

Abstract
Small interfering RNA (siRNA) has significant potential to evolve into a new class of pharmaceutical inhibitors, but technologies that enable robust, tissue-specific intracellular delivery must be developed before effective clinical translation can be achieved. A pH-responsive, smart polymeric nanoparticle (SPN) with matrix metalloproteinase (MMP)-7-dependent proximity-activated targeting (PAT) is described here. The PAT-SPN was designed to trigger cellular uptake and cytosolic delivery of siRNA once activated by MMP-7, an enzyme whose overexpression is a hallmark of cancer initiation and progression. The PAT-SPN is composed of a corona-forming PEG block, an MMP-7-cleavable peptide, a cationic siRNA-condensing block, and a pH-responsive, endosomolytic terpolymer block that drives self-assembly and forms the PAT-SPN core. With this novel design, the PEG corona shields cellular interactions until it is cleaved in MMP-7-rich environments, shifting SPNζ-potential from +5.8 to +14.4 mV and triggering a 2.5 fold increase in carrier internalization. The PAT-SPN exhibited pH-dependent membrane disruptive behavior that enabled siRNA escape from endo-lysosomal pathways. Efficient intracellular siRNA delivery and knockdown of the model enzyme luciferase in R221A-Luc mammary tumor cellssignificantly depended on MMP-7 pre-activation. These combined data indicate that the PAT-SPN provides a promising new platform for tissue-specific, proximity-activated siRNA delivery to MMP-rich pathological environments.
Keywords: Proximity-activated targeting, environmental targeting, smart polymer, gene delivery, RNA interference, endosome escape, MMP-7, pH responsive, Matrix metalloproteinase-responsive
1. Introduction
Nanocarrier-mediated delivery of siRNA for silencing of genes that inhibit apoptosis, promote oncogenesis, or induce multidrug resistance is an exciting new paradigm for anti-cancer therapies.[1] Ideally, cell- and tissue-specific siRNA delivery is necessary in tumor tissues in order to avoid off-target effects likely to compromise therapeutic index and clinical applications.[2] Cancer targeting of nanoparticulate drug carriers typically involves optimizing carrier biophysical properties for nonspecific enhanced permeation and retention (EPR) tumor accumulation[3] and/or through biochemical targeting of cell surface receptors that are overexpressed on cancer cells.[4-7] For example, in an exciting recent clinical breakthrough, phase I human trial data demonstrated safe siRNA-mediated gene silencing in human melanoma patients using nanoparticles molecularly targeted to cancer cells that overexpress the transferrin receptor.[8]
Alternatively, pathological environmental signals and enzymatic activity can also be exploited to trigger changes in drug carrier properties, exposing latent nanocarrier functionalities that can increase their internalization by target cells or trigger drug payload release in pathologic sites.[9] For example, the tumor microenvironment is mildly acidic, which has been exploited by a variety of nanocarrier designs.[10, 11] We and others have also leveraged peptides that are cleaved by naturally-occurring proteases for “proximity activation” that uncovers latent targeting ligands on inorganic nanoparticle image contrast agents.[12-17] In an analogous approach, Harishima and co-authors have developed a multifunctional envelope-type nano device (MEND) for siRNA delivery that incorporates a lipid with a matrix metalloproteinase (MMP)-cleavable PEG layer that blocks liposomal uptake until it is enzymatically removed.[18, 19]
MMPs are a family of zinc-dependent proteases that degrade extracellular matrix[20] and are known to be important in the establishment and growth of primary breast tumors and metastatic lesions.[21, 22] MMP-7 is secreted even in the earliest stages of breast tumor development. Small, benign “precancerous” lesions, for example, secrete MMP-7 early in disease progression, before any of the lesions would be detectable clinically.[22, 23] Furthermore, MMP-7 expression is predominantly localized to cells of epithelial origin, in contrast to the expression of most MMP family members in cells of connective tissue origin.[24] These critical characteristics motivate the development of activity probes that respond to MMP-7 activity with changes in their properties that lead to drug release, for eventual applicaton in breast cancers.
Here, a novel, pH-responsive micellar nanocarrier capable of MMP-7 proximity-activated targeting for siRNA delivery is reported. The base delivery system consists of a smart polymeric nanoparticle (SPN) that is capable of packaging and delivering siRNA cargo.[25-28] This SPN consists of a positively-charged dimethylaminoethyl methacrylate (DMAEMA) corona that functions as a siRNA-condensing block and a pH-responsive, core-forming terpolymer block that mediates endosomal disruption and cytosolic delivery of the siRNA payload.[29] The DMAEMA block effectively mediates siRNA delivery, but, like other polycationic delivery vehicles, it is internalized indiscriminately by cells due to its interaction with negatively charged elements of the cell membrane.[28-30] Furthermore, polycationic carriers typically suffer from low circulation half-lives and can cause undesirable interactions among cells and other blood constituents.[31, 32] PEGylation can make cationic polymer-based systems more stealthy in the systemic circulation by shielding these carrier-cell interactions, reducing clearance by the reticuloendothelial system (RES), and extending blood circulation half-lives.[33-36] However, stable PEGylation can also mask underlying functionalities that drive cell uptake and/or endosomolytic activity.[37, 38]
The novel SPN design presented here incorporates surface PEGylation that undergoes MMP-7-triggered shedding, or PAT, of the outer PEG layer, which uncovers the underlying cationic corona and triggers cellular uptake. This PAT-SPN was designed to be sensitive to MMP-7 because, along with other MMPs such as MMP-2 and MMP-9, high levels of MMP-7 activity have been correlated with invasive and metastatic cancer phenotypes that yield poor outcomes in breast cancer patients.[39-43] Importantly, the current design is modular in nature, and the sensitivity and specificity of MMP cleavage can be tuned by altering the MMP-sensitive peptide used in the PAT element.[44] The ability to alter the proteolytically sensitive peptide enables flexible selection of the most appropriate sequence that can be selectively cleaved by the anticipated or known MMP type(s) in the target tissue. This study focused on proof-of-concept synthesis and characterization of MMP-7-sensitive PAT-SPNs, and our data indicate that this delivery platform offers significant potential for environmentally-targeted tissue specific siRNA delivery.
2. Results
2.1. PAT-SPN synthesis and characterization
The MMP-7-cleavable peptide (H-VPLSLYSGCG-OH)[44] was made using solid phase peptide synthesis and purified by high performance liquid chromatography (HPLC), as confirmed by liquid chromatography-mass spectrometry (LC-MS) (Supporting Information, Figs. S1-S2). Proteolytic susceptibility of this sequence to cleavage by human MMP-7 was verified using SDS-PAGE (Supporting Information, Fig. S3). Peptide PEGylation was carried out by reacting a 5 kDa maleimide end-functionalized PEG with the MMP-7 cleavable peptide through its C-terminal, cysteine residue as shown in Scheme 1. GPC analysis confirmed the expected peak retention time shift between the PEG precursor and the PEG-peptide conjugate (Supporting Information, Fig. S4). An N-hydroxysuccinimide-functionalized RAFT chain transfer agent (CTA), NHS-ECT, synthesized in-house (1H-NMR spectra in Supporting Information, Fig. S5 for ECT and Fig. S6 for NHS-ECT) was coupled with the peptide N-terminus. Successful coupling of NHS-ECT was confirmed using UV-vis spectroscopy based on the characteristic spectrophotometric absorbance of the trithiocarbonate group in ECT at 320 nm (Supporting Information, Fig. S7).
Scheme 1.
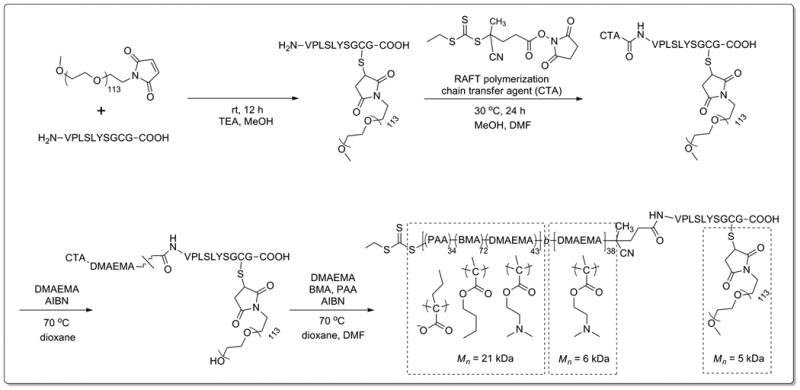
Synthetic scheme of MMP-7-responsive, PEG-pep-pD-pDPB (forming PAT-SPN) polymer construct.
A 6 kDa pDMAEMA homopolymer was extended from the PEG-peptide macroCTA via RAFT polymerization. Conversion was calculated from 1H-NMR spectroscopy analysis of the monomeric form of DMAEMA before and after polymerization relative to the internal index trioxane. Retention of the trithiocarbonate was confirmed in the resulting PEG-peptide-pDMAEMA (PEG-pep-pD) by UV-vis spectrophotometric absorbance at 320 nm. GPC analysis confirmed successful polymerization of a 21 kDa butyl methacrylate (BMA), propyl acrylic acid (PAA), and DMAEMA copolymer block from the PEG-pep-pD macroCTA, resulting in PEG-peptide-pDMAEMA-p(DMAEMA-co-PAA-co-BMA) (PEG-pep-pD-pDPB) (Scheme 1). The full PEG-pep-pD-pDPB polymer construct had a Mn of 33 kDa and a PDI of 1.41. Using the GPC-determined Mn and 1H-NMR spectroscopy, the pDPB block was determined to be composed of 28.6 mol% DMAEMA, 21.7 mol% PAA, and 49.6 mol% BMA (1H-NMR in Supporting Information, Fig. S8).
The PEG-pep-pD-pDPB polymer spontaneously formed micellar nanoparticles upon dropwise addition from ethanol into PBS (pH 7.4). The resulting hydrodynamic particle diameter of 46 nm, as determined by dynamic light scattering (DLS) (Fig. 1a), represents an optimal size for cellular uptake while minimizing non-specific uptake by macrophages of the RES.[17, 29] TEM imaging of the dehydrated micelles counter-stained with uranyl acetate further confirmed the formation of the PAT-SPNs (Fig. 1b). Notably, sizing of dried micelles via TEM typically leads to smaller measured diameters relative to sizing via DLS, due to dehydration and collapse of the particle corona.
