

Abstract
Fragile X syndrome, a common form of inherited mental retardation, is caused by loss of the fragile X mental retardation protein (FMRP). As a selective RNA-binding protein, FMRP is localized predominately in cytoplasm, where it regulates translational control. However, there is a small portion of FMRP present in the nucleus, and its function there has been elusive. Here, we show that Drosophila dFMR1 in nucleus is required for replication stress-induced H2Av phosphorylation in the DNA damage response (DDR). Replication stress could induce the expression of dFmr1 and promote the nuclear accumulation of dFMR1. We show that, upon the stimulation of replication stress, dFMR1 is associated with chromatin in a domain-specific manner, which is essential for its ability to induce the phosphorylation of H2Av. These results together reveal an unexpected nuclear role of FMRP in DDR and uncover a feed-forward mechanism by which dFmr1 and early DDR induced by replication stress reciprocally regulate each other, thereby synergistically triggering activity of the DDR signaling cascade.
INTRODUCTION
Fragile X syndrome (FXS), one of the most common forms of inherited mental retardation, is caused by loss of the fragile X mental retardation protein (FMRP) (1–3). FMRP is widely expressed in fetal and adult tissues, with the most abundant expression in brain and testis (4). FMRP, along with its autosomal paralogs the fragile X-related proteins, FXR1P and FXR2P, make up a small family of RNA-binding proteins (RBPs; the fragile X-related gene family) (5–7). These proteins share >60% amino acid identity and contain two types of RNA-binding motif: two ribonucleoprotein K homology domains (KH domains) and a cluster of arginine and glycine residues (the RGG box) (8,9). The fragile X-related gene family is well conserved throughout evolution; there are orthologs of FMR1, FXR1 and FXR2 in mouse, chicken and Xenopus; Drosophila, however, contains just a single fragile X-related gene, dFmr1 (10).
FMRP associates with polyribosomes in an RNA-dependent manner through messenger ribonucleoprotein (mRNP) particles, and it can suppress translation both in vitro and in vivo (11–14). Extensive studies have shown that FMRP can regulate synaptic plasticity by regulating the synthesis of a set of plasticity-related proteins posttranscriptionally (15). Although FMRP is predominantly localized in the cytoplasm, both functional nuclear localization signal (NLS) and nuclear export signal (NES) are also found within FMRP (16,17). Whereas NLS is located in the N-terminus of FMRP, the NES of FMRP closely resembles the NES motifs described for HIV1 Rev and protein kinase inhibitor and is sufficient to direct the nuclear export of a microinjected protein conjugate. Very recently FMRP was also identified as a chromatin-binding protein that functions in the DNA damage response, suggesting that nuclear FMRP could regulate genomic stability at the chromatin interface and may impact gametogenesis and some developmental aspects of fragile X syndrome (18).
Drosophila has proved to be an excellent model for the dissection of FMRP-regulated biological pathways (19). Consistent with FMRP function in mammals, dFMR1 regulates the translation of its mRNA targets, including futsch (Drosophila ortholog of MAP1B), chickadee and Rac1 (20–22). dFMR1-deficient flies also display abnormalities in neural architecture, courtship behavior, synaptogenesis and spermatogenesis (21,23–25). In particular, both the neural architecture of the mushroom bodies in fly brain and disturbances in male courtship behavior have been considered significant phenotypic readouts that may parallel defects in learning capacity and social behavior in humans with FXS. Besides neuronal functions, dFMR1 is also required for maintenance of germline stem cells in ovary (26, 27).
DNA lesions are continuously generated in living cells as a result of replication errors and oxidative metabolism (28). They also arise as a consequence of exposure to environmental agents (e.g. ultraviolet, ionizing radiation), radiation therapy and chemotherapeutic drugs (29). It is therefore crucial for the cell to detect DNA damage, signal its presence, and effect DNA repair, cell cycle arrest, and ultimately cell fate decisions, which together are called the DNA damage response (DDR) (29). Intriguingly, recent large-scale genetic and molecular analyses have identified RBPs as major players in the prevention of genome instability (29). The proposition is that upon DNA damage, RBPs coordinately regulate various aspects of both RNA and DNA metabolism.
Here, we show that Drosophila dFMR1 is required for chemical mutagen-induced H2Av phosphorylation in Drosophila germline, which is one of the earliest responses to either double-strand break (DSB) formation or replication stress. We find that dFMR1 specifically participates in the replication stress-induced DDR. Replication stress could induce the expression of dFmr1 and promote the nuclear accumulation of dFMR1. We show that dFMR1 is associated with chromatin in a domain-specific manner. dFMR1 association with chromatin requires both the agenet and KH1 domains, which are essential for dFMR1’s ability to induce the phosphorylation of H2Av. These results together reveal an unexpected nuclear role of FMRP in DDR and uncover a feed-forward mechanism by which dFmr1 and early DDR induced by replication stress reciprocally regulate each other, thereby synergistically triggering activity of the DDR signaling cascade.
RESULTS
dFMR1 is required for chemical mutagen-induced H2Av phosphorylation in Drosophila germline
To determine whether dFMR1 has a role in the regulation of DDR, we employed the Drosophila early germline as an in vivo DDR model and performed immunostaining assays to examine one of the earliest responses to DSB formation, the expression pattern of the phosphorylated form of the Drosophila histone H2AX variant (γ-H2Av), which induces γ-H2Av to accumulate at DNA break sites (30,31). Immunostaining analyses on wild-type ovary indicated a mild signal of γ-H2Av foci that are restricted to of the germaria, where meiotic DSBs were formed. We found no significant difference in pattern between wild-type and dfmr1 mutant germaria, although the intensity of the γ-H2Av foci was found to be relatively lower in a portion of dfmr1 mutant germaria, suggesting that dFMR1 does not play a significant role in the meiotic DSB-induced phosphorylation of H2Av (Fig. 1A and B).
