, when a mutant version of the hinge motion protein calmodulin was used as a cross-linker in a poly(ethylene glycol)-based hydrogel network37. The network was formed via a Michael-type addition reaction between a dicysteine mutant of calmodulin and a 4-arm poly(ethylene glycol)-acrylate. Calmodulin molecules in the resulting hydrogel were able to bind to specific biochemical ligands, leading to collapse of the calmodulin hinge and significant decreases in the hydrogel volume. The volume decreases, though modest in magnitude (~15% decrease in the hydrogel volume), could be attributed to nanometre-scale changes in calmodulin conformation. Two subsequent studies extended this approach to achieve much larger, tunable changes in material properties (Fig. 1). In particular, Sui et al. demonstrated that poly(ethylene glycol)–calmodulin conjugates could be photo-polymerized to form dynamic hydrogels. The photopolymerized hydrogels showed a maximum volume decrease of ~80%,38 and the response magnitude could be tuned by varying hydrogel polymerization conditions (e.g. calmodulin concentration, total polymer concentration, polymer molecular weight).39 These early studies using calmodulin as a dynamic unit established sets of design rules for synthesis of dynamic, protein-based hydrogels, which may be useful in future embodiments of this approach with other hinge motion proteins.
In a separate set of studies, investigators have created networks of catalytic enzymes as a mechanism to create dynamic materials (Fig. 3). Kopecek and co-workers40 created hydrogels that incorporated adenylate kinase, a protein that undergoes pronounced hinge motion in the presence of ATP. Specifically, the sulfhydryl groups of an adenylate kinase mutant (C77S, A55C, V169C) were reacted with pendant maleimide groups incorporated into N-(2-hydroxypropyl)methacrylamide (HPMA) in the presence of DTT. Upon exposure to ATP these hydrogels underwent volume decreases of up to 15%. As in the case of the calmodulin-based hydrogels described above, the adenylate kinase study demonstrated that a nanometre-scale hinge motion could be translated into macroscopic hydrogel dynamics. However, the results of Kopecek and co-workers further suggested that a broad range of other enzymes that undergo hinge motion as part of their catalytic function could be useful as dynamic components in materials. This is an important suggestion, as enzymes tend to undergo hinge and/or shear motions during the catalytic cycle. Specific examples of hinge motion in enzymes that could be employed in dynamic materials using the Kopecek et al. approach include citrate synthase and alcohol dehydrogenase.
Daunert et al. have also recently harnessed a hinge motion in a study with particular biomedical relevance (Fig. 2). They generated a di-cysteine mutant of glucose-binding protein (GBP), which transitions from an extended conformation to a collapsed conformation upon glucose binding.41 The mutant GBP was then derivatized with allyl groups and incorporated as a functional unit into poly(acrylamide) hydrogels, which underwent reversible volume changes in the presence of glucose. This study is of particular significance, as glucose responsiveness has implications in diabetes in terms of both glucose biosensing and glucose-responsive insulin delivery.
Fig. 2.
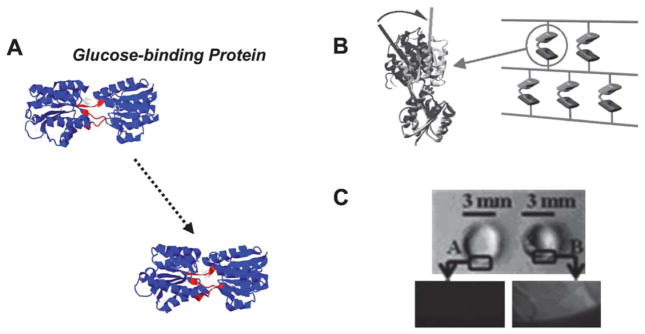
(A) Glucose binding protein (GBP) crystal structures before and after binding to glucose (PDB: 2fw0 → 2fvy). A central region is shown in red as a point of reference to demonstrate the hinge motion. (B) Schematic representing design of poly(acrylamide) hydrogels that include GBP. (C) Top view of a cylindrical hydrogel before and after glucose binding, demonstrating GBP’s dynamic function (B and C from Ehrick et al.41, copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission).
