

Abstract
A lack of X-ray or nuclear magnetic resonance structures of proteins inhibits their further study and characterization, motivating the development of new ways of analyzing structural information without crystal structures. The combination of hydrogen–deuterium exchange mass spectrometry (HDX-MS) data in conjunction with homology modeling can provide improved structure and mechanistic predictions. Here a unique diheme cytochrome c (DHCC) protein from Heliobacterium modesticaldum is studied with both HDX and homology modeling to bring some definition of the structure of the protein and its role. Specifically, HDX data were used to guide the homology modeling to yield a more functionally relevant structural model of DHCC.
Cytochrome c’s are essential metalloproteins in the electron-transfer chain of most living organisms, including all photosynthetic taxa. Metalloproteins move electrons around the cell to produce energetic compounds that drive cellular metabolism. Many of the photosynthetic electron-transfer proteins of interest belong to the cytochrome c family. Understanding their structure and function will contribute not only to understanding photosynthesis, its evolution, and its role in early Earth history but also to harnessing solar energy.
Cytochrome c’s are electron-transport hemoproteins that are covalently bound through two thioether bonds between the vinyl groups of a heme macrocycle and the sulfhydryls of a CXXCH heme-binding motif.1 Two heme propionate groups are exposed and can participate in hydrogen bonding. The main function is to mediate single-electron-transfer reactions between protein electron donors and acceptors via reversible oxidation/reduction of Fe2+ and Fe3+. The axial coordination of the heme iron and its effect on other parts of the larger protein environment play key roles in determining redox, electron-transfer, and other properties of cytochrome c.2,3
Prokaryotic diheme cytochrome c’s, as representatives of electron-transfer proteins with two iron centers, have been studied mainly via various spectroscopic methods.4−6 For example, cytochrome c4 from Pseudomonas stutzeri, which shares the same origin as DHCC and has a sequence similar to that of DHCC,7 possesses a strong hydrogen bond involving the heme propionate groups, and the protein facilitates intramolecular, interheme electron transfer.8 Both hemes contain hexacoordinated iron involving histidine and methionine axial ligands. The hydrogen bond in P. stutzeri closes one edge of the heme, allowing it to act as the donor and the other as the acceptor.
DHCC from Heliobacterium modesticaldum, an early evolving, Gram-positive phototrophic anaerobic prokaryote, plays a role as the terminal electron acceptor in the high-potential electron-transfer chain of the cytochrome bc complex. This complex participates in the photosynthetic electron-transport cycle by oxidizing the quinone pool, sending electrons to the reaction center, and pumping protons to establish an electrochemical gradient that drives ATP production. DHCC replaces the canonical monoheme cytochrome c1 in the cytochrome bc1 complex or cytochrome f in the cytochrome b6f complex.9,10 DHCC has two c-type hemes that are predicted to reside in similar c-type cytochrome folds. The heme 1 domain is closer to the N-terminal helix, whereas the heme 2 domain begins approximately halfway through the sequence. The unusual diheme architecture is an interesting contrast with those of the more common monoheme cytochromes. In general, an electron transfer of one metal center induces conformational changes in the site of the other, and that usually enhances the rates of subsequent steps.11 Studying the structure, function, and redox properties of this protein is crucial to the understanding of the function of the entire heliobacterial cytochrome bc complex. Furthermore, an understanding of the Heliobacterium electron-transport chain (ETC), which is simpler and smaller than later evolved organisms, can provide clues about how life and photosynthesis evolved on Earth and possibly on other planets. Similarly, the simple nature of the ETC makes it an attractive target for biomimicry in solar energy production.12−15
Redox-dependent conformations of cytochrome c’s have been studied extensively for decades by using various biochemical and biophysical approaches.16−23 Although some comparative X-ray crystallographic studies of tuna heart cytochromes c show little or no difference between the backbone structures,24 almost all solution-based studies show a clear conformational change between the two redox states.25−27 Specifically, in the case of horse heart cytochrome c, the radius of the oxidized state is significantly larger than that of the reduced state.25 Despite the success of these approaches, there is a need for other methods. For example, many biophysical techniques usually require a large amount of sample,28−30 which is not always available, and their structural resolution is relatively low.28 In contrast, mass spectrometry (MS) approaches are highly sensitive and exhibit moderate structural resolution. For studying protein conformations, hydrogen–deuterium exchange (HDX) coupled with MS is an effective and now commonly employed approach.31−35 The exchange rates for HDX are good readouts for hydrogen bonding and solvent accessibility at global, peptide, and even amino acid levels.36 It is also possible to use HDX data to adjudicate protein subunit docking and ligand-binding structural models.37−39
In HDX studies, one often compares the exchange rate of the same protein regions for two different states that are produced by some protein perturbation (e.g., change in redox states or formation of a complex). Although each amino acid has different intrinsic deuterium exchange rate constants, this causes no problem because the experimental exchange rates between two states of the protein are compared. Previously, HDX coupled with infrared spectroscopy was used to study the conformations of reduced and oxidized horse heart cytochrome c.40 Viala and co-workers41 reported an HDX-MS study of the same system, indicating a more open structure in the oxidized state. Thus far, however, there are no HDX-MS studies of the DHCC system. To test our proposed mechanism of the DHCC subunit of the heliobacterial cytochrome bc complex, which involves a more closed conformation upon reduction,42 we designed an HDX study of both the reduced and oxidized states of the heliobacterial DHCC in an effort to achieve peptide level resolution of structural changes.
Homology modeling (HM) is another common approach to protein structure and function, especially when data from other structural approaches [e.g., X-ray crystallography and nuclear magnetic resonance (NMR)] are not available. Although HM statistics for assigning model quality do not necessarily reflect the in vivo structure of the protein, the approach should be more accurate when coupled with experimental approaches. HDX-MS with HM was used previously to investigate docking and binding studies in combination with electron microscopy.43−46 These semiquantitative approaches rely on heavy computational techniques and assess the quaternary structure of the protein. Here, we are using HDX-MS results to direct HM to obtain a low-resolution understanding of protein tertiary structure and to predict a reaction mechanism for the complex. Our method relies on readily available applets for modeling and HDX for making a qualitative structural distinction between a buried or exposed region to adjudicate the models, quite unlike the previous studies that use HDX to study the docking of ligands or subunits. Success will provide insight into DHCC and establish the utility of the combined approach for other protein systems.
Materials and Methods
Protein Expression and Purification
The diheme cytochrome c subunit from the H. modesticaldum bc complex was expressed in Escherichia coli with the cytochrome c insertion plasmid and purified as previously described, yielding only the holoprotein.42 The N-terminal transmembrane helix region was truncated to express only the fully soluble portion of the protein. Postprocessing did not eliminate the hemes, both being covalently bound to the protein.
