

Abstract
α1-Antitrypsin deficiency (ATD) is a common genetic disorder that can lead to end-stage liver and lung disease. Although liver transplantation remains the only therapy currently available, manipulation of the proteostasis network (PN) by small molecule therapeutics offers great promise. To accelerate the drug-discovery process for this disease, we first developed a semi-automated high-throughput/content-genome-wide RNAi screen to identify PN modifiers affecting the accumulation of the α1-antitrypsin Z mutant (ATZ) in a Caenorhabditis elegans model of ATD. We identified 104 PN modifiers, and these genes were used in a computational strategy to identify human ortholog–ligand pairs. Based on rigorous selection criteria, we identified four FDA-approved drugs directed against four different PN targets that decreased the accumulation of ATZ in C. elegans. We also tested one of the compounds in a mammalian cell line with similar results. This methodology also proved useful in confirming drug targets in vivo, and predicting the success of combination therapy. We propose that small animal models of genetic disorders combined with genome-wide RNAi screening and computational methods can be used to rapidly, economically and strategically prime the preclinical discovery pipeline for rare and neglected diseases with limited therapeutic options.
INTRODUCTION
α1-Antitrypsin (AT)/SERPINΑ1 is the prototypical member of the serpin superfamily and a major anti-protease in the circulation and extracellular fluids (1). The function of AT is to protect tissues from collateral damage by neutralizing leukocyte-derived peptidases (2,3). Decreased circulating AT levels, as occurs in the classical form of α1-antitrypsin deficiency (ATD), predisposes adults to developing emphysema and chronic obstructive pulmonary disease (4–6). In addition, ATD patients homozygous for the most common mutation, Z (E342K), are at increased risk of developing liver disease throughout their lifetime (7,8).
Hepatocytes are the major biosynthetic source of AT, where the protein normally enters the constitutive secretory pathway (9). However, the Z mutation delays native folding and impairs secretion, which leads to polymerization and aggregation of the mutant protein (ATZ) by a domain swapping mechanism (10). Consequently, ATZ is retained within the endoplasmic reticulum (ER) as large inclusions that cause fibrosis/cirrhosis and hepatocellular carcinoma (11–17). In ATD patients, therefore, a loss of serpin inhibitory activity underlies the lung disease, whereas a toxic gain-of-function triggers liver disease.
The only treatment currently available for the complications of ATD liver disease is transplantation (18). A number of therapeutic strategies are being developed, including cell-based strategies (19–21), gene therapy strategies (22–24) small molecules originating from structural analysis (25–28) and drugs that are predicted to capitalize on endogenous proteostasis mechanisms (29,30). Although these diverse approaches are in various stages of evolution towards clinical application, there is still a great need for small-molecule drugs to enter the discovery pipeline for this relatively common genetic disorder (1 in 2000–5000 individuals of Northern European decent) (19). Until recently, this unmet clinical need stemmed partly from the lack of both an ATD model amenable to high-throughput genetic modifier and drug-discovery campaigns. To overcome this problem, we developed a small animal model of ATD using transgenic C. elegans strains expressing either wild-type AT (ATM) or mutant ATZ fused C-terminal to sGFP (s indicates signal peptide). sGFP::ATZ, but not the control sGFP::ATM, expressing strains accumulated aggregated protein within their ER and this accumulation resulted in systemic toxicity manifested by decreased brood sizes, growth and longevity. Moreover, the sGFP::ATZ-expressing animals were easily distinguished from sGFP::ATM-expressing animals, which facilitated the development of an automated high-throughput screening (HTS), high-content screening (HTS/HCS) assay for drugs that modulate the disposition of ATZ in live animals (20). Using a modification of this technology, we report here a semi-automated, genome-wide RNAi screen for proteostasis network (PN) disease modifiers of sGFP::ATZ accumulation. By using these PN modifiers to query target–ligand interaction databases, we developed a computational approach to identify new compounds that were effective in reducing ATZ accumulation in the C. elegans and a mammalian cell line overexpressing ATZ. This study demonstrated that a combination of genome-wide RNAi screens and in silico drug-discovery strategies provided a rapid and economical means for preclinical drug repurposing for common as well as rare and neglected diseases, such as ATD.
RESULTS
A genome-wide RNAi screen for genes that modify sGFP::ATZ accumulation
Genome-wide RNAi screens are powerful means of systematically investigating genes that modulate biological processes (21). Previously, we developed a high-quality, HTS/HCS protocol for drugs that effect sGFP::ATZ accumulation in C. elegans (20). We adapted the assay to perform a semi-automated, genome-wide RNAi screen using the Arhinger feeding library (21). Our copy of the library contains Escherichia coli strains expressing double-stranded RNAs for 16 256 genes (∼500 of the original clones were eliminated due to annotation or technical errors (22)), which covers ∼85% of the C. elegans genome. The RNAi library was re-arrayed by chromosome number into 203 deep-well 96-well plates and an aliquot was removed for overnight growth and IPTG induction. One hundred transgenic sGFP::ATZ animals with similar fluorescence intensity were sorted into each well of a 96-well optical bottom assay plate along with a single bacterial RNAi clone for 48 h. Animals were immobilized and imaged utilizing the ArrayScan VTI. A flowchart outlining RNAi screen workflow, ArrayScan VTI images and development of a high quality single-well screening assay are summarized in Supplementary Material, Fig. S1A–K. Image capture and data analysis required 60–80 min per 96-well plate. Assuming a 40 h workweek, a single person could screen the entire library of ∼17 000 clones in ∼20 days.
For each RNAi sample on a plate, a z-score was calculated using a single plate sample-based method (see Materials and Methods). A total of 255 RNAi clones exceeded the arbitrary threshold of an absolute z-score >2.35 (corresponding to a P < 0.05) and were selected for verification by a second round of testing (Fig. 1A and B). The second round of screening was completed in liquid culture as in the primary screen, except that: (i) RNAi samples were tested in triplicate wells (n = 100 animals/well), (ii) several vector(RNAi) control wells were included on each individual plate and (iii) each RNAi sample was assayed independently on 2 or more separate days. A two-tailed t-test identified individual RNAi sample well averages on each plate that were significantly different (P < 0.05) from that of the corresponding vector(RNAi) controls (Fig. 1C–E). A total of 104 RNAi clones passed the secondary screen with 100 increasing and four decreasing sGFP::ATZ accumulation (Supplementary Material, Table S1). Since we inferred that the action of a wild-type gene was opposite to that of the RNAi effect (i.e. if gene A was a proteostasis enhancer and normally decreased sGFP::ATZ accumulation, gene A(RNAi) increased sGFP::ATZ accumulation), we arbitrarily classified the two groups of 100 and 4 PN modifier genes as ‘PN enhancers’ and ‘PN inhibitors’, respectively (Supplementary Material, Table S1).