Esser-Kahn et al. have used metallothionein conformational changes to generate hydrogels that respond to heavy metal ions42 (Fig. 4). Metallothionein undergoes collapse from an extended coil to a more globular morphology upon binding of various toxic heavy metal ions, such as mercury and cadmium. This motion is somewhat distinct from those discussed thus far in this section, as it is not a classical hinge motion. It instead represents an example of protein collapse upon multivalent binding to target molecules. When incorporated into poly(acrylamide) hydrogels, metallothionein’s conformational shift translated into up to 80% decreases in the hydrogel volume. In turn, the volume shift was used to sense the presence of heavy metals and to generate a heavy metal-sensitive valve for contaminated slough water. Importantly, this study suggests that protein motions within hydrogel networks can be used not only for biosensing and bio-responsiveness, but also for simultaneous molecular sequestering.
Fig. 4.

Collapse of metallothionein in the presence of heavy metals can also result in significant decreases in the hydrogel volume. (A) Structural representations of metallothionein before and after metal binding. (B) A novel chemistry was used to end-functionalize the protein and incorporate it as a hydrogel cross-linker. (C) The resulting hydrogels underwent measurable volume decreases in the presence of heavy metals, such as Cd2+. (Reprinted with permission from Esser-Kahn et al.42, copyright 2008 American Chemical Society).
As noted above, motor proteins have been used as active transport components in a variety of nanotechnology applications.28–33 One can also envision that motor proteins could be used to form materials with inherent dynamic properties, and a recent study demonstrates a critical initial step in this direction. Specifically, Diehl et al.43 created cooperative motor assemblies by linking kinesin 1 motors to synthetic protein scaffolds using a protein engineering approach (Fig. 6A and B). The investigators engineered a kinesin 1 motor linked to a leucine zipper motif, and allowed the resulting molecule to interact with a co-polymer containing a complementary leucine zipper motif spaced by an elastin-like sequence. The resulting motor assemblies had controllable spacing between kinesin monomers, and displayed enhanced ATP hydrolysis activity and microtubule gliding velocity when compared to monomeric motors. This study demonstrates the intriguing potential to harness cooperative protein motors, and suggests that future studies may extend the approach to higher order assemblies. For example, well-defined placement of kinesin motors within a three-dimensional protein network using design rules derived from the Diehl et al. study could result in a hydrogel with intriguing cooperative dynamic components. Future seminal studies will be needed to explore the potential role of kinesin, and other motor proteins such as myosin and F1 ATPase, as functional components of dynamic biomaterials. Notably, another recent study from Tao et al. has demonstrated assembly of F1 ATPase into self-assembled, micro-scale complexes, which disassemble upon activation of F1 ATPase motor activity.44 Again, these studies suggest that motor proteins may ultimately be useful as functional components of bio-responsive polymer networks.
Fig. 6.
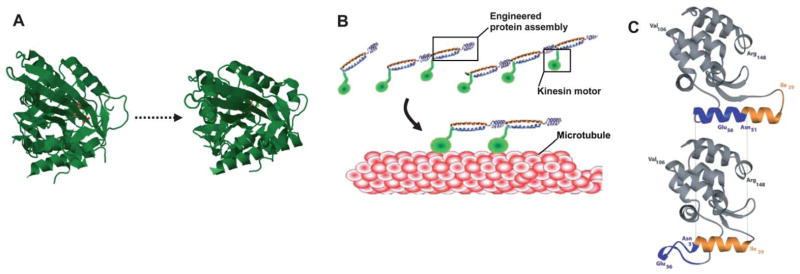
Emerging approaches to exploit new dynamic proteins for biomaterial applications. (A) Several well-characterized protein motions have been used in some bioengineering applications, but have not yet been used to generate dynamic hydrogels. One specific example is the kinesin-1 motor. Shown here are crystal structures for the K1F1 monomeric kinesin motor in open (PDB: 1vfv) and closed (PDB: 1i5s) conformation. (B) A previous study formed a truncated version of the kinesin motor into cooperative assemblies, suggesting the potential to generate dynamic, macroscopic materials with motor proteins (from Diehl et al.43 Reprinted with permission from AAAS). (C) Protein engineering has been used to confer dynamic, ligand responsive properties into the protein lysozyme—shown here are structures of engineered lysozyme in its guanidinium-bound (upper) and guanidinium-free (lower) states (from Yousef et al.53, copyright 2004 National Academy of Sciences, U.S.A.). This strategy may be used to broaden the capabilities of existing protein motions.
