

Abstract
Dendritic cell-specific intracellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN) is a C-type lectin highly expressed on the surface of antigen-presenting dendritic cells. DC-SIGN mediates interactions among dendritic cells, pathogens, and a variety of epithelia, myeloid cells, and endothelia by binding to high mannose residues on pathogenic invaders or fucosylated residues on the membranes of other immune cells. Although these interactions are normally beneficial, they can also contribute to disease. The structural characterization of binding geometries is therefore of interest as a basis for the construction of mimetics that can mediate the effects of abnormal immune response. Here, we report the structural characteristics of the interaction of the DC-SIGN carbohydrate recognition domain (CRD) with a common fucosylated entity, the LewisX trisaccharide (LeX), using NMR methods. Titration of the monomeric DC-SIGN CRD with LeX monitored by 2D NMR revealed significant perturbations of DC-SIGN cross-peak positions in 1H–15N heteronuclear single quantum coherence (HSQC) spectra and identified residues near the binding site. Additionally, saturation transfer difference (STD) and transferred nuclear Overhauser effect (trNOE) NMR experiments, using a tetrameric form of DC-SIGN, identified binding epitopes and bound conformations of the LeX ligand. The restraints derived from these multiple experiments were used to generate models for the binding of LeX to the DC-SIGN CRD. Ranking of the models based on the fit of model-based simulations of the trNOE data and STD buildup curves suggested conformations distinct from those seen in previous crystal structures. The new conformations offer insight into how differences between binding of LewisX and mannose-terminated saccharides may be propagated.
Dendritic cells (DCs) are the first participants in the long series of events in host–pathogen interactions leading to activation of specific T-cells.1,2 Dendritic cell-specific ICAM-3 grabbing nonintegrin (DC-SIGN) is a C-type lectin present mainly on the surface of immature dendritic cells.3 It is responsible for the binding and uptake of a multitude of pathogens, such as HIV-1,3 ebola virus,4 hepatitis C virus,5Candida albicans,6 and Mycobacterium tuberculosis(7) via oligomannose-dependent interactions. Upon recognition of LewisX carbohydrates, DC-SIGN also associates with distinct signaling molecules to induce differential production of cytokines that in turn lead to enhancement or suppression of proinflammatory responses.8 The mechanisms by which these diverging signals are generated are poorly understood, although it has been suggested that the molecular structure of DC-SIGN might be altered differently upon binding to the two different classes of carbohydrates. Thus, the characterization of the bound carbohydrate geometry is fundamental to understanding the interactions among DC-SIGN, other elements of immune cells, and pathogens.
In addition to its normal defensive role, DC-SIGN plays a role in the actual infection processes of some pathogens, including HIV, SIV, and hepatitis C.3,9 Therefore, many groups are developing strategies to block the sugar binding site within the DC-SIGN CRD to prevent its use by pathogens.10−13 Others are using pathogen glycan recognition to deliver materials to dendritic cells in order to harness the immune response for anticancer therapy.14 Development of these disease-related reagents also benefits from a better understanding of ligand recognition by DC-SIGN.
Structurally, DC-SIGN is a type II transmembrane protein with a short cytosolic region, a transmembrane segment, and an extended extracellular domain (ECD).15−17 This extracellular domain is divided into two structurally and functionally distinct regions: a neck region, involved in the tetramerization of the receptor,18−20 and a calcium-dependent carbohydrate recognition domain (CRD), which is at the heart of the molecular recognition processes mediated by DC-SIGN. This CRD is responsible for the interaction with highly glycosylated structures present at the surface of pathogens, mediating internalization and presentation as a part defense against invasion.1 DC-SIGN interactions also have a higher level of complexity in that they include multipoint attachment, made possible by DC-SIGN’s tetrameric state and its organization into clustered patches at the cell membrane.20−22 However, all of these begin with some fundamental difference in glycan interaction at the CRD level, and understanding this may lead to new ways of mediating the effects of autoimmune and inflammatory disease.