Figure 1.

RNAi screen for sGFP::ATZ PN modifiers. (A) A schematic showing how the final list of 104 PN modifiers were obtained from 16 256 RNAi clones. (B) Summary of the genome-wide RNAi screen. Graph shows z-scores for each RNAi treatment. A positive or negative z-score indicates that the treatment either increased or decreased ATZ accumulation, respectively. (C–E) Sample fluorescence well images. Images show animals treated with an RNAi that had no effect (C), decreased (D) or increased (E) ATZ accumulation. Numbers at the bottom represent actual z-scores.
Comparison with other RNAi screens directed against C. elegans models of protein misfolding disorders
Transgenic C. elegans strains have been used to model aspects the cellular pathology of protein misfolding disorders associated with polyglutamine (polyQ) repeat sequences (e.g. Huntington's disease) (23), α-synuclein mutants (e.g. Parkinson's-like diseases) (24), tau mutants (e.g. frontotemporal dementia) (25) and superoxide dismutase (SOD1) mutants (e.g. amyotrophic lateral sclerosis) (26) (summarized in Supplementary Material, Table S2). Selected subset or genome-wide RNAi screens were conducted for PN modifiers, with the majority of RNAi clones exacerbating abnormal phenotypes or increasing the accumulation of the misfolded proteins (see Silva et al., for a notable exception 27). Surprisingly, there was little overlap among PN modifier sets identified by these studies, although many of the studies identified genes associated with common GO biological processes (vide infra).
To determine whether our set of PN modifiers overlapped with those from the other C. elegans screens, we employed a modified Fisher's exact test (28). None of the gene lists from these RNAi screens showed a statistically significant overlap with our dataset. However, a total of eight PN modifiers from this study were detected in a least one of the three genome-wide RNAi screens for PN modifiers (Fig. 2; Supplementary Material, Table S2). These screens included two for polyQ-induced pathology in either body wall muscle or neurons (23,29), and one for mutant α-synuclein expression in neurons (26) (Fig. 2; Supplementary Material, Table S2). Notable in this group was genes involved in the RNAi pathway and protein synthesis.
Figure 2.
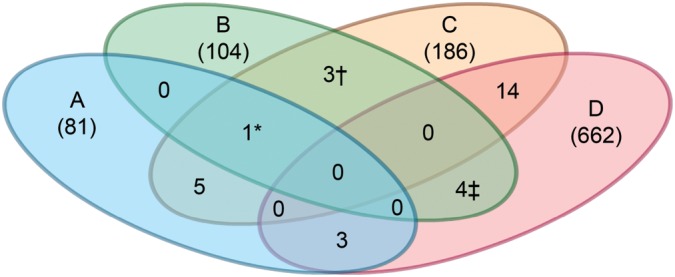
Comparison of RNAi screens for modifiers of protein misfolding diseases. A Venn diagram highlighting overlapping hits from other RNAi screens using models for diseases caused by protein aggregation or misfolding. Wang et al. (26) study (A), current study (B), Nollen et al. (22) study (C) and Lejune et al. (29) study (D). The details of each screen are summarized in Supplementary Material, Table S2. Numbers within parenthesis represent the total number of hits reported. Numbers not in parentheses represent number of overlapping hits with other screens. *F48F7.1 (alg-1); †C14B9.7 (rpl-21), F42C5.1 (rpl-8), Y46G5.4 (phi-10); ‡C07B5.5 (nuc-1), Y47D3B.2 (hum-5), ZC395.10.
As a final test to ensure that common PN modifiers had not been overlooked due to the initial single-well assay format, we retested 30 different RNAi clones that were identified in at least two of the historical RNAi screens (Supplementary Material, Fig. S2). However, none of these RNAi clones altered steady-state levels of sGFP::ATZ. Taken together, these studies suggested that the PN modifiers regulating the cellular response to sGFP::ATZ were different from those associated with other aggregation prone-proteins, or that the PN pathways vary to a certain extent by the cellular (e.g. intestine, neuronal and muscle) and subcellular sites (e.g. ER, cytosolic) of misfolded protein accumulation.
Gene ontology analysis
Enrichment of gene ontology (GO) terms significantly overrepresented in the 104 C. elegans PN modifier genes was assessed using the GOrilla web-based application (http://cbl-gorilla.cs.technion.ac.il/) (30). We found no significant enrichment for any of the molecular function terms. Among the cellular component terms, only the intracellular part (GO:0044424) was overrepresented (1.7-fold) in comparison to a genome-wide control group of 11870 C. elegans genes with associated GO terms. However, the majority of enriched terms were associated with different developmental pathways, as might be expected from the high degree of development-related annotation in WormBase. To overcome this potential bias and to obtain data relevant to mammalian systems, the 104 C. elegans PN modifier genes were assigned human orthologs (Fig. 3A) utilizing two independent methods. First, we used WormBase (http://www.WormBase.org; referential freeze WS236) to query the sequence names of the 104 PN modifier genes and identify the corresponding human orthologs with the highest pBLAST score and/or best predicted human ortholog (based on curated data from TreeFam, Inparanoid, Panther, EnsEMBL-compara ortholog prediction programs) (Supplementary Material, Table S1). Using this method, we found 77% (80 of 104) of the C. elegans genes to be orthologous to human genes (Fig. 3A and B). This high percentage of orthologous genes appeared to be typical (48–61%) of the gene sets accrued from RNAi PN modifier screens using other C. elegans models of proteotoxicity (24–27). Second, we utilized the more stringent OrthoList compiled by Shaye and Greenwald (31), which is now available as an online tool via WormBase. The OrthoList was derived from a meta-analysis of 4 independent prediction methods, in order to generate a human ortholog list of 7663 C. elegans genes. Of the 104 C. elegans PN modifiers, 55 were found in OrthoList, yielding a match of 53% (Fig. 3A and B). While this was a lower matching rate compared with WormBase, it was still higher than the 35–38% human orthologs predicted to be present in the C. elegans genome (31). Comparison of both the WormBase- and OrthoList-assigned human orthologs showed ∼90% overlap with a combined set of 85 genes (Fig. 3B; Supplementary Material, Table S1). These findings suggested that the RNAi screen for PN modifiers identified a proportionally higher percentage of evolutionary conserved genes.
Figure 3.

Human orthologs and drug–target interaction prediction. (A) A flow chart summarizing the in silico approach used to identify human drug–targets from the 104 C. elegans PN modifiers. (B) A Venn diagram showing the overlap between human orthologs identified by OrthoList and WormBase. (C) DAVID analysis comparing the WormBase (outer ring) and OrthoList (inner ring) assigned orthologs to the original C. elegans protein profile (middle ring). (D) Final list of targets and interacting drugs identified using STITCH and Metacore.