4. Applying protein motions: toward bio-responsive drug delivery
Specific bio-responsiveness is among the most pressing needs in emerging applications of dynamic hydrogels. Bio-responsiveness can be defined as a change in material properties as a direct consequence of the presence of a specific biochemical molecule. This is in contrast to traditional dynamic hydrogels, which are designed to respond to changes in physicochemical inputs (e.g. pH, temperature). Drug delivery may be a particularly relevant application of emerging bio-responsive materials, as an ideal drug carrier would deliver a needed drug in response to specific biomarkers that signal the need for the drug. The potential of this approach is underscored by the recent emphasis on identifying early biomarkers for a variety of disease states, including cancer,45 heart disease,46 and neurological disorders.47
Recent studies provide early examples of bio-responsive drug release mediated by protein motion. King et al.48 showed that the release rate of the therapeutic protein vascular endothelial growth factor (VEGF) could be varied in response to trifluoperazine, an FDA approved drug that induces calmodulin’s hinge motion. Specifically, VEGF was absorbed into hydrogel matrices, in which calmodulin was a functional component. Calmodulin’s hinge motion in response to trifluoperazine led to a decrease in the hydrogel volume (40–80%, depending on hydrogel cross-linking conditions), and a concomitant increase in the release rate of VEGF. Increased VEGF release was likely a combined result of fluid exclusion from the hydrogel and a decrease in the surface charge of calmodulin in the network after hinge motion, which each would provide a driving force for VEGF transport out of the hydrogel matrix. Importantly, this study demonstrated that the hydrogel characteristics, including the polymer volume fraction and the degree of dynamic response, could be varied to tune VEGF release. The importance of VEGF and other growth factors during tissue formation processes combined with the need for temporal regulation of these factors19,49 suggests that bio-responsive hydrogel matrices could be an important component of emerging tissue engineering strategies.
Calmodulin-based hydrogels have also recently been used as an injectable microsphere formulation to trigger drug release (Fig. 5). King et al.50–52 encapsulated or absorbed growth factors within hydrogel microspheres, in which calmodulin was included as a functional component. The release rate of VEGF or bone morphogenetic protein-2 (BMP-2) from these microspheres was minimal in the absence of the trifluoperazine ligand. However, in the presence of trifluoperazine the microspheres underwent a rapid (~30 seconds) collapse to 40% of their initial volume, which was accompanied by an 8-fold increase in VEGF or BMP-2 release. Importantly, the approach used for dynamic, protein-based microsphere synthesis has recently been shown to produce microspheres with controllable size ranges (1–50 μm diameter) using multiple protein components.51,52 Collectively, these studies provide a first demonstration of triggered drug release as a result of a protein conformational change, and a similar mechanism could potentially be applied using dynamic hydrogels that undergo protein motion in response to other biochemical triggers, including glucose,41 metal ions,42 and ATP.40
Fig. 5.

Hydrogels synthesized with dynamic protein units can be used for triggered drug delivery. (A) Calmodulin’s conformational change can trigger hydrogel volume decreases and, in turn, release of drugs from hydrogel microspheres, shown schematically here (from King et al.50, copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission). (B) Calmodulin-based microspheres undergo substantial decreases in volume in response to the specific biochemical ligands chlorpromazine (CPZ) and trifluoperazine (TFP). (C) Volume changes can be used to trigger release of growth factors, including vascular endothelial growth factor (VEGF, top) and bone morphogenetic protein-2 (BMP-2, bottom) at T =0 or T =24 h. (D) Growth factor release can also be “pulsed” at multiple times via repeated ligand-induced conformational shifts. The ubiquity of protein conformational changes in nature suggests that these initial demonstrations can potentially be extended to a broad range of specific biochemical “triggers” (reprinted from King et al.51, with permission from Elsevier).