Figure 1. Confirmation of PAT-SPN formation.

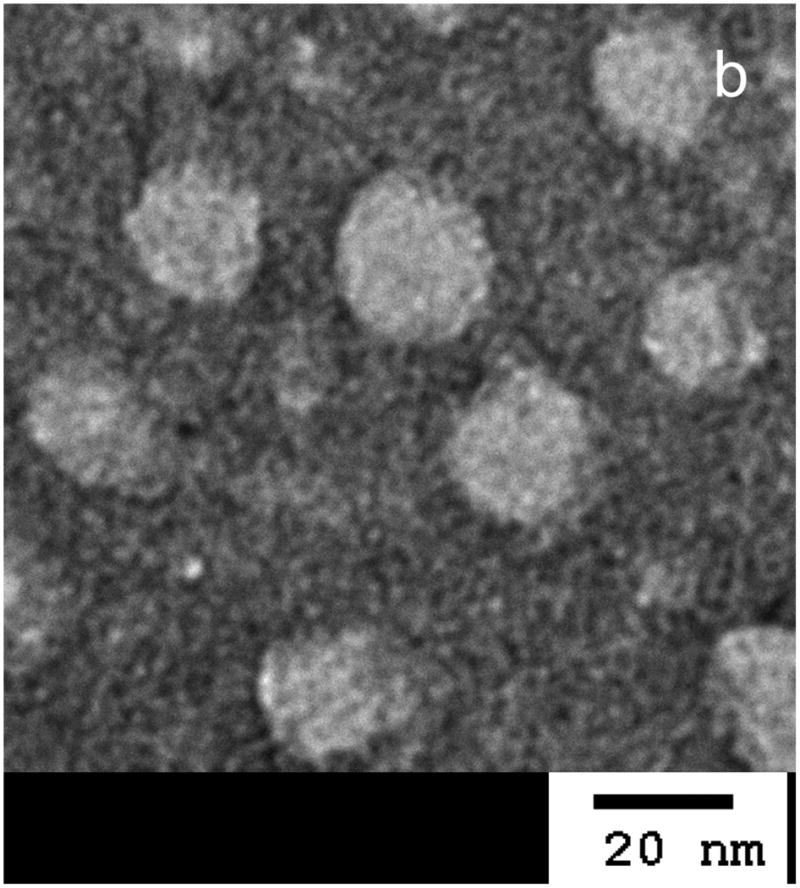
(a) Dynamic light scattering (DLS) (left) suggested that PAT-SPNs exhibit an average hydrodynamic diameter of ∼46 nm. (b) Transmission electron microscopy (TEM) (right) of dried PAT-SPN nanoparticles on TEM grids counterstained with 3% uranyl acetate confirmed self-assembly of PEG-pep-pD-pDPB polymer into micelles with average dehydrated diameters of 20 nm.
2.2. Assessment of responsiveness of PAT-SPN nanoparticles to MMP proteolysis
GPC, ζ-potential, and DLS confirmed MMP-7 proteolytic cleavage of self-assembled PAT-SPNs. PAT-SPNs were treated with 50 nM MMP-7 in order to mimic the metastatic tumor microenvironment.[45] The resultant product was re-isolated and analyzed by GPC with DMF as the mobile phase. The small shift in the peak elution time of the PEG-pep-pD-pDPB after treatment with MMP-7 is consistent with the expected drop in molecular weight due to proteolytic removal of the PEG segment. The sensitivity of the PAT-SPN to MMP-7 treatment was further supported by the appearance of a peak eluting at 25 min (Fig. 2), which aligns with the elution time of the predicted 5 kDa PEG cleavage product. Untreated, control PAT-SPN micelles had a ζ-potential of +5.8 mV. Within 10 min of MMP-7 addition, the ζ–potential increased from +5.8 to +7.2 mV, and it approached +13.8 mV after 3 h (Fig. 3). Exposure to 1-5 nM concentrations of MMP-7 relevant to normal, healthy tissue generated a slower rate of increase in ζ-potential following treatment. Over 3 h, with the MMP-7-concentration dependent change in ζ-potential was +0.26, +1.07, and +7.97 mV for 1, 5, and 50 nM MMP-7, respectively (Supporting Information, Fig. S9). As expected based on the promiscuity of the VPLSLYSCG peptide, PAT-SPN micelles were also activatable by MMP-2 or MMP-9, with consequent increase in zeta potential and decrease in hydrodynamic diameter as observed for MMP-7 (Supporting Information, Fig. S10). Increased ζ-potential upon treatment with pathophysiologic concentrations of MMP is consistent with detachment of the peptide-tethered PEG chains from micelle surface, unveiling the cationic pDMAEMA layer that forms the corona of the underlying base SPN. These data support efficient PAT properties of the siRNA nanocarrier.
Figure 2. GPC analysis of RAFT polymerization from PEG-peptide macro-CTA and PAT-SPN construct responsiveness to MMP-7.

PEG-pep-pD-pDPB generated from PEG-peptide macro-CTA (dash-dot) after two successive RAFT polymerizations was confirmed by the shift in GPC elution time (dash-dot versus solid line traces). Elugrams of PEG-pep-pD-pDPB treated with MMP-7 (dash) was consistent with cleavage of the distal PEG segment as indicated by the shift of the primary elution peak (solid versus dashed traces at ∼23 min) and the appearance of a shoulder peak at ∼25 min that corresponds to the PEG-peptide macro-CTA (dash-dot).
Figure 3. MMP-7-dependent physicochemical switch in PAT-SPNs.

Nanocarrier zeta potential increased from +5.8 to +14.4 mV over 6.5 h of treatment with a physiologically-relevant MMP-7 concentration (50 nM).
2.3. Particle diameter and ζ-potential evaluations of PAT-SPN/siRNA treated with MMP-7
PAT-SPNs loaded with siRNA across a range of N(+)/P(-) charge ratios exhibited an increase in ζ–potential and a decrease in hydrodynamic particle diameter following treatment with MMP-7, indicating that siRNA loading did not adversely affect MMP-7 responsiveness (Table 1). Note that efficient PAT-SPN siRNA loading occurred across a range of N(+)/P(-) charge ratios of 2 or greater (Supporting Information, Fig. S11).
Table 1.
MMP-7 dependent modulation of zeta potential and hydrodynamic diameter of siRNA-loaded PAT-SPNs.
| PAT-SPN/siRNA | PAT-SPN/siRNA+MMP-7 | |||
|---|---|---|---|---|
| Formula | Particle diameter (nm) | Zeta (mV) | Particle diameter (nm) | Zeta (mV) |
| No siRNA | 46 ± 6 | 5.8 ± 0.0 | 42 ± 6 | 14.4 ± 0.0 |
| N(+)/P(-) = 12 | 45 ± 6 | 7.5 ± 6.4 | 42 ± 7 | 14.5 ± 6.9 |
| N(+)/P(-) = 8 | 44 ± 5 | 7.0 ± 5.8 | 42 ± 7 | 14.4 ± 7.6 |
| N(+)/P(-) = 6 | 44 ± 6 | 6.6 ± 7.0 | 40 ± 4 | 15.1 ± 6.3 |
| N(+)/P(-) = 4 | 44 ± 5 | 6.8 ± 6.4 | 41 ± 6 | 15.9 ± 5.5 |
| N(+)/P(-) = 2 | 42 ± 4 | 6.2 ± 5.6 | 37 ± 4 | 12.0 ± 6.1 |
All data are representative of repeated measures of two samples. MMP-7 treatment significantly reduced particle diameter (p = 0.02) and significantly increased ζ-potential (p < 0.0001) as determined by paired t-test.
2.4. PAT-SPN/siRNA serum stability and siRNA protection
The ability of the PAT-SPN/siRNA formulations to remain stable under serum conditions and to protect siRNA from degradation was demonstrated by a Förster (Fluorescence) resonance energy transfer (FRET)-based assay (Fig. 4) and agarose gel electrophoresis (Supporting Information, Fig. S12). In both of these experiments, incubation of PAT-SPN/siRNA in 50% fetal bovine serum at 37 °C was performed to simulate in vivo conditions. For the serum particle stability assay, dsDNA molecules labeled with FAM (DNAFAM) or Cy5 (DNACy5) were co-loaded onto PAT-SPN/DNAFAM/DNACy5. These dsDNA were 21 base pairs and served as model siRNA molecules. The FAM/Cy5 fluorophore pair have minimal emission overlap and make a good donor/acceptor pair for use with FRET. When simply mixed together in solution (with no PAT-SPN formulation), the FRET-paired dsDNA did not produce a FRET signal. However, a robust FRET signal was seen when the FRET-paired dsDNA were co-loaded into PAT-SPN/DNAFAM/DNACy5. Furthermore, the FRET signal of the PAT-SPN/DNAFAM/DNACy5 formulations did not significantly change over a 24 h incubation in 50% serum (student's t-test, p> 0.05 all time points relative to 0 h baseline sample, Fig. 4). Because the FRET signal is dependent on close packing of the two fluorophores, these data suggest that siRNA loading onto PAT-SPNs is serum stable.
Figure 4. FRET-based demonstration of PAT-SPN/DNAFAM/DNACy5 serum stability.

PAT-SPN/DNAFAM/DNACy5 incubated at 37 °C for 2, 4, 8, and 24 h in the presence of 50% fetal bovine serum. FRET was measured with excitation at 485 ± 20 nm using FAM as a donor and emission at 670 ± 25 nm using Cy5 as the acceptor and indicated that the PAT-SPN/DNAFAM/DNACy5 formulation was serum-stable over 24 h. Data presented as mean +/- standard error with n = 3. *p<0.0001 compared with PAT-SPN/DNAFAM/DNACy5 in PBS (no serum).