To compare the profiles of C. elegans PN modifiers to the human orthologs derived from WormBase and OrthoList, all three datasets were analyzed using the Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics Resource v6.7 (32). A functional annotation chart was generated using a term-centric singular enrichment analysis so that the protein profiles could be directly compared from C. elegans to humans (Fig. 3C). The corresponding donut chart shows overlap of the protein functional profiles, between C. elegans and humans. Comparison of functional categories between C. elegans PN modifiers and WormBase- and OrthoList-assigned genes showed 11/16 (68%) and 7/16 (43%) overlap, respectively. Consistent with ∼90% overlap in genes, overlap in 13/16 (81%) functional categories was seen between WormBase- and OrthoList-assigned human orthologs.
Computational identification of potential drug–target interactions using human homologs/orthologs to c. elegans hits
Based on the examination of our PN modifier set using comparative analysis to other RNAi screens, pathway analysis and ortholog searches; no PN master gene set emerged with exception of a few known PN modifiers such as age-1, ire-1 and daf-16. Rather than further investigate the biologic activity of each new PN modifier, we sought to utilize the gene set as whole to serve as potential drug–target list and search for potential compounds that would be effective in decreasing sGFP::ATZ accumulation. The advantage of this approach is (i) prior knowledge of the gene function was not required, just whether the gene functioned as a PN enhancer or inhibitor in order to select an agonist or antagonistic compound, respectively; (ii) the low cost and high processivity of screening and validation in C. elegans; (iii) selection of druggable targets from a gene set based on phenotype; (iv) the identification of drugs that could be tested rapidly for efficacy in other types of protein misfolding disorders; and (v) acceleration of the drug-discovery process by repurposing of FDA-approved drugs that also prove to be effective in vertebrate models of misfolded protein disorders.
We investigated the possibility of using a computational approach to identify potentially therapeutic drugs that are predicted or known to target proteins encoded by the PN modifiers. We searched STITCH (Search Tool for Interactions of Chemicals; www.stitch.embl.de) and METACORE by Thomson Reuters (http://thomsonreuters.com/products_services/science/science_products/a-z/metacore/) databases for relevant drug–target interactions (33,34). STITCH currently contains information on >2 million interactions between >300 000 chemicals and 2.6 million proteins from >1100 organisms. MetaCore is a systems biology platform for pathway analysis and drug-discovery. STITCH primarily uses keyword mining of the literature and experimental data to predict protein–drug interactions. A confidence score (ranging from 0 to 0.999) is provided to indicate the probability that the predicted interaction exists. For the purpose of this study, only protein–drug interactions with a confidence score of ≥0.9 were investigated. MetaCore uses manually curated literature searches, and links drug–target information to the original references. Unlike STITCH, no confidence score is provided. Of the 85 human PN modifiers queried, 12 drug–target interactions were identified using either STITCH or MetaCore. These data are captured in expandable online archive and can be used to identify all potential drug–target interactions (Supplementary Material, Fig. S3, http://www.ccbb.pitt.edu/faculty/bahar/hitanalysis/). In some instances, PN modifiers had multiple (>75) predicted drug interactions. Conversely, we found some drugs to have multiple predicted targets. For example, midostaurin, a synthetic indolocarbazole kinase inhibitor, was predicted to interact with several targets including ABL, VEGFR2, PDGFR, AKT-1, PIK3, MAPK10, ERN1, FLT3 and PRKAΑ1. To increase stringency, drugs with multiple or non-specific target interactions were omitted from further analysis. Moreover, only drug–target interactions predicted by both STITCH and MetaCore were chosen for further investigation. Since some drugs were not readily available due to licensing restrictions or excessive cost, we tested only those compounds that were found in Library of Pharmacologically Active Compounds (LOPAC). In total, eight drugs targeting four PN modifiers (PI3 K, TTR, ABC and OPRL-1) met our criteria for further investigation (Fig. 3D).
Drug analysis
To determine whether any of the eight compounds were potentially therapeutic, sGFP::ATZ animals were treated for 24 h and misfolded protein accumulation was measured using the ArrayScan VTI. Fluphenazine was identified in a previous small molecule screen to reduce sGFP::ATZ accumulation and was included as a positive control (20). Average results from three independent experiments showed that wortmannin, fluspirilene, fluoxetine and amiodarone significantly decreased sGFP::ATZ accumulation in a dose-dependent fashion (12.5–100 µm) compared with the DMSO control (Fig. 4A and B). Based on these findings, we selected one of these compounds, fluspirilene and tested it on a mammalian cell line expressing ATZ. As shown with C. elegans, fluspirilene showed a dose dependent decrease in ATZ accumulation in ATZ-inducible HeLa cell line, HTO/Z (Fig. 4C).
Figure 4.
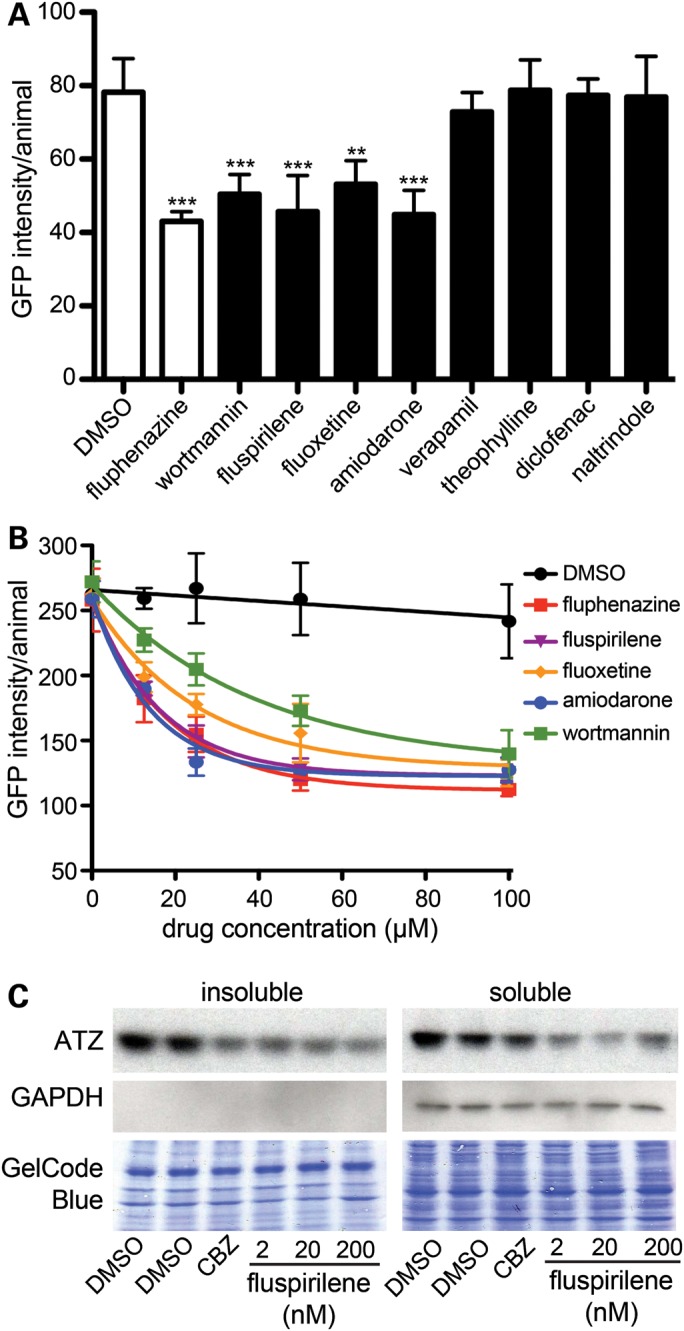
Drug–target analysis. (A) L4 GFP::ATZ animals were treated with 100 µm of each drug for 24 h and analyzed using the ArrayScan VTI. (B) Drug–dose response curves. The experiment was repeated three times, and a representative experiment shown. The error bars represent the SD of five replicate wells (n > 150 animals/treatment). Statistical significance was determined by using a Student's t-test. ***P < 0.001, **P < 0.01. (C) Effect of fluspirilene on steady-state levels of ATZ in a cell line model of ATZ. HeLa cells engineered to express ATZ (HTO/Z) were treated with DMSO, carbamazepine (CBZ) (positive control) or fluspirilene for 48 h. Lysates were prepared and separated into soluble and insoluble fractions. Samples were analyzed by immunoblotting with antibodies against AT (top) and GAPDH (middle). GAPDH is cytosolic marker and its absence in the insoluble fraction indicates correct fractionation. The blots were also stained with GelCode Blue (bottom) to demonstrate equal sample loading in each well.