Interestingly, Daunert and coworkers demonstrated that the presence of a biochemical trigger (glucose) could also result in decreased protein transport in protein-based hydrogels, in this case GBP-based hydrogels41 (Fig. 7B). Although this is generally in contrast to the triggered release of growth factors from calmodulin-based hydrogels described above, bio-responsive decreases in drug release could also be important in drug delivery applications. Indeed, one could envision the need to block release of a drug in the presence of a specific biomarker associated with drug toxicity or when drug release is no longer required. Ideally, an “intelligent” drug release system might combine the ability to trigger release in response to a specific biomarker with the ability to block release in response to a second biomarker.
Fig. 7.
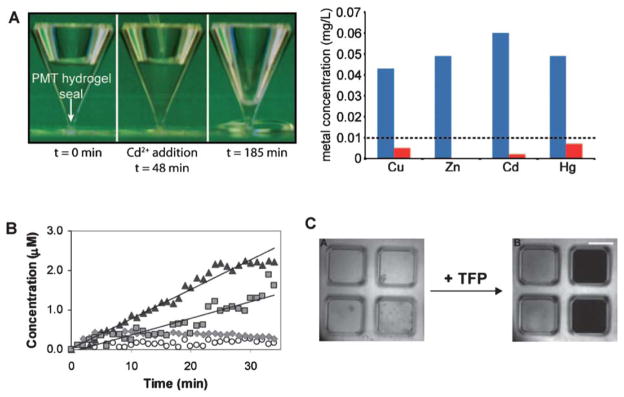
Emerging applications of dynamic, soft materials based on protein conformational changes. (A) Metallothionein changes its conformation in response to heavy metal binding (left), which can be used as a mechanism to create valves for toxin sequestering and water purification (right, blue bars indicate [metal] prior to hydrogel addition, red bars after addition) (reprinted with permission from Esser-Kahn et al.42, copyright 2008 American Chemical Society). (B) Glucose-binding protein motion can be used to create a molecular sieve that allows for faster transport of small molecules such as vitamin B12 (triangle) and 10 kD dextran (square), and slower transport of larger molecules such as BSA (circle) and 200 kD dextran (diamond) (from Ehrick et al.41, copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission). (C) Hinge motion of the protein calmodulin in response to trifluoperazine (TFP) translates into tunable changes in optical transparency, which can be used as a mechanism for label-free biosensing (from Sui et al.39, copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission).
The bio-responsive protein motions described in the previous sections, coupled with the recent use of protein motions to trigger or mitigate drug release, demonstrate early feasibility of bio-responsive drug delivery. However, the goal of developing dynamic hydrogels that can be tailored to deliver a drug in response to any desired biochemical input remains elusive, and the use of engineered, recombinant proteins as functional material components in drug delivery applications presents practical challenges. Synthesis of sufficient quantities of dynamic proteins for biomedical applications may be difficult or expensive in some cases, and engineered versions of native proteins may be antigenic in vivo. In addition, proteolytic degradation of protein-based materials would be an important consideration when designing a pharmaceutical formulation. Finally, the regulatory approval process may be less clear for a drug delivery system that includes a recombinant protein component when compared to existing drug carriers, which are often based on naturally derived proteins or synthetic polymers. Future studies will be required to fully explore the potential of protein motions in drug delivery, and to address potential challenges of this emerging approach.