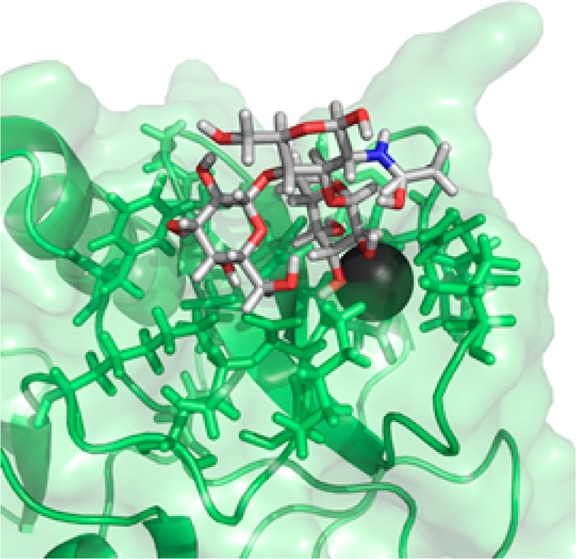
Natural ligands of DC-SIGN consist of mannose oligosaccharides often found on pathogens or fucose-containing Lewis-type determinants common to humans. In all cases, the binding occurs in a Ca2+-dependent manner.23−25 Many crystal structures of DC-SIGN bound to carbohydrate ligands have provided data on bound conformations of both mannose- and fucose-containing ligands.24,26 For Lewis oligosaccharides, most data suggest the structure to be rigid and compact, with the fucose ring stacked on top of the galactose residue.27 The conformation of LewisX carbohydrate determinants bound to antibodies was, for example, found to be compact and extremely similar to that observed for the free oligosaccharides in solution. This could suggest that the recognition and binding of the LewisX carbohydrates by their protein partners does not induce significant conformational changes.27 However, in at least one case, the general structure of the LewisX motif bound to DC-SIGN departs to some extent from that observed in solution or in antibody complexes, the crystal structure of DC-SIGN with lacto-N-fucopentose III (LNFP III, a pentasaccharide containing LewisX at the nonreducing end), raising the possibility that DC-SIGN selects a somewhat different conformation (PDB ID: 1SL5). There is also the possibility that this difference is induced by more remote interactions; the structure shows a distance of 3 Å between the O2 of the LewisX galactose and the Glu286 side chain oxygen of a DC-SIGN molecule in an adjacent unit cell (Figure 1), indicating a potential hydrogen-bonding interaction that allows the ligand to bridge between two DC-SIGN molecules.24 Given the interest in using structures of native ligands in their bound conformation to develop therapeutic mimics of carbohydrates, further investigation of bound LewisX seems to be justified.
Figure 1.
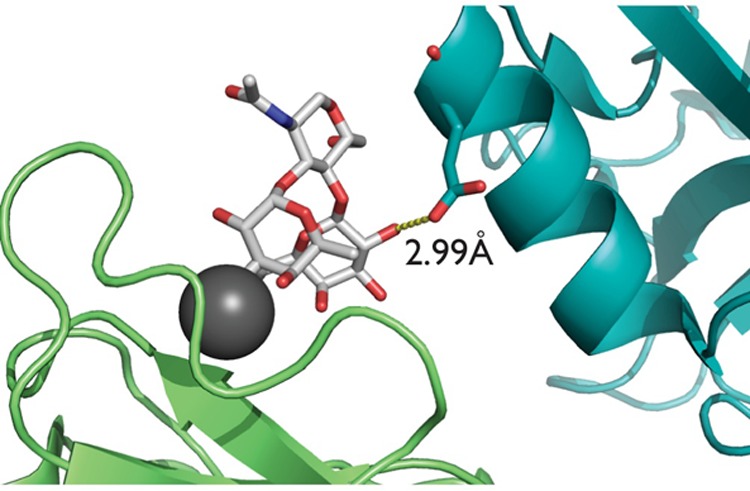
Crystal structure of DC-SIGN with a LewisX derivative shows a potential hydrogen bond between the galactose O2 and the carboxylate oxygen of Glu286 of the DC-SIGN in an adjacent unit cell (PDB ID: 1SL5).
NMR provides an alternative approach to structural investigation of protein–ligand complexes in solution and free from effects that could arise in a crystal environment. Using NMR, we present a comprehensive model of the DC-SIGN–LewisX binding interaction. To obtain an extended picture of the LewisX–DC-SIGN interaction in solution, we performed a full analysis of the interaction with the CRD by NMR spectroscopy and computational techniques. Ligand binding was analyzed during titration using cross-peaks in 1H–15N HSQC spectra to identify protein residues involved in binding and saturation transfer difference (STD) NMR to identify binding epitopes on the ligand. Transfer nuclear Overhauser effect spectroscopy (trNOESY) experiments provided additional information about the conformation of the ligand when bound to the CRD. The experimental data were used with the HADDOCK ligand docking software28 to generate ligand–protein structures, and the CORCEMA-ST protocol29 was used to predict STD intensities from the atomic coordinates of models for the ligand–protein complex and to score the various LewisX–DC-SIGN structures. These studies demonstrated that the fucose residue of LewisX strongly interacts with DC-SIGN, the galactose and fucose residues are stacked more tightly than reported in crystal structure, but the free and bound LewisX conformations still differ significantly.24,30
Materials and Methods
All enzymes and chemicals were purchased from Sigma-Aldrich unless otherwise noted. Uniformly labeled 13C-d-glucose, 15N-ammonium chloride, 15N-Ala, Phe, and Lys, and deuterium oxide (D2O) were purchased from Cambridge Isotope Laboratories. LewisX was purchased from CarboSynth.