Drug–target validation
We used a genetic approach to obtain insight into drug–target interactions. Wortmannin is a fungal steroid metabolite that inhibits mostly Classes I and III phosphatidylinositol 3-kinases (PI3Ks) (35). In a C. elegans model of hypoxic injury, 100 µm wortmannin blocks autophagy by inhibition of the class III PI3K, VPS-34 (36). Since autophagy inhibition enhances sGFP::ATZ accumulation, wortmannin was more likely to inhibit the class I PI3K, AGE-1, which would phenocopy the effects of reduced insulin/insulin-like signaling (IIS), rather than VPS-34 (37). To determine whether AGE-1 was the target of wortmannin in this model, we first crossed sGFP::ATZ animals with age-1(hx546) mutants. As expected, the loss of AGE-1 activity resulted in a marked, but not complete, decrease in ATZ accumulation (Fig. 5A). If the effects of wortmannin and age-1(hx546) on sGFP::ATZ accumulation were in the same or different pathways, then treatment of sGFP::ATZ; age-1(hx546) animals with an effective, but not maximal, dose of wortmannin (Fig. 5B) would be expected to have no or an additive effective, respectively. No additive effect was detected (Fig. 5C), despite GFP(RNAi) demonstrating that the sGFP::ATZ levels were not below the ArrayScan VTI level of detection (Fig. 5B and C). Loss of AGE-1 activity, activates a downstream FOXO transcription factor, DAF-16, which leads to decreased sGFP::ATZ accumulation (Fig. 5A). Thus, if wortmannin inhibits AGE-1, a daf-16 loss-of-function mutation should suppress the protective effects of the drug. This was the case as sGFP::ATZ; daf-16(m26) animals were resistant to the effects of the drug, although sGFP::ATZ accumulation could still be modulated with GFP(RNAi) treatment (Fig. 5D). Interestingly, the other three compounds isolated via the in silico screen, as well as the fluphenazine positive control, reduced sGFP::ATZ accumulation in sGFP::ATZ; daf-16(m26) animals (Fig. 5E). Taken together, these studies strongly suggested that wortmannin inhibited the class I PI3K, AGE-1 and that the other compounds were active on other target pathways. If this were the case, then combination therapy between wortmannin and one of the other compounds should be feasible. To test this hypothesis, we treated sGFP::ATZ animals with equal amounts of wortmannin and fluphenazine at three different concentrations. In all cases, combination therapy decreased sGFP::ATZ accumulation more than either monotherapy (Fig. 5F).
Figure 5.

Drug–target validation. (A) Steady-state expression levels of sGFP::ATZ in the N2, age-1(hx546) and daf-16(m26) backgrounds. Data are normalized to N2;sGFP::ATZ worms. (B–D) Effect of wortmannin on steady-state levels of sGFP::ATZ. N2;sGFP::ATZ (B), sGFP::ATZ;age-1(hx546) (C) and sGFP::ATZ;daf-16(m26) (D) animals were treated with wortmannin (100 µm) for 24 h and analyzed using the ArrayscanVTI. GFP(RNAi) treatment was included as a control to show that ATZ levels could be further reduced in each line. Note wortmannin reduced the sGFP::ATZ level in the wild-type N2 but not in age-1(hx546) or daf-16(m26) mutant backgrounds. (E) Effect of various drugs on sGFP::ATZ;daf-16(m26) animals. Of the drugs known to decrease sGFP::ATZ levels in the N2 background, only wortmannin failed to reduce sGFP::ATZ in the daf-16(m26) background. (F) ATZ::GFP animals were treated with 5, 12.5 or 50 μm of fluphenazine and wortmannin, either singly or in combination. The data were normalized to the untreated DMSO control within each experiment. All experiments were repeated at least three times with n > 150 animals/treatment. Error bars represent SD (A–E) or SEM (F). Statistical significance was determined using the Student's t-test. ***P < 0.001, **P < 0.01, *P < 0.05.
DISCUSSION
We developed a semi-automated high-content genome-wide RNAi screening strategy that employs three crucial components to greatly streamline the screening process: the COPAS™ BIOSORT to select for homogenous fluorescing populations of transgenic animals (decreases assay variability), the culturing of 100 animals in liquid media of each well of a 96-well plate (higher-order plates with this number of animals did not provide enough capacity for media and bacteria to conduct a typical 48 h RNAi assay), and the automated ArrayScan VTI image acquisition platform to identify 104 PN modifiers affecting the accumulation of sGFP::ATZ. These methodological improvements to genome-wide RNAi screening eliminated the labor intensiveness, subjective analysis and operator fatigue associated past screens and permitted one person to complete the screen in ∼20 days (23–27).