5. Emerging directions: mimicking and exploiting nature’s protein motions
Materials design
The studies performed to date using protein motions as building blocks in dynamic materials have just begun to scratch the surface of what is possible. There are hundreds of protein motions that have been structurally characterized or modeled via predictive algorithms,7,8 and many of these motions could be harnessed in future studies to build dynamic materials using design rules established in early studies to date.38–42,48,50–52 However, it is noteworthy that the majority of these proteins are not ideal components of putative synthetic materials. Many natural dynamic proteins are too large to readily synthesize recombinantly, require multimerization for their dynamic function, or respond to a biochemical trigger only under very specific circumstances (e.g. in the presence of specific cofactors).7,8 Therefore, each candidate protein requires a challenging, unique, optimized synthetic approach for incorporation into a material. These protein-specific challenges become particularly obvious when one considers synthesis of materials with multiple dynamic units for multiplexed bio-responsiveness. In view of these challenges, there is a tremendous need for new, “generic” protein synthesis strategies that can tailor protein units to respond predictably to any desired biochemical trigger. Therefore, one important emerging direction in dynamic materials design may involve expanding beyond nature’s dynamic protein toolkit.
Common proteins can be modified using recombinant DNA technologies, with the goal of customizing protein responsiveness. For example, Yousef et al. showed that the protein lysozyme could be modified to respond to the presence of a guanidinium ion53 (Fig. 6C). Specifically, this group created a modified version of T4 lysozyme that included a duplicated α-helix, which was then destabilized via an amino acid point mutation. The helix predictably re-stabilized in the presence of a soluble guanidinium ion, leading to a 20° translocation of the helix. These studies demonstrate that a protein can be genetically modified using standard recombinant techniques to “design-in” a new bio-responsiveness, which is a critical step toward custom design of dynamic proteins.
The understanding of natural protein motions and the predictive capabilities of emerging computer algorithms have also progressed toward a paradigm in which de novo design of dynamic protein units may become realistic. For example, the properties of metastable linkers that connect distinct domains in large proteins have been well characterized, and the correlations between linker peptide sequences and linker stability are becoming more well-defined.15,16 One could envision ultimately using or mimicking these linkers to create metastable hinges in synthetic proteins, which might mimic the ligand-induced hinge motions in proteins like calmodulin and GBP. A similar approach could be used to generate synthetic proteins that collapse due to multivalent binding to target molecules in a manner that mimics the function of metallothionein. These general approaches, combined with identification of ligand-binding moieties using phage display or directed evolution, could ultimately lead toward de novo design of custom bio-responsive proteins.
Applications
As described above, bio-responsive drug delivery is a clear emerging application for dynamic materials based on protein motions. A long-term goal of this general approach could involve developing custom dynamic hydrogels, which would be capable of triggering release of a drug in response to virtually any desired input. Interestingly, the same fundamental capabilities that make bio-responsive materials attractive for drug delivery also apply to biosensing, actuation, and tissue engineering applications. Biosensing may be a particularly attractive emerging direction, based on the high sensitivity of some ligand-protein interactions. For example, a class of G-protein-coupled receptors known as “olfactory receptors” undergoes a conformational shift upon binding to odorant molecules. Human olfactory receptors can recognize up to 1000 distinct odorants, and the odorant-receptor binding affinity is often in the parts per billion range.54 Therefore, although the conformational changes involved in olfactory receptor function remain incompletely understood, they may be attractive components of future biosensing strategies. Importantly, biosensing based on protein motion has been demonstrated to some extent in early studies (Fig. 7). For example, metallothionein-based hydrogels have been used as valves to modulate fluid flow and, in turn, indicate the presence of heavy metal ion contaminants in water samples42 (Fig. 7A). Ehrick et al. quantified glucose concentration-depending displacement of GBP-based hydrogels in response to a range of glucose concentrations and found measurable hydrogel displacement (Fig. 2) and changes in transport properties (Fig. 7B) with as low as 0.1 mM [glucose].41 In addition, Sui et al. demonstrated that calmodulin-based hydrogels undergo tunable and readily detectable changes in optical properties as well as volume shifts in the presence of the anti-psychotic drugs trifluoperazine and chlorpromazine39 (Fig. 7C). These approaches demonstrate the potential of protein-based hydrogels to serve as “label-free” biosensors for heavy metal ions, glucose, and anti-psychotic drugs, respectively, though in each case further optimization would be needed to create practical biosensing formats.