Figure 1.
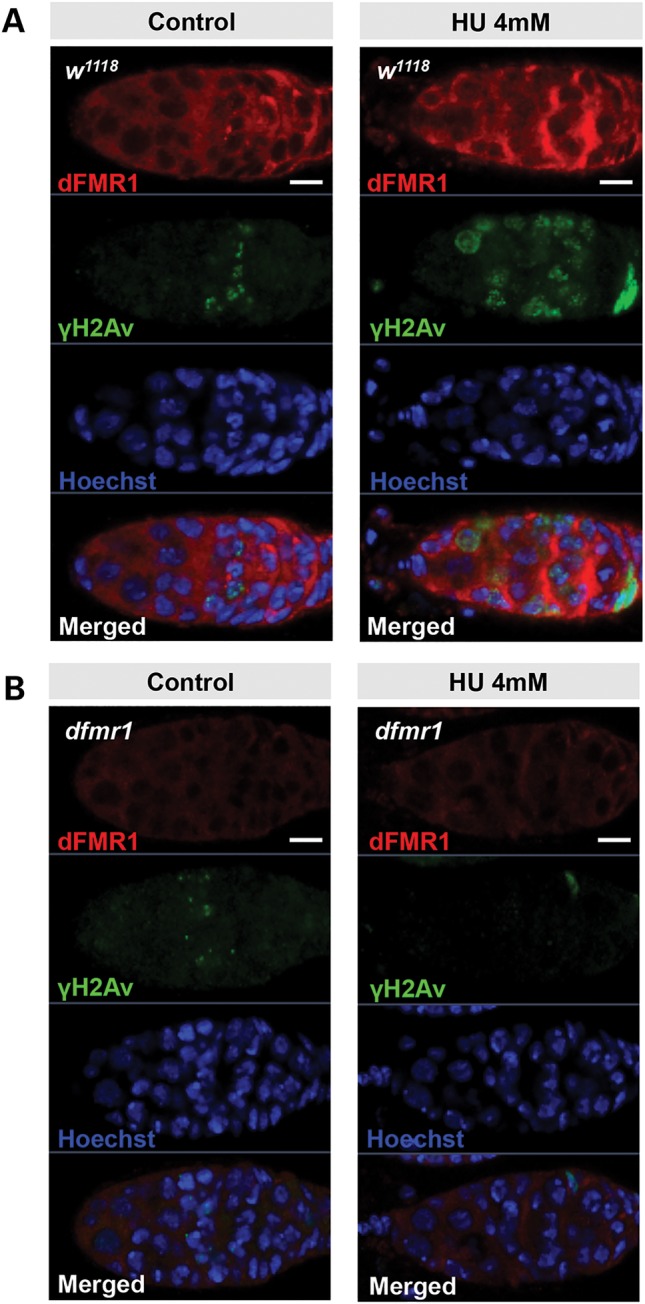
dFmr1 is required for H2Av phosphorylation generated by replication stress but not double-strand breaks in germline. Three- to 4-day-old w1118 (A) or dfmr1 (B) ovaries treated with or without 4 mm HU for 3 h were stained with anti-dFMR1 (red), anti-γ-H2Av (green) and Hoechst (blue).
Although we know that both ataxia telangiectasia-mutated (ATM) and ataxia telangiectasia-related (ATR) kinases phosphorylate H2AV (γ-H2Av), ATM and ATR in Drosophila have distinct roles in the regulation of the DDR (32,33). ATM plays specific roles in the repair of DSBs, whereas ATR is primarily required for checkpoint activity in DDR (32). To further test whether dFmr1 has role(s) in replication stress-induced DDR, we pretreated wild-type and dfmr1 mutant ovaries with hydroxyurea (HU), a chemical DNA damage inducer for replication stress, and then performed immunostaining assays using the specific anti-γ-H2Av antibody. As shown in Figure 1, in wild-type ovaries, the signal of γ-H2Av foci significantly increases in germaria and persists in intensity as cysts mature in early egg chambers in ovaries compared with the wild-type control without HU pretreatment. In contrast, the γ-H2Av foci signal was apparently reduced or undetectable in dfmr1 mutant ovaries pretreated with HU. This points to a potential role for dFMR1 in regulating the DDR induced by the chemical DDR inducer.
dFMR1 specifically participates in replication stress-induced DDR
To further investigate the general role of dFMR1 in DDR, we sought to employ the RNAi strategy to knockdown dFmr1 in Drosophila S2 cells and performed cell-based assays using γ-H2Av as a reporter. To this end, we designed two dFmr1 dsRNAs that target to the ORF and 3′ UTR regions, respectively, of the dFmr1 gene. As shown in a western blot assay, these dsRNAs efficiently knocked down dFmr1 expression in S2 cultured cells (Fig. 2A). We then examined the γ-H2Av protein abundance in wild-type S2 cells and dFmr1-knockdown S2 cells that were treated with or without X-ray radiation, which generates DSB, or HU, which chemically produces replication stress. As shown in Figure 2, there was no significant difference in the level of γ-H2Av expression between wild-type and dFmr1-knockdown S2 cells when treated with X-ray radiation (Fig. 2B–D). In contrast, we found that the level of γ-H2Av was dramatically reduced in dFmr1-knockdown S2 cells compared with control when treated with HU (Fig. 2A, C and D). These results suggest that dFMR1 may be involved in replication stress-induced DDR. Similar results were obtained when UV radiation, another replication stress inducer, was used to treat S2 cells (Fig. 2A, C and D). To further confirm the specific role of dFmr1 in replication stress-induced DDR, we performed rescue experiments. As shown in Figure 2E, exogenous expression of dFmr1 could restore the reduction of the γ-H2Av protein caused by a dFmr1 dsRNA targeting to the 3′ UTR of dFmr1 gene, when the cells were stimulated with replication stressors.