Analysis of genome-wide of RNAi screens for PN modifiers in other C. elegans models of misfolded protein accumulation failed to identify any genes in common to all the models, which included the expression polyglutamine (polyQ) repeats, α-synuclein, tau or superoxide dismutase (SOD G85R) (23–27). Thus, it was not surprising that few of the 104 modifiers of this RNAi screen were found in common with the others. While variations in experimental designs and selection criteria may account for this apparent discrepancy, it also conceivable that the types of PN modifiers modulating misfolded proteins localizing to the ER lumen (e.g. ATZ) are different from those restricted to the cytosol (e.g. polyQs, SOD), much as ER-associated degradation and heat-shock response pathways regulate protein misfolding in the ER and cytosol, respectively (38). The lack of overlap between the results from different RNAi screens might also be due to the nature of the transgene product itself (e.g. polyQ versus ATZ) or cell types expressing the misfolded protein (e.g. muscle versus intestine).
The molecular mechanisms or pathways associated with 104 PN modifiers that modulate sGFP::ATZ disposition are still poorly defined and merit further investigation. However, the major advantage of this study was the determination that this type of RNAi screen could be used to rapidly identify potential drug targets using computational approaches, even in the absence of extensive knowledge about target functions, other than their effects on SGFP::ATZ accumulation. We employed two independent, but complementary programs, STITCH and MetaCore, to identify chemical/drug and protein interactions (33,34). Of the 85 human PN modifiers queried, a total of eight compounds (two directed against each of four targets) were selected as a proof-of principal for this strategy. Remarkably, one compound for each of the four targets showed a dose-dependent decrease sGFP::ATZ accumulation in C. elegans. Failure of the other four compounds to have the predicted effects in C. elegans were not investigated, but might be due to differences in pharmacokinetics, pharmacodynamics or target-binding site homology between the C. elegans and mammalian systems. While the overall success rate of 50% was encouraging, small numbers preclude the calculation of a meaningful positive predictive value. The results do, however, underscore the great potential for combining genome-wide RNAi screens with computational drug-discovery methodologies. The demonstration that one of the compounds, fluspirilene, was also effective in reducing ATZ accumulation in a mammalian cell line lends additional support for further development of this rapid preclinical drug discovery/repurposing strategy.
A second advantage of this drug-discovery strategy was the use of facile genetic techniques in C. elegans to determine whether the observed drug effect was due to activity within the predicted target pathway or to an off-target effect. Wortmannin was identified in the screen as a potential inhibitor of the type I PI3K kinase, AGE-1. AGE-1 functions downstream of the sole insulin-like receptor, DAF-2, and inhibition of this IIS pathway suppresses the proteotoxic effects of sGFP::ATZ in this C. elegans model, as well as other C. elegans models of misfolded protein accumulation (39). However, wortmannin also inhibits the class III PI3K, VPS-34, which blocks autophagy in C. elegans and mammals as well (36). Since autophagy was an important means of reducing sGFP:: ATZ accumulation (40), suppression of this pathway would be deleterious to these animals. Treatment of the animals with wortmannin decreased sGFP::ATZ accumulation, and this effect was neither enhanced in age-1 mutants nor effective in animals with a mutation in the downstream AGE-1 target gene, daf-16. Taken together, we concluded that the effects of wortmannin at the concentrations used in these animals were via inhibition of AGE-1 and not VPS-34 or some off-target pathway. The large collection of C. elegans single gene mutants, combined with a simple quantitative readout system using fluorescent fusion proteins, makes this system ideal for identifying potential drug targets or target pathways after phenotype-based drug screening. While this technology was not meant to replace target identification by the gold-standard of drug–ligand binding measurements in vitro, it does provide the rationale for embarking upon more detailed kinetic or structural studies with purified reagents or expensive development of a lead series by exploring structure–activity relationships in vitro.
A third advantage of this drug-discovery strategy was the ability to test for the efficacy of combinational therapy. Due to their toxicity, drugs like wortmannin have been largely abandoned as therapeutics in humans. One means to lower toxicity is to use different delivery systems, such as microspheres, to directly deliver lower concentrations of a drug directly to the tissue of interest (41). Another means to avoid toxicity is to utilize lower concentration of drug by combining it with other therapeutics directed at different targets or target pathways. By using the genetic methods outlined above, we showed that unlike wortmannin, none of the other three candidates exerted their effect via the IIS pathway. This observation was validated by using the C. elegans model to show that comparable reductions of sGFP::ATZ accumulation could be achieved at lower doses of wortmannin when it was combined with one of the other drugs. This effect underscores the ability of this experimental system to both identify and test the efficacy of complementary therapeutics. In conclusion, these studies showed that by utilizing the hits from a genome-wide RNAi screen, computational methods could be used to rapidly and strategically develop compounds to prime the preclinical drug-discovery pipeline for rare or neglected diseases lacking effective treatments.
MATERIALS AND METHODS
Worm strains and culture conditions
Strains VK694 (vkIs694 [Pnhx-2::sGFP::ATZ;Pmyo-2::mRFP]) and VK695 (vkIs695 [Pnhx-2::sGFP::ATZ;Pmyo-2::mRFP]) were generated by co-injecting young adults with the plasmids Pnhx-2::sGFP::ATZ and Pmyo-2::mRFP at a final concentration of 70 and 10 ng/µl, respectively. The extra-chromosomal array was integrated by gamma irradiation as described (42). VK2026 (age-1(hx546);vkIs695 [Pnhx-2::sGFP::ATZ;Pmyo-2::mRFP]) and VK757 (daf-16(m26);vkIs695 [Pnhx-2::sGFP::ATZ;Pmyo-2::mRFP]) were generated by crossing VK695 with strains TJ1052 and DR26, respectively. All animals were maintained at 22°C on nematode growth medium (NGM) plates seeded with E. coli OP50 (NGM/OP50) (43).
Preparation of animals for RNAi screening
Twelve to 15 adult sGFP::ATZ transgenic animals were placed on five 10 cm NGM/OP50 plates. Approximately 7 days later, early-staged larval animals were isolated by differential sedimentation and transferred to ten 50 cm NGM/OP50 plates. The larvae were incubated at 22°C until the majority of the animals were at the L4 larval stage, ∼48 h later. Animal sorting using the COPAS™ BIOSORT. As previously described (20), the COPAS™ BIOSORT (Union Biometric, Holliston, MA, USA) reduced assay variability by sorting animals into a more homogeneous population based upon size and fluorescence intensity. Animals were cultivated as described above and sorted using previously described parameters, except that PBS was used for both washing and sorting (sheath fluid) (20). One hundred L4/young-adult animals were sorted into each well of a 96-well optical bottom plate (Nunc MicroWell 96). Approximately 50 000 transgenic animals were required for each 96-well plate and the sorting time was ∼45 min/plate. RNAi cultures prepared as outlined below and added to the wells immediately upon completion of the sort.