Initial demonstrations of triggered drug delivery and label-free biosensing likely represent only a glimpse of the future of this new class of materials. Indeed, each of the biomedical applications explored to date using traditional dynamic hydrogels such as poly(NiPAAm) can potentially be explored using dynamic, protein-based materials. Thus, one can speculate that emerging applications may include cell sheet engineering,55 dynamic ligand presentation,24 adaptive optics,56 and bio-actuation.57 Importantly, unique capabilities may become possible in these applications based on the fundamentals of protein motions, perhaps including multiplexed responsiveness with specificity, “intelligent” regulation of biological systems, and highly specific therapeutic/diagnostic (“theranostic”) strategies.
Acknowledgments
The author is grateful to the National Science Foundation for a CAREER award (0745563) and the National Institutes of Health (R01HL093282).
Biography

Murphy is an Associate Professor of Biomedical Engineering, Pharmacology, and Orthopedics & Rehabilitation at the University of Wisconsin, where he has been since 2004. He received his PhD in Biomedical Engineering from the University of Michigan in 2002, and was a postdoctoral fellow in Chemistry at the University of Chicago from 2002–2004. Murphy’s research interests focus on designing “bioinspired” materials that mimic and exploit biological systems. Examples of mimicking biology include biomaterials that translate nature’s molecular dynamics into controllable drug delivery. Examples of exploiting biology include biomaterials that leverage specific molecules and stem cells already present in the body to promote tissue regeneration. He has published over 50 manuscripts and 15 patents.
References and notes
- 1.Qiu Y, Park K. Adv Drug Delivery Rev. 2001;53:321–339. doi: 10.1016/s0169-409x(01)00203-4. [DOI] [PubMed] [Google Scholar]
- 2.de Las Heras Alarcon C, Pennadam S, Alexander C. Chem Soc Rev. 2005;34:276–285. doi: 10.1039/b406727d. [DOI] [PubMed] [Google Scholar]
- 3.Jeong B, Gutowska A. Trends Biotechnol. 2002;20:305–311. doi: 10.1016/s0167-7799(02)01962-5. [DOI] [PubMed] [Google Scholar]
- 4.Spudich JA, Sivaramakrishnan S. Nat Rev Mol Cell Biol. 2010;11:128–137. doi: 10.1038/nrm2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yon JM, Perahia D, Ghelis C. Biochimie. 1998;80:33–42. doi: 10.1016/s0300-9084(98)80054-0. [DOI] [PubMed] [Google Scholar]
- 6.Harrison SC, Olson AJ, Schutt CE, Winkler FK, Bricogne G. Nature. 1978;276:368–373. doi: 10.1038/276368a0. [DOI] [PubMed] [Google Scholar]
- 7.Gerstein M, Krebs W. Database of Macromolecular Motions. doi: 10.1093/nar/26.18.4280. http://molmovdb.org/ [DOI] [PMC free article] [PubMed]
- 8.Gerstein M, Krebs W. Nucleic Acids Res. 1998;26:4280–4290. doi: 10.1093/nar/26.18.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wriggers W, Chakravarty S, Jennings PA. Biopolymers. 2005;80:736–746. doi: 10.1002/bip.20291. [DOI] [PubMed] [Google Scholar]
- 10.Mohammed JS, Murphy WL. Adv Mater. 2009;21:2361–2374. [Google Scholar]
- 11.Zhang S. Nat Biotechnol. 2003;21:1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- 12.Lu Y, Liu J. Acc Chem Res. 2007;40:315–323. doi: 10.1021/ar600053g. [DOI] [PubMed] [Google Scholar]
- 13.Gerstein M, Levitt M. Proc Natl Acad Sci U S A. 1997;94:11911–11916. doi: 10.1073/pnas.94.22.11911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levitt M, Gerstein M. Proc Natl Acad Sci U S A. 1998;95:5913–5920. doi: 10.1073/pnas.95.11.5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.George RA, Heringa J. Protein Eng. 2002;15:871–879. doi: 10.1093/protein/15.11.871. [DOI] [PubMed] [Google Scholar]
- 16.Gerstein M, Echols N. Curr Opin Chem Biol. 2004;8:14–19. doi: 10.1016/j.cbpa.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 17.Gerstein M, Lesk AM, Chothia C. Biochemistry. 1994;33:6739–6749. doi: 10.1021/bi00188a001. [DOI] [PubMed] [Google Scholar]
- 18.More specifically, the torsion angle changes in the flexible linker region are nearly parallel to the axis of protein rotation, which allows the local hinge to translate directly into protein motion.
