

Abstract
Engineered nanoparticles are endowed with very promising properties for therapeutic and diagnostic purposes. This work describes a fast and reliable method of analysis by flow cytometry to study nanoparticle interaction with immune cells. Primary immune cells can be easily purified from human or mouse tissues by antibody-mediated magnetic isolation. In the first instance, the different cell populations running in a flow cytometer can be distinguished by the forward-scattered light (FSC), which is proportional to cell size, and the side-scattered light (SSC), related to cell internal complexity. Furthermore, fluorescently labeled antibodies against specific cell surface receptors permit the identification of several subpopulations within the same sample. Often, all these features vary when cells are boosted by external stimuli that change their physiological and morphological state. Here, 50 nm FITC-SiO2 nanoparticles are used as a model to identify the internalization of nanostructured materials in human blood immune cells. The cell fluorescence and side-scattered light increase after incubation with nanoparticles allowed us to define time and concentration dependence of nanoparticle-cell interaction. Moreover, such protocol can be extended to investigate Rhodamine-SiO2 nanoparticle interaction with primary microglia, the central nervous system resident immune cells, isolated from mutant mice that specifically express the Green Fluorescent Protein (GFP) in the monocyte/macrophage lineage. Finally, flow cytometry data related to nanoparticle internalization into the cells have been confirmed by confocal microscopy.
Keywords: Immunology, Issue 85, Flow cytometry, blood leukocytes, microglia, Nanoparticles, internalization, Fluorescence, cell purification
Introduction
Engineered nanomaterials are nowadays inspiring the interest of life scientists for potential applications to biomedicine1. A wide variety of inorganic and organic materials can be used to produce nanostructures with different shapes, physical and chemical features. Among these structures, engineered nanoparticles of spherical shape demonstrated great potential for diagnostic and translational medicine2. Their core and surface design are driven by a possible application and it implies a profound study of target cell responses following nanoparticle contact and interaction. Nanoparticles that are thought to be deliberately administered to human subjects will come in direct contact with several types of immune cells. Their responsibility to maintain the body integrity makes them a crucial topic of investigation in nanomedicine3.
The cellular part of the innate immune system is mainly represented by phagocytes. Among them, the monocyte/macrophage lineage derived cells, including the central nervous system resident microglia, play a key role in the immune defense4,5. They are able to trigger protective responses within few hours after encountering with foreign bodies. Moreover, the monocytic cells coordinate and instruct the adaptive immune response through the release of cytokines. All these events occur even in presence of engineered materials, which are largely perceived as "nonself" by the immune system6.
Among the methods historically used in immunology for cell analysis, flow cytometry represents one of the most powerful tools. Furthermore, the availability of technologies for the identification or purification of a specific immune subpopulation (often exploiting the exclusion or the presence of one single membrane protein) allows a precise investigation of the effects of a certain nanoparticle on that particular primary cell type 7. However, cells may present physiological and morphological alterations after exposure to nanoparticles. As well, nanoparticles may interfere with specific optical parameters, such as absorption or emission of light at defined wavelengths, influencing the obtained results8. So, limits of use and eventual adaptations of classical immunological assays to the study of novel materials should be considered.
This work concerns the detection of nanoparticle interactions with primary immune cells by flow cytometry. To address this issue, 50 nm FITC-SiO2 nanoparticles were employed as a model nanomaterial to describe the method. Silica particles can be produced in a very precise manner in the nano-metric scale. Size, shape, and surface properties, such as charge or hydrophobicity, can be finely tuned to increase their biocompatibility9. Many features of SiO2 nanoparticles allow them to be used as a model for drug delivery particles10. Furthermore, fluorescent dyes or quantum dots can be entrapped or linked to these particles offering useful nano-tools for imaging purposes11.
Protocol
ETHICS STATEMENT
Human and animal samples were processed following the guidelines of the Italian Ministry of Health, the law 116/92 and the European Communities Council Directive 86/609/EEC.
1. Monocyte Cell Cultures
Culture human THP-1 cells in T-75 flasks containing RPMI-1640 medium supplemented with 10% human serum, 50 U/ml penicillin and 50 mg/ml streptomycin, 0.05 mM β-mercaptoethanol at 37 °C in 5% CO2.
Split cultures when confluent (every 3-5 days).