Hydrogen–Deuterium Exchange (HDX) of Reduced and Oxidized Diheme Cytochrome c Samples
Differential, solution HDX experiments were performed at 4 °C. DHCC in a 20 mM Tris-HCl buffer (pH 7.5) solution was incubated with either sodium dithionite (reductant) or potassium ferricyanide (oxidant) at a final concentration of 25 μM for both the protein and the reductant or oxidant. The samples were equilibrated for 1 h prior to the initiation of HDX. Continuous labeling was initiated by incubating 2 μL of the mixture with 18 μL of D2O for seven predetermined times (0.17, 0.5, 1, 2, 15, 60, and 240 min). The exchange reaction was quenched by mixing the D2O solution with 30 μL of 3 M urea and 1% trifluoroacetic acid (TFA) at 0 °C. The quenched solution was then flash-frozen in liquid nitrogen and stored at −80 °C for <48 h prior to analysis.
The quenched sample was then digested by being passed through a custom-packed pepsin column (2 mm × 2 cm) at a rate of 200 μL/min; the peptides were captured on a C8 trap column (2 mm × 1 cm, Agilent Inc., Santa Clara, CA) and desalted with a 3 min flow. Peptides were then separated by using a C18 column (2.1 mm × 5 cm, 1.9 μm, Hypersil Gold, Thermo Fisher Scientific, Waltham, MA) with a 5 min linear gradient from 4 to 40% CH3CN in 0.1% formic acid. Protein digestion and peptide separation were conducted in an ice–water bath to minimize back exchange. All analyses were conducted with a hybrid LTQ Orbitrap (Thermo Fisher Scientific, San Jose, CA) at an ESI capillary temperature of 225 °C. Each experiment was performed in duplicate.
Peptide Identification and HDX Data Processing
MS/MS experiments were performed on the same mass spectrometer. Product-ion spectra were acquired in a data-dependent mode, and the six most abundant ions were selected for product ion analysis. The data files were converted using MassMatrix Mass Spectrometric Data File Conversion Tools (version 3.9) and then submitted to MassMatrix (version 2.4.2) for peptide identification.47 Peptides used for HDX data analysis had a MassMatrix score of ≥10, with peptide and MS/MS tolerances for m/z of 10 ppm and 0.8 Da, respectively. The MS/MS MassMatrix search was also performed against a decoy (reverse) sequence, and ambiguous identifications were ruled out. The product-ion spectra of all peptide ions from the MassMatrix search were manually inspected, and only those manually inspected were used in the following steps. The centroid masses of isotopic envelopes were calculated with HDX WorkBench:48 deuterium level (%) = {[m(P) – m(N)]/[m(F) – m(N)]} × 100%, where m(P), m(N), and m(F) are centroid masses of partially deuterated peptides, undeuterated peptides, and fully deuterated peptides, respectively. To accommodate the situation in which a fully deuterated control was not available, m(F) was determined with the equation m(F) = m(N) + (n – p – 2)/z, where n and p represent the total number of amino acids and prolines, respectively, and z represents the charge. The first two amino acids that do not retain deuterium were not taken into consideration (the value “2” in the equation). No correction was made for back exchange because all values are used in a relative way (used in a two-state comparison) and susceptible to the same back exchange processes.
Homology Modeling and Model Refinement
Homology modeling of DHCC was conducted with three different modeling platforms. The first was MODELLER49 version 9.10 and version 9.12 using alignment algorithms ClustalW2 and ClustalΩ, respectively, with both Protein Data Bank (PDB) entries 1ETP(8) and 3MK7c(50) generating 100 models from each run. The best models from each run were selected on the basis of the molpdf, GA341, and DOPE scores. The best model from each run was then tested by taking the experimental HDX data for each peptide as a measure of the secondary and tertiary structure of that portion of the protein. The amount of exchange in each section was compared to the secondary and tertiary structure predicted by the model. For regions that disagreed, the template to DHCC alignment was manually adjusted and another round of models with the new alignment was generated until the best match was obtained with the HDX data.
The second approach used models that were generated using the Phyre251 and I-TASSER52 online modeling suites. The models generated from these two suites were compared to each other because they do not allow for manual adjustment of the alignments or parameters or further analysis options. Disorder prediction was run on the best model in Phyre2. The top resulting model was compared to the HDX data and to the MODELLER-generated models.
The third approach was built on previous work10 in which a complete model of the cytochrome bc complex was built using the Phyre2 server to analyze the whole complex structure and predict the mechanism for electron transfer within the complex.
Assessment of the Solvent-Accessible Surface Area (SASA) of Homology Models
The SASA was calculated for all four homology models using GetArea.53 The SASA for the backbone only was chosen as the value to be used in our assessment because the only HDX that was counted was of the backbone hydrogens, not the whole residue. The backbone SASA was calculated for each residue and is reported in square angstroms. For comparison to the HDX data, the SASA was averaged for all residues in each of the MS peptic peptides. The percent of deuterium uptake was calculated by taking the average of the initial and final point.
Results and Discussion
Prior to conducting HDX, we tested whether charge-state distributions of the intact protein could provide coarse structural data to distinguish the reduced and oxidized states of DHCC. Experiments revealed that the conditions of native ESI are sufficiently oxidative to preclude obtaining mass spectra for the pure reduced species. Expression of the protein with the covalently bound heme precluded any comparisons with the apo state. Thus, we turned to HDX footprinting to obtain structural data for the DHCC protein.
HDX Shows the Oxidized Form of DHCC Is More Flexible
An online pepsin digestion of expressed and purified DHCC afforded hundreds of peptides, of which 69 detected with a high signal/noise ratio and validated MS/MS information were chosen for the subsequent peptide mapping. The coverage is 95% (shown in Figure 1), allowing us to assess nearly fully the protein in both of the redox states. As mentioned above, no correction for back exchange of the protein was made because all data were used in two-state comparisons of the redox-state samples under identical conditions. All samples were subjected to back exchange after quenching and processing, and this converted any side chain deuteriums (e.g., on -NH2) back to hydrogens. Thus, it is the backbone deuteriums that remain on the polypeptide for the LC–MS analysis. Changes in protein dynamics, as reflected by HDX, reflect changes to the protein induced by reduction. The resulting perturbations alter the conformation of the protein backbone and its H bonding as reflected by changes in HDX. Therefore, all fluctuations in HDX rates reflect only reduction-induced changes. For example, we expect that less constrained regions should exhibit relatively fast HDX, whereas structured and buried regions exchange slowly.
Figure 1.

HDX peptide coverage map for DHCC showing 95% coverage. The same peptides with different charge states are also shown.