Protein Expression
For the uniformly 15N-labeled samples, a pET28a plasmid (Novagen) containing the coding region for amino acids 250–404 of the DC-SIGN CRD was transformed into BL21(DE3) gold competent cells (Stratagene) and expressed in M9 media containing kanamycin with 15N-NH4Cl (1 g) as the sole nitrogen source at 37 °C. Once an OD600 of ∼0.5 was reached, expression was induced by addition of isopropyl-β-d-thiogalatopyranoside (IPTG) to the culture at a final concentration of 1 mM. After 6 h, the cells were harvested by centrifugation at 5000g. A 13C,15N-labeled DC-SIGN CRD sample was prepared similarly using both 15N-NH4Cl (1 g) and U-13C-glucose (2 g) as the sole sources of nitrogen and carbon, respectively.
For the 15N sparsely labeled samples, overexpression of the protein was also done using Escherichia coli BL21(DE3) gold competent cells (Stratagene) in 1 L of M9 minimal medium containing 1 g of NH4Cl and 20 mL of 20% glucose at natural abundance. All 19 amino acids except for the label of interest were added as natural abundance materials at 0.1 g/L of culture. Growth was started with a seed culture using 20 mL of the M9 minimal medium in a 125 mL Erlenmeyer flask in a 250 rpm shaker at 37 °C overnight. The following day, the seed culture was inoculated in 1 L of M9 minimal medium in a 2.8 L culture flask, and growth was continued in a 250 rpm shaker at 37 °C. At an optical cell density (OD600) ∼ 0.3, 0.1 g of the 15N amino acid of choice was added to the medium. At OD600 ∼ 0.6, 1 mL of 1 mM IPTG was added to induce protein expression. The culture was then grown overnight to OD600 ∼ 1.4 at 18 °C. The cells were harvested by centrifugation at 5000g.
Protein Purification and On-Column Refolding
Cell pellets were resuspended in pH 8.0 buffer containing 50 mM Tris and 150 mM potassium chloride and stored at −20 °C. The resuspended cells were thawed and lysed by three passages through a french pressure cell at 18 000 psi. Inclusion bodies containing DC-SIGN CRD were then isolated by centrifugation at 45 000g for 1 h at 4 °C, and the supernatant was discarded. The insoluble inclusion body pellet was washed twice with 25 mL of pH 8.0 buffer containing 50 mM Tris, 10 mM EDTA, 2% Triton X-100, 500 mM sodium chloride, and 2 M urea and once with 25 mL of pH 8.0 buffer containing 50 mM Tris and 10 mM EDTA. Each time, the suspension was homogenized, and inclusion bodies were recollected by centrifugation at 45 000g and 4 °C for 30 min. The washed inclusion bodies were resuspended in 50 mM Tris, 10 mM imidazole, 300 mM sodium chloride, 6 M guanidine hydrochloride, and 10 mM β-mercaptoethanol to a concentration of ∼5 mg/mL. One milliliter of the protein solution was then added dropwise to 20 mL of slowly stirring Ni-NTA resin. The resin was packed in a 3 × 18 cm column, and the resin-bound protein was subjected to an on-column refolding procedure as described by Veldkamp et al. with one minor modification.31 The second wash step of the refolding procedure was made a two-step process to more gradually change the redox potential by adjusting the l-glutathione reduced (GSH) to l-glutathione oxidized (GSSG) ratios from 0.5 mM GSH and 1 mM GSSG to 1 mM GSH and 0.5 mM GSSG. The protein, eluted with 25 mM Tris, 300 mM sodium chloride, 300 mM imidazole, and 2.5 mM calcium chloride, was then dialyzed twice into 1 L of pH 5 buffer containing 20 mM MES, 100 mM sodium chloride, and 2.5 mM calcium chloride to precipitate the misfolded DC-SIGN CRD. The dialyzed protein was centrifuged at 5000g for 10 min, and the soluble, properly folded protein was recovered and subsequently concentrated, giving an overall yield of ∼0.5–0.7 mg. Refolding was repeated with additional aliquots of resuspended inclusion bodies to obtain sufficient protein for the experiments.