2. Isolation of Peripheral Blood Mononuclear Cells (PBMCs) from Buffy Coats
Before starting the isolation protocol, prepare a solution of phosphate buffered saline (PBS) pH 7.2, 2 mM ethylenediaminetetraacetic acid (EDTA), 0.5% bovine serum albumin (BSA), adding 5 ml BSA Stock Solution to 95 ml Rinsing buffer (1:20 dilution). Degas the buffer and keep it cold (2-8 °C). IMPORTANT: Failure to degas the buffer may result in less than optimal results because bubbles may block the isolation column (see step 3.2). Note: Anticoagulant citrate dextrose formula-A (ACD-A) or citrate phosphate dextrose (CPD) can be alternatively used. Conversely, other albumin proteins or sera from other species can replace BSA. Do not use buffers containing Ca2+ or Mg2+.
Obtain buffy coats from healthy donors (aged 20-60 years old).
Prepare 50 ml conical tubes for density gradient centrifugation. Determine the number of tubes required for processing the blood (each tube can process 35 ml of diluted blood) and add 15 ml of Ficoll-Paque to each empty tube.
Dilute 8 ml of blood with 24 ml of PBS/BSA/EDTA solution (1:4 dilution).
Carefully layer the diluted solution on top of Ficoll-Paque (density = 1.077 g/ml) in each of the 50 ml conical tubes. Do not mix the blood and Ficoll-Paque. IMPORTANT: To avoid mixing of the blood and Ficoll-Paque, hold the tube at a 45° angle and layer the blood mixture slowly.
Centrifuge at 400 x g for 30 min at 4 °C. Mononuclear cells (MNCs) will remain at the plasma-Ficoll-Paque interface, whereas granulocytes and erythrocytes sediment due to higher density at the osmotic pressure of Ficoll-Paque.
Transfer the interphase cells (Peripheral blood mononuclear cells, PBMCs) to a new 50 ml tube filled with PBS/BSA/EDTA and centrifuge at 300 x g for 10 min at 4 °C.
Wash the pellet twice with PBS/BSA/EDTA to remove platelets. Spin at 200 x g for 10 min at 4 °C.
Culture the cells in complete RPMI-1640 with 10% human serum and antibiotics or proceed to monocytes purification.
3. Purification of Primary Monocytes from PBMCs
- Isolate monocytes from PBMCs by magnetic bead separation using Human Pan monocyte isolation kit:
- Pass cells through a 30 µm Nylon mesh (preseparation filters, 30 µm) to remove possible cell clumps.
- Determine cell number using a hemocytometer.
- Centrifuge cell suspension at 300 x g for 10 min at room temperature (RT). Aspirate supernatant completely.
- Resuspend cell pellet in 30 µl of PBS/EDTA/BSA buffer per 107 total cells.
- Add 10 µl FcR Blocking Reagent per 107 total cells.
- Add 10 µl of Biotin-Antibody Cocktail per 107 total cells.
- Mix well and incubate for 5 min at 2-8 °C.
- Add 30 µl of buffer per 107 total cells.
- Add 20 µl of Anti-Biotin MicroBeads per 107 total cells.
- Mix well and incubate for 10 min at 2-8 °C.
- Resuspend up to 108 cells in 500 µl of PBS/EDTA/BSA buffer.
- Magnetic separation
- Accordingly to the total cell number, choose an appropriate MACS Column and MACS Separator (see MACS Column datasheet).
- Place the column in the magnetic field of the MACS Separator.
- Prepare column by rinsing with the appropriate amount of PBS/EDTA/BSA buffer (MS columns: 500 µl; LS columns: 3 ml). Note: Columns are "flow stop" and do not run dry .
- Apply cell suspension onto the column. Collect flow-through containing unlabeled cells, representing the enriched monocyte fraction.
- Wash column 3x with PBS/EDTA/BSA buffer (500 µl for MS and 3 ml for LS per wash).
- Collect unlabeled cells that pass through, representing the enriched monocyte cells, and add to the flow-through from section 3.2.4. Note: Wash the column by adding PBS/EDTA/BSA buffer aliquots only when the column reservoir is empty.
- (Optional) Remove column from the separator and place it on a suitable collection tube. Pipette the buffer onto the column (MS columns: 1 ml; LS columns: 5 ml). Immediately flush out the magnetically labeled nonmonocyte cells by firmly pushing the plunger into the column.