Agarose gel electrophoresis was utilized to confirm PAT-SPN protection of siRNA from degradation. Free siRNA was significantly degraded within 15 minutes of incubation in 50% serum, as evidenced by the disappearance of the bright band corresponding to free siRNA (Fig. S12). However, when SDS was utilized to release the siRNA from PAT-SPN/siRNA following serum incubation, the siRNA was found to remain relatively intact over the time course of the experiment, though there was some visible decrease in stability at 24 h (Fig. S12). These data indicate that PAT-SPN loading improved the stability of siRNA in a model in vivo environment.
2.5. Cytotoxicity assessment
The high cytocompatibility of the PAT-SPNs loaded with siRNA was in agreement with previous studies using the base SPN carrier.[28, 29] Cell viability of R221A-Luc mammary tumor cells treated with PAT-SPN/siRNA over a range of N(+)/P(-) ratios indicated that significant cytotoxicity was apparent only at 12:1, which was the highest N(+)/P(-) ratio tested (Fig. 5). No statistically significant cytotoxicity was measured at the other charge ratios (2:1, 4:1, 6:1, or 8:1) examined. This data confirmed that appropriately formulated PAT-SPNs can serve as cytocompatible siRNA nanocarriers.
Figure 5. PAT-SPN cytocompatibility across a range of N(+)/P(-) ratios assessed on luciferase-expressing R221A-Luc tumor cells.

Cell viability was assessed by treating luciferase expressing R221A-Luc mammary tumor cells with PAT-SPNs loaded with 50 nM scrambled siRNA at N(+)/P(-) ratio of 2:1, 4:1, 6:1, 8:1, and 12:1 in the presence of MMP (50 nM) and supplemented with Zn2+ (50 μM) (MMP+Zn). Cell viability was reported relative to non-treated cells (NT). Significant cytotoxicity was observed relative to NT at a charge ratio of 12:1 (58 ± 9% viability) but not at lower charge ratios. Data presented as mean +/- standard error with n=3 from three independent experiments. *p<0.05 compared with NT.
2.6. PAT-SPN cell uptake
Flow cytometry and confocal microscopy data demonstrated that internalization of PAT-SPN/siRNA was significantly increased in MDA-MB-231 breast cancer cells following MMP-7 treatment. Cellular internalization of PAT-SPN siRNA nanocarriers activated by MMP-7 was 2.5-fold greater than for unactivated controls (Figs. 6a, b; mean intracellular uptake increased from 178±9 (relative fluorescence units) before MMP-7 activation to 447±41 (relative fluorescence units) following MMP-7 activation). Confocal microscopy images also supported the flow cytometry findings that cell internalization of the nanocarriers was enhanced following MMP-7 proximity activation (Fig. 6c). Furthermore, uptake was found to remain low at both 50 and 100 nM siRNA doses in PAT-SPNs without MMP-7 activation, while the level of uptake was dose-dependent for PAT-SPNs that had been activated with MMP-7.
Figure 6. Enhanced PAT-SPN cell internalization following exposure to MMP-7.

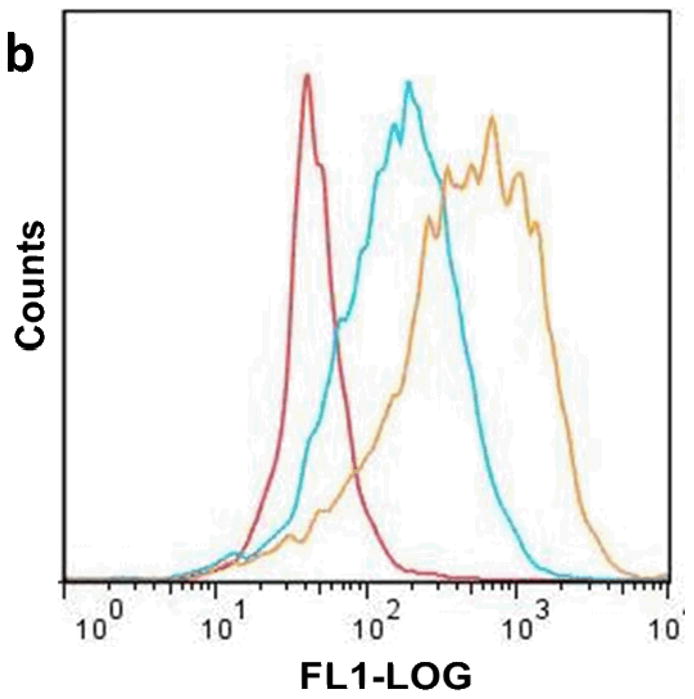
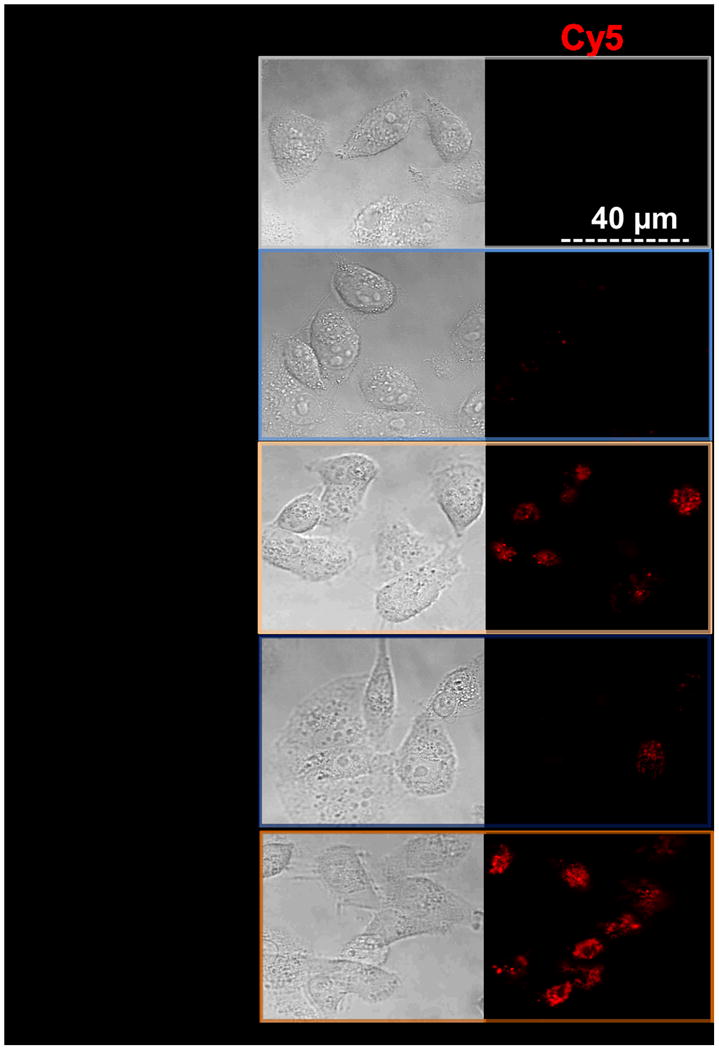
(a-b) Flow cytometry analysis of DNAFAM delivered to MDA-MB-231 breast cancer cells using intact PAT-SPNs (PAT-SPN/DNAFAM, blue), MMP-7-pretreated PAT-SPNs (MMP7 pretreated PAT-SPN/DNAFAM, yellow), or no treatment (NT) (red). Mean FAM fluorescence was significantly higher in cells treated with PAT-SPN/DNAFAM proximity activated by MMP-7 compared to all other treatment groups. Data presented as mean +/- standard error with n=3. *p < 0.05 compared with NT and #p < 0.05 compared with PAT-SPN/FAM-DNA. (c) Confocal microscopy images of MDA-MB-231 breast cancer cells treated with PAT-SPN/DNACy5 nanocarriers (at 50 and 100 nM DNA concentration) with or without MMP-7 treatment correlated with quantitative flow cytometry data. PAT-SPN/DNA complexes of N(+)/P(-) = 6 were applied.
2.7 PAT-SPN pH-responsiveness and endosomal escape
The pDPB block endows the PAT-SPNs with the ability to undergo pH-dependent destabilization of phospholipid membranes[25, 26, 28], and this behavior was confirmed for the PAT-SPN formulations using the red blood cell hemolysis assay[25, 26] (Fig. 7a). Minimal hemolysis was measured at extracellular pH (7.4), and a sharp increase in hemolysis was apparent in the endo-lysosomal pH range (≤6.2) suggesting that the PAT-SPNs can facilitate disruption and escape from endosomal vesicles without causing significant cytotoxicity due to disruption of the outer cell membrane. In agreement with the pH-responsive hemolysis activity, MMP-7 pre-activated PAT-SPNs facilitated enhanced escape of complexed DNACy5 from the endo-lysosomal trafficking pathway, as visualized by confocal microscopy (Fig. 7b). In this experiment, the presence of yellow signal (co-localization of the DNACy5 in red, and LysoTracker in green) was used to indicate endo-lysosomal entrapment. It was found that significant free DNACy5 (red fluorescence) could be visualized intradcellularly, indicating that a portion of the DNA cargo had escaped the endo-lysosomal pathway.
Figure 7. pH-dependent membrane disruption and endo-lysosomal escape of PAT-SPNs.