Tetramer Sample Preparation
Additional preparations of unlabeled constructs forming a tetramer were made in the Warwick laboratory of Daniel A. Mitchell as previously described.18 Soluble recombinant DC-SIGN tetramers corresponding to the full extracellular domain were expressed in BL21/DE3 cells containing modified DC-SIGN cDNA fragments cloned into the pT5T vector and induced via the T7 promoter using 100 mg/L IPTG. Following growth at 37 °C for 150 min, cells were recovered via centrifugation and sonicated. Inclusion bodies were recovered via centrifugation and solubilized in 6 M guanidine hydrochloride, 100 mM Tris, pH 7.0, 0.01% v/v β-mercaptoethanol and centrifuged at 100g for 30 min to remove membranous debris. Protein was allowed to refold via dialysis in loading buffer (10 mM Tris, pH 7.8, 1 M NaCl, 5 mM CaCl2) with three buffer changes. Refolded protein was isolated via affinity chromatography using mannose-Sepharose, with washing in 10 column volumes of loading buffer and eluting in 10 mM Tris, 150 mM NaCl, 2.5 mM EDTA. Further purification was performed via anion-exchange chromatography using a Mono-Q column and AKTA liquid chromatography system (GE Healthcare) with loading in low-salt buffer (10 mM Tris, pH 7.8, 2.5 mM EDTA) and eluting in a linear 0–500 mM NaCl gradient with high-salt buffer (10 mM Tris, pH 7.8, 2.5 mM EDTA, 1 M NaCl). The protocol for the generation of the DC-SIGN extracellular domain, containing seven tandem neck repeats, consistently yields tetrameric complexes, as determined via equilibrium ultracentrifugation and homobifunctional cross-linking with bis-sulfosuccinimidyl suberate.17 Ultracentrifugation studies indicate that tetramer stability is high across a broad concentration range.17,18
NMR Spectroscopy
NMR spectroscopy was carried out on spectrometers operating at 21.1 or 14.0 T. The 21.1 T instrument was equipped with a Varian VNMRS console and 5 mm cryogenically cooled triple resonance probe. The 14.0 T instrument was equipped with Varian Inova console and 5 mm cryogenically cooled triple resonance probe. Once the instrument temperature had equilibrated and the optimization of field homogeneity was completed, acquisition was initiated. 2,2-Dimethyl-2-silapentane-5-sulfonate (DSS) was included as an internal reference in each sample. NMR samples consisted of 1–2 mg of protein (specific concentrations given in descriptions of individual experiments below) in 100% or 10% D2O buffer containing 25 mM Tris, 100 mM sodium chloride, 2.5 mM calcium chloride, and 50 μM DSS, pH 7.5, for the STD/trNOE and titration/assignment experiments, respectively.
Sequential backbone assignments of the U-15N,13C-labeled DC-SIGN CRD (500 μM) were made at 37 °C using three-dimensional intra- and inter-residue coherence transfer experiments, HNCA, HNCB (HNCACB with tauCC = 7 ms), and HN(CO)CACB.32−34 The assigned 1H–15N-HSQC of the DC-SIGN CRD (BMRB 19931) was used to identify residues involved in ligand binding during the titration.
The DC-SIGN CRD (100 μM) was titrated with increasing concentrations of LewisX (500 μM to 2 mM in steps of 500 μM and 5 mM to 20 mM in steps of 5 mM) and monitored by 1H–15N HSQC NMR. Total chemical shift change for each protein residue was calculated using the following formula35
Dissociation constants were determined by fitting chemical shift curves as described in Barb et al.36 and by surface plasmon resonance experiments (Supporting Information). Additionally, where significant line broadening could be observed, the off-rate for LewisX was estimated using the following formula, where Δυmax is the chemical shift change on full complexation and Δυ1/2 is the change in line width at 50% complexation37
Saturation transfer difference (STD) NMR experiments on the DC-SIGN tetramer interacting with LewisX were performed with a 50 μM monomer (12.5 μM tetramer) and a 1:100 protein/ligand ratio. The sample was irradiated at both 0 and 8 ppm, and saturation times were incremented from 1 to 4 s in steps of 1 s. Transferred nuclear Overhauser effect spectroscopy (trNOESY) experiments of the DC-SIGN tetramer with LewisX were performed with a 250 μM monomer (62.5 μM tetramer) and 1:20 protein/ligand ratio and a mixing time of 150 ms. All data were processed using NMRpipe38 and analyzed using Sparky39 and NMRViewJ.40,41 The α-anomer proved to bind more tightly, and the NOEs for this anomer were calibrated using the 2.39 Å as the N-acetyl-glucosamine (GlcNAc) H1–H2 distance. Methyl NOEs for the fucose H6 and GlcNAc N-acetyl were scaled by a factor of 3 prior to distance calculation.