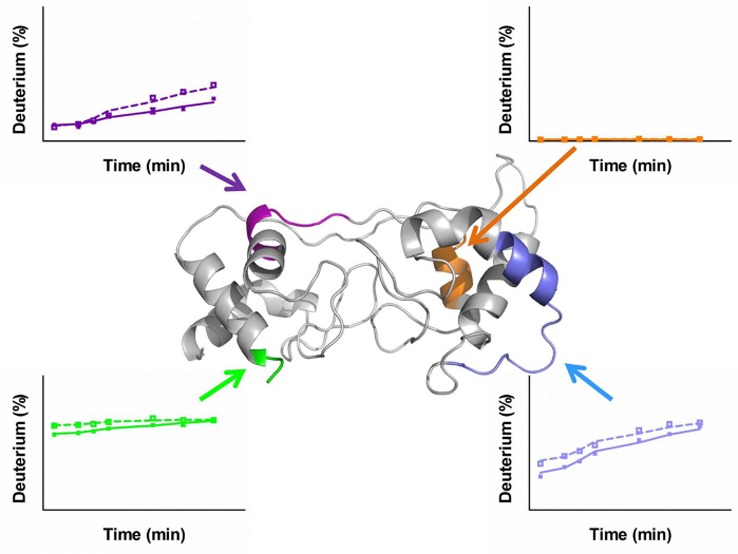
We plotted the HDX kinetic curves for each of the peptides from both the reduced (Figure 2, solid lines, for a representative set and Figure S1 of the Supporting Information, solid lines, for the remainder) and oxidized (Figure 2 and Figure S1 of the Supporting Information, dashed lines) states of the DHCC. When peptides with different charges covered the same region, we found that the results were nearly identical, indicating that the HDX results have good precision. More importantly, we conducted each experiment in duplicate and show the error bars (average deviations) in each plot. To show more clearly the precision, we enlarged one of the typical HDX plots to show the precision of the experiments (Figure S2 of the Supporting Information). In summary, all the HDX experiments afforded a 0–3% relative average deviation for duplicate determinations.
Figure 2.

Kinetic curves of selected peptides used for HDX mapping for reduced (—) and oxidized (---) states of the DHCC. Numbers in parentheses with a plus sign are the charge states of the peptides.
The extents of HDX for most regions of the protein are different for the two states (oxidized and reduced), indicating that the structural conformation of DHCC is redox-state-dependent. For those regions that show differences, the oxidized state always shows a level of deuterium uptake greater than that of the reduced state, indicating that the oxidized state is more flexible (less structurally constrained). This is consistent with other horse heart cytochrome c HDX studies that show that the oxidized state is more dynamic and flexible than the reduced state.41
The largest differences in HDX kinetics occur near the heme-binding CXXCH motif (i.e., for peptides 1–29 and 109–127) (Figure 2). These differences indicate that conformational opening in the heme-binding region accompanies oxidation. This result suggests that redox-dependent conformational changes begin at the metal center. Comparing the changes in HDX for the oxidized and reduced states at the two heme-binding pockets, we find similar outcomes. Specifically, the oxidized protein undergoes between 30 and 50% HDX at the two sites, whereas the reduced protein undergoes between 20 and 40% exchange (Figure 2). Our previous reported potentiometric titration of DHCC42 shows a single observable midpoint potential, which is consistent with the conclusion that the two heme-binding pockets have similar chemical environments.
HDX Reflects Secondary Structure
Although four regions display no changes upon reduction [e.g., 45–49, 130–138, 143–149, and 169–172 (Figure 2 and Figure S1 of the Supporting Information)], they can be viewed as controls because they indicate that regions insensitive to oxidation do not show differences in HDX. Importantly, these regions also show no detectable HDX as a function of time, indicating that these regions are heavily H-bonded and/or buried in the protein. We identify these four regions as being involved in tertiary structural helix bundles (category I). We take this result to indicate that the core structural components of DHCC do not change dramatically upon reduction, whereas flexible regions are more likely to be affected and participate in the conformational changes that accompany redox changes. In contrast, region 173–208 undergoes extensive exchange, even at short times (Figure 2). We categorize regions of this type to be a part of loops (category II).
Regions that constitute a third category also show an increasing level of HDX with time but to a more modest extent (none showed ≥90% deuterium uptake). This lower level of HDX occurs in part because the peptides are long and likely cover regions containing both H-bonded helices and flexible loops, thus reporting an “average” of these structural elements. For example, the peptides for region 78–108 show a level of deuterium uptake ranging from 20 to 50% for the reduced state and from 30 to 60% for the oxidized state (Figure 2). The helix region undergoes less HDX, whereas the peptide bonds in the flexible loop exchange more readily. Similarly, regions 78–91 and 92–108 also comprise helix and loop structures and, thus, show an intermediate level of deuterium uptake (∼60%). This intermediate exchange also pertains to regions 109–127 and 150–168.
We also identified a fourth category in which the level of HDX at the shortest times is near zero but increases slowly with time (Figure 2 and Figure S1 of the Supporting Information). We identified these regions, including 38–44, 50–57, and 128–142, as being likely helices but not part of buried helix bundles.
Homology Model Building and Assessment
A series of homology models were generated with MODELLER and PHYRE2 (Figure 3 and Table 1). To begin, we chose template 1ETP because it is a bacterial cytochrome c4 protein, and the evolutionary origin of DHCC may be the cytochrome c4 protein family.42 Likewise, 1ETP undergoes electron transfer from a donor to an acceptor protein.8 We selected 3MK7c as the second template on the basis of bioinformatic analysis. A BLAST search of the Protein Data Bank with DHCC’s sequence returns 3MK7c as the most similar protein by alignment statistics. Functionally, 3MK7c is a soluble member of a protein complex but resides in the cytoplasm and accepts electrons from a cytoplasmic donor and shuttles them into the complex, the opposite direction from which DHCC should function.50 Both 1ETP and 3MK7c have hemes with two different redox potentials. Although the generated homology models do not incorporate the cofactor heme by nature of the program, the templates we chose do include the cofactors within the protein. Even though we did not add the hemes in the HM modeling, the protein structure should still be representative. Functional considerations of the template are a way to assess model quality.
Figure 3.
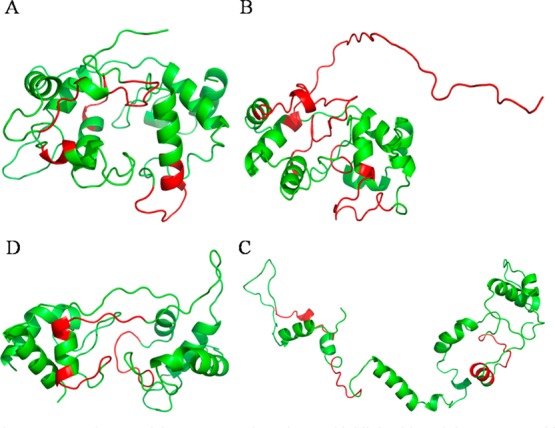
Homology models A–D. Regions that are colored red do not agree with HDX results.