RNAi bacterial preparation and induction
RNAi clones (E. coli strain HT115 (DE3) containing a PCR-generated genomic DNA fragment ligated into the EcoRV site of plasmid L4440) were obtained from the Ahringer RNAi feeding Library (Geneservice Limited, Cambridge, UK) (44). A sterile pinning device was used to inoculate each 96-well deep-well plate containing 400 µl of Luria Bertani (LB) broth (10 g tryptone, 5 g yeast extract, 10 g NaCl and 50 µg/ml ampicillin/l) (45). Each plate was sealed with ThinSeal (Sigma–Aldrich, St. Louis, MO, USA) film and incubated overnight at 37°C with shaking. The next morning IPTG was added to each well to a final concentration of 4 mm. The plates were then resealed and incubated for 1 h with shaking to induce production of dsRNA. Plates were then centrifuged at 3500 RPM for 5 min to pellet the bacteria. Supernatant was carefully decanted and the bacterial pellet was resuspended with 400 µl of LB ampicillin/IPTG.
RNAi assay procedure
To each well of a 96-well plate seeded with ∼100 animals, 40 µl of the induced RNAi culture was added. Next, 5-fluorodeoxyuridine (FUDR) was added to each well to prevent eggs from developing. Plates were incubated in a 22°C shaking incubator for 48 h. To prevent evaporation, plates were placed into moist sealed plastic containers.
Image acquisition
Animals in a 96-well plate were anesthetized with 4 mm levamisole or 50 mm sodium azide prior to image capture. Images were acquired with the ArrayScan VTI HCS Reader (Cellomics, ThermoFisher, Pittsburgh, PA, USA) fitted with a 2.5× objective and a 0.63× coupler. The images were captured utilizing a two-channel (TRITC and GFP) assay previously described (20). Data were normalized by dividing the total GFP area by the number of animals per well.
Identification of initial hits
To maintain well alignment with the RNAi library, each plate contained 96-samples and no additional RNAi controls. As most RNAi samples were expected to have a minimal effect on ATZ expression, data from each assay plate served as its own control. This sample-based, rather than a control-based, normalization has been recommended for RNAi screening by Birmingham et al. (46). However, for each batch of plates run on a single day we included an additional plate containing GFP(RNAi) and vector(RNAi) as RNAi positive and negative controls, respectively. These controls ensured that the assay was functioning properly (e.g. animals were viable, growth medium was adequate), but these data were not used for normalization and were not factored into the z-score determinations. For each RNAi sample, a z-score was calculated by: z = (x − µ)/ σ, where x, sample raw score; µ, mean of the samples on the plate; σ, the standard deviation of samples on the plate (46). We arbitrarily selected z-score above or below 2.35 as potential hits for verification as it corresponds to P < 0.05.
Secondary screening: hit verification
Utilizing the methods described above, RNAi clones yielding absolute z-score of ≥2.35 were verified by repeating the assay three times (n = 300 animals). These values were then compared with the transgenic strain fed on control RNAi (L4440 feeding vector) for statistical significance through unpaired, Student's two-tailed t-test (P < 0.05). RNAi clones that did not show statistically significant data in two of three experiments were excluded from further analysis. The identity of the positive RNAi clones were confirmed by DNA sequencing.
Gene ontology analysis
The WormBase ortholog list was generated by entering the 104 PN modifier gene sequence names individually into the search box, (http://www.WormBase.org; referential freeze WS236), selecting the ‘for a gene’ search option, and clicking on the homology section on the left side tool bar. The corresponding human orthologs with the highest pBLAST score and/or best predicted human ortholog (based on curated data from TreeFam, Inparanoid, Panther and ENSEMBL-compara ortholog prediction programs)were then selected. The corresponding ENSEMBL protein ID and gene names were compiled in Supplementary Material, Table S1. To generate the OrthoList ortholog list, the PN modifiers were cross-referenced with the published compendium OrthoList compiled by Shaye and Greenwald (31).The OrthoList was derived from a meta-analysis of four independent prediction methods, in order to generate a human ortholog list of 7663 C. elegans genes. The corresponding ENSEMBL gene ID and gene name were compiled in Supplementary Material, Table S1.
To compare the profiles of C. elegans PN modifiers to the human orthologs derived from WormBase and OrthoList, all three datasets were analyzed using The DAVID Bioinformatics Resource v6.7 (32). The functional annotation option was selected and the ENSEMBL Gene ID was uploaded for the OrthoList and WormBase lists (WormBase protein ID was first converted to gene ID via ENSEMBL website http://useast.ensembl.org) and the human background selected for analysis. For the C. elegans dataset, the WormBase gene ID was uploaded and C. elegans background selected (Supplementary Material, Table S1), respectively. Functional Annotation was selected from the tool bar, and SP-PIR-Keywords under the Functional categories section. A functional annotation chart was generated detailing the category descriptors, number of genes in this category and the associated P-value. To compare the three datasets, the category descriptors were cross referenced, and a donut chart generated. Each colored section represents the genes in that category, the larger the section, the more genes represented. Note a single gene may have many associated keywords and therefore may be present in many categories. The unique category represents non-overlapping categories.
Computational identification of potential drug–target interactions using human homologs–orthologs to C. elegans hits
For the MetaCore analysis, the ENSBL identifier list for both WormBase and OrthoList was uploaded into MetaCore (from Supplementary Material, Table S1), and the ‘Drug look-up’ option selected, under the 1-click analysis section. This generated a list of 12 genes with 69 drug interactors, and 18 genes with 147 drugs interactors for the OrthoList and WormBase, respectively.
For STITCH analysis, the search by name tab was selected and the gene names entered individually from a composite WormBase, OrthoList ortholog list of 85 genes (Supplementary Material, Table S1). This generated a list of 23 genes with 41 drugs, and 36 genes with 61 drugs for OrthoList and WormBase, respectively. The drug lists were then cross-referenced against the LOPAC library, consolidating the OrthoList to 11 genes with 17 potential drug interactors, and the WormBase to 10 genes with 44 drug interactors and these data shown in Supplementary Material, Figure S3.
Drug analysis (combination drug analysis)
Animals were synchronized by placing ∼3000 adult worms onto a fresh 15 cm NGM agar plate seeded with a lawn of OP50, and allow egg-laying to occur for 4 h. At that point the adults were removed by washing in PBS, and only freshly laid eggs were left. Eggs were allowed to hatch and larvae removed to fresh seeded plates after 24 h. After 48 h, the worms were then in L4 stage and harvested for the experiment.
The plate was set up by making a 2× assay solution consisting of 150 l of OP50 working stock, 800 m FUDR, 2× ABAM, in PBS. This was aliquoted into 200 µl aliquots. The drugs were reconstituted to 20 mm in DMSO, aliquoted and stored at −80°C. A fresh aliquot was obtained for each experiment. Drugs were added to the aliquoted assay solution, to a maximal DMSO concentration of 0.5%. Thirty microliters of this solution was then dispensed into five replicate wells of the 384-well optical bottom plate.