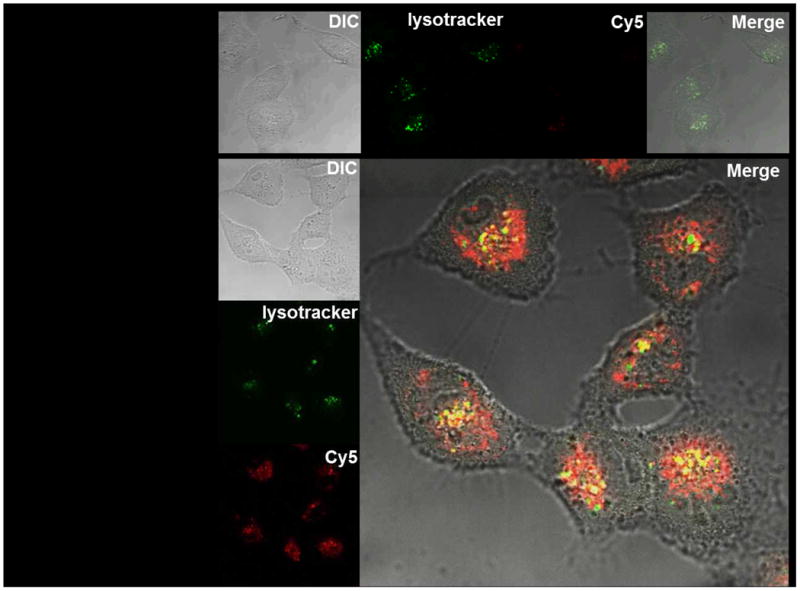
(a) pH-dependent hemolysis of erythrocytes was determined for intact PAT-SPN and MMP-7 pretreated PAT-SPN at 1, 5, 15, and 30 μg/mL relative to 1% Triton X-100 as a positive control (100% lysis). Hemolysis was pH-dependent, with approximately 0% at extracellular pH 7.4 to more than 90% at pH 5.6. Hemolysis mediated by PAT-SPN is essentialy unchanged following MMP-7 proteolytic activation. Error bars indicate standard deviation of n = 4. (b) Confocal imaging of MDA-MB-231 cells treated with MMP-7 pretreated PAT-SPN/DNACy5 and stained with LysoTracker. Control cells only stained by LysoTracker are shown as a reference (upper panel). Intracellular DNACy5 delivered by the MMP-7 pretreated PAT-SPNs is predominantly distributed outside the lysosomes (red) with minimal colocalization with the LysoTracker signal (yellow).
2.8. PAT-SPN reduction in luciferase activity in R221A-Luc mammary tumor cells
To examine the ability of PAT-SPNs to facilitate functional, siRNA-mediated gene knockdown, anti-luciferase siRNA was delivered to R221A-Luc mammary tumor cells, which constitutively express luciferase. siRNA delivered by MMP-7-pretreated PAT-SPNs facilitated significantly greater luciferase knockdown, versus the control PAT-SPN/siRNA group delivered without MMP-7 pre-treatment (p=0.0007; post-hoc two-way ANOVA). The reduction in luciferase activity was significantly greater in the MMP-7-activated PAT-SPNs for all charge ratios compared to controls receiving no treatment (Fig. 8a). Because the PAT-SPNs are inherently cationic, we also quantified the ability of intact, untreated PAT-SPNs to facilitate the knockdown of luciferase activity over a range of N(+)/P(-) ratios. At the lower N(+)/P(-) ratios, no significant reduction of luciferase activity relative to untreated controls was apparent. However, at an N(+)/P(-) charge ratio of 6:1, significant reduction in luciferase activity was observed without MMP-7-pretreatment of PAT-SPNs. Importantly, following MMP-7 treatment of PAT-SPNs, activated PAT-SPNs enhanced luciferase silencingto a level that was not statistically different from that facilitated through the commercial transfection reagent Lipofectamine™. However, all formulations of PAT-SPNs that were not pre-treated with MMP-7 produced statistically less luciferase knockdown than Lipofectamine™. An overview of the pairwise statistical comparisons between treatment groups appears as Fig. 8b. On this plot, positive values represent significant differences, while negative values represent the lack of statistically significant differences. These data suggest that PAT-SPN mediated siRNA delivery will induce significant gene silencing in MMP-7-rich environments and, depending on the precise formulation and exposure, have little or no effect in MMP-7-deficient regions.
Figure 8. Effect of MMP-7 pre-treatment on luciferase knockdown by PAT-SPNs.


(a) Protein level gene silencing readouts were acquired in R221A-Luc mammary tumor cells that constitutively express firefly luciferase, following treatment with luciferase-targeted siRNA complexed into PAT-SPNs at N(+)/P(-) charge ratios of 2, 4, and 6. Lipofectamine (Lipofec) was used as a positive control, and luciferase activity was reported relative to untreated cells (NT). (Data presented as mean +/- standard error with n=3). *p < 0.05 compared with NT, #p < 0.05 compared with Lipofec, and $p < 0.001 between the intact and the MMP-7 pretreated PAT-SPN groups. (b) Variance analysis by ANOVA(one way Tukey's range test) on knockdown results of PAT-SPN/siRNA versus no treatment (NT) (left) or versus Lipofectamine™/siRNA (Lipofec) (right). Formulations with variance > 0 are significantly different to NT (left) and to Lipofec (right)
2.9. PAT-SPN activation by endogenously-produced MMPs
The invasive breast cancer cell line MDA-MB-231, which has been documented to overexpress several MMPs including MMP-7[46, 47], was used to confirm that cell secreted MMPs triggered activation and enhanced cell uptake of the PAT-SPN/DNA. This claim was supported by the confocal microsopy observation that decreased cellular internalization of PAT-SPN/DNAcy5 was observed in cells with media that was supplemented with a broad-spectrum MMP inhibitor (Fig S13b). Cell-mediated and MMP-dependent activation of the PAT mechanism was further confirmed by temporal analyisis of PAT-SPN ζ-potential following exposure to MDA-MB-231 conditioned media in the presence or absence of MMP inhibitor. In the presence of the MMP inhibitor, the ζ–potential was relatively stable over time, while exposure to the conditioned media without the MMP inhibitor triggered a +6.6 mV increase after incubation for 18 hours. Under these treatment conditions, the ζ-potential value was comparable to that of PAT-SPN/DNA treated with exogenous MMP-7 (Fig S13a). These results suggest that a significant increase in PAT-SPN ζ-potential can be triggered by physiologically-relevant MMPs endogenously produced within breast tumor microenvironments.
3. Discussion
While siRNA-based therapies are promising, significant delivery barriers prevent the clinical translation of this new class of therapies. The novel PAT-SPN disclosed herein was primarily motivated by the desire to integrate a mechanism for tumor-specific accumulation with other advanced polymer functionalities in order to overcome each of the primary challenges to siRNA delivery: siRNA protection, stealth in the circulation, site-specific biodistribution and cell uptake, and endosomal pathway escape.
Proximity-activated targeting was chosen due to the potential shortcomings of cell surface receptor targeting. Cell surface targeting of drugs is likely to achieve diminishing effects upon repeat treatment and risks potentially selecting for a population cancer cells that do not highly express the targeted receptor. This could lead to incomplete tumor eradication and/or recurrence. Also, because many of the commonly-targeted receptors that are overexpressed by cancer cells are also present on some healthy cells (e.g., folate and transferrin), systemic delivery of these molecularly-targeted nanocarriers requires finely-tuned ligand presentation on the nanoparticle surface in order to achieve sufficient selectivity, reducing the potential for off-target side effects.[48] Furthermore, molecular targeting to an overexpressed receptor does not always enhance tumor biodistribution following intravenous injection of nanoparticles. For example, systemically-injected transferrin-targeted nanoparticles formed with cyclodextrin-containing polycations and siRNA produced biodistribution and tumor accumulation properties similar to nontargeted control particles; instead, receptor targeting improved tumor cell internalization and bioactivity of the nanoparticles.[49] These interesting results suggest that nanocarrier physicochemical properties should be optimized for EPR-driven tumor accumulation, and that additional targeting strategies may be best motivated by improving tumor retention and cell uptake.
PEGylation is important for incorporating particle stealth, increasing circulation time, and improving EPR-driven tumor biodistribution, but conventional, permanent PEGylation can inhibit interaction and uptake by cells in pathological tissues. In order to increase carrier uptake and local retention following EPR-driven distribution to tumor sites, the current PAT-SPN was designed to be sensitive to MMP-7, an abundant constituent of many solid tumors.[42, 50-56] Here MMP-7-dependent cleavage of the PEG corona exposed the underlying, cationic SPN and consequently triggered a significant increase in particle ζ–potential (Fig. 3). Although pH-dependent PEG shedding from polymeric nanocarriers, has been described,[57] this is, to our knowledge, the first report of polymeric nanocarrier utilizing tissue-specific enzymatic modulation of surface charge as an approach for environmentally-triggered siRNA uptake. Reversible PEGylation removable by acidic conditions has been shown to decrease cytotoxicity relative to non-PEGylated carriers and to improve bioactivity of delivered nucleotides relative to stable PEGylation for a variety of applications using cationic polymers such as PEI.[57, 58] The proteolytically-meditated PEG deshielding demonstrated here provides a potentially more broadly-applicable platform because it is not limited to local environments of reduced pH and is able to accommodate essentially any short peptide that can be enzymatically cleaved.
The modular nature of our design is derived from the synthetic scheme that incorporates the proteolytically-cleavable PAT functionality onto the base SPNs through site-specific orthogonal conjugation chemistries and by leveraging the ability of reversible addition fragmentation chain transfer (RAFT) polymerization to synthesize well-defined block copolymer architectures. The presented strategy for pre-conjugation of the CTA with the PAT components prior to conducting RAFT polymerization of the SPN was used to achieve efficient substitution of the PAT component onto the RAFT polymers and a high density of PEG chains on the final PAT-SPN. This “grafting-from” strategy for the RAFT polymerizations also avoids inefficient reactions between large biomacromolecules and potentially cumbersome product purification.[59, 60]
Once this synthesis scheme was successfully performed, it was important to verify that the resulting PAT-SPNs would be selectively activated in tumor tissue in vivo. MMP activity levels vary among tissues, and the elevated concentration of MMPs in tumors has been estimated to be as high as 1 mM.[19, 61] In our studies, we utilized 50 nM MMP, which falls at the lower end of the range of MMP concentrations that other researchers typically use to study the kinetics of cleavage of MMP-responsive biomaterial sunder conditions that mimic the tumor microenvironment.[14, 19, 45, 62] We primarily assessed cleavage of our construct in response to MMP-7, although we also studied responsiveness to MMP-2 and MMP-9. The chosen substrate VPLSLYSG can be cleaved by all three of these MMPs and has a mid-rate kcat/KM for MMP-7 cleavage of 22,000 ± 3,000 (M-1s-1) [44], though it is anticipated that presence of the PEG corona distal to the peptide sterically limits access of MMPs to the peptide substrate and slows its cleavage. Treating the PAT-SPN formulations with 1, 5, and 50 nM MMP-7 produced a concentration-dependent rate of increase in ζ-potential, suggesting that the PAT-SPN design is tuned to be activated under pathophysiologically-relevant MMP-7 concentrations (Supporting information, Fig. S9). The rates of cleavage observed were also comparable to results previously reported using the RPLALWRS peptide in a PAT quantum dot system.[62]
While the VPLSLYSG sequence appears optimal for our current breast cancer application, the modular nature of the PAT-SPN design can be leveraged through substitution with peptides possessing different proteolytic cleavage characteristics and/or different PEG molecular lengths. The VPLSLYSG peptide used here is cleavable by multiple MMPs in addition to MMP-7, including MMP-2 (kcat/KM = 61,000 ± 4,000) and MMP-9 (kcat/KM = 49,000 ± 3,000).[44] We have confirmed that PAT-SPNs based on the VPLSLYSG peptide are also responsive to MMP-2 and MMP-9 exposure, displaying simultaneous increase in zeta potential and decrease in hydrodynamic diameter upon treatment with these MMPs (Supporting Information, Fig. S10). The tissue selectivity and efficiency of PAT-SPN activity is predicted to be enhanced in environments simultaneously expressing multiple MMPs having proteolytic activity against VPLSLYSG.