Figure 2.

dFmr1 is required for H2Av phosphorylation generated by chemical mutagen or UV irradiation but not double-strand breaks in S2 cells. (A and B) S2 cells were treated with indicated dsRNAs for 72 h and then with 2 mm HU for 12 h, or irradiated with a dose of 50 J/m2 UV (A) or 6 Gy X-ray (B). Western blots were performed to detect the expressions of dFMR1, α-tubulin and γ-H2Av. S2 cells treated without (C) or with (D) dFmr1 dsRNA were incubated with 2 mm HU for 12 h or irradiated with a dose of 50 J/m2 UV or 6 Gy X-ray. Immunostaining was performed to show the expressions of γ-H2Av (green) and dFMR1 (red). (E) S2 cells were incubated with dsRNA targeting dFmr1 3′ UTR for 72 h, followed by transfection with indicated vector, and then irradiated with UV. Western blots were performed to show the level of dFmr1, α-tubulin and γ-H2Av.
Replication stress promotes dFmr1 expression at both mRNA and protein levels
Because we noted that overexpression of dFmr1 does not significantly affect γ-H2Av levels upon UV or HU treatment (Fig. 3A), we reasoned that the levels or activity of endogenous dFmr1 are near saturation status in the regulation of γ-H2Av. To test this idea, we next examined the dynamics of endogenous dFmr1 expression upon replication stress treatments in S2 cells by performing both quantitative RT-PCR (qRT-PCR) and western blot analyses. As shown in qRT-PCR assay, the dFmr1 mRNA level in S2 cells increased and reached a peak at the 3-h time point after UV treatment (Fig. 3B). Similarly, western blot analysis revealed that the levels of dFmr1 protein were progressively elevated upon stimulation by UV, reaching a peak at a time point similar to post-HU-treatment (Fig. 3C). Results were consistent with cells treated with HU since the levels of dFmr1 protein also increased in response to HU treatment (Fig. 3D). These results indicate that dFmr1 expression itself is positively regulated by replication stressors. Thus, our findings reveal a feed-forward mechanism involving dFmr1 to control the replication stress-mediated DDR.
Figure 3.

Replication stress promotes the expression of dFmr1. (A) S2 cells were transfected with the DNA vector to overexpress dFmr1, and then treated with HU or UV. Western blots were performed to show the level of dFmr1, α-tubulin and γ-H2Av. (B) The mRNA levels of dFmr1 in S2 cells at 5 min, 1, 2 and 3 h after UV irradiation were determined by qRT-PCR. (C) Western blots were performed to detect the protein levels of dFMR1 in S2 cells at 5 min, 1 h, 2 h and 3 h after UV irradiation. (D) Western blots were performed to detect the protein levels of dFmr1 in w1118 ovaries after 4-h treatment with or without HU.
Replication stress promotes dFMR1 nuclear accumulation
The feed-forward mechanism further emphasizes the important role of dFmr1 in replication stress-induced DDR. To understand how dFmr1 and replication stress-induced DDR signaling reciprocally regulate each other, we next sought to investigate the dynamics of the subcellular localization of dFMR1 in DDR induced by HU or UV radiation treatment. FMRP in both Drosophila and mammals is known to be localized predominately in the cytoplasmic region as an RBP to regulate translational control; however, a very small portion (∼4%) of FMRP is reported to be present in the nucleus, where its function has remained largely unknown. We therefore carried out biochemical assays to separate nucleus and cytosol from S2 cell lysates, and then performed western blot analyses. As shown in Figure 4A, both nuclear and cytosolic fractions could be clearly separated, as indicated by the presence of GAPDH, a marker for cytosol, and Otefin, a marker for nucleus, respectively. We noted that a small portion of dFMR1 was apparent in the nuclear fraction (Fig. 4A), suggesting that, like its counterpart FMRP in mammals, dFMR1 is also present in the nucleus. We then treated S2 cells with HU or UV radiation and found that the abundance of dFMR1 in nucleus increased significantly upon HU or UV treatment (Fig. 4A), suggesting that replication stress could cause dFMR1 to accumulate in the nucleus.
Figure 4.
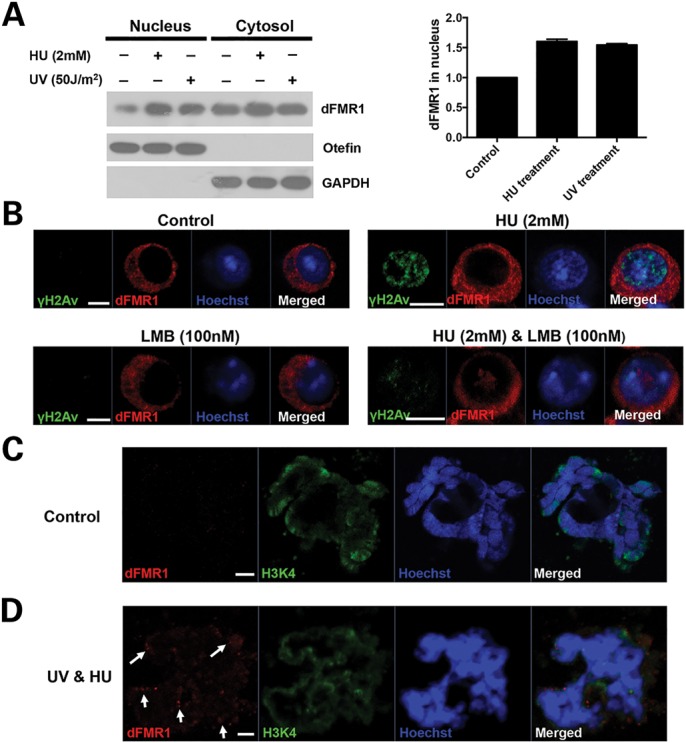
Replication stress promotes the nuclear accumulation of dFMR1 and its association with chromatin. (A) S2 cells were treated with HU or irradiated with UV, and then nucleus and cytosols were fractionated. Western blot was performed to show the levels of dFmr1 (left panel). GAPDH and Otefin were used as markers of cytosolic and nuclear fraction, respectively. Right panel shows the quantitative intensity of bands boxed in the left panel. (B) S2 cells without treatment, or treated with HU alone, or with LMB alone, or with both HU and LMB were stained by anti-dFMR1 (red), anti-γ-H2Av (green) antibodies and Hoechst (blue). Polytene chromosomes from third-instar larvae without treatment (C) or irradiated with 50 J/m2 UV, and then treated with 4 mm HU (D) were stained for dFMR1 (red) and H3K4me1 (green) and Hoechst (blue).