Animals were harvested and the L4 population selected by differential sedimentation, and washed 3× in PBS. Using the COPAS™ BIOSORT (Union Biometrica, Holliston, MA, USA), 35 worms of similar age and GFP expression were sorted into wells as described (20), diluting the drug solution to 1×. The plate was then covered in Breathe-EASIER™ film, incubated for 24 h at 20°C with shaking. After 24 h, the animals were anesthetized with 50 mm sodium azide prior to image capture. Images were acquired with the ArrayScan VTI HCS Reader previously described (20). Data were normalized by dividing the total GFP intensity by the total area of the mRFP head marker per well.
Fluspirilene treatment of HTO/Z cells
The human epidermal HeLa cell line with doxycycline-regulated expression of ATZ (HTO/Z) has been described previously (47). For experiments with Fluspirilene, the Tet-off inducible cell lines were cultured in the absence of doxycycline for about 4 weeks to ensure expression of AT. The cells were then subcultured into separate monolayers in fresh complete growth medium and incubated for 48 h in the absence or presence of fluspirilene or carbamazepine. After the incubation, cells were homogenized and cell homogenates separated into insoluble and soluble fractions according to our previously established technique (48). Samples of 10 μg each were subjected to immunoblot analysis for AT and GAPDH. The blots were also stained with GelCode Blue (Pierce) so that loading of insoluble fractions could be assessed.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This study was supported by grants from The Hartwell Foundation (G.A.S.), and the National Institutes of Health (DK079806 and DK081422 to G.A.S., DK084512 and DK096990 to D.H.P., GM103712 to I.B. and DK086112 to M.T.M.). Some nematode strains used in this work were provided by the Caenorhabditis Genetics Center (CGC), which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Supplementary Material
REFERENCES
- 1.Silverman G.A., Whisstock J.C., Bottomley S.P., Huntington J.A., Kaiserman D., Luke C.J., Pak S.C., Reichhart J.M., Bird P.I. Serpins flex their muscle: I. Putting the clamps on proteolysis in diverse biological systems. J. Biol. Chem. 2010;285:24299–24305. doi: 10.1074/jbc.R110.112771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huber R., Carrell R.W. Implications of the three-dimensional structure of a 1- antitrypsin for structure and function of serpins. Biochemistry. 1989;28:8951–8966. doi: 10.1021/bi00449a001. [DOI] [PubMed] [Google Scholar]
- 3.Perera N.C., Schilling O., Kittel H., Back W., Kremmer E., Jenne D.E. NSP4, an elastase-related protease in human neutrophils with arginine specificity. Proc. Natl. Acad. Sci. USA. 2012;109:6229–6234. doi: 10.1073/pnas.1200470109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crystal R.G. Alpha 1-antitrypsin deficiency, emphysema, and liver disease. Genetic basis and strategies for therapy. J. Clin. Invest. 1990;85:1343–1352. doi: 10.1172/JCI114578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janoff A. Elastases and emphysema. Current assessment of the protease-antiprotease hypothesis. Am. Rev. Respir. Dis. 1985;132:417–433. doi: 10.1164/arrd.1985.132.2.417. [DOI] [PubMed] [Google Scholar]
- 6.Janus E.D., Phillips N.T., Carrell R.W. Smoking, lung function, and alpha 1-antitrypsin deficiency. Lancet. 1985;1:152–154. doi: 10.1016/s0140-6736(85)91916-6. [DOI] [PubMed] [Google Scholar]
- 7.Eriksson S., Larsson C. Purification and partial characterization of pas-positive inclusion bodies from the liver in alpha 1-antitrypsin deficiency. N. Engl. J. Med. 1975;292:176–180. doi: 10.1056/NEJM197501232920403. [DOI] [PubMed] [Google Scholar]
- 8.Jeppsson J.O., Larsson C., Eriksson S. Characterization of alpha1-antitrypsin in the inclusion bodies from the liver in alpha 1-antitrypsin deficiency. N. Engl. J. Med. 1975;293:576–579. doi: 10.1056/NEJM197509182931203. [DOI] [PubMed] [Google Scholar]
- 9.Perlmutter D.H., Silverman G.A. Hepatic fibrosis and carcinogenesis in alpha1-antitrypsin deficiency: a prototype for chronic tissue damage in gain-of-function disorders. Cold Spring Harb. Perspect. Biol. 2011;3:181–194. doi: 10.1101/cshperspect.a005801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamasaki M., Sendall T.J., Pearce M.C., Whisstock J.C., Huntington J.A. Molecular basis of alpha1-antitrypsin deficiency revealed by the structure of a domain-swapped trimer. EMBO Rep. 2011;12:1011–1017. doi: 10.1038/embor.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perlmutter D.H. The cellular response to aggregated proteins associated with human disease. J. Clin. Invest. 2002;110:1219–1220. doi: 10.1172/JCI16780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perlmutter D.H. Pathogenesis of chronic liver injury and hepatocellular carcinoma in alpha-1-antitrypsin deficiency. Pediatr. Res. 2006;60:233–238. doi: 10.1203/01.pdr.0000228350.61496.90. [DOI] [PubMed] [Google Scholar]
- 13.Perlmutter D.H. In: Molecular and Cellular Aspects of the Serpinopathies and Disorders in Serpin Activity. Silverman G.K., Lomas D.A., editors. World Scientific Publishing Co; 2007. pp. 483–508. [Google Scholar]
- 14.Perlmutter D.H. Alpha-1-antitrypsin deficiency: importance of proteasomal and autophagic degradative pathways in disposal of liver disease-associated protein aggregates. Annu. Rev. Med. 2011;62:333–345. doi: 10.1146/annurev-med-042409-151920. [DOI] [PubMed] [Google Scholar]
- 15.Perlmutter D.H. In: Molecular Pathology of Liver Diseases. Monga S.P.S., editor. New York: Springer; 2011. pp. 683–700. ISBN: 10.1007/978-1-4419-7107-4. [Google Scholar]
- 16.Rudnick D.A., Perlmutter D.H. Alpha-1-antitrypsin deficiency: a new paradigm for hepatocellular carcinoma in genetic liver disease. Hepatology. 2005;42:514–521. doi: 10.1002/hep.20815. [DOI] [PubMed] [Google Scholar]
- 17.Eriksson S., Carlson J., Velez R. Risk of cirrhosis and primary liver cancer in alpha 1-antitrypsin deficiency. N. Engl. J. Med. 1986;314:736–739. doi: 10.1056/NEJM198603203141202. [DOI] [PubMed] [Google Scholar]
- 18.Maurice N., Perlmutter D.H. Novel treatment strategies for liver disease due to alpha1-antitrypsin deficiency. Clin. Transl. Sci. 2012;5:289–294. doi: 10.1111/j.1752-8062.2011.00363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Serres F.J. Worldwide racial and ethnic distribution of alpha1-antitrypsin deficiency: summary of an analysis of published genetic epidemiologic surveys. Chest. 2002;122:1818–1829. doi: 10.1378/chest.122.5.1818. [DOI] [PubMed] [Google Scholar]