In addition to targeted delivery, key design criteria considered in development of the PAT-SPNs were stable packaging and protection of the siRNA cargo, stealth in the circulation, and endosomal pathway escape. As confirmed by gel electrophoresis and a FRET-based assay, the PAT-SPNs were able to efficiently package siRNA into serum-stable particles that also helped to decrease siRNA susceptibility to degradation (Supporting Information, Figs. S11 and S12). Furthermore, PEGylation of the particle surface is known to impart nanocarrier stealth that reduces reticuloendothelial clearance and aggregation with blood cells.[33, 34] In combination with control of nanoparticle size, PEGylation may also facilitate nanocarrier accumulation in vascularized tumor tissues via the EPR effect. To this end, all of the PAT-SPN formulations tested had diameters of 40-50 nm, which is known to be the optimal size for the EPR-driven tumor accumulation, while minimizing RES-mediated clearance of the nanocarriers.[17, 25, 29] Furthermore, the pH-dependent membrane-disruptive behavior of the terpolymer that forms the SPN core[29] was found to be retained in the PAT-modified SPN design, ensuring endosomolytic release of the siRNA into the cytosol where it functions (Fig. 7).[63, 64]
4. Conclusions
A multifunctional, ‘smart’ siRNA carrier (PAT-SPN) intended for specific activation in vascularized tumors and metastases was successfully constructed, characterized and demonstrated. Activation of these nanocarriers by MMP-7 both increased intracellular delivery and enabled RNA interference and protein-level knockdown by siRNA. The combined results are consistent with the design concept that MMP-7-dependent removal of an outer PEG corona increased PAT-SPN ζ-potential, which consequently increased rate of uptake into tumor cells. Once internalized, the pH-responsive core of the PAT-SPN was activated and facilitated pH-dependent siRNA endosomal escape and bioactivity. Overall, evidence suggests that this novel nanoscale vehicle has important advantages in targeted and effective therapeutic delivery to MMP-rich cancer tissues for gene-specific protein silencing. It is anticipated that this approach will be useful for the development of effective stand-alone therapies and/or as an adjuvant used to sensitize cells to traditional chemotherapies. The promising proof-of-concept data reported here inform our ongoing work to optimize environmentally-triggered targeting of efficient siRNA nanocarriers.
5. Experimental
Materials
Chemicals and materials were purchased by Sigma-Aldrich or Fisher Scientific and used without purification unless otherwise specified. DMAEMA monomers were purified by distillation under vacuum prior to use in polymerizations and BMA was passed through basic alumina prior to use. 2,2′-Azobis(2-methylpropionitrile) (AIBN) was recrystallized twice from methanol. siRNA was purchased from Ambion and ssDNA from Sigma Aldrich. Two 21mer ssDNAs with complementary sequences were dissolved in nuclease-free water, and a stock concentration of 50 μM dsDNA was annealed by three cycles of heating to 94 °C and cooling to 50 °C. PD-10 desalting columns were purchased from from GE Healthcare, Lipofectamine™ high efficiency transfection reagent were purchased from Invitrogen (Carlsbad, CA), and active human MMPs (including MMP-7, MW=19 kDa; MMP-2, MW=66 kDa; and MMP-9, MW=83 kDa) were purchased from EMD Chemicals. PAA monomer was synthesized following a protocol adapted from previous literature.[65]
Synthesis of 4-cyano-4-(ethylsulfanylthiocarbonyl) sulfanylpentanoic acid (ECT)
ECT was synthesized following protocols previously described by Convertine et al. and Moad et al.[25, 66] Briefly, ethanethiol (4.72 g, 76 mmol) was reacted with carbon disulfide (6.00 g, 79 mmol) in the presence of sodium hydride (3.15 g, 79 mmol) in diethyl ether for 1 h. The resulting sodium S-ethyl trithiocarbonate was further reacted with iodine (6.3 g, 25 mmol) to obtain bis(ethylsulfanythiocarbonyl) disulfide, which was then refluxed with 4,4′-azobis(4-cyanopentanoic acid) in ethyl acetate for 18 h to yield ECT. The crude ECT was purified by column chromatography using silica gel as the stationary phase and ethyl acetate:hexane (50:50) as the mobile phase. 1H NMR (400 MHz, CDCl3): 3.35 (q, 2H, S-CH2-CH3), 2.69 (t, 2H, -CH2-CH2-COOH), 2.36-2.58 (m, 2H, -CH2-CH2-CH3), 1.88 (s, 3H, CH3-C-CN), 1.36 (t, 3H, S-CH2-CH3)
Synthesis of the N-hydroxyl succinimide-functionalized ECT (NHS-ECT)
Purified ECT (1.25 g, 4.75 mmol) and N-hydroxylsuccinimide (0.55 g, 4.75 mmol) were added to a round-bottom flask and dissolved in dichloromethane (DCM, 45 mL). The mixture was held at 4 °C and purged with N2 for 30 min. A solution of N,N′-dicyclohexylcarbodiimide (DCC) (0.98 g, 4.75 mmol) in DCM (5 mL) was injected, and the mixture was purged with N2 for 30 min. The reaction mixture was kept at 4° C for 2 h and continued at room temperature for 22 h. The resulting solution was filtered through a Buchner funnel, and the solvent was evaporated under vacuum. The crude NHS-ECT was purified by column chromatography on silica gel with methanol as mobile phase to rapidly elute unreacted ECT, followed by ethyl acetate to elute purified NHS-ECT. The product was dried under vacuum to yield a solid pale yellow powder. 1H NMR (400 MHz, CDCl3): 3.36 (q, 2H, -S-CH2-CH3), 2.94 (m, 2H, -C-CH2-CH2-), 2.86 (s, 4H, -CO-CH2-CH2-CO-), 2.71-2.50 (m, 2H, -CH2-CH2-COOH), 1.89 (s, 3H, CH3-C-CN), 1.37 (t, 3H, -S-CH2-CH3).
Synthesis of PEG-peptide RAFT macro chain transfer agent
The MMP-7-cleavable peptide (H-VPLS↓LYSGCG-OH; arrow indicates expected MMP-7 cleavage site) was synthesized through standard Fmoc-based solid phase techniques on a PS3 synthesizer (Protein Technologies, Tucson, AZ). The peptide was purified using an HPLC (Waters Breeze system) with a gradient mobile phase consisting of methanol and water, both containing 0.05% formic acid, through a Phenomenex Luna semi-prep 5U C18 (2) column (250 × 10 mm). Peptide sensitivity to MMP-7 was confirmed on a 16.5% tris-tricine polyacrylamide gel, containing 12% glycerol. Samples were separated in tris-tricine running buffer under 150 V volt-constant current. Cleavage of the peptide (5 mg/mL) by MMP-7 (1 μg/mL) in PBS buffer (pH 7.4) was tested before and after the removal of the protecting group on the amino acid side chains. The peptide with side chains that had not been deprotected was used as a control since the tert-butyl protecting groups block MMP cleavage of the peptide.
After validation of MMP-7 peptide cleavability, peptide (208 mg, 0.21 mmol) and maleimide-PEG (mal-PEG 5 kDa, 520 mg, 0.11 mmol) (Laysan Bio, Inc.) were added in a 50 mL round-bottom flask and purged with N2 for 30 min before dissolving into N2 pre-purged methanol. Triethylamine (TEA) (24.2 mg, 0.24 mmol) was then added. The reaction was stirred at room temperature for 12 h. PEG-peptide was purified from unreacted peptide using PD-10 columns (GE Healthcare Life Sciences), and the purified product was lyophilized. Purified PEG-peptide (270 mg, 0.045 mmol) and NHS-ECT (162 mg, 0.45 mmol) were dissolved in a co-solvent of 33 mL MeOH and 15 mL DMF and purged with N2 for 30 min. The reaction was stirred at 30 °C for 24 h and then dialyzed against methanol (MWCO = 3000 Da, Fisher Scientific) to remove DMF. Methanol was subsequently removed by rotary evaporation. Further purification was completed by precipitating the PEG-peptide RAFT macro-CTA two times from chloroform into ether.
Synthesis of pDMAEMA with PEG-peptide RAFT macro-CTA
The RAFT polymerization of DMAEMA was conducted at 70 °C under a nitrogen atmosphere. DMAEMA (420 mg, 2.67 mmol), PEG-peptide macroCTA (186 mg, containing 0.02 mmol of CTA functionality, as determined by its UV-Vis spectroscopy extinction coefficient at 320nm), AIBN (0.42 mg, 0.002 mmol), and trioxane (10 mg) were dissolved in 1.23 mL dioxane in a 5 mL flask equipped with a magnetic stir bar. Initial monomer to CTA ([M]0/[CTA]0) and CTA to initiator molar ([CTA]0/[I]0) ratios were 133/1 and 10/1. Kinetic samples were removed periodically by nitrogen-purged syringe to monitor monomer conversion by 1H NMR spectroscopy. The polymerization was quenched after 8.6 h (29% conversion) by opening the vial to air and cooling the reaction in an ice bath. The resultant PEG-peptide-pDMAEMA (PEG-pep-pD) product was isolated by precipitation into cold 40/60 vol.% diethyl ether/pentane. The resultant polymer was redissolved in deionized water, further purified using PD-10 desalting columns, and lyophilized.