To further confirm these observations, we next performed immunostaining assays. As shown in Figure 4B, dFMR1 could not be detected in the nucleus of S2 cells, even when treated with leptomycin B (LMB), an inhibitor of nuclear protein export (16). In contrast, we found that in the presence of LMB, dFMR1 forms dot-structure aggregates that could be detected easily in the nucleus of the HU-treated S2 cells. Together, our results demonstrate that replication stress could promote the nuclear accumulation of dFMR1.
dFMR1 associates with chromatin in a domain-specific manner upon the stimulation of replication stress
Mounting evidence suggests that proteins involved in DDR are often recruited and associate with chromatin upon the stimulation of DNA damage stress (32). To determine whether dFMR1 could be loaded onto chromosome and associate with chromatin in response to replication stress, we employed the Drosophila larval salivary gland system and performed immunostaining assays using anti-H3K4 and anti-dFMR1 antibodies to analyze the potential localization of dFMR1 on polytene chromosomes. As shown in Figure 4C, in the absence of replication stress, there was no detectable dFMR1 colocalized with H3K4 staining on the polytene chromosomes isolated from the third-star larva. Interestingly, dFMR1 could be reproducibly detected to associate with H3K4 staining on polytene chromosomes when the UV radiation-stimulated third-star larvae were further treated with 2 mm HU on the medium for 2 h (Fig. 4D). These findings suggest that replication stress could promote dFMR1 loading onto chromosomes. To confirm this, we performed chromatin fractionation assays to determine whether dFMR1 associates directly with chromatin. We noted that dFMR1 was significantly enriched in the chromatin fraction upon treatment with HU or UV radiation (shown in Fig. 5D), indicating replication stress promotes dFMR1 association with chromatin.
Figure 5.

Both the agenet and KH domains are important for dFMR1 to associate with chromatin. (A) Schematic drawings of dFmr1 and its deletion mutants. (B and C) S2 cells expressing Flag-dFmr1 ΔKH1 (B) or Flag-dFmr1 ΔAgenet (C) without treatment (left) or treated with both HU and LMB (right) were stained with anti-Flag (green), anti-dFMR1 (red) antibodies and Hoechst (blue). (D) S2 cells expressing the indicated proteins were irradiated with or without UV, and the chromosome fractions were separated. Western blots with anti-FLAG, anti-GAPDH, anti-H2B and anti-α-tubulin antibodies were performed to show the levels of indicated proteins.
To better understand the molecular basis of the dFMR1-chromatin association, we generated a series of mutant forms of dFMR1, including dFMR1ΔAgenet and dFMR1ΔKH1, based on the different domains of dFMR1 (Fig. 5A). Using an immunostaining assay, we found that full-length dFMR1 could be easily detected in the nucleus of the HU-treated cells, whereas the deletion of the first KH domain (KH1), dFMR1ΔKH1, significantly reduced its presence in nucleus (Fig. 5B). Furthermore, we could detect no dFMR1ΔAgenet signal in the nucleus in the treated cells (Fig. 5C). These findings indicate that the agenet domain is essential for dFMR1 nuclear localization, whereas KH domains are required for efficient dFMR1 nuclear localization.
To further confirm these observations, we performed a chromatin fractionation experiment and found that the lack of the agenet domain could completely abolish the association of dFMR1 with chromatin, and the removal of two KH domains dramatically reduced its association with chromatin (Fig. 5D). Taken together, our results suggest dFMR1 associates with chromatin in a domain-specific manner in response to replication stress.
Both the agenet and KH domains are important for dFMR1-mediated regulation of H2Av phosphorylation in DDR
The action of dFMR1 in regulating DDR could either be direct, via its nuclear localization and binding to chromatin, or indirect, via its localization in cytoplasm. To determine this, we performed rescue experiments using wild-type dFMR1, dFMR1ΔAgenet and dFMR1ΔKH1. We expressed these dFMR1 variants in dFmr1 knockdown S2 cells that were treated by HU or UV radiation. As shown in Figure 6, while wild-type dFMR1 could significantly restore the induction of H2Av phosphorylation in the dfmr1 knockdown S2 cells treated with HU or UV radiation, the mutant forms of dFMR1ΔAgenet and dFMR1ΔKH could not, suggesting that both the agenet and KH domains, which are required for dFMR1 binding to chromatin, are important for dFMR1 activity in promoting the phosphorylation of H2Av.
Figure 6.

Both the agenet and KH domains are important for dFMR1 to promote phosphorylation of H2Av. S2 cells were incubated with dsRNA targeting dFmr1 3′ UTR for 72 h, followed by transfection with indicated vectors and then irradiated with UV. Western blots were performed to show the level of dFMR1, α-tubulin and γ-H2Av.