- Culture purified monocytes in complete RPMI-1640 with 10% human serum and antibiotics.
4. Purification of Primary Monocytes from Whole Blood
Purify monocytes from whole blood using whole blood monocyte isolation kit following manufacturer’s instructions.
5. Nanoparticle Internalization in Blood Leukocytes and Purified Monocytes
Plate 5 x 105 cells/ml (1 ml/well) of the isolated PBMCs or the purified CD14 positive monocytes in a 12-well plate.
Vortex FITC-SiO2 nanoparticle stock solution and resuspend in complete medium to a working concentration of 100 nM.
Add 10 µl of working nanoparticle suspension (100 nM) for treated samples to reach a nanoparticle final concentration of 1 nM. Add the same volume of complete medium for untreated controls to each well.
After 1 hr internalization collect the samples in a 1.5 ml polypropylene tube and centrifuge them for 3 min at 6,000 x g at RT.
Wash the pellet with 1 ml of running buffer 3 min at 6,000 x g at RT.
Resuspend the pellet in 200 µl of running buffer. The cells are now ready for reading by flow cytometry.
6. Isolation of Primary Mixed Glial Cultures (See Bertero et al.7)
7. Replating a Mixed Glial Confluent Cell Culture
Wash a T-75 flask once with 10 ml of PBS (w/o Ca2+/Mg2+), then add 10 ml of Versene and incubate at 37 °C for 5-7 min.
Pipette the supernatant up and down several times in order to detach the cells from the T-75 surface and dissolve cell aggregates.
Transfer the cell suspension in a 15 ml polypropylene tube. Collect a small amount of cellular suspension to count the cells with a hemocytometer.
Centrifuge the cell suspension for 5 min at 300 x g at RT.
Remove the supernatant and resuspend the pellet at a final concentration of 5 x 105 cells/ml in glial complete culture medium.
Plate 0.5 ml/well in a 24-well plate and let the cells settle for 2-4 hr before nanoparticle treatment.
8. Nanoparticle Internalization in Microglia
Add 500 µl of complete glial culture medium (for untreated controls) or 500 µl of a 2x Rhodamine-SiO2 nanoparticle suspension in complete glial culture medium (for treated samples) to each well.
Pipette the cells to allow the detachment of nonrapid-adhering cells at the chosen time points and collect them in 2 ml polypropylene tubes.
Add 500 µl of Versene to each well and incubate the plate at 37 °C for 5 min, then detach the remaining cell fraction and collect each sample with the corresponding nonadhering cells of the previous step.
Centrifuge the samples for 3 min at 300 x g at RT.
Resuspend the pellet in 100 µl of AutoMACS Running buffer.
9. Staining with CD11b-VioBlue
Add 10 µl of VioBlue-CD11b human/mouse antibody to 100 µl of cell suspension (1:11 ratio) in running buffer and incubate 10 min at 4 °C.
Add 1 ml of running buffer and mix the cell suspension.
Centrifuge each sample for 3 min at 300 x g.
Discard the supernatant and resuspend the pellet in 100-150 µl of running buffer.
Read the samples to the cytometer (store the samples at 4 °C between the last two steps). IMPORTANT: before analyzing the samples, proceed to compensation of overlapping signals in emission spectra observed between different fluorochromes (fluorochrome-conjugated nanoparticles or antibodies).
10. Spectral Spill Over Compensation
Open the "Instrument settings" box (present in all the instrument software programs). Note: If using a different instrument from the one reported in the Material Table, please refer to the instrument specific manual for the acquisition details.
Acquire untreated sample (without nanoparticles) at low fluidic speed (if allowed by the instrument). During the acquisition, manually adjust forward and side scattering (FSC and SSC, respectively) by regulating the voltage channels. Note: Modify the SSC and FSC axis scales in order to best visualize the complete cell population in the plot. An instrument setting template for each cell type of interest can be created, saved and used for future acquisitions.
Open the specific "region" tab in the software. Draw an appropriate large region ("gate") around the desired population. Note: Internalization of nanoparticles induces a SSC shift of the cells. If the gate is too closely restricted around the cell population of interest, the acquired data will probably miss part of the cells to be detected.
Use two unstained samples, one for use as blank sample and one for compensation against PI staining (optional), one single-stained sample with the appropriate fluorochrome for each fluorescence channel to be compensated. Note: Use a different 1.5 ml sample tube for each fluorochrome-conjugated antibody to be used in the experiment labeling.