- 20.Gosai S.J., Kwak J.H., Luke C.J., Long O.S., King D.E., Kovatch K.J., Johnston P.A., Shun T.Y., Lazo J.S., Perlmutter D.H., et al. Automated high-content live animal drug screening using C. elegans expressing the aggregation prone serpin α1-antitrypsin Z. PLoS ONE. 2010;5:e15460. doi: 10.1371/journal.pone.0015460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamath R.S., Fraser A.G., Dong Y., Poulin G., Durbin R., Gotta M., Kanapin A., Le Bot N., Moreno S., Sohrmann M., et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- 22.Qu W., Ren C., Li Y., Shi J., Zhang J., Wang X., Hang X., Lu Y., Zhao D., Zhang C. Reliability analysis of the Ahringer Caenorhabditis elegans RNAi feeding library: a guide for genome-wide screens. BMC Genomics. 2011;12:170. doi: 10.1186/1471-2164-12-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nollen E.A., Garcia S.M., van Haaften G., Kim S., Chavez A., Morimoto R.I., Plasterk R.H. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. USA. 2004;101:6403–6408. doi: 10.1073/pnas.0307697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Ham T.J., Thijssen K.L., Breitling R., Hofstra R.M., Plasterk R.H., Nollen E.A. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 2008;4:e1000027. doi: 10.1371/journal.pgen.1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kraemer B.C., Burgess J.K., Chen J.H., Thomas J.H., Schellenberg G.D. Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum. Mol. Genet. 2006;15:1483–1496. doi: 10.1093/hmg/ddl067. [DOI] [PubMed] [Google Scholar]
- 26.Wang J., Farr G.W., Hall D.H., Li F., Furtak K., Dreier L., Horwich A.L. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 2009;5:e1000350. doi: 10.1371/journal.pgen.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silva M.C., Fox S., Beam M., Thakkar H., Amaral M.D., Morimoto R.I. A genetic screening strategy identifies novel regulators of the proteostasis network. PLoS Genet. 2011;7:e1002438. doi: 10.1371/journal.pgen.1002438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kao C.Y., Los F.C., Huffman D.L., Wachi S., Kloft N., Husmann M., Karabrahimi V., Schwartz J.L., Bellier A., Ha C., et al. Global functional analyses of cellular responses to pore-forming toxins. PLoS Pathog. 2011;7:e1001314. doi: 10.1371/journal.ppat.1001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lejeune F., Mesrob L., Parmentier F., Bicep C., Vazquez R., Parker A., Vert J.P., Tourette C., Neri C. Large-scale functional RNAi screen in C. elegans identifies genes that regulate the dysfunction of mutant polyglutamine neurons. BMC Genomics. 2012;13:91. doi: 10.1186/1471-2164-13-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eden E., Navon R., Steinfeld I., Lipson D., Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaye D.D., Greenwald I. OrthoList: a compendium of C. elegans genes with human orthologs. PLoS ONE. 2011;6:e20085. doi: 10.1371/journal.pone.0020085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 33.Kuhn M., Szklarczyk D., Franceschini A., von Mering C., Jensen L.J., Bork P. STITCH 3: zooming in on protein-chemical interactions. Nucleic Acids Res. 2012;40:D876–D880. doi: 10.1093/nar/gkr1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nikolsky Y., Ekins S., Nikolskaya T., Bugrim A. A novel method for generation of signature networks as biomarkers from complex high throughput data. Toxicol. Lett. 2005;158:20–29. doi: 10.1016/j.toxlet.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 35.Knight Z.A. Small molecule inhibitors of the PI3-kinase family. Curr. Top. Microbiol. Immunol. 2010;347:263–278. doi: 10.1007/82_2010_44. [DOI] [PubMed] [Google Scholar]
- 36.Samokhvalov V., Scott B.A., Crowder C.M. Autophagy protects against hypoxic injury in C. elegans. Autophagy. 2008;4:1034–1041. doi: 10.4161/auto.6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kimura K.D., Tissenbaum H.A., Liu Y., Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- 38.Buchberger A., Bukau B., Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol. Cell. 2010;40:238–252. doi: 10.1016/j.molcel.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 39.Cohen E., Bieschke J., Perciavalle R.M., Kelly J.W., Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 40.Hidvegi T., Ewing M., Hale P., Dippold C., Beckett C., Kemp C., Maurice N., Mukherjee A., Goldbach C., Watkins S., et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–232. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- 41.Karve S., Werner M.E., Sukumar R., Cummings N.D., Copp J.A., Wang E.C., Li C., Sethi M., Chen R.C., Pacold M.E., et al. Revival of the abandoned therapeutic wortmannin by nanoparticle drug delivery. Proc. Natl. Acad. Sci. USA. 2012;109:8230–8325. doi: 10.1073/pnas.1120508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mello C., Fire A. DNA transformation. Methods Cell Biol. 1995;48:451–482. [PubMed] [Google Scholar]
- 43.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ashrafi K., Chang F.Y., Watts J.L., Fraser A.G., Kamath R.S., Ahringer J., Ruvkun G. Genome-wide RNAi analysis of Caenorhabditis elegans fat regulatory genes. Nature. 2003;421:268–272. doi: 10.1038/nature01279. [DOI] [PubMed] [Google Scholar]
- 45.Lehner B., Crombie C., Tischler J., Fortunato A., Fraser A.G. Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat. Genet. 2006;38:896–903. doi: 10.1038/ng1844. [DOI] [PubMed] [Google Scholar]
- 46.Birmingham A., Selfors L.M., Forster T., Wrobel D., Kennedy C.J., Shanks E., Santoyo-Lopez J., Dunican D.J., Long A., Kelleher D., et al. Statistical methods for analysis of high-throughput RNA interference screens. Nat. Methods. 2009;6:569–575. doi: 10.1038/nmeth.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hidvegi T., Schmidt B.Z., Hale P., Perlmutter D.H. Accumulation of mutant alpha1-antitrypsin Z in the endoplasmic reticulum activates caspases-4 and -12, NFkappaB, and BAP31 but not the unfolded protein response. J. Biol. Chem. 2005;280:39002–39015. doi: 10.1074/jbc.M508652200. [DOI] [PubMed] [Google Scholar]
- 48.Schmidt B.Z., Perlmutter D.H. Grp78, Grp94, and Grp170 interact with alpha1-antitrypsin mutants that are retained in the endoplasmic reticulum. Am. J. Physiol. Gastrointest. Liver Physiol. 2005;289:G444–G455. doi: 10.1152/ajpgi.00237.2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.