DISCUSSION
FXS, the most common cause of inherited mental retardation, results from the loss of functional FMRP (5). Since the FMR1 gene was first cloned in 1991, most studies have focused on understanding the role of FMRP as an RBP in posttranscriptional regulation. FMRP plays important role(s) in synaptic plasticity via the regulation of mRNA transport and translation, particularly local protein synthesis in the dendrites (15). Although a small portion of FMRP is known to be present in nucleus, its nuclear function has remained elusive. Here, we show that Drosophila dFMR1 in nucleus is required for replication stress-induced H2Av phosphorylation in the DDR. Replication stress could induce the expression of dFmr1 and promote the nuclear accumulation of dFMR1. We show that, upon the stimulation of replication stress, dFMR1 is associated with chromatin in a domain-specific manner, which is essential for its ability to induce the phosphorylation of H2Av. These results together reveal an unexpected nuclear role of FMRP in DDR.
Previous research demonstrated the presence of FMRP in the nucleus, and the suggestion is that nuclear FMRP could be involved in the initial assembly of the mRNP complex, despite the lack of experimental data supporting this notion (16,17). Indeed, whether the presence of FMRP in the nucleus plays any physiological role at all has not been clear. Our results here demonstrate that dFMR1 does have a functional role(s) in the nucleus and is involved in the replication stress-induced DDR by stimulating H2Av phosphorylation. More intriguing still, dFMR1 carries out this function through its association with chromatin. Our deletion analyses suggest that the nuclear function of dFMR1 requires its N-terminal agenet domain, which is distinct from the well-established role of FMRP in translational control. The agenet domain is a previously identified double-tudor domain that belongs to the Royal family of chromatin-binding proteins (34,35). The agenet domain is also important for the dFMR1-mediated regulation of H2Av phosphorylation in DDR. Our results are consistent with the recent observations on the role of mammalian FMRP in DDR (18). It would be important for future research to determine whether specific motif(s) or chromatin signature(s) are associated with the genomic regions bound by dFMR1.
Our in vivo and in vitro studies point to a potential role for FMRP in DDR. It is well known that DNA lesions are continuously generated in living cells as a result of replication errors and oxidative metabolism (28). The accumulation of DNA insults is associated with multiple diseases, from neurodegenerative disorders to cancers. It is therefore crucial for the cell to detect DNA damage, signal its presence, and effect DNA repair, cell cycle arrest and ultimately cell fate decisions, which are together called the DDR. Previous work identified a novel protein that interacts with FMRP, nuclear FMRP-interacting protein (NUFIP), which was found to be colocalized with nuclear isoforms of FMRP in a dot-like pattern (36). Furthermore, NUFIP was found to interact with BRCA1, a major DDR protein, to activate transcription by RNA polymerase II (37). It would be interesting to take this further and examine whether NUFIP is involved in the FMRP-mediated modulation of DDR.
There has been some evidence linking FMRP to cancer. It has been reported that FMRP is overexpressed in hepatocellular carcinoma cells (38,39). Moreover, patients with FXS have a decreased risk of cancer, and one FXS patient showed an unusual decrease in brain tumor invasiveness (40,41). More recent work has revealed that FMRP levels correlate with prognostic indicators of aggressive breast cancer, probability of lung metastases and triple-negative breast cancer (42). FMRP overexpression in murine breast primary tumors is also found to enhance lung metastasis, while its reduction has the opposite effect on cell spreading and invasion (42). It is therefore likely that FMRP-mediated modulation of the activity of the DDR signaling cascade could contribute to the role of FMRP in tumorigenesis.
Since the major phenotype associated with the loss of FMRP is intellectual disability, further investigation of whether the role FMRP plays in DDR could contribute to the molecular pathogenesis of fragile X syndrome is critical. Our earlier work demonstrated that FMRP regulates the proliferation and differentiation of adult neural stem cells (adult neurogenesis) (43). The loss of FMRP leads to increased proliferation and decreased neuronal differentiation, but increased glial differentiation. Altered adult neurogenesis is a major contributor to the impaired learning and memory caused by the loss of FMRP (44). Meanwhile, components of the DDR are known to safeguard replicative fidelity and control neuronal differentiation in neural progenitor cells (45). It would be interesting to test the possibility that an altered DDR response contributes to the increased proliferation of adult neural stem cells caused by the loss of FMRP.
In summary, here we demonstrate an unexpected nuclear role of FMRP in DDR and uncover a feed-forward mechanism by which dFmr1 and early DDR induced by replication stress reciprocally regulate each other, thereby synergistically triggering the activity of the DDR signaling cascade. Determining whether this nuclear function of FMRP could affect neurodevelopment and whether the loss of FMRP has any DDR consequences in patients with fragile X syndrome will be critical areas for future research.
MATERIALS AND METHODS
Drosophila stocks
All fly stocks were maintained under standard culture conditions. The w1118 was used as the wild-type strain. dfmr1delta50 and dfmr1delta113 were described previously (21,25).
Immunohistochemistry and microscopy
Immunohistochemistry and microscopy of ovaries were prepared for reaction with antibodies as described previously (27). For S2 cell immunofluorescence, cells were transferred onto a poly-l-lysine-treated coverslip 12 h after transfection. Twenty-four hours later, the cells were fixed with 4% formaldehyde in PBS containing 0.1% Tween 20 for 20 min, and then blocked with PBS containing 1.5% BSA and 0.3% Tween-20 for 1 h at room temperature. Cells were then incubated with primary antibody and secondary antibody sequentially.