Open the "Compensation tab" in the "Instrument settings" box. Increase or decrease the compensation factor during the acquisition until the fluorescence intensity in the spillover channel is roughly the same for the fluorochrome positive and negative population.
Acquire nanoparticle-treated samples after the compensation has been adjusted.
Representative Results
Flow cytometry is a useful tool to identify and characterize different cells and it is the technique of choice to identify specific immune cells, such as monocytes, granulocytes, T cells, B cells, natural killer (NK) cells, dendritic cells (DCs), and other subpopulations of leukocytes.
In the effort to better characterize white blood cells behavior in response to nanoparticles, we performed internalization assays with primary leukocytes isolated from the blood of healthy donors (Figures 1 and 2) and with human monocyte cell line (THP-1 cells, Figure 3).
As reported in Figure 1A, the three major blood leukocyte subpopulations were clearly identified by forward and side scattering after PBMCs isolation. Moreover, after FITC-SiO2 treatment, lymphocytes (grey), monocytes (blue) and granulocytes (red) have a different nanoparticle internalization rate as shown by green fluorescence intensity (Figure 1B). The described protocol allows the purification of primary CD14 positive monocytes from PBMCs. Figure 2A reports the flow cytometry dot plot of CD14+ monocytes in presence of FITC-SiO2. Figure 2B shows FITC-SiO2 internalization quantified in the same cells and expressed in logarithmic scale histogram.
Similar internalization experiments were performed on THP-1 monocytes treated with increasing concentrations of FITC-SiO2 nanoparticles. Untreated cells were used as negative control. Figure 3A shows a dose-dependent increase in side scattering with an unchanged forward scattering in THP-1 cell line. In Figure 3B, together with the histograms of SSC and FSC at each FITC-SiO2 nanoparticle concentration tested, mean fluorescence intensity (MFI) quantification is presented. This data suggest that treatment with FITC-SiO2 nanoparticles induces a dose-dependent internalization in monocytes highlighted by the enhancement of intracellular granularity (side scattering) and fluorescence (green channel).
To gain further insights into the type of interaction between immune cells and nanoparticles, primary mixed glial cultures were isolated and microglia, the central nervous system resident immune cells, was purified. The use of a transgenic mouse model expressing green fluorescent microglia permits the visualization of different neuro-inflammatory mechanisms. The transgenic mouse B6.129P-CX3CR1tm1Litt/J used in this work expresses the green fluorescent protein (GFP) under the control of CX3CR1 promoter12. After 7 days in vitro (DIV), fluorescence microscopy shows a mixed primary glial culture with a large number of astrocytes (GFP negative adherent cells) and some green cells (GFP positive, Figure 5A). In this mouse model, three glial subpopulations can be distinguished by flow cytometry with a single CD11b-antibody staining: the first CD11b-GFP- (astrocytes and other glial cells), a second distinct group of microglial CD11b+GFP+ cells, and a third CD11b+GFP- subpopulation (Figure 4A). These two latter subpopulations are both able to internalize nanoparticles with a slight increased efficiency by the GFP+ population (representing the patrolling immature microglia by the transcription of CX3CR1 promoter), as shown by flow cytometry analysis (Figure 4B). The occurred internalization can be further verified by confocal microscopy using the same final concentration of Rhodamine-SiO2 nanoparticles as shown in Figure 5B.
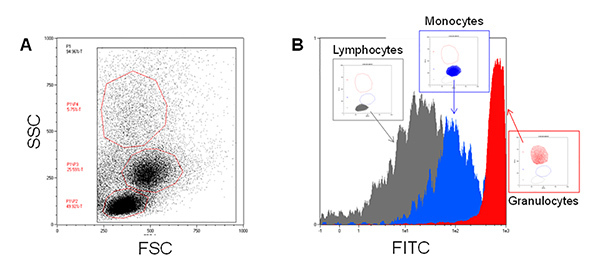 Figure 1. FITC-SiO2 nanoparticle internalization in isolated blood leukocytes.A) Representative forward scattering (FSC) versus side scattering (SSC) flow cytometry dot plot of Ficoll-Paque isolated blood leukocytes. B) Green fluorescence overlay histogram plot of the three major blood leukocyte cell subpopulations in presence of 1 nM FITC-SiO2 nanoparticles (+45 mV) for 1 hr. Please click here to view a larger version of this figure.