Computational Docking
Models of LewisX bound to DC-SIGN were generated using HADDOCK.28 Ambiguous interaction restraints were defined for active residues identified as involved in binding using the NMR titration (protein) and STD data (ligand). Distance restraints based on trNOESY data were used to constrain the conformation of the bound ligand. Additional distance restraints based on the observed coordination of the fucose O3 and O4 to the Ca2+ ion in other DC-SIGN or DC-SIGNR structures were also included. Initially, 1000 structures were determined through rigid-body docking. Simulated annealing was performed with the 200 lowest-energy structures followed by water refinement using default force field parameters except that the radius parameter for the Ca2+ ion was increased to produce typical oxygen coordination distances. During the flexible docking, the ligand was allowed to be fully flexible. The beta strands of the protein (residues 355–367) were specified as semiflexible, and the loops of the protein near the binding pocket (residues 311–316, 344–354, and 368–374) were set as fully flexible. Semiflexible residues are allowed to introduce side chain flexibility during the third stage of simulated annealing and backbone flexibility during the fourth stage. Fully flexible residues are allowed to have backbone and side chain flexibility throughout all four stages of simulated annealing. The best structures were chosen from the refined structures based on lowest energy and fewest distance restraint violations.
Evaluation of Docked Structures
The best four DC-SIGN–LewisX complexes and the crystal structure (PDB ID: 1SL5) were evaluated using the CORCEMA-ST protocol to simulate STD build-up curves.29,42 Input parameters for CORCEMA-ST reflected the experimental conditions and also included the NMR-determined association rate constant (kon = 105 M–1 s–1), SPR-determined dissociation constant (1 mM), and estimates of the free and bound ligand correlation times (0.25 × 10–9 and 80 × 10–9, respectively). The tetramer was considered to be symmetric, and no corrections for the effects of anisotropic tumbling were applied. In order to account for selective saturation effects, proton chemical shifts of the aromatic and methyl resonances of DC-SIGN CRD were predicted using SHIFTX2.43 The resonances excited by the saturation pulse were limited to those within 10 Å of the binding pocket and with predicted chemical shifts within ±1 ppm of the saturation frequency. Experimental data were normalized relative to the intensity of a 1D 1H experiment collected immediately preceding the STD data. After normalization, the RMSD between experimental and simulated points for each resonance was calculated using the 1–4 s STD intensities. The best overall complex was determined to be the one with no RMSDs more than three times the average RMSD (of all four structures) for any given residue and the lowest overall RMSD for the entire ligand at both saturation frequencies.
Results
Chemical Shift Titration
Chemical shift perturbation of protein resonances provides a qualitative indication of residues involved in ligand interaction. Figure 2A shows a portion of the 1H–15N HSQC spectrum of the DC-SIGN CRD monomer in the presence of 2.5 mM calcium chloride. Cross-peaks are superimposed for the same DC-SIGN CRD sample with 2 mM (blue) and 20 mM (red) LewisX added. Fitting a protein chemical shift vs ligand concentration curve for DC-SIGN with LewisX (as described in the Materials and Methods) as well as SPR data indicates a binding affinity of around 1 mM (Figure S1). 1H–15N-HSQC peaks for the backbone resonances were assigned using a 13C,15N-labeled sample and traditional triple resonance experiments (BMRB 19931). Three additional samples prepared with labeling of single amino acids (15N-Lys, 15N-Phe, or 15N-Ala) facilitated assignments through association of cross-peaks with these specific amino acid types. The total chemical shift perturbation for each DC-SIGN residue is plotted in Figure 2C. The protein residues with chemical shift changes > 0.075 as well as those that disappeared upon adding ligand were deemed to be involved in binding for the purposes of docking experiments (Figure 2B). Interestingly, Val351, which has been previously implicated in LewisX binding,44 did not appear to be affected.
Figure 2.

(A) A portion of the 1H–15N-HSQC spectra for DC-SIGN with no ligand (black) or with 2 mM (blue) or 20 mM (red) LewisX are overlaid. Shifted resonances are presumed to be close to the binding site. Peaks that disappear or have the largest total chemical shift perturbation are labeled. Fitting of titration data gives a ∼1 mM dissociation constant. (B) A representation of the crystal structure of the DC-SIGN CRD (PDB ID: 1SL5), with residues perturbed by titration with LewisX identified. Residues with the largest perturbation (>0.075 ppm) are indicated in burgundy, smaller perturbations (>0.05 ppm) are indicated in yellow, and residues with peaks that disappear are indicated in blue. The residues shown in burgundy and blue (labeled) were used as ambiguous interaction restraints in HADDOCK. (C) The total chemical shift perturbation for each residue is plotted. Residues with peaks that disappear upon ligand binding are indicated by a light gray bar marked with an asterisk.