For immunostaining of polytene chromosomes, salivary glands from third-instar larvae were dissected in PBS and incubated in fixing solution (45% glacial acetic acid, 1.85% formaldehyde) for 10 min on a coverslip. We then took up the coverslip with a poly-l-lysine-treated slide and tapped the coverslip to break the cells and spread the chromosomes. After freezing in liquid nitrogen, the slides were washed with PBS twice (10 min each time), and then with PBS containing 1% Triton X-100 for 10 min. After incubating in blocking solution (PBS containing 5% non-fat dry milk) for 1 h at room temperature, the slides were then incubated with primary antibody and secondary antibody sequentially.
The following antibodies were used in this study: rabbit anti-FLAG (1:2000, Sigma), rabbit anti-γ-H2Av (1:2000, Cell Signaling Technology), mouse anti-dFMR1 (1:2000, Sigma) and rabbit anti-H3K4me1 (1:1000, Abcam). Secondary antibodies used were goat anti-mouse Alexa 555, goat anti-rabbit Alexa 488 (Molecular Probes), all at 1:2000. All samples were examined by Zeiss Microscope, and images were captured using the Zeiss LSM 710 META laser scanning confocal system.
Cell culture, transfection and RNAi knockdown
S2 cells were cultured in Schneider's Drosophila medium (Sigma) at 27°C. Transfection was performed using the calcium phosphate transfection method as described previously (26). All dsRNAs were synthesized according to the standard protocol. For RNAi knockdown, S2 cells were first incubated in serum-free medium containing dsRNAs for 30 min, and then FBS was added to a 10% final concentration and incubated for a further 72 h.
UV, X-ray irradiation and HU treatment
S2 cells were irradiated with a dose of 50 J/m2 UV or 6 Gy X-ray and harvested after 2 h (UV) or 1 h (X-ray) to perform immunohistochemistry or western blot. S2 cells were treated with HU at a 2 mm final concentration and incubated for 12 h and then harvested. Ovaries were dissected in PBS and incubated in S2 cell culture medium with 4 mm HU for 3 h.
Western blot
Western blots were performed as described previously (26). The following antibodies were used: mouse anti-dFMR1 antibody (Sigma, 1:2000), rabbit anti-γ-H2Av antibody (Cell Signaling Technology, 1:1000), rabbit anti-Otefin (46), mouse anti-α-tubulin antibody (Sungene Biotech, 1:5000), rabbit anti-GAPDH (Sungene Biotech, 1:2000) and rabbit anti-H2B antibody (Santa Cruz, 1:500). The quantitation of band intensity in Figure 2C was measured using ImageJ software.
Quantitative RT-PCR
S2 cells were irradiated without or with a dose of 50 J/m2 UV and harvested after 5 min, 1 h, 2 h and 3 h to extract total RNA with Trizol (Invitrogen). cDNAs were synthesized according to the standard protocol. Real-time PCR was performed via standard methods using SYBR, and each sample was repeated in triplicate. The relative enrichment was calculated by normalizing the quantity of Rp49. Primers used in this study are shown below.
Nucleus and chromosome fraction
Harvested S2 cells were washed twice with 1× PBS and then resuspended with three volumes of hypotonic buffer [1.5 mm MgCl2, 10 mm KCl, 10 mm HEPES (pH 7.5), protease inhibitor cocktail (Roche)] and incubated for 20 min on ice. The resuspended cells were then transferred to the 1-ml daunce and homogenized with tight stroke 100 times. The homogenates were then centrifuged at 1000 g at 4°C. The supernatants were collected as the cytosolic fractions, and the pellets were washed twice with hypotonic buffer and collected as nuclear fractions. Nuclei were collected as described above, and then lysed in the buffer [0.2 mm EGTA, 3 mm EDTA and protease inhibitor cocktail (Roche)] for 20 min. Chromatins were collected by centrifugation at 1800 g at 4°C for 10 min.
Primer sequences
Primers for qPCR
dFmr1 rt1—forward: TGAACAGGATCAGAACATACCA
dFmr1 rt1—reverse: TTCTACTTCCTTCAGGTGCG
dFmr1 rt2—forward: GTGTTTCGAATCAAGATCGCT
dFmr1 rt2—reverse: TGTTCTACTTCCTTCAGGTGC
rp49—forward: AAGATGACCATCCGCCCAGCATAC
rp49—reverse: ACGCACTCTGTTGTCGATACCCTTG
Primers for synthesis of dsRNA
ds dFmr1—forward: GAGAAGATGGAGATTGATCAGC
ds dFmr1—reverse: GATAGTCCCTGCCATTCTGC
Primers for synthesis of dfmr1 3′ UTR
dFmr1 3′ UTR-s: GGATCCTAATACGACTCACTATAGGgccgagtggccaaagcgg
dFmr1 3′ UTR-as: GGATCCTAATACGACTCACTATAGGgttattgaagcttttttagtttca
Conflict of Interest statement. None decalred.
FUNDING
This study was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA01010306), National Basic Research Program of China (2013CB945002), Natural Science Foundation of China (91019022 and 31130036), National Institutes of Health (NS079625 to P.J.) and March of Dimes (FY13-354 to P.J.).