Figure 1. FITC-SiO2 nanoparticle internalization in isolated blood leukocytes.A) Representative forward scattering (FSC) versus side scattering (SSC) flow cytometry dot plot of Ficoll-Paque isolated blood leukocytes. B) Green fluorescence overlay histogram plot of the three major blood leukocyte cell subpopulations in presence of 1 nM FITC-SiO2 nanoparticles (+45 mV) for 1 hr. Please click here to view a larger version of this figure.
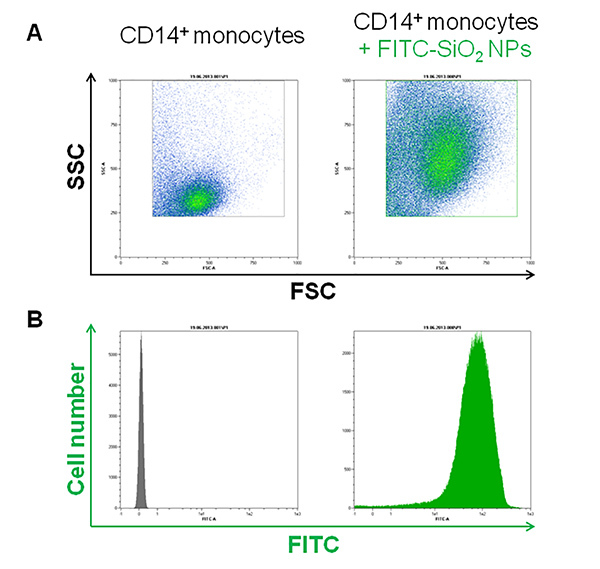 Figure 2. FITC-SiO2 nanoparticle internalization in CD14+ purified monocytes.A) Representative forward scattering (FSC) versus side scattering (SSC) flow cytometry dot plot of purified CD14 positive monocytes. B) Green fluorescence histogram plot of the purified monocyte subpopulation in presence of 1nM FITC-SiO2 nanoparticles (+45 mV) for 1 hr. Please click here to view a larger version of this figure.
Figure 2. FITC-SiO2 nanoparticle internalization in CD14+ purified monocytes.A) Representative forward scattering (FSC) versus side scattering (SSC) flow cytometry dot plot of purified CD14 positive monocytes. B) Green fluorescence histogram plot of the purified monocyte subpopulation in presence of 1nM FITC-SiO2 nanoparticles (+45 mV) for 1 hr. Please click here to view a larger version of this figure.
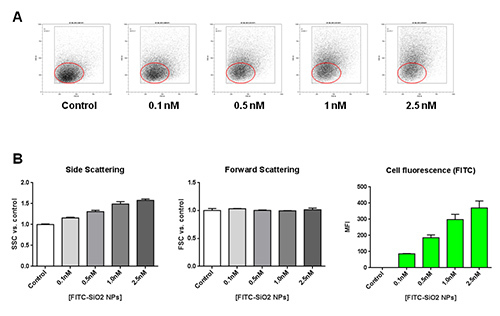 Figure 3. Effects of FITC-SiO2 nanoparticle internalization on THP-1 cells.A) Representative forward scattering (FSC) versus side scattering (SSC) flow cytometry dot plot of THP-1 monocyte cell line, following 1 hr exposure of FITC-SiO2 nanoparticles increasing concentration. B) Concentration-dependent variation of the side scattering (SSC), forward scattering (FSC) and green fluorescence in presence of FITC-SiO2 nanoparticles (+45 mV) for 1 hr. Please click here to view a larger version of this figure.
Figure 3. Effects of FITC-SiO2 nanoparticle internalization on THP-1 cells.A) Representative forward scattering (FSC) versus side scattering (SSC) flow cytometry dot plot of THP-1 monocyte cell line, following 1 hr exposure of FITC-SiO2 nanoparticles increasing concentration. B) Concentration-dependent variation of the side scattering (SSC), forward scattering (FSC) and green fluorescence in presence of FITC-SiO2 nanoparticles (+45 mV) for 1 hr. Please click here to view a larger version of this figure.