Transferred Nuclear Overhauser Effect
Unlike many ligands investigated by NMR methods, glycans can be quite flexible. Primary degrees of freedom are the torsion angles about the glycosidic bonds connecting both the galactose (Gal) and fucose (Fuc) to N-acetyl-glucosamine (GlcNAc), and these can be altered on interaction with the protein. Transferred nuclear Overhauser effect spectroscopy (trNOESY) provides insight into the protein bound ligand conformation because of the heavy weighting of NOEs by the longer correlation time of the complexed ligand compared to that of the free ligand. To improve reporting, we have used the more slowly tumbling tetrameric (CRD plus stem) form of DC-SIGN with an effective molecular weight of 160 kDa as opposed to 20 kDa for the DC-SIGN CRD. In Table 1, we report distances between pairs of protons affected by glycosidic torsion angles derived from trNOEs and compare them to the distances seen in the crystal structure of 1SL5(24) and in the conformations believed to exist in free solution.30 There is some disagreement between the distances derived for the bound LewisX in the crystal structure and the distances indicated by trNOE measurements (Table 1). In particular, a long-range NOE was measured between galactose H2 and fucose H6, and the derived distance (4.7 Å) was found to be larger than that in the crystal structure (3.9 Å). Also, the trNOE-derived distance of 2.6 Å between fucose H1 and GlcNAc N-acetyl is much shorter than the crystal structure (3.7 Å). The NOEs measured across the glycosidic linkages also indicate some deviation from angles in the crystal structure. The trNOE distances determined for a bound LewisX conformation also differ significantly from the free conformation recently measured by solution NMR by Zierke et al. (Table 1), indicating preferred binding of a conformer different from the dominant solution conformer.30 These experimental observations provided additional restraints on poses determined by docking algorithms.
Table 1. Transglycosidic Distances Measured in Crystal Structure 1SL5,24 Free LewisX,30 and by trNOEa.
| residue and atom | residue and atom | PDB (Å) | trNOE (Å) | free (Å) |
|---|---|---|---|---|
| Fuc H1 | GlcNAc H2 | 3.5 | ||
| Fuc H1 | GlcNAc H3 | 2.6 | 2.7 (+0.75) | 2.8 |
| Fuc H1 | GlcNAc H4 | 4.5 | ||
| Fuc H1 | GlcNAc HNAc | 3.7 | 2.6 (+0.75) | 3.8 |
| Fuc H5 | GlcNAc H3 | 3.1 | 3.0 (+0.75) | |
| Fuc H6 | GlcNAc H3 | 4.4 | 3.1 (+1.50) | |
| Fuc H6 | Gal H2 | 3.6 | 4.7 (+1.75) | 2.8 |
| Gal H1 | GlcNAc H3 | 4.5 | 2.7 (+0.75) | |
| Gal H1 | GlcNAc H4 | 2.4 | 2.5 (+0.75) | 2.3 |
| Gal H1 | GlcNAc H5 | 3.8 |
trNOE distances were used in HADDOCK docking of LewisX to the DC-SIGN CRD. In HADDOCK, the lower error for all trNOE distances was defined to make the minimum distance 1.80 Å; upper errors are given in parentheses.
Saturation Transfer Difference NMR
Saturation transfer difference spectra can give a qualitative identification of binding epitopes on ligands of interest. During the experiment, saturation of magnetization of protons on the protein is transferred to protons of the bound ligand in roughly a 1/r6 fashion. On returning to the solution state, the ligand carries this saturation information, where it can be observed as a reduction in ligand resonance intensities, even with ligand in great excess. Frequently, steady-state reductions in ligand resonance intensities are taken to reflect proximity to the protein surface, with the larger intensity loss taken to correspond to a smaller distance between particular ligand protons and the surface of the protein. However, a number of other factors can affect the intensity of ligand resonances, including the time the ligand remains bound to the protein and the actual distribution of transferring protons on the protein surface. Longer residence times allow diffusion of the saturation throughout the ligand, making identification of binding epitopes less definitive (this makes tight-binding complexes ill-suited to this technique). Proton-poor regions of the protein binding site may also produce less intensity loss on the ligand. Such effects make it important to consider interpretation of STDs at both qualitative and more quantitative levels.
STD intensity loss for various LewisX protons at a series of saturation times are given in Figure 3A. To allow a qualitative interpretation, the STD values at the longest time (4 s) were mapped onto the LewisX structure as shown in Figure 3B, where red indicates large STD intensity loss and purple indicates low STD intensity loss. These results indicate that the fucose residue is most proximate to the protein surface, with H2 experiencing the strongest perturbation of any part of the ligand and H1 and H4 also experiencing strong perturbations. The N-acetyl group of the GlcNAc also demonstrates close proximity to the protein surface, whereas the remainder of GlcNAc and the galactose residue experience smaller perturbations. This qualitative identification of interacting parts of the ligand provides another source of information for docking. However, because of the complexities mentioned above, more detailed analysis of STD data was implemented as a means of scoring docked poses.
Figure 3.