Synthesis of p(DMAEMA-co-PAA-co-BMA) terpolymer block from PEG-pep-pD macro-CTA
Stoichiometric quantities of DMAEMA, PAA, and BMA (28/20/52 mol%), PEG-pep-pD macroCTA (200 mg, containing 0.01 mmol of CTA functionality, as determined by its UV-Vis spectroscopy extinction coefficient at 320nm), and AIBN (0.27 mg, 0.0017 mmol) were dissolved in 1.3 mL cosolvent of dioxane:DMF=67:33 vol.%. The [M]0/[CTA]0 and [CTA]0/[I]0 ratios were 285/1 and 5.6/1. The reaction mixture was purged with nitrogen for 20 min and allowed to react at 70 °C for 20 h. The resultant PEG-peptide-pDMAEMA-b-p(DMAEMA-co-PAA-co-BMA) (PEG-pep-pD-pDPB) crude product was isolated by precipitation into cold 50/50 diethyl ether/pentane twice. The resultant polymer was resuspended in water, dialyzed against water overnight, and then lyophilized.
Polymer characterization
Polymer products were dissolved in deuterated methanol (MeOD) and analyzed by 1H-NMR spectroscopy using a 9.3T Oxford magnet equipped with a Bruker DRX console operating at 400 MHz. The PEG-pep-pD composition was determined from the resonance of DMAEMA methylene. Composition of the completed PAT-SPN polymer (PEG-pep-pD-pDPB) was determined from the resonances associated with the DMAEMA methylene, the BMA methylene, the PAA methylene, and the PEG backbone.
Molecular weight and polydispersity of the copolymers were characterized by GPC using a mobile phase of N,N-dimethylformamide (DMF) with 100 mM LiBr at 60 °C using a flow rate of 1.0 mL min-1 (Columns: Tosoh Biosciences 1x TSKGel Alpha4000, 2x TSKGel Alpha3000 in series). Chromatograms were recorded with a Shimadzu SPD-10A UV detector, a Shimadzu RID-10A refractive index detector (Shimadzu Scientific Instruments, Columbia, MD), and a Wyatt miniDAWN Treos multi-angle light scattering detector (MALS; Wyatt Technology, Santa Barbara, CA). Absolute molecular weight was determined from first principles via this GPC-MALS setup by measuring the polymer dn/dc using known concentrations of the purified PEG-pep-pD-pDPB polymer sample.
Nanoparticle assembly and size measurement by DLS and TEM
PEG-pep-pD-pDPB polymer was dissolved in ethanol at a concentration of 20 mg/mL. This freshly prepared stock solution in ethanol was then added dropwise into a 20 fold volume excess of 1x phosphate buffer saline (PBS) at pH 7.4 under stirring. This process triggered spontaneous self-assembly of micellar PAT-SPNs at 1 mg/mL. PAT-SPNs were diluted with PBS to 0.20 mg/mL for particle diameter measurement on DLS. Samples were also prepared for transmission electron microscopy (TEM) imaging using a Philips CM20 system. Carbon film-backed copper grids (Electron Microscopy Sciences, Hatfield, PA) were sequentially dipped into ethanol for 30 seconds, then 0.15 mg/mL nanoparticle suspension for 1 min, and finally 3% uranyl acetate in water. Following each dipping step, grids were gently blotted dried and further dried in a vacuum desiccator for 6 h prior to imaging.
pH-dependent membrane disruption hemolysis measurement
Hemolysis was measured according to a reported method[67, 68] to determine the membrane-disruptive activity of intact PAT-SPN and MMP-7 pretreated PAT-SPN nanoparticle at pH values that mimic intracellular trafficking. Following Vanderbilt University Institutional Review Board approval of human subject protocols, whole human blood was collected, centrifuged, plasma aspirated, and washed three times in 150 mM NaCl to isolate the red blood cells (RBC). RBCs were then resuspended in phosphate buffer (PB) in 96 well plates at pH 7.4, 6.8, 6.2 or 5.6 in the presence of the polymers at 1, 5, 15, 30 μg/mL and incubated at 37 °C for 1 h. Samples were centrifuged to pellet intact RBCs, and percent hemolysis relative to 1% Triton X-100 was determined based on absorbance of the supernatants at 450 nm.
Assessment of siRNA loading by gel electrophoresis
Agarose gel electrophoresis was used to determine siRNA loading capacity and siRNA protection of the nanoparticles. The siRNA (21 base pairs, 0.04 nmol in each sample) was complexed with varied amounts of PAT-SPNs to achieve N(+)/P(-) charge ratios of 1, 2, 4, and 8, and free siRNA and the siRNA-loaded PAT-SPN formulations were run on a 2% agarose gel. The N(+)/P(-) charge ratios were calculated using the positively-charged DMAEMA block, which is estimated to be 50% protonated at pH=7.4 (forming –+NH-(CH2)2), divided by the negatively-charged phosphate groups in the siRNA.[29] The equation is expressed as:
where Molpolymer is the number of mol of the complete PEG-pep-pD-pDPB polymer, RUDMAEMA is the number of repeating units of DMEAMA in the pDMAEMA block, MolsiRNA is the number of mol of siRNA, and bpsiRNA is the base pair number in the siRNA. This equation was also used for studies in which fluorophore-labeled dsDNA is used since the charge per base pair is the same as for siRNA.
For the assessment of siRNA protection from degradation in serum, formulations of N(+)/P(-) = 6 calculated from the above equation were applied. PAT-SPN/siRNA were incubated in 50% FBS for 1, 4, 8, or 24 h. Afterward, 1% SDS was added to each sample to disrupt the PAT-SPN/siRNA and release the complexed siRNA. Free siRNA was incubated in 50% serum for 0.25, 0.5, 1, 2, and 4 h. Free siRNA and the siRNA-loaded PAT-SPN formulations were run on 2% agarose gels.
Nanoparticle stability study by FRET
Equal moles of DNAFAM and DNACy5 were mixed and incubated at room temperature for 1 h before loading onto PAT-SPNs at room temperature for 1 h. PAT-SPN/DNAFAM/DNACy5 formulations of N(+)/P(-) = 6 were incubated (125 nM DNAFAM/DNACy5 in 200 μL medium) at 37 °C for 2, 4, 8, and 24 h in the presence of 50% fetal bovine serum. FRET signal was determined relative to PAT-SPN/DNAFAM/DNACy5 in PBS (lack of serum and without incubation). Samples containing 50% serum were background-corrected to account for any FRET signal resulting from 50% serum in PBS. FRET signal was measured on an Infinite F500 TECAN plate reader with excitation at 485 ± 20 nm using FAM as a donor and emission at 670 ± 25 nm using Cy5 as the acceptor.
Assessment of PAT-SPN MMP-7 responsiveness
PAT-SPN proximity activation by MMP-7 was tested in vitro by GPC and through physicochemical assessments of particle size and surface charge. PAT-SPNs in PBS (0.9 mg/mL) were treated with 1 μg/mL (50 nM) active human MMP-7 (EMD chemicals) in the presence of 50 μM Zn2+ ions or just PBS as a control. PAT-SPNs were incubated with MMP-7 at 50 nM at 37 °C, and, at various time points, samples were diluted to 0.2 mg/mL with deionized water and used to measure size and ζ-potential on a Malvern Zetasizer Nano-ZS. The change in ζ-potential for PAT-SPNs incubated with MMP-7 at 1 and 5 nM at 37 °C was also measured with the same method. An aliquot of the 1 mg/mL sample after 6 h of MMP-7 exposure was dialyzed against deionized water for one day to remove buffer salts, lyophilized, and analyzed by GPC.
Cell Culture
MDA-MB-231 breast cancer cells (ATCC, Manassas, VA) and R221A-Luc mammary tumor cells that constitutively express luciferase were cultured on standard tissue culture-treated polystyrene surfaces in an incubator maintained at 37°C / 5% CO2. MDA-MB-231 cells were maintained in growth media consisting of Dulbecco's Modified Eagle Medium (Gibco) supplemented with 10% fetal bovine serum (Gibco) and 50 μg/mL gentamicin (Mediatech). R221A-Luc cells were grown in media consisting of Dulbecco's Modified Eagle Medium (Gibco) supplemented with 2 mM glutamine (Mediatech) and 5 μg/mL puromicin dihydrochloride.
Flow cytometry and confocal microscopy cell uptake measurements
Flow cytometry was utilized to quantify intracellular delivery of PAT-SPNs. In these studies, DNAFAM was used as a model siRNA molecules and was pre-complexed with PAT-SPN at N(+)/P(-) = 6 for 1 h. MDA-MB-231 breast cancer cells were seeded at 5 × 104 cells/cm2 in 24 well plates (Costar) and allowed to adhere overnight. PAT-SPNs were pre-treated with 50 nM MMP-7 and 50 μM ZnSO4, in the absence of serum, for 6 h prior to use in cell experiments. PAT-SPN/FAM-labeled dsDNA complexes with or without MMP-7 pre-treatment were then added to the cell wells with 50 nM FAM-labeled dsDNA in DMEM medium supplemented with 10% FBS and 50 μg/mL gentamicin. After 6 h incubation, cells prepared for flow cytometry were washed with PBS, trypsinized, and resuspended in PBS (containing 0.04% trypan blue to quench extracellular fluorescence). Cellular internalization of the PAT-SPNs was quantified by fluorescence measurement of FAM label in the cell samples using a BD FACSCalibur flow cytometer (San Jose, CA).