Table 1. Homology Modeling Data.
| model | template | alignment algorithm | percent similarity | percent identity | modeling software | Molpdf score |
|---|---|---|---|---|---|---|
| A | 1ETP | Clustal W2 | 49% (100/205) | 15% (32/205) | MODELLER version 9.10 | 0.24 (0.49, highest) |
| B | 1ETP | Clustal Omega | 45% (92/205) | 16% (33/205) | MODELLER version 9.10 and version 9.12 | 0.12 (0.28, highest) |
| C | 3MK7c | Clustal W2 | 57% (116/205) | 28% (68/205) | MODELLER version 9.10 | 0.46 (highest) |
| D | 3MK7c | Phyre2 | 57% (116/205) | 28% (58/205) | I-TASSER and PHYRE2 | 3.2 Å resolution N/A |
Homology modeling (HM) statistics assess model quality in a more quantitative way. The percent similarity and percent identity examine the alignment of the template and model sequences. Amino acid residues that align are examined to see if they are identical or in the same category. The percent identity increases from model A to model B as a result of manual alignment changes implemented on the basis of the HDX data. Further increases in both percent similarity and percent identity occur for models C and D, which use 3MK7 as a template (Table 1). The higher these values, the more similar a model is to the template and the more faithful the homology model. The 28% identity achieved for models C and D is just under that preferred for HM but is sufficient for further consideration. We also view it as acceptable because there are so few diheme cytochromes with determined structures in the PDB. After HM is run in MODELLER, score reports are generated for each model. The molpdf score can be used to compare models across runs. Model C has the highest molpdf score, indicating it is the better homology model. Model B may be a better model functionally as discussed below, but it has a lower molpdf score because manual alignment changes were made (Table 1). Homology modeling data indicate that models C and D are better models; therefore, 3MK7c is the better template. This result is based on only alignment and threading algorithms, however, and does not speak to function or biochemical properties of the models and templates. Thus, biochemical and/or biophysical data are needed to assess these models further.
HDX Analysis of DHCC with Its Homology Model
HDX data generated by MS can provide biophysical data that, when mapped onto the four homology models, should be able to test the correctness of secondary and tertiary structures generated by the models (Figure 3 and Table 2). For model A, most of the regions that show discrepancies between the HDX results and the HM outcomes are the helical regions (Figure 3A and Table 2). For example, peptides in category I show almost no deuterium uptake in either of the states (Figure 2 and Figure S1 of the Supporting Information), suggesting they participate in the formation of tertiary structure, most likely a helix bundle. In model A, however, only region 143–149 has the helix bundle motif. Peptides 45–49 and 130–138 are modeled to be a loop or a singular helix structure. After the alignment is manually adjusted on the basis of this observation, models B and C show better fits for fragments 45–49, 143–149, and 169–172, as these regions are predicted to be helices in helix bundles. Furthermore, model A HM did not identify the second heme-binding motif, and the alignment and subsequent modeling are poor in this region, 109–127.
Table 2. Comparison of HDX Data and Homology Models.
| HDX agreement
on the modelsd |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| regiona | categoryb | model Ac | model B | model C | model D | A | B | C | D |
| 1–29 | III | h/l | h/l | h/l | h/l | ** | ** | ** | ** |
| 33–37 | III | h/l | l | l | l | ** | * | ||
| 38–44 | IV | h bundle | h/l | h/l | h/l | * | |||
| 45–49 | I | h | h bundle | h/l | h bundle | ** | ** | ||
| 50–57 | IV | l | l | h bundle | h/l | * | |||
| 58–65 | II | l | l | l | l | ** | ** | ** | ** |
| 78–91 | III | h/l | h/l | h/l | h/l | ** | ** | ** | ** |
| 92–108 | III | h/l | h/l | h/l | h/l | ** | ** | ** | ** |
| 109–127 | III | l | l | h/l | h/l | ** | ** | ||
| 128–142 | IV | h/l | l | l | l | * | * | ||
| 143–149 | I | h bundle | h bundle | h | h bundle | ** | ** | * | ** |
| 150–168 | III | h/l | h/l | h/l | h/l | ** | ** | ** | ** |
| 169–172 | I | h | h bundle | h bundle | h bundle | ** | ** | ** | |
| 173–208 | II | h/l | l | h/l | h/l | ** | ** | ** | ** |
Regions determined by HDX. Most of the regions are covered by multiple peptides, and they give similar results.
Different regions are assigned to four categories on the basis of their HDX behavior.
Secondary and tertiary structure elements in each model. h stands for helix, l for loop, h/l for a mixture of helix and loop, and h bundle for a helix bundle.
Measures of degrees of agreement between HDX and modeling results. Two asterisks denote agreement. One asterisk denotes agreement with restrictions (information from order prediction by Phyre2). Blank cells denote a lack of agreement.
To resolve regions for which there is a lack of agreement, we generated model B (Figure 3B and Table 2) by manually adjusting the alignment on the basis of the results of model A as evaluated by HDX. Model B showed improved agreement between HDX and the HM structure. The helix bundle and second heme regions predicted by model B are consistent with the experimental results, but achieving these improvements sacrificed other regions that had agreed with model A. For example, region 33–37 shows as a loop, but it takes up only ∼50% of the possible deuterium. On the other hand, a mostly loop region, 173–208, does show ∼70% HDX. In addition, residues 173–205 show the poorest agreement with HDX results compared to other models, as the sequence does not align with the template. 1ETP is a smaller protein, and in the adjustment for the heme regions, residues at the end of DHCC were left without template residues. As a result, this region is unstructured according to model B. Even though this region shows the highest level of HDX for the whole protein (∼70%), it still should have some secondary structure. Despite these areas of disagreement, model B is better than model A because the active regions of the protein are consistent with the outcomes of both HDX and HM. Overall, the transition from model A to model B demonstrates that HDX data can inform the manual refinement and improve homology models.
Model C (Figure 3C and Table 2) was developed in the same manner as models A and B but used 3MK7c as a template. Model C gives better agreement for the second heme-binding region, 109–127, than do models A and B (Table 2). It also decreased the number of regions of disagreement. The model points to a highly open structure, however, and it does not form the expected two cytochrome c-type folds as seen in the other models. Thus, model C, although affording better agreement in the second heme region, is not a good model overall for DHCC.
Model D (Figure 3D and Table 2) was generated from the online suites I-TASSER and Phyre2, and nearly identical results were obtained after running both I-TASSER and Phyre2. Both packages identified 3MK7c as the best template and gave highly similar alignments and consequently identical models. Model D is presented as that obtained by using Phyre2. It is consistent with the HDX results in all regions of the subunit except regions 38–44, 50–57, and 130–138 (Figure 3D and Table 2). These regions are loops in model D; however, they undergo a lower level of HDX, more typical of a rigid region. The Phyre2 software includes a disorder prediction, identifying secondary and tertiary structure (Figure 4), and a disorder prediction analysis was performed on model D. More order is predicted for all three regions of disagreement, suggesting that, although they are not α helices, there is more order in the region than can be explained by a loop. The order in these regions is likely caused by the interaction of the chains across the domains, as these regions are in the middle where the two domains interact. Thus, the regions of disagreement are actually areas of partial agreement. The region containing residues 130–138 is the only region that is inconsistent with all four models and across the three software packages, suggesting that the region is different from that seen in the templates. Taking all the evidence together, we conclude that model D best agrees with the HDX data.
Figure 4.
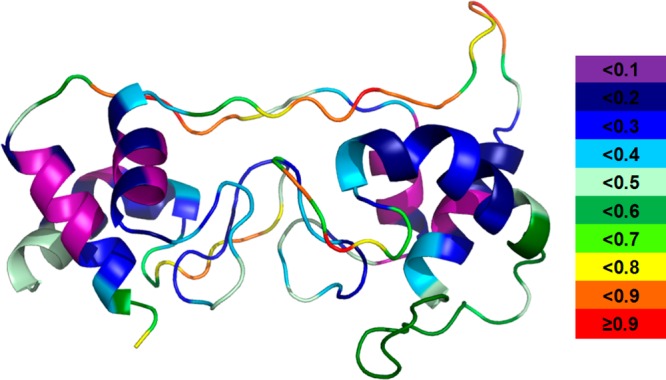
Model D with disorder prediction data. A warmer color and a larger number indicate a more disordered region. Color codes are shown on the right.