(A) Experimental STD build up for saturation at both 0 and 8 ppm with saturation times from 1 to 4 s. (B) Representation of LewisX with the average intensity of maximum STD transfer (normalized relative to a 1D 1H experiment) shown on a color scale with red as the most intense and purple as the least. (C) RMSD between the simulated and the normalized experimental STD intensities for each resolved resonance.
Computational Docking
The determination of the structure for the DC-SIGN CRD–LewisX complex was accomplished using the docking software, HADDOCK.28 This package is well-suited to this type of problem because it allows use of more qualitative data, such as those coming from STD-based identification of binding epitopes on the ligand and chemical shift-based identification of residues in the protein binding pocket. In addition, it allows entry of pairwise distance restraints from trNOE data. One additional piece of information was used. The location of the fucose within the binding pocket was further constrained (2.5 ± 0.2 Å) using well-established distances for the fucose O3 and O4 to the Ca2+ ion. These are constant through many of the structures involving fucose binding and are not likely to change significantly in solution. The data entered into the HADDOCK procedure are indicated in Table 1 and Figure 2. Procedures for docking are described in the Materials and Methods section.
Evaluation of Docked Poses
HADDOCK provides scoring of various poses based on interaction energy and clustering of repeatedly recurring poses. However, once a set of proposed structures for the complex are provided, STD data can be turned to a more quantitative use by simulating STD build-up curves based on the exact location of protein and ligand protons in each pose. We used simulations generated using the CORCEMA-ST package to do additional scoring.29 Figure 3C compares back-calculated STD build ups using various poses to experimental data. Figures displaying resulting structures were generated using Chimera (http://www.cgl.ucsf.edu/chimera).45
Simulation of STD intensities based on proton positions for the protein and LewisX in the crystal structure indicated reasonable agreement between the crystal structure and the experimental data, as characterized by low overall RMSD between the normalized observed and simulated STD intensities measured for 1–4 s saturation times. However, the RMSDs for fucose H1 and H2 during saturation at 0 ppm were quite large (Figure 3C). Four HADDOCK structures with no distance restraint violations greater than 0.5 Å and total energies and HADDOCK scores in the lowest 15% were chosen for STD simulation. Although all four HADDOCK structures have significantly improved fit to the fucose H1 and H2 data, only structures 1, 3, and 4 have low overall RMSD between the experimental and simulated STD data. Structures 1 and 3 have outliers more than three times the average RMSD (Figure 3C). Furthermore, structure 3 has an unusual LewisX conformation missing the favorable fucose–galactose stacking interaction. We regard the remaining HADDOCK structure (structure 4) as our best representation of the solution-bound structure.
Discussion
To date, there have been many crystal structures of the DC-SIGN CRD binding to both oligomannose and LewisX ligands,16,24 as well as some showing binding to the CRD plus stalk fragments, that are oligomeric.18,26 These structures have demonstrated that both mannoside and LewisX ligands bind in the same pocket of the DC-SIGN CRD but interact with different sets of residues within the binding pocket. However, potential interactions between the ligand and the DC-SIGN molecule in an adjacent unit cell have been observed in many of these structures, raising questions about the possible influence of these interactions on the conformations seen in the binding site. In this study, we used NMR titration and STD to probe the ligand–protein binding interface as it exists in solution and combined information from those experiments with bound ligand conformation information from trNOE experiments. Using these restraints, we were able to model the DC-SIGN–LewisX complex using HADDOCK28 and validate these structures against the experimental STD data using CORCEMA-ST.29
C-type lectins, which contain an EPN (Glu-Pro-Asn) tripeptide motif, are known to preferentially bind mannose, fucose, N-acetyl-glucosamine, and glucose, but they usually have little preference for larger oligosaccharides.46,47 DC-SIGN has an EPN motif and binds some of these monosaccharides, but, like several other EPN-containing C-type lectins, it has enhanced binding for certain higher-order oligosaccharides. In particular, it binds 2α-mannobiose in preference to mannose; it also has a preference for the LewisX trisaccharide over fucose, whereas other C-type lectins prefer the fucose monosaccharide. This indicates that DC-SIGN may also be interacting with the branching GlcNAc or even terminal galactose despite the absence of a common galactose interaction motif (Gln-Pro-Asp).48,49
Crystal structures of the DC-SIGN CRD with LewisX bound have consistently shown the fucose residue to be bound to the Ca2+ ion within the binding pocket. Binding to Ca2+ via fucose is not surprising since there is evidence that this association is present even for free fucose in solution.50 In most cases, interactions with other sugars in fucose-containing oligosaccharides appear to be minor. There is one LewisX case in which the galactose is near the protein surface, but the GlcNAc in this structure (PDB ID: 1SL5) is oriented away from the protein.24 However, this crystal structure (1SL5) also shows a distance of only 3 Å between galactose O2 of the LewisX and the Glu286 side chain oxygen of an adjacent DC-SIGN.24 This close contact is typical of hydrogen bonding and could indicate distortion of the preferred binding orientation and conformation of the ligand.