- 19.King WJ, Murphy WL. Polym Chem. 2011 doi: 10.1039/C0PY00244E. [DOI] [Google Scholar]
- 20.Noppe W, Vanhoorelbeke K, Galaev IY, Mattiasson B, Deckmyn H. J Dairy Sci. 2004;87:3247–3255. doi: 10.3168/jds.S0022-0302(04)73461-X. [DOI] [PubMed] [Google Scholar]
- 21.Chattopadhyaya R, Meador WE, Means AR, Quiocho FA. J Mol Biol. 1992;228:1177–1192. doi: 10.1016/0022-2836(92)90324-d. [DOI] [PubMed] [Google Scholar]
- 22.Vandonselaar M, Hickie RA, Quail JW, Delbaere LT. Nat Struct Biol. 1994;1:795–801. doi: 10.1038/nsb1194-795. [DOI] [PubMed] [Google Scholar]
- 23.Muller CW, Schlauderer GJ, Reinstein J, Schulz GE. Structure. 1996;4:147–156. doi: 10.1016/s0969-2126(96)00018-4. [DOI] [PubMed] [Google Scholar]
- 24.Hoffman AS, Stayton PS. Prog Polym Sci. 2007;32:922–932. [Google Scholar]
- 25.Vale RD. Cell. 2003;112:467–480. doi: 10.1016/s0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- 26.Seidel R, Bloom JG, van Noort J, Dutta CF, Dekker NH, Firman K, Szczelkun MD, Dekker C. EMBO J. 2005;24:4188–4197. doi: 10.1038/sj.emboj.7600881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirokawa N, Nitta R, Okada Y. Nat Rev Mol Cell Biol. 2009;10:877–884. doi: 10.1038/nrm2807. [DOI] [PubMed] [Google Scholar]
- 28.Burghard M. Angew Chem, Int Ed. 2008;47:8565–8566. doi: 10.1002/anie.200803021. [DOI] [PubMed] [Google Scholar]
- 29.Song W, Mohwald H, Li J. Biomaterials. 2010;31:1287–1292. doi: 10.1016/j.biomaterials.2009.10.026. [DOI] [PubMed] [Google Scholar]
- 30.Diez S, Reuther C, Dinu C, Seidel R, Mertig M, Pompe W, Howard J. Nano Lett. 2003;3:1251–1254. [Google Scholar]
- 31.van den Heuvel MG, Dekker C. Science. 2007;317:333–336. doi: 10.1126/science.1139570. [DOI] [PubMed] [Google Scholar]
- 32.Bohm KJ, Stracke R, Muhlig P, Unger E. Nanotechnology. 2001;12:238–244. [Google Scholar]
- 33.Fischer T, Agarwal A, Hess H. Nat Nanotechnol. 2009;4:162–166. doi: 10.1038/nnano.2008.393. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki H, Yamada A, Oiwa K, Nakayama H, Mashiko S. Biophys J. 1997;72:1997–2001. doi: 10.1016/S0006-3495(97)78844-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mansson A, Sundberg M, Balaz M, Bunk R, Nicholls IA, Omling P, Tagerud S, Montelius L. Biochem Biophys Res Commun. 2004;314:529–534. doi: 10.1016/j.bbrc.2003.12.133. [DOI] [PubMed] [Google Scholar]
- 36.Korten T, Mansson A, Diez S. Curr Opin Biotechnol. 2010;21:477–488. doi: 10.1016/j.copbio.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 37.Murphy WL, Dillmore WS, Modica J, Mrksich M. Angew Chem, Int Ed. 2007;46:3066–3069. doi: 10.1002/anie.200604808. [DOI] [PubMed] [Google Scholar]