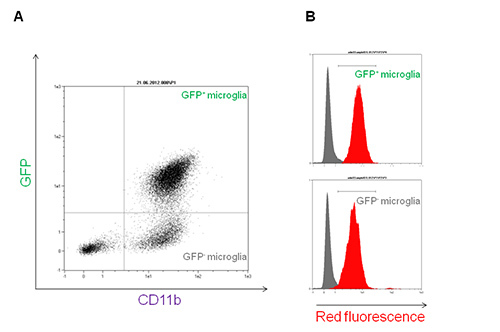 Figure 4. Rhodamine-SiO2 nanoparticle internalization into primary microglia isolated from B6.129P-CX3CR1tm1Litt/J mice.A) Green Fluorescent protein (GFP) versus CD11b-VioBlue flow cytometry dot plot of primary mixed glia isolated from B6.129P-CX3CR1tm1Litt/J mice. CD11b+GFP+ and CD11b+GFP- populations are shown in the upper right and lower right quadrants, respectively. B) Red fluorescence overlay histogram plot of microglia subpopulations in presence of 1 nM Rhodamine-SiO2 nanoparticles (+45 mV) for 30 min (red histogram) versus control (grey histogram). Please click here to view a larger version of this figure.
Figure 4. Rhodamine-SiO2 nanoparticle internalization into primary microglia isolated from B6.129P-CX3CR1tm1Litt/J mice.A) Green Fluorescent protein (GFP) versus CD11b-VioBlue flow cytometry dot plot of primary mixed glia isolated from B6.129P-CX3CR1tm1Litt/J mice. CD11b+GFP+ and CD11b+GFP- populations are shown in the upper right and lower right quadrants, respectively. B) Red fluorescence overlay histogram plot of microglia subpopulations in presence of 1 nM Rhodamine-SiO2 nanoparticles (+45 mV) for 30 min (red histogram) versus control (grey histogram). Please click here to view a larger version of this figure.
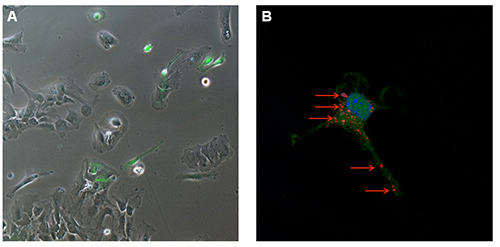 Figure 5. Visualization of GFP+-microglia. (A) Fluorescence microscopy at 7 DIV and (B) Confocal microscopy of Rhodamine-SiO2 nanoparticle internalization (red arrows) in GFP+-microglia.Please click here to view a larger version of this figure.
Figure 5. Visualization of GFP+-microglia. (A) Fluorescence microscopy at 7 DIV and (B) Confocal microscopy of Rhodamine-SiO2 nanoparticle internalization (red arrows) in GFP+-microglia.Please click here to view a larger version of this figure.
Discussion
The experimental protocol presents very crucial points to be taken into account. It is really important to work at 4 °C (on ice) and possibly in the dark during all the staining steps, because higher temperatures and lights may negatively affect the staining yield. Nanoparticles could be sonicated to be better resuspended just before use.
A correct flow cytometry analysis requires a correct calibration in the different channels. Calibration of the instrument should be performed before every experimental session. Besides technical issues with the instrumentation, there can be also problems with the antibody labeling. It is mandatory to use the antibody in an appropriate concentration. If the concentration is too high or too low, dissatisfying signal intensities can be the consequence.
The disadvantages of this technique concern the need of working with monodisperse samples, the inability to localize the site of origin of the signal (i.e. different cellular compartments). There are also some limits in the choice of fluorochromes to be used in combination: the wavelength of the excitation and the emission bands must be sufficiently separated to allow their appropriate measurement. If the used antibody spectra overlap, a correct compensation is required.
Flow cytometry is a powerful method for cell analysis in presence or absence of nanoparticles. This technique permits a multiparametric study of a cell, a high number of events examined, rapidity of analysis (more than 1,000 cells/sec), reproducibility and statistical readings. Samples can be processed without losing cell viability.
By using fluorescently labeled nanoparticles it is possible to qualify and quantify their internalization in cell subpopulations, which are identified by specific markers exposed on the cell membrane. Cell parameters can change in presence of specific nanoparticles. Depending on the aim of the research, these variations can be used to identify a specific phenomenon, such as side scattering of cells that proportionally increases with the increasing nanoparticle internalization rate.