SASA Calculation Shows Model D Correlates Best with the HDX Data
A correlation of SASA at the peptide level (calculated as an average of the SASA for the individual amino acids comprising the region) and the average extents of HDX for the corresponding peptide regions confirmed our assignment of model D as the best homology model of DHCC (Figure 5 and Figure S3 of the Supporting Information). This model gave the best correlation coefficient in comparison to correlations with models A–C. In fact, SASA analysis disagreed in only one area with model D. Model C has the most open structure and, thus, larger SASA values. Overall, SASA analysis is a more quantitative assessment to illustrate that model D is the best model presented thus far.
Figure 5.
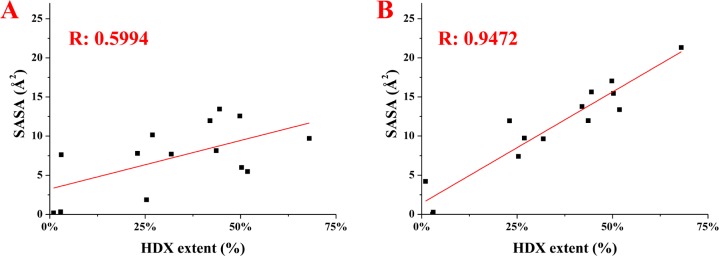
Correlation of HDX and SASA data for both model A (A) and model D (B). The Pearson correlation coefficients (R) are colored red. Similar plots for the other models are given in the Supporting Information.
Mapping of HDX Data onto Model D
To view the two redox states at the protein level, we mapped the HDX differences between the two redox states of DHCC taken as an average of the differences for each time point onto homology model D (Figure 6). The most prominent difference occurs for the short helix in the heme 2 domain (colored blue in Figure 6). In agreement with the kinetic traces, most helical and loop regions around the heme sites have larger differences, indicating that structural changes accompany the redox chemistry. Adding credibility to this conclusion is the fact that the cores of the two helix bundles show no significant changes.
Figure 6.
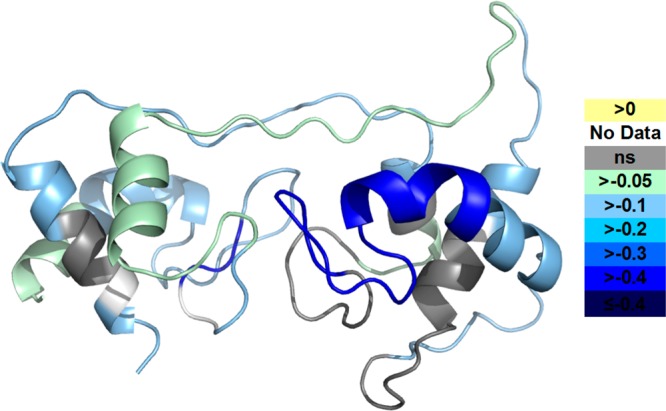
Differences in deuterium uptake between the oxidized and reduced states of DHCC are mapped onto model D. Color codes show the differences (oxidized state subtracted from reduced state).
We also mapped the extent of HDX for all the peptide regions onto our best model, model D, with a color scheme indicating the amount of exchange at each time point (Figure 7). Instead of an overall picture of kinetics information as in Figure 6, these maps show the dynamics of the protein at each time point. By comparing the same state at different times, we see that most of the regions of the oxidized state are dynamic, acquiring more and more deuterium as seen by the increasingly warmer colors.
Figure 7.

Deuterium uptake levels for all peptides are mapped with color for each exchange point onto model D. The figures for the oxidized state are in panel A and those for the reduced state in panel B. Color codes are shown at the right.
General features of the protein’s secondary structure are seen by the different HDX extents. Loop regions achieve either a level of HDX higher than that of helices or a similar level at an earlier time. Those loops undergoing less HDX are more ordered and constrained by interactions within the protein. The core regions within the helix bundle are always seen as cold colors, indicating they are so inflexible that the level of HDX is low even after incubation for 4 h. The data reveal important trends about the impact of changing redox states as well as the secondary and tertiary structural and solvent accessibility changes of DHCC.
Proposed Function of DHCC
HDX provides kinetics and dynamics information about the peptide level of DHCC in both reduced and oxidized states, whereas HM provides a potential protein structure even in the absence of a crystal structure. HM can yield only models that are as good as the information entered, and they cannot provide the high-resolution structural information from a crystal structure. The approach described here takes HM statistics and complements them with HDX results to assess model agreement. We used HDX data to refine model A and correct areas of disagreement to generate model B. Likewise, we used HDX to determine if model B were more informative, and we found that it is in better agreement with HDX than model A, demonstrating the utility of HDX data for testing and refining a homology model. Comparing all four models, we found that model D had the highest HM statistics and the best agreement with HDX, including a good correlation with SASA. We subsequently selected it as the best model for the protein structure.
Functionally, the choice of templates will bias the predicted model. The DHCCs 1ETP and 3MK7c are the best templates for structural alignments and are both soluble proteins that contain two c-type hemes. Neither protein has been chemically oxidized or reduced; thus, the template structure is probably a mix of both forms. 1ETP, from P. stutzeri, is a periplasmic diheme cytochrome c4. It does not belong to a complex but instead shuttles electrons between complexes in an electron-transport chain. The two hemes are located in different protein environments, yielding two distinct redox potentials and allowing for interheme electron transfer. Protein 3MK7c, from P. stutzeri, is a subunit of the cbb3 cytochrome oxidase. Like DHCC, 3MK7c is a soluble subunit with an N-terminal anchor. The cytochrome cbb3 complex, however, moves electrons in the opposite direction of the cytochrome bc complex. As for 1ETP, 3MK7c has two distinct redox potentials, but they differ to a greater extent than do those of 1ETP. Interheme electron transfer is still observed. On the basis of their biological roles, 3MK7c is closer functionally to DHCC. This agrees with the observation that model D accommodates better the HM parameters and the HDX data.
Numerous solution-based spectroscopic and computational studies have shown that there is a conformational change between the reduced and oxidized states of horse heart cytochrome c.40,41 These results are in apparent conflict with high-resolution crystallographic data that show the crystal structures of the two states are similar.54 One explanation is that the solid-state X-ray structure does not reflect the solution structure, which is likely to be dynamic. It was proposed that the oxidized state undergoes lower-frequency and larger-amplitude motions than the reduced state.55 Our HDX results for DHCC suggest that there are significant structural differences between the two different redox states. It is also possible that both X-ray structures represent the reduced form of the protein, produced by X-ray irradiation.
Just as the HDX kinetics suggest a redox-dependent conformational change for DHCC, they also indicate areas that are likely more solvent-accessible (less H-bonded). The heme 2 domain particularly exhibits faster exchange. It follows that the heme 2 domain interacts with the soluble electron acceptor cytochrome c553. Similarly, in vivo, the heme 1 domain would have an N-terminal helix anchoring the protein to the b and IV subunits of the bc complex. Heme 1 would also then interact with the Rieske subunit. These subunit–subunit interactions would lower the HDX rates in the heme 1 region, which is observed.