REFERENCES
- 1.Oberle I., Rousseau F., Heitz D., Kretz C., Devys D., Hanauer A., Boue J., Bertheas M.F., Mandel J.L. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–1102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- 2.Verkerk A.J., Pieretti M., Sutcliffe J.S., Fu Y.H., Kuhl D.P., Pizzuti A., Reiner O., Richards S., Victoria M.F., Zhang F.P. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 3.Kremer E.J., Pritchard M., Lynch M., Yu S., Holman K., Baker E., Warren S.T., Schlessinger D., Sutherland G.R., Richards R.I. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science. 1991;252:1711–1714. doi: 10.1126/science.1675488. [DOI] [PubMed] [Google Scholar]
- 4.Hinds H.L., Ashley C.T., Sutcliffe J.S., Nelson D.L., Warren S.T., Housman D.E., Schalling M. Tissue specific expression of FMR-1 provides evidence for a functional role in fragile X syndrome. Nat. Genet. 1993;3:36–43. doi: 10.1038/ng0193-36. [DOI] [PubMed] [Google Scholar]
- 5.Warren S.T., Sherman S.L. In: The Metabolic & Molecular Bases of Inherited Disease. Syndrome F.X., Scriver C.R., Beaudet A.L., Valle D., Childs B., Kinzler K.W., Vogelstein B., editors. Vol. I. New York: McGraw-Hill Companies; 2001. pp. 1257–1290. [Google Scholar]
- 6.Siomi M.C., Siomi H., Sauer W.H., Srinivasan S., Nussbaum R.L., Dreyfuss G. FXR1, an autosomal homolog of the fragile X mental retardation gene. EMBO J. 1995;14:2401–2408. doi: 10.1002/j.1460-2075.1995.tb07237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y., O'Connor J.P., Siomi M.C., Srinivasan S., Dutra A., Nussbaum R.L., Dreyfuss G. The fragile X mental retardation syndrome protein interacts with novel homologs FXR1 and FXR2. EMBO J. 1995;14:5358–5366. doi: 10.1002/j.1460-2075.1995.tb00220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashley C.T., Jr., Wilkinson K.D., Reines D., Warren S.T. FMR1 protein: conserved RNP family domains and selective RNA binding. Science. 1993;262:563–566. doi: 10.1126/science.7692601. [DOI] [PubMed] [Google Scholar]
- 9.Siomi H., Siomi M.C., Nussbaum R.L., Dreyfuss G. The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell. 1993;74:291–298. doi: 10.1016/0092-8674(93)90420-u. [DOI] [PubMed] [Google Scholar]
- 10.Jin P., Alisch R.S., Warren S.T. RNA and microRNAs in fragile X mental retardation. Nat. Cell Biol. 2004;6:1048–1053. doi: 10.1038/ncb1104-1048. [DOI] [PubMed] [Google Scholar]
- 11.Feng Y., Absher D., Eberhart D.E., Brown V., Malter H.E., Warren S.T. FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol. Cell. 1997;1:109–118. doi: 10.1016/s1097-2765(00)80012-x. [DOI] [PubMed] [Google Scholar]
- 12.Todd P.K., Mack K.J., Malter J.S. The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc. Natl. Acad. Sci. USA. 2003;100:14374–14378. doi: 10.1073/pnas.2336265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown V., Jin P., Ceman S., Darnell J.C., O'Donnell W.T., Tenenbaum S.A., Jin X., Feng Y., Wilkinson K.D., Keene J.D. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- 14.Darnell J.C., Jensen K.B., Jin P., Brown V., Warren S.T., Darnell R.B. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell. 2001;107:489–499. doi: 10.1016/s0092-8674(01)00566-9. [DOI] [PubMed] [Google Scholar]
- 15.Bassell G.J., Warren S.T. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eberhart D.E., Malter H.E., Feng Y., Warren S.T. The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum. Mol. Genet. 1996;5:1083–1091. doi: 10.1093/hmg/5.8.1083. [DOI] [PubMed] [Google Scholar]
- 17.Feng Y., Gutekunst C.A., Eberhart D.E., Yi H., Warren S.T., Hersch S.M. Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J. Neurosci. 1997;17:1539–1547. doi: 10.1523/JNEUROSCI.17-05-01539.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alpatov R., Lesch B.J., Nakamoto-Kinoshita M., Blanco A., Chen S., Stutzer A., Armache K.J., Simon M.D., Xu C., Ali M., et al. A Chromatin-Dependent Role of the Fragile X Mental Retardation Protein FMRP in the DNA Damage Response. Cell. 2014;157:869–881. doi: 10.1016/j.cell.2014.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao F.B. Understanding fragile X syndrome: insights from retarded flies. Neuron. 2002;34:859–862. doi: 10.1016/s0896-6273(02)00740-7. [DOI] [PubMed] [Google Scholar]
- 20.Lee A., Li W., Xu K., Bogert B.A., Su K., Gao F.B. Control of dendritic development by the Drosophila fragile X-related gene involves the small GTPase Rac1. Development. 2003;130:5543–5552. doi: 10.1242/dev.00792. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y.Q., Bailey A.M., Matthies H.J., Renden R.B., Smith M.A., Speese S.D., Rubin G.M., Broadie K. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107:591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]
- 22.Tessier C.R., Broadie K. Drosophila fragile X mental retardation protein developmentally regulates activity-dependent axon pruning. Development. 2008;135:1547–1557. doi: 10.1242/dev.015867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wan L., Dockendorff T.C., Jongens T.A., Dreyfuss G. Characterization of dFMR1, a drosophila melanogaster homolog of the fragile X mental retardation protein [In Process Citation] Mol. Cell Biol. 2000;20:8536–8547. doi: 10.1128/mcb.20.22.8536-8547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morales J., Hiesinger P.R., Schroeder A.J., Kume K., Verstreken P., Jackson F.R., Nelson D.L., Hassan B.A. Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain. Neuron. 2002;34:961–972. doi: 10.1016/s0896-6273(02)00731-6. [DOI] [PubMed] [Google Scholar]