For confocal microscopy experiments, PAT-SPN/DNACy5 of N(+)/P(-) = 6 was prepared as for the flow cytometry studies. MDA-MB-231cells were seeded at a density of 5 × 104 cells/cm2 in 8-well chamber slides (Nunc / Thermo-Fisher). Cells were treated with PAT-SPN/DNACy5 with or without MMP-7 pre-activation (at 50 and 100 nM DNA concentrations), or PBS in DMEM medium supplemented with 10% FBS and 50 μg/mL gentamicin for 6 h. After washing twice with PBS and replaced with fresh medium, the live cells were imaged on a Zeiss LSM 510 confocal microscope. For LysoTracker co-localization imaging, some cells were further incubated for 4 h, stained with 75 nM LysoTracker (Invitrogen) for 30 min, washed twice with PBS, and imaged on a Zeiss LSM 510 confocal microscope.
Assessment of PAT-SPN cytocompatibility and siRNA gene knockdown
PAT-SPNs were screened for cytocompatibility and gene silencing bioactivity using R221A-Luc mammary cancer cells that constitutively express luciferase. Luciferase activity in cell samples was confirmed to have a linear relationship with cell number (Supporting information, Fig. S14). In gene silencing experiments, R221A-Luc were seeded at a density of 5 × 104 cells/cm2 in 96 well plates and allowed to adhere overnight. Different formulations of PAT-SPNs were made by complexing with luciferase siRNA (Ambion) at N(+)/P(-) ratios of 2, 4, and 6. Some PAT-SPNs were pre-activated with MMP-7 for 6 h as discussed for uptake experiments. Lipofectamine dose (1 μL Lipofectamine in 200 μL medium per 96 plate well) was chosen based on the manufacturer instructions and used as a comparison with PAT-SPNs. Cells were treated for 6 h with 50 nM siRNA concentration for all formulations. Afterward, the cells were given fresh media and incubated for 18 more h. To measure luciferase gene silencing, luminescence in each sample was quantified on the IVIS-100 (Caliper Life Sciences) after adding 0.15 mg/mL luciferin, and luciferase activity was normalized to the total protein content measured from the cell lysate of each well using the BCA assay.
For nanoparticle cytotoxicity assessment, a scrambled siRNA (proven in other experiments to have no luciferase silencing activity) was complexed with the PAT-SPNs at charge ratios of 2, 4, 6, and 8. The treatment protocol was the same as described above, and the relative cell number was determined by quantification of luciferase activity in each well. Each group was tested in triplicate and normalized to cells receiving no treatment. Results from three independent experiments were averaged to generate the final data reported here.
Confirmation of PAT-SPN activation by endogenously-produced MMPs
The ability of cell-secreted MMPs to cleave the PAT component and trigger cell uptake was assessed in confocal microscopy cell uptake experiments using the MDA-MB-231 cell line. MDA-MB-231 cells were seeded at a density of 5 × 104 cells/cm2 in 8-well chamber slides (Nunc / Thermo-Fisher) and allowed to adhere for 6 h in DMEM with 10% FBS before replacing the media with serum-free DMEM supplemented with 50 μg/mL gentamicin. The cells were then incubated for 12 h to allow for cell secretion of MMPs. At that time, the existing media in a subset of wells was supplemented with 25 μM broad-spectrum MMP inhibitor GM 6001. Cells were then incubated for an additoinal 2 h, after which the the conditioned media on all cells (with or without prior GM6001 addition) was supplemented with PAT-SPN/DNACy5 N(+)/P(-) = 6 (50 nM of DNACy5). After 5 h of incubation, the samples were washed twice with PBS, supplied fresh media, and the live cells were imaged on a Zeiss LSM 510 confocal microscope.
The MMP-dependent increase in PAT-SPN/DNA ζ-potential following exposure to the conditioned media of breast cancer cells was also characterized. MDA-MB-231 breast cancer cells were seeded at 2 × 105 cells/cm2 in 12 well plates in 10% FBS, and after 6 h, the media was replaced with serum-free and phenol red-free RPMI 1640. After 24 h, the media was collected, centrifuged, and aliquoted into 50 μL samples. GM 6001 was added at 25 μM into a subset of the samples, and all conditioned media samples were incubated at 37°C for 2 h. PAT-SPN/DNA samples (0.5 mg/mL of polymer, N(+)/P(-) = 6) were then treated with a 50% dilution of these conditioned media samples. To assess PAT-SPN/DNA cleavage by the MMPs secreted into the conditioned media, samples were taken after 0 h, 6 h, and 18 h of treatment, and the ζ-potential was measured following sample dilution to 0.1 mg/mL with deionized water. Conditioned media containing GM6001 and MMP-7 precleaved PAT-SPN/DNA nanocarriers were used as controls to confirm MMP-dependency of ζ– potential changes and to benchmark ζ-potential changes against exogenous MMP-7 treatment.
Statistical analysis
All data are reported as mean and standard error of the mean (SEM). Analysis of Variance (ANOVA) was used to establish statistical significance, and p < 0.05 was considered significant.
Supplementary Material
Acknowledgments
This research was supported by grants from the Department of Defense Congressionally Directed Medical Research Programs (W81XWH-10-1-0445 and W81XWH-10-1-0446) and NSF REU Grant DMR-1005023. Dynamic light scattering, spectrofluorimetry, and TEM were conducted through the use of the core facilities of the Vanderbilt Institute of Nanoscale Sciences and Engineering (VINSE). Confocal microscopy was performed in the VUMC Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, HD15052, DK59637 and EY08126). The authors would like to give thanks to E. Duco Jansen and Mark A. Mackanos for kindly providing IVIS(R) 100 imaging system for luminescence analysis, to Donald Stec for extensive and discussions about 1H-NMR spectrometry analysis, and to M. Wade Calcutt for technical support in regard to mass spectrometry analysis.
Footnotes
Supporting Information: Supporting Information is available online from Wiley Inter Science or from the author.
The authors declare that they have no competing interests.
Contributor Information
Dr. Hongmei Li, Department of Biomedical Engineering, Vanderbilt University, VU Station B, Box 351631, Nashville, TN, USA; Vanderbilt Institute of Nanoscale Science and Engineering, Vanderbilt University, Nashville, TN, USA
Shann S. Yu, Department of Biomedical Engineering, Vanderbilt University, VU Station B, Box 351631, Nashville, TN, USA
Martina Miteva, Department of Biomedical Engineering, Vanderbilt University, VU Station B, Box 351631, Nashville, TN, USA.
Christopher E. Nelson, Department of Biomedical Engineering, Vanderbilt University, VU Station B, Box 351631, Nashville, TN, USA
Thomas Werfel, Vanderbilt Institute of Nanoscale Science and Engineering, Vanderbilt University, Nashville, TN, USA; Department of Engineering and Physics, Murray State University, Murray, KY, USA.
Todd D. Giorgio, Email: todd.d.giorgio@vanderbilt.edu, Department of Biomedical Engineering, Vanderbilt University, VU Station B, Box 351631, Nashville, TN, USA; Department of Cancer Biology, Department of Chemical and Biomolecular Engineering, Vanderbilt University, Nashville, TN, USA.
Craig L. Duvall, Email: craig.duvall@vanderbilt.edu, Department of Biomedical Engineering, Vanderbilt University, VU Station B, Box 351631, Nashville, TN, USA; Vanderbilt Institute of Nanoscale Science and Engineering, Vanderbilt University, Nashville, TN, USA.