For monoheme cytochromes, electron transfer is a two-step process. After the electron donor binds with and reduces the cytochrome, the cytochrome binds the electron acceptor and passes the electron to it. DHCC, as other diheme cytochrome c’s, adds one more step, the intramolecular interheme electron transfer. In DHCCs from other species, the two hemes usually possess two distinct macroscopic redox potentials (e.g., 240 and 330 mV for cytochrome c4 from P. stutzeri). The values are consistent with the negative and positive electrostatic charges of the two domains.11 The direction of the electrostatic field within the protein gives rise to the distinct heme redox potentials and directs the flow of the electron from the lower- to higher-potential heme. The DHCC from the Heliobacterium cytochrome bc complex, as studied here, however, has only one redox midpotential,42 indicating similar chemical environments around the two heme-binding pockets. The HDX kinetic data are consistent with this observation. The significant conformational changes upon reduction, as revealed by HDX, may work to facilitate an interheme electron transfer despite the observation of a single midpoint potential in vitro. Our results point to formation of a closed structure upon reduction of the protein, providing a shorter distance between the two hemes. In P. stutzeri, the interheme electron transfer occurs upon hydrogen bond formation between the heme propionate groups and a surrounding hydrogen-bond network established by a conformational change to the reduced state, producing the shorter heme–heme distance and an altered heme environment. Such alterations change the redox potential and facilitate interheme electron transfer. It is likely that only one redox potential is observed in vitro because the heme environments are sensitive to the truncated N-terminal helix and subunit interactions that are missing in the expressed DHCC. Thus, our prediction is consistent with the in vitro data.
On the basis of the evidence discussed above, we can propose a potential complex mechanism for the H. modesticaldum bc complex (Figure 8). As for other bc1 or b6f complexes, menaquinol enters at the Qo site.56,57 Two electrons and two protons are removed, one at a time in a bifurcated electron transfer. The first electron is transferred quickly to the Rieske iron–sulfur cluster, reducing the Rieske cluster and causing it to move toward the periplasm, preventing rapid back transfer of the electron to Qo. The Rieske cluster is then in position to transfer an electron to heme 1 of DHCC. Docking of the Rieske cluster is probably the first step in inducing the redox-based conformational changes of DHCC. On the basis of the mechanism of interheme electron transfer within other diheme cytochrome c’s, reducing heme 1 changes the conformation and causes formation of a hydrogen-bonding network that permits rapid interheme electron transfer. Heme 2 then donates the electron to soluble cytochrome c553, which departs, allowing the Rieske to return to Qo. The second electron would then travel down the b heme pathway, reducing menaquinone at the Qi site. Another menaquinol docks, and the process is repeated, completing one turnover of the bc complex and translocating a net of two protons to the periplasm.
Figure 8.
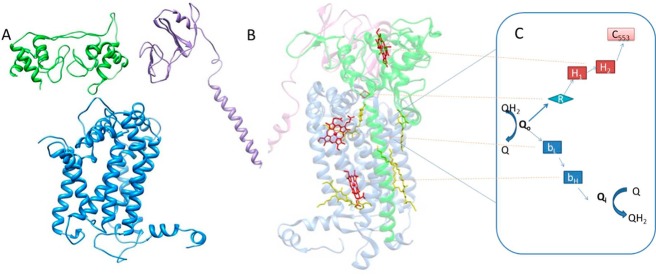
(A and B) Assumed dimer of the H. modesticaldum cytochrome bc complex: blue for the cytochrome b subunit, purple for the Fe–S subunit, and green for the cytochrome f subunit. (A) Imagined left half of dimer shown as homology-modeled subunits of the H. modesticaldum cytochrome bc complex. (B) Right side of the dimer shown as the determined crystal structure of the cytochrome bc1 complex from Rhodobacter sphaeroides used as the template for Figure 8A. Heme cofactors are colored red. (C) Proposed H. modesticaldum cytochrome bc complex bifurcated electron-transfer steps and mechanism: Q for quinones, Qo for the quinone oxidation site, Qi for the quinone reduction site, R for Rieske Fe–S, bL,H for b-type hemes, H1,2 for DHCC c-type hemes, and c553 for electron acceptor soluble cytochrome c553.
Such a mechanism is efficient for many species with a monoheme but not with a diheme cytochrome. Dihemes often result from gene duplication that could be random.6 However, multiheme systems in nature serve to transport electrons rapidly or to store electrons.58 In the mechanism proposed here, the diheme is fulfilling both of these functions. The interheme electron transfer has been observed to be very rapid compared to transfer to a donor and can be used to prevent back reaction with the Rieske cluster. Additionally, if cytochrome c553 is scarce or turning over at a rate slower than that of the bc complex, the diheme can store an electron and wait for cytochrome c553 without significantly hindering the turnover rate of the bc complex. Further study, particularly crystallographic information and inhibitor studies, will clarify these mechanistic proposals.
Conclusions
HDX data, when used in conjunction with HM and verified by SASA, improve protein structure models and allow mechanistic predictions. For DHCC, HDX kinetic analysis reveals that the oxidized state is more dynamic and open than the reduced state. This is seen in loop regions that generally undergo more extensive HDX at shorter times. The bundle core regions do not show significant differences between the redox states, suggesting that they are stable and undergo little conformational change during redox cycling. Redox-dependent conformational change facilitates the formation of potential hydrogen bonds, permitting interheme electron transfer. The dynamics data reveal faster uptake on the heme 2 domain, suggesting that it is the site of donation of electrons to cytochrome c553. This evidence is used to predict the mechanism for the bc complex of H. modesticaldum.
The study provides insight into the relation of conformational change to the binding of the electron donor Rieske subunit and soluble electron acceptor, cytochrome c553, and the function of the DHCC subunit within the bc complex of the H. modesticaldum photosynthetic electron-transfer chain. In addition, we believe that the combined approach of modeling and protein footprinting will be useful for other problems in structural biology in which protein crystal or NMR structures are lacking. Information at the amino acid level, such as what can be provided by HDX with electron capture dissociation or electron-transfer dissociation or by OH radical footprinting (e.g., FPOP), will improve the ability to distinguish models.
Acknowledgments
We acknowledge helpful discussions with and support in our utilization of HDX WorkBench from Dr. Bruce Pascal at The Scripps Research Institute (Jupiter, FL). We also acknowledge Dr. Manolo D. Plasencia for help in the calculation of SASA data.
Glossary
Abbreviations
- HDX-MS
hydrogen–deuterium exchange mass spectrometry
- HM
homology modeling
- DHCC
diheme cytochrome c
- TFA
trifluoroacetic acid.