Nanoparticle-induced modifications of cell surface can also represent a limitation of the technique. For this reason, cell membrane receptors turnover must be always taken in consideration and possibly known in advance to precisely characterize the cell population of interest. Extreme impairments of membrane osmosis in nanoparticle overloaded samples can lead to cell death.
Nanomaterial dose-dependent toxicity should be empirically tested for each cell population. Clearly defined dead cells must be excluded from the fluorescence quantification. For instance, Annexin V/PI staining is one of the several methods usually used to detect both necrotic and apoptotic cells.
GFP-expressing primary cells are also a powerful tool to select a certain cell subpopulation and collect data without prelabeling. The combination with complementary fluorescent nanoparticles allows a very fast and precise quantification of cell-nanoparticle interaction. Drug or gene delivery is thought to be improved in the future by the application of nanoparticles able to release a specific drug load into selected tissues.
Employment of nanoparticles as specific delivery carriers and/or immune modulators for pharmaceutics requires the knowledge of the biological environment (i.e. through flow cytometry) to study cell-nanoparticle interactions.
Disclosures
The authors declare that Miltenyi Biotech GmbH has sponsored the submission of this manuscript. HiQ-Nano Company (http://www.hiq-nano.com/index.html) is a nanotechnology spin off company arisen from IIT.
Acknowledgments
This work was supported by Fondazione Istituto Italiano di Tecnologia.The authors would like to acknowledge Miltenyi Biotec GmbH (Bergisch Gladbach, Germany) for the sponsorship of this manuscript, Dr. Paolo Petrucciani (Department of Immuno-hematology and Transfusion Service, Hospital Lotti Pontedera, Pisa, Italy) for providing human buffy coats and Prof. Massimo Pasqualetti (Department of Biology - Unit of Cellular and Developmental Biology, University of Pisa, Pisa, Italy) for housing the mouse colony.
References
- Zhang XQ, et al. Interactions of nanomaterials and biological systems: Implications to personalized nanomedicine. Adv. Drug Deliv. Rev. 2012;64:1363–1384. doi: 10.1016/j.addr.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radad K, Al-Shraim M, Moldzio R, Rausch WD. Recent advances in benefits and hazards of engineered nanoparticles. Environ. Toxicol. Pharmacol. 2012;34:661–672. doi: 10.1016/j.etap.2012.07.011. [DOI] [PubMed] [Google Scholar]
- Dobrovolskaia MA, McNeil SE. Immunological properties of engineered nanomaterials. Nat. Nanotechnol. 2007;2:469–478. doi: 10.1038/nnano.2007.223. [DOI] [PubMed] [Google Scholar]
- Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell JA, Thomas SN, Swartz MA. Materials engineering for immunomodulation. Nature. 2009;462:449–460. doi: 10.1038/nature08604. [DOI] [PubMed] [Google Scholar]
- Bertero A, et al. Surface functionalisation regulates polyamidoamine dendrimer toxicity on blood-brain barrier cells and the modulation of key inflammatory receptors on microglia. Nanotoxicology. 2013;8:158–168. doi: 10.3109/17435390.2013.765054. [DOI] [PubMed] [Google Scholar]
- Kroll A, Pillukat MH, Hahn D, Schnekenburger J. Interference of engineered nanoparticles with in vitro toxicity assays. Arch. Toxicol. 2012;86:1123–1136. doi: 10.1007/s00204-012-0837-z. [DOI] [PubMed] [Google Scholar]
- Malvindi MA, et al. SiO2 nanoparticles biocompatibility and their potential for gene delivery and silencing. Nanoscale. 2012;4:486–495. doi: 10.1039/c1nr11269d. [DOI] [PubMed] [Google Scholar]
- Bardi G. SiO2 NPs: Promising Candidates for Drug and Gene Delivery. Drug Deliv. Lett. 2011;1:9–12. [Google Scholar]
- Bardi G, et al. The biocompatibility of amino functionalized CdSe/ZnS quantum-dot-Doped SiO2 nanoparticles with primary neural cells and their gene carrying performance. Biomaterials. 2010;31:6555–6566. doi: 10.1016/j.biomaterials.2010.04.063. [DOI] [PubMed] [Google Scholar]
- Jung S, et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion andgreen fluorescent protein reporter gene insertion. Mol. Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]