References
- 1.Kanasty RL, Whitehead KA, Vegas AJ, Anderson DG. Mol Ther. 2012;20:513. doi: 10.1038/mt.2011.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burnett JC, Rossi JJ. Chem Biol. 2012;19:60. doi: 10.1016/j.chembiol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsumura Y, Maeda H. Cancer Res. 1986;46:6387. [PubMed] [Google Scholar]
- 4.Guo M, Que C, Wang C, Liu X, Yan H, Liu K. Biomaterials. 2011;32:185. doi: 10.1016/j.biomaterials.2010.09.077. [DOI] [PubMed] [Google Scholar]
- 5.Narayanan S, Binulal NS, Mony U, Manzoor K, Nair S, Menon D. Nanotechnology. 2010;21:285107. doi: 10.1088/0957-4484/21/28/285107. [DOI] [PubMed] [Google Scholar]
- 6.Lee ES, Na K, Bae YH. J Control Release. 2003;91:103. doi: 10.1016/s0168-3659(03)00239-6. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Q, Liu F, Nguyen KT, Ma X, Wang X, Xing B, Zhao Y. Adv Funct Mater. 2012 doi: 10.1002/adfm.201201316. [DOI] [Google Scholar]
- 8.Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A. Nature. 2010;464:1067. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gullotti E, Yeo Y. Mol Pharm. 2009;6:1041. doi: 10.1021/mp900090z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao W, Chan JM, Farokhzad OC. Mol Pharm. 2010;7:1913. doi: 10.1021/mp100253e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barick KC, S S, NV J, D B, Pandey BN, Hassan PA. Adv Funct Mater. 2012 doi: 10.1002/adfm.201201140. [DOI] [Google Scholar]
- 12.Smith R, Sewell SL, Giorgio TD. Int J Nanomedicine. 2008;3:95. doi: 10.2147/ijn.s2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuhn SJ, Finch SK, Hallahan DE, Giorgio TD. Nano Lett. 2006;6:306. doi: 10.1021/nl052241g. [DOI] [PubMed] [Google Scholar]
- 14.Harris TJ, von Maltzahn G, Lord ME, Park JH, Agrawal A, Min DH, Sailor MJ, Bhatia SN. Small. 2008;4:1307. doi: 10.1002/smll.200701319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.von Maltzahn G, Harris TJ, Park JH, Min DH, Schmidt AJ, Sailor MJ, Bhatia SN. J Am Chem Soc. 2007;129:6064. doi: 10.1021/ja070461l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sewell SL, Giorgio TD. Cancer Research. 2009;69:348s. [Google Scholar]
- 17.Yu SS, Lau CM, Thomas SN, Jerome WG, Maron DJ, Dickerson JH, Hubbell JA, Giorgio TD. Int J Nanomedicine. 2012;7:799. doi: 10.2147/IJN.S28531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hatakeyama H, Akita H, Ito E, Hayashi Y, Oishi M, Nagasaki Y, Danev R, Nagayama K, Kaji N, Kikuchi H, Baba Y, Harashima H. Biomaterials. 2011;32:4306. doi: 10.1016/j.biomaterials.2011.02.045. [DOI] [PubMed] [Google Scholar]
- 19.Hatakeyama H, Akita H, Kogure K, Oishi M, Nagasaki Y, Kihira Y, Ueno M, Kobayashi H, Kikuchi H, Harashima H. Gene Ther. 2007;14:68. doi: 10.1038/sj.gt.3302843. [DOI] [PubMed] [Google Scholar]
- 20.McIntyre JO, Matrisian LM. J Cell Biochem. 2003;90:1087. doi: 10.1002/jcb.10713. [DOI] [PubMed] [Google Scholar]
- 21.McCawley LJ, Matrisian LM. Mol Med Today. 2000;6:149. doi: 10.1016/s1357-4310(00)01686-5. [DOI] [PubMed] [Google Scholar]
- 22.Sternlicht MD, Bergers G. Expert Opin Ther Targets. 2000;4:609. [Google Scholar]
- 23.Crawford HC, Scoggins CR, Washington MK, Matrisian LM, Leach SD. J Clin Invest. 2002;109:1437. doi: 10.1172/JCI15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson CL, Matrisian LM. Int J Biochem Cell Biol. 1996;28:123. doi: 10.1016/1357-2725(95)00121-2. [DOI] [PubMed] [Google Scholar]
- 25.Convertine AJ, Benoit DS, Duvall CL, Hoffman AS, Stayton PS. J Control Release. 2009;133:221. doi: 10.1016/j.jconrel.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duvall CL, Convertine AJ, Benoit DS, Hoffman AS, Stayton PS. Mol Pharm. 2010;7:468. doi: 10.1021/mp9002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nelson CE, Gupta MK, Adolph EJ, Shannon JM, Guelcher SA, Duvall CL. Biomaterials. 2012;33:1154. doi: 10.1016/j.biomaterials.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Convertine AJ, Diab C, Prieve M, Paschal A, Hoffman AS, Johnson PH, Stayton PS. Biomacromolecules. 2010;11:2904. doi: 10.1021/bm100652w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Convertine AJ, Benoit DS, Duvall CL, Hoffman AS, Stayton PS. J Control Release. 2009;133:221. doi: 10.1016/j.jconrel.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu C, Jung S, Luo S, Meng F, Zhu X, Park TG, Zhong Z. Biomaterials. 2010;31:2408. doi: 10.1016/j.biomaterials.2009.11.077. [DOI] [PubMed] [Google Scholar]
- 31.Li W, Nakayama M, Akimoto J, Okano T. Polymer. 2011;52:3783. [Google Scholar]
- 32.Dobrovolskaia MA, Patri AK, Simak J, Hall JB, Semberova J, De Paoli Lacerda SH, McNeil SE. Mol Pharm. 2012;9:382. doi: 10.1021/mp200463e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verbaan FJ, Oussoren C, Snel CJ, Crommelin DJ, Hennink WE, Storm G. J Gene Med. 2004;6:64. doi: 10.1002/jgm.475. [DOI] [PubMed] [Google Scholar]
- 34.Vader P, van der Aa LJ, Engbersen JF, Storm G, Schiffelers RM. Pharm Res. 2012;29:352. doi: 10.1007/s11095-011-0545-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Varkouhi AK, Mountrichas G, Schiffelers RM, Lammers T, Storm G, Pispas S, Hennink WE. Eur J Pharm Sci. 2012;45:459. doi: 10.1016/j.ejps.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 36.Crownover E, Duvall CL, Convertine A, Hoffman AS, Stayton PS. J Control Release. 2011;155:167. doi: 10.1016/j.jconrel.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chaudhari KR, Ukawala M, Manjappa AS, Kumar A, Mundada PK, Mishra AK, Mathur R, Monkkonen J, Murthy RS. Pharm Res. 2011;29:53. doi: 10.1007/s11095-011-0510-x. [DOI] [PubMed] [Google Scholar]
- 38.Murthy N, Campbell J, Fausto N, Hoffman AS, Stayton PS. J Control Release. 2003;89:365. doi: 10.1016/s0168-3659(03)00099-3. [DOI] [PubMed] [Google Scholar]
- 39.Talvensaari-Mattila A, Paakko P, Turpeenniemi-Hujanen T. Br J Cancer. 2003;89:1270. doi: 10.1038/sj.bjc.6601238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Talvensaari-Mattila A, Turpeenniemi-Hujanen T. Cancer Lett. 2005;217:237. doi: 10.1016/j.canlet.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 41.Garcia MF, Gonzalez-Reyes S, Gonzalez LO, Junquera S, Berdize N, Del Casar JM, Medina M, Vizoso FJ. Int J Exp Pathol. 2010;91:324. doi: 10.1111/j.1365-2613.2010.00709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsunezumi J, Higashi S, Miyazaki K. J Cell Biochem. 2009;106:693. doi: 10.1002/jcb.22062. [DOI] [PubMed] [Google Scholar]
- 43.Beeghly-Fadiel A, Long JR, Gao YT, Li C, Qu S, Cai Q, Zheng Y, Ruan ZX, Levy SE, Deming SL, Snoddy JR, Shu XO, Lu W, Zheng W. Cancer Res. 2008;68:6453. doi: 10.1158/0008-5472.CAN-08-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turk BE, Huang LL, Piro ET, Cantley LC. Nat Biotechnol. 2001;19:661. doi: 10.1038/90273. [DOI] [PubMed] [Google Scholar]
- 45.McIntyre JO, Fingleton B, Wells KS, Piston DW, Lynch CC, Gautam S, Matrisian LM. Biochem J. 2004;377:617. doi: 10.1042/BJ20030582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kousidou OC, Roussidis AE, Theocharis AD, Karamanos NK. Anticancer Res. 2004;24:4025. [PubMed] [Google Scholar]
- 47.Shan L. Molecular Imaging and Contrast Agent Database (MICAD) Bethesda (MD): 2004. [PubMed] [Google Scholar]
- 48.Low PS, Henne WA, Doorneweerd DD. Acc Chem Res. 2008;41:120. doi: 10.1021/ar7000815. [DOI] [PubMed] [Google Scholar]
- 49.Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Proc Natl Acad Sci U S A. 2007;104:15549. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beeghly-Fadiel A, Shu XO, Long J, Li C, Cai Q, Cai H, Gao YT, Zheng W. Int J Cancer. 2009;124:208. doi: 10.1002/ijc.23859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koskensalo S, Louhimo J, Nordling S, Hagstrom J, Haglund C. Tumour Biol. 2011;32:259. doi: 10.1007/s13277-010-0080-2. [DOI] [PubMed] [Google Scholar]
- 52.Koskensalo S, Mrena J, Wiksten JP, Nordling S, Kokkola A, Hagstrom J, Haglund C. Tumour Biol. 2010;31:149. doi: 10.1007/s13277-010-0020-1. [DOI] [PubMed] [Google Scholar]
- 53.Liu H, Zhang T, Li X, Huang J, Wu B, Huang X, Zhou Y, Zhu J, Hou J. Cancer Sci. 2008;99:2185. doi: 10.1111/j.1349-7006.2008.00922.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luukkaa H, Klemi P, Hirsimaki P, Vahlberg T, Kivisaari A, Kahari VM, Grenman R. Acta Oncol. 2010;49:85. doi: 10.3109/02841860903287197. [DOI] [PubMed] [Google Scholar]
- 55.Wang FQ, So J, Reierstad S, Fishman DA. Int J Cancer. 2005;114:19. doi: 10.1002/ijc.20697. [DOI] [PubMed] [Google Scholar]
- 56.Yi YC, Chou PT, Chen LY, Kuo WH, Ho ES, Han CP, Yang SF. Clin Chem Lab Med. 2010;48:337. doi: 10.1515/CCLM.2010.082. [DOI] [PubMed] [Google Scholar]
- 57.Yang XZ, Du JZ, Dou S, Mao CQ, Long HY, Wang J. ACS Nano. 2012;6:771. doi: 10.1021/nn204240b. [DOI] [PubMed] [Google Scholar]
- 58.Sethuraman VA, Na K, Bae YH. Biomacromolecules. 2006;7:64. doi: 10.1021/bm0503571. [DOI] [PubMed] [Google Scholar]
- 59.Li M, Li H, De P, Sumerlin BS. Macromol Rapid Commun. 2011;32:354. doi: 10.1002/marc.201000619. [DOI] [PubMed] [Google Scholar]
- 60.Li H, Li M, Yu X, Bapat AP, Sumerlin BS. Polym Chem. 2011;2:1531. [Google Scholar]
- 61.Duffy MJ, Maguire TM, Hill A, McDermott E, O'Higgins N. Breast Cancer Res. 2000;2:252. doi: 10.1186/bcr65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith R, Sewell SL, Giorgio TD. Int J Nanomedicine. 2008;3:95. doi: 10.2147/ijn.s2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Foster S, Duvall CL, Crownover EF, Hoffman AS, Stayton PS. Bioconjug Chem. 2010 doi: 10.1021/bc100204m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duvall CL, Convertine AJ, Benoit DS, Hoffman AS, Stayton PS. Mol Pharm. 2010;7:468. doi: 10.1021/mp9002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ferrito M, Tirrell DA. Macromol Synth. 1992;11:59. [Google Scholar]
- 66.Moad G, Chong YK, Postma A, Rizzardo E, Thang SH. Polymer. 2005;46:8458. [Google Scholar]
- 67.Plank C, Oberhauser B, Mechtler K, Koch C, Wagner E. J Biol Chem. 1994;269:12918. [PubMed] [Google Scholar]
- 68.Evans BC, Nelson CE, Yu SS, Kim AJ, Li H, Nelson HM, Giorgio TD, Duvall CL. J Vis Exp. 2012 doi: 10.3791/50166. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.