Supporting Information Available
Figures S1–S3. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This research was supported by National Institute of General Medical Sciences Grant 8 P41 GM103422-35 to M.L.G.. H.Y. was supported by NASA Exobiology Grant NNX12AD85G to R.E.B. An Olin foundation fellowship supported E.L.-W.M.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Margoliash E. (1963) Primary Structure and Evolution of Cytochrome C. Proc. Natl. Acad. Sci. U.S.A. 50, 672–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace C. J.; Clark-Lewis I. (1992) Functional role of heme ligation in cytochrome c. Effects of replacement of methionine 80 with natural and non-natural residues by semisynthesis. J. Biol. Chem. 267, 3852–3861. [PubMed] [Google Scholar]
- Wuttke D. S.; Gray H. B. (1993) Protein Engineering as a Tool for Understanding Electron-Transfer. Curr. Opin. Struct. Biol. 3, 555–563. [Google Scholar]
- Van Beeumen J. (1991) Primary structure diversity of prokaryotic diheme cytochromes c. Biochim. Biophys. Acta 1058, 56–60. [DOI] [PubMed] [Google Scholar]
- Di Rocco G.; Battistuzzi G.; Bortolotti C. A.; Borsari M.; Ferrari E.; Monari S.; Sola M. (2011) Cloning, expression, and physicochemical characterization of a new diheme cytochrome c from Shewanella baltica OS155. JBIC, J. Biol. Inorg. Chem. 16, 461–471. [DOI] [PubMed] [Google Scholar]
- Gibson H. R.; Mowat C. G.; Miles C. S.; Li B. R.; Leys D.; Reid G. A.; Chapman S. K. (2006) Structural and functional studies on DHC, the diheme cytochrome c from Rhodobacter sphaeroides, and its interaction with SHP, the sphaeroides heme protein. Biochemistry 45, 6363–6371. [DOI] [PubMed] [Google Scholar]
- Baymann F.; Nitschke W. (2010) Heliobacterial Rieske/cytb complex. Photosynth. Res. 104, 177–187. [DOI] [PubMed] [Google Scholar]
- Kadziola A.; Larsen S. (1997) Crystal structure of the dihaem cytochrome c4 from Pseudomonas stutzeri determined at 2.2 Å resolution. Structure 5, 203–216. [DOI] [PubMed] [Google Scholar]
- Xiong J.; Inoue K.; Bauer C. E. (1998) Tracking molecular evolution of photosynthesis by characterization of a major photosynthesis gene cluster from Heliobacillus mobilis. Proc. Natl. Acad. Sci. U.S.A. 95, 14851–14856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducluzeau A. L.; Chenu E.; Capowiez L.; Baymann F. (2008) The Rieske/cytochrome b complex of Heliobacteria. Biochim. Biophys. Acta 1777, 1140–1146. [DOI] [PubMed] [Google Scholar]
- Raffalt A. C.; Schmidt L.; Christensen H. E.; Chi Q.; Ulstrup J. (2009) Electron transfer patterns of the di-heme protein cytochrome c4 from Pseudomonas stutzeri. J. Inorg. Biochem. 103, 717–722. [DOI] [PubMed] [Google Scholar]
- Sattley W. M.; Madigan M. T.; Swingley W. D.; Cheung P. C.; Clocksin K. M.; Conrad A. L.; Dejesa L. C.; Honchak B. M.; Jung D. O.; Karbach L. E.; Kurdoglu A.; Lahiri S.; Mastrian S. D.; Page L. E.; Taylor H. L.; Wang Z. T.; Raymond J.; Chen M.; Blankenship R. E.; Touchman J. W. (2008) The genome of Heliobacterium modesticaldum, a phototrophic representative of the Firmicutes containing the simplest photosynthetic apparatus. J. Bacteriol. 190, 4687–4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattley W. M.; Blankenship R. E. (2010) Insights into heliobacterial photosynthesis and physiology from the genome of Heliobacterium modesticaldum. Photosynth. Res. 104, 113–122. [DOI] [PubMed] [Google Scholar]
- Tang K. H.; Yue H.; Blankenship R. E. (2010) Energy metabolism of Heliobacterium modesticaldum during phototrophic and chemotrophic growth. BMC Microbiol. 10, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrou I.; Khan Z.; Cowgill J.; Lin S.; Brune D.; Romberger S.; Golbeck J. H.; Redding K. E. (2012) Purification of the photosynthetic reaction center from Heliobacterium modesticaldum. Photosynth. Res. 111, 291–302. [DOI] [PubMed] [Google Scholar]
- Margoliash E.; Schejter A. (1966) Cytochrome c. Adv. Protein Chem. 21, 113–286. [DOI] [PubMed] [Google Scholar]
- Salemme F. R. (1977) Structure and function of cytochromes c. Annu. Rev. Biochem. 46, 299–329. [DOI] [PubMed] [Google Scholar]
- Mines G. A.; Pascher T.; Lee S. C.; Winkler J. R.; Gray H. B. (1996) Cytochrome c folding triggered by electron transfer. Chem. Biol. 3, 491–497. [DOI] [PubMed] [Google Scholar]
- Pascher T.; Chesick J. P.; Winkler J. R.; Gray H. B. (1996) Protein folding triggered by electron transfer. Science 271, 1558–1560. [DOI] [PubMed] [Google Scholar]
- Feinberg B. A.; Liu X.; Ryan M. D.; Schejter A.; Zhang C.; Margoliash E. (1998) Direct voltammetric observation of redox driven changes in axial coordination and intramolecular rearrangement of the phenylalanine-82-histidine variant of yeast iso-1-cytochrome c. Biochemistry 37, 13091–13101. [DOI] [PubMed] [Google Scholar]
- Baddam S.; Bowler B. E. (2006) Tuning the rate and pH accessibility of a conformational electron transfer gate. Inorg. Chem. 45, 6338–6346. [DOI] [PubMed] [Google Scholar]
- Bandi S.; Baddam S.; Bowler B. E. (2007) Alkaline conformational transition and gated electron transfer with a Lys 79 → His variant of iso-1-cytochrome c. Biochemistry 46, 10643–10654. [DOI] [PubMed] [Google Scholar]
- Bandi S.; Bowler B. E. (2008) Probing the bottom of a folding funnel using conformationally gated electron transfer reactions. J. Am. Chem. Soc. 130, 7540–7541. [DOI] [PubMed] [Google Scholar]
- Takano T.; Dickerson R. E. (1981) Conformation change of cytochrome c. II. Ferricytochrome c refinement at 1.8 Å and comparison with the ferrocytochrome structure. J. Mol. Biol. 153, 95–115. [DOI] [PubMed] [Google Scholar]
- Trewhella J.; Carlson V. A.; Curtis E. H.; Heidorn D. B. (1988) Differences in the solution structures of oxidized and reduced cytochrome c measured by small-angle X-ray scattering. Biochemistry 27, 1121–1125. [DOI] [PubMed] [Google Scholar]
- Calvert J. F.; Hill J. L.; Dong A. (1997) Redox-dependent conformational changes are common structural features of cytochrome c from various species. Arch. Biochem. Biophys. 346, 287–293. [DOI] [PubMed] [Google Scholar]
- Oellerich S.; Wackerbarth H.; Hildebrandt P. (2002) Spectroscopic characterization of nonnative conformational states of cytochrome c. J. Phys. Chem. B 106, 6566–6580. [Google Scholar]
- Kelly S. M.; Jess T. J.; Price N. C. (2005) How to study proteins by circular dichroism. Biochim. Biophys. Acta 1751, 119–139. [DOI] [PubMed] [Google Scholar]
- Storaska A. J.; Neubig R. R. (2013) NMR methods for detection of small molecule binding to RGS4. Methods Enzymol. 522, 133–152. [DOI] [PubMed] [Google Scholar]