- 25.Dockendorff T.C., Su H.S., McBride S.M., Yang Z., Choi C.H., Siwicki K.K., Sehgal A., Jongens T.A. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron. 2002;34:973–984. doi: 10.1016/s0896-6273(02)00724-9. [DOI] [PubMed] [Google Scholar]
- 26.Yang L., Duan R., Chen D., Wang J., Jin P. Fragile X mental retardation protein modulates the fate of germline stem cells in Drosophila. Hum. Mol. Genet. 2007;16:1814–1820. doi: 10.1093/hmg/ddm129. [DOI] [PubMed] [Google Scholar]
- 27.Yang Y., Xu S., Xia L., Wang J., Wen S., Jin P., Chen D. The bantam microRNA is associated with drosophila fragile X mental retardation protein and regulates the fate of germline stem cells. PLoS Genet. 2009;5:e1000444. doi: 10.1371/journal.pgen.1000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aguilera A., Garcia-Muse T. Causes of genome instability. Annu. Rev. Genet. 2013;47:1–32. doi: 10.1146/annurev-genet-111212-133232. [DOI] [PubMed] [Google Scholar]
- 29.Dutertre M., Lambert S., Carreira A., Amor-Gueret M., Vagner S. DNA damage: RNA-binding proteins protect from near and far. Trends Biochem. Sci. 2014;39:141–149. doi: 10.1016/j.tibs.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 30.Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 31.Zhou B.B., Elledge S.J. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 32.Ciccia A., Elledge S.J. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Joyce E.F., Pedersen M., Tiong S., White-Brown S.K., Paul A., Campbell S.D., McKim K.S. Drosophila ATM and ATR have distinct activities in the regulation of meiotic DNA damage and repair. J. Cell Biol. 2011;195:359–367. doi: 10.1083/jcb.201104121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maurer-Stroh S., Dickens N.J., Hughes-Davies L., Kouzarides T., Eisenhaber F., Ponting C.P. The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem. Sci. 2003;28:69–74. doi: 10.1016/S0968-0004(03)00004-5. [DOI] [PubMed] [Google Scholar]
- 35.Ramos A., Hollingworth D., Adinolfi S., Castets M., Kelly G., Frenkiel T.A., Bardoni B., Pastore A. The structure of the N-terminal domain of the fragile X mental retardation protein: a platform for protein-protein interaction. Structure. 2006;14:21–31. doi: 10.1016/j.str.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 36.Bardoni B., Schenck A., Mandel J.L. A novel RNA-binding nuclear protein that interacts with the fragile X mental retardation (FMR1) protein. Hum. Mol. Genet. 1999;8:2557–2566. doi: 10.1093/hmg/8.13.2557. [DOI] [PubMed] [Google Scholar]
- 37.Cabart P., Chew H.K., Murphy S. BRCA1 cooperates with NUFIP and P-TEFb to activate transcription by RNA polymerase II. Oncogene. 2004;23:5316–5329. doi: 10.1038/sj.onc.1207684. [DOI] [PubMed] [Google Scholar]
- 38.Li Y., Tang Y., Ye L., Liu B., Liu K., Chen J., Xue Q. Establishment of a hepatocellular carcinoma cell line with unique metastatic characteristics through in vivo selection and screening for metastasis-related genes through cDNA microarray. J. Cancer Res. Clin. Oncol. 2003;129:43–51. doi: 10.1007/s00432-002-0396-4. [DOI] [PubMed] [Google Scholar]
- 39.Liu Y., Zhu X., Zhu J., Liao S., Tang Q., Liu K., Guan X., Zhang J., Feng Z. Identification of differential expression of genes in hepatocellular carcinoma by suppression subtractive hybridization combined cDNA microarray. Oncol. Rep. 2007;18:943–951. [PubMed] [Google Scholar]
- 40.Schultz-Pedersen S., Hasle H., Olsen J.H., Friedrich U. Evidence of decreased risk of cancer in individuals with fragile X. Am. J. Med. Genet. 2001;103:226–230. [PubMed] [Google Scholar]
- 41.Kalkunte R., Macarthur D., Morton R. Glioblastoma in a boy with fragile X: an unusual case of neuroprotection. Arch. Dis. Child. 2007;92:795–796. doi: 10.1136/adc.2006.103382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luca R., Averna M., Zalfa F., Vecchi M., Bianchi F., La Fata G., Del Nonno F., Nardacci R., Bianchi M., Nuciforo P. The fragile X protein binds mRNAs involved in cancer progression and modulates metastasis formation. EMBO Mol. Med. 2013;5:1523–1536. doi: 10.1002/emmm.201302847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luo Y., Shan G., Guo W., Smrt R.D., Johnson E.B., Li X., Pfeiffer R.L., Szulwach K.E., Duan R., Barkho B.Z. Fragile x mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet. 2010;6:e1000898. doi: 10.1371/journal.pgen.1000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo W., Allan A.M., Zong R., Zhang L., Johnson E.B., Schaller E.G., Murthy A.C., Goggin S.L., Eisch A.J., Oostra B.A. Ablation of Fmrp in adult neural stem cells disrupts hippocampus-dependent learning. Nat. Med. 2011;17:559–565. doi: 10.1038/nm.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frontini M., Kukalev A., Leo E., Ng Y.M., Cervantes M., Cheng C.W., Holic R., Dormann D., Tse E., Pommier Y. The CDK subunit CKS2 counteracts CKS1 to control cyclin A/CDK2 activity in maintaining replicative fidelity and neurodevelopment. Dev. Cell. 2012;23:356–370. doi: 10.1016/j.devcel.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang X., Xia L., Chen D., Yang Y., Huang H., Yang L., Zhao Q., Shen L., Wang J. Otefin, a nuclear membrane protein, determines the fate of germline stem cells in Drosophila via interaction with Smad complexes. Dev. Cell. 2008;14:494–506. doi: 10.1016/j.devcel.2008.02.018. [DOI] [PubMed] [Google Scholar]