- 38.Sui ZJ, King WJ, Murphy WL. Adv Mater. 2007;19:3377–3380. [Google Scholar]
- 39.Sui ZJ, King WJ, Murphy WL. Adv Funct Mater. 2008;18:1824–1831. [Google Scholar]
- 40.Yuan W, Yang J, Kopeckova P, Kopecek J. J Am Chem Soc. 2008;130:15760–15761. doi: 10.1021/ja805634x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ehrick JD, Luckett MR, Khatwani S, Wei Y, Deo SK, Bachas LG, Daunert S. Macromol Biosci. 2009;9:864–868. doi: 10.1002/mabi.200800337. [DOI] [PubMed] [Google Scholar]
- 42.Esser-Kahn AP, Iavarone AT, Francis MB. J Am Chem Soc. 2008;130:15820–15822. doi: 10.1021/ja807095r. [DOI] [PubMed] [Google Scholar]
- 43.Diehl MR, Zhang K, Lee HJ, Tirrell DA. Science. 2006;311:1468–1471. doi: 10.1126/science.1122125. [DOI] [PubMed] [Google Scholar]
- 44.Tao N, Cheng J, Wei L, Yue J. Langmuir. 2009;25:5747–5752. doi: 10.1021/la804083f. [DOI] [PubMed] [Google Scholar]
- 45.Madu CO, Lu Y. J Cancer. 2010;1:150–177. doi: 10.7150/jca.1.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dekker MS, Mosterd A, van ‘t Hof AW, Hoes AW. Heart. 2010;96:1001–1010. doi: 10.1136/hrt.2009.189886. [DOI] [PubMed] [Google Scholar]
- 47.Zetterberg H, Mattsson N, Shaw LM, Blennow K. Biomarkers Med. 2010;4:91–98. doi: 10.2217/bmm.09.80. [DOI] [PubMed] [Google Scholar]
- 48.King WJ, Mohammed JS, Murphy WL. Soft Matter. 2009;5:2399–2406. [Google Scholar]
- 49.Fischbach C, Mooney DJ. Biomaterials. 2007;28:2069–2076. doi: 10.1016/j.biomaterials.2006.12.029. [DOI] [PubMed] [Google Scholar]
- 50.King WJ, Pytel NJ, Ng K, Murphy WL. Macromol Biosci. 2010;10:580–584. doi: 10.1002/mabi.200900382. [DOI] [PubMed] [Google Scholar]
- 51.King WJ, Toepke MW, Murphy WL. Acta Biomater. 2010 doi: 10.1016/j.actbio.2010.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.King WJ, Toepke MW, Murphy WL. Chem Commun. 2011;47:526–528. doi: 10.1039/c0cc02716b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yousef MS, Baase WA, Matthews BW. Proc Natl Acad Sci U S A. 2004;101:11583–11586. doi: 10.1073/pnas.0404482101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J, Luthey-Schulten ZA, Suslick KS. Proc Natl Acad Sci U S A. 2003;100:3035–3039. doi: 10.1073/pnas.262792899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nagase K, Kobayashi J, Okano T. J R Soc, Interface. 2009;6(suppl 3):S293–S309. doi: 10.1098/rsif.2008.0499.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dong L, Agarwal AK, Beebe DJ, Jiang H. Nature. 2006;442:551–554. doi: 10.1038/nature05024. [DOI] [PubMed] [Google Scholar]
- 57.Tondu B, Emirkhanian R, Mathe S, Ricard A. Sens Actuators, A. 2009;150:124–130. [Google Scholar]