- Davis A. M.; Teague S. J.; Kleywegt G. J. (2003) Application and limitations of X-ray crystallographic data in structure-based ligand and drug design. Angew. Chem., Int. Ed. 42, 2718–2736. [DOI] [PubMed] [Google Scholar]
- Chalmers M. J.; Busby S. A.; Pascal B. D.; West G. M.; Griffin P. R. (2011) Differential hydrogen/deuterium exchange mass spectrometry analysis of protein-ligand interactions. Expert Rev. Proteomics 8, 43–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltashov I. A.; Bobst C. E.; Abzalimov R. R. (2009) H/D exchange and mass spectrometry in the studies of protein conformation and dynamics: Is there a need for a top-down approach?. Anal. Chem. 81, 7892–7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roder H.; Elove G. A.; Englander S. W. (1988) Structural characterization of folding intermediates in cytochrome c by H-exchange labelling and proton NMR. Nature 335, 700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wales T. E.; Engen J. R. (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 25, 158–170. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Smith D. L. (1993) Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Sci. 2, 522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amon S.; Trelle M. B.; Jensen O. N.; Jorgensen T. J. (2012) Spatially resolved protein hydrogen exchange measured by subzero-cooled chip-based nanoelectrospray ionization tandem mass spectrometry. Anal. Chem. 84, 4467–4473. [DOI] [PubMed] [Google Scholar]
- Liu T.; Pantazatos D.; Li S.; Hamuro Y.; Hilser V. J.; Woods V. L. Jr. (2012) Quantitative assessment of protein structural models by comparison of H/D exchange MS data with exchange behavior accurately predicted by DXCOREX. J. Am. Soc. Mass Spectrom. 23, 43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamuro Y.; Burns L.; Canaves J.; Hoffman R.; Taylor S.; Woods V. (2002) Domain organization of D-AKAP2 revealed by enhanced deuterium exchange-mass spectrometry (DXMS). J. Mol. Biol. 321, 703–714. [DOI] [PubMed] [Google Scholar]
- Hsu Y. H.; Burke J. E.; Li S.; Woods V. L. Jr.; Dennis E. A. (2009) Localizing the membrane binding region of Group via Ca2+-independent phospholipase A2 using peptide amide hydrogen/deuterium exchange mass spectrometry. J. Biol. Chem. 284, 23652–23661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmer D. D.; Kagi J. H. (1968) Hydrogen-deuterium exchange of cytochrome c. I. Effect of oxidation state. Biochemistry 7, 2710–2717. [DOI] [PubMed] [Google Scholar]
- Nabedryk-Viala E.; Thiery C.; Calvet P.; Thiery J. M. (1976) Hydrogen-isotope exchange of oxidized and reduced cytochrome c. A comparison of mass spectrometry and infrared methods. Eur. J. Biochem. 61, 253–258. [DOI] [PubMed] [Google Scholar]
- Yue H.; Kang Y.; Zhang H.; Gao X.; Blankenship R. E. (2012) Expression and characterization of the diheme cytochrome c subunit of the cytochrome bc complex in Heliobacterium modesticaldum. Arch. Biochem. Biophys. 517, 131–137. [DOI] [PubMed] [Google Scholar]
- Anand G. S.; Law D.; Mandell J. G.; Snead A. N.; Tsigelny I.; Taylor S. S.; Ten Eyck L. F.; Komives E. A. (2003) Identification of the protein kinase A regulatory RIα-catalytic subunit interface by amide H/2H exchange and protein docking. Proc. Natl. Acad. Sci. U.S.A. 100, 13264–13269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms M. J.; Eick G. N.; Goswami D.; Colucci J. K.; Griffin P. R.; Ortlund E. A.; Thornton J. W. (2013) Biophysical mechanisms for large-effect mutations in the evolution of steroid hormone receptors. Proc. Natl. Acad. Sci. U.S.A. 110, 11475–11480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu S.; Kim Y.; Li S.; Durrant E. S.; Pace R. M.; Woods V. L. Jr.; Gentry M. S. (2009) Structural insights into glucan phosphatase dynamics using amide hydrogen-deuterium exchange mass spectrometry. Biochemistry 48, 9891–9902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble A. J.; Zhang Q.; O’Donnell J.; Hariri H.; Bhattacharya N.; Marshall A. G.; Stagg S. M. (2013) A pseudoatomic model of the COPII cage obtained from cryo-electron microscopy and mass spectrometry. Nat. Struct. Mol. Biol. 20, 167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.; Freitas M. A. (2007) A mass accuracy sensitive probability based scoring algorithm for database searching of tandem mass spectrometry data. BMC Bioinf. 8, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal B. D.; Willis S.; Lauer J. L.; Landgraf R. R.; West G. M.; Marciano D.; Novick S.; Goswami D.; Chalmers M. J.; Griffin P. R. (2012) HDX workbench: Software for the analysis of H/D exchange MS data. J. Am. Soc. Mass Spectrom. 23, 1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., and Sali A. (2006) Comparative protein structure modeling using Modeller. Current protocols in bioinformatics, Chapter 5, Unit 5, 6, Wiley, New York. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschmann S.; Warkentin E.; Xie H.; Langer J. D.; Ermler U.; Michel H. (2010) The structure of cbb3 cytochrome oxidase provides insights into proton pumping. Science 329, 327–330. [DOI] [PubMed] [Google Scholar]
- Kelley L. A.; Sternberg M. J. (2009) Protein structure prediction on the Web: A case study using the Phyre server. Nat. Protoc. 4, 363–371. [DOI] [PubMed] [Google Scholar]
- Roy A.; Kucukural A.; Zhang Y. (2010) I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraczkiewicz R.; Braun W. (1998) Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comput. Chem. 19, 319–333. [Google Scholar]
- Rackovsky S.; Goldstein D. A. (1984) On the redox conformational change in cytochrome c. Proc. Natl. Acad. Sci. U.S.A. 81, 5901–5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden D.; Matthew J. B.; Rosa J. J.; Richards F. M. (1982) Increase in apparent compressibility of cytochrome c upon oxidation. Proc. Natl. Acad. Sci. U.S.A. 79, 815–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malnoe A.; Wollman F. A.; de Vitry C.; Rappaport F. (2011) Photosynthetic growth despite a broken Q-cycle. Nat. Commun. 2, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan S. S.; Yamashita E.; Baniulis D.; Cramer W. A. (2013) Quinone-dependent proton transfer pathways in the photosynthetic cytochrome b6f complex. Proc. Natl. Acad. Sci. U.S.A. 110, 4297–4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burggraf F.; Koslowski T. (2014) Charge transfer through a cytochrome multiheme chain: Theory and simulation. Biochim. Biophys. Acta 1837, 186–192. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.