The best HADDOCK structure, based on docking and STD scoring, is overlaid with the crystal structure in Figure 4A. There are many similarities, but there are important differences. The HADDOCK structure brings the galactose residue down into the binding pocket, which also pulls the GlcNAc down closer to the protein. As mentioned earlier, one of the key disagreements between the simulated STD data for the crystal structure and the experimental STD data is a large RMSD for fucose H1 and H2. In particular, the simulated values were much higher than those observed experimentally. In the HADDOCK structure, the fucose has been tilted slightly away from the protein, resulting in lower simulated STD intensities in much better agreement with the STD data.
Figure 4.
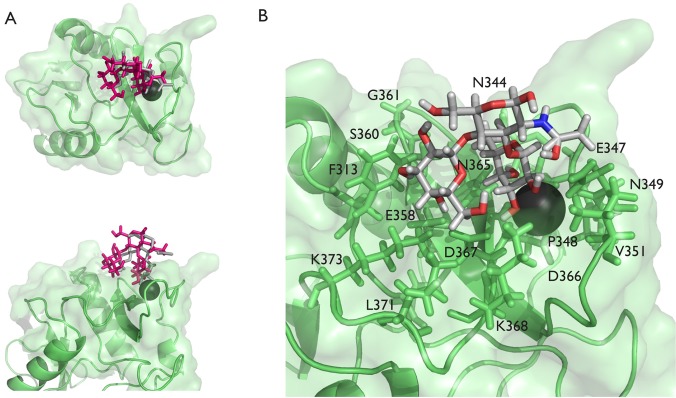
(A) Crystal structure of the DC-SIGN CRD (green) with LewisX (gray) bound (PDB ID: 1SL5). The conformation of the bound LewisX in the best HADDOCK structure (pink) is overlaid into the binding site. (B) Expanded view of the binding pocket for the best HADDOCK structure with interacting residues within 0.4 Å of van der Waals contact shown as sticks and labeled. The fucose of LewisX is the ligand residue closest to the calcium ion, the GlcNAc is above the fucose, and the galactose is to the left.
Figure 4B shows an expanded version of the binding pocket with primary interacting residues highlighted. Despite the altered positioning, several potential favorable interactions are suggested. Many of these involve hydrogen-bonding contacts, for example, between fucose hydroxyl protons (HO4 and HO3) to carboxylate or amide oxygens of D367, N365, and E347. Side chain terminal protons of K368, N349, and N365 are also in a position to hydrogen bond with fucose oxygens, O2, O3, or O4. The slight move of the GlcNAc residue toward the surface of the protein does suggest a better hydrophobic contact between the N-acetyl methyl of GlcNAc and a methyl of V351 (2.4 Å proton to proton). Although we did not see a chemical shift perturbation of the backbone amide resonances of V351, close contact does not necessarily produce shift changes. Additionally, the galactose, which appeared to be interacting with an adjacent DC-SIGN molecule in the crystal structure, is now well anchored into the protein surface. Important contacts include possible hydrogen bonds between Gal O6, O4, and HO4 and side chain protons of K368, K373, and D367 respectively. Although not oriented optimally for hydrophobic contacts, the side chain of F313 makes close contacts on one side of the galactose, whereas the side chain of L371 makes probable hydrophobic contacts on the other side (C6 methylene protons). These favorable contacts may help to explain why DC-SIGN preferentially binds to the LewisX trisaccharide over the fucose monosaccharide. The other protein residues identified as being involved in binding by a large chemical shift perturbation or disappearance are also in close proximity to the LewisX.
As shown in Table 1, the conformation of the bound LewisX molecule is also significantly different from what is observed in solution. Compared to the conformation of free LewisX determined by Zierke et al. (also based on solution NMR data), our distances between fucose H1 and the N-acetyl methyl of GlcNAc are shorter, and our distances between fucose H6 and Gal H2 are longer.30 The latter deviation is seen in the crystal structure as well. There are other solution studies of LewisX-containing ligands bound to proteins that suggest only minor variations in bound geometry from solution geometry. Studies of sialyl-LewisX binding to P-, E-, and L-selectin, for example, show only the glycosidic linkage of the sialic acid to galactose to change upon binding.51 However, the interaction of selectins with sialyl-LewisX is influenced strongly by sialic acid binding, in contrast to the interaction directly with the fucose in the DC-SIGN–LewisX case.
We cannot exclude the existence of other bound conformations, including that found in solution or that shown in the crystal structure. Much of the binding data is qualitative, and significant deviations between the more quantitative STD data and its simulation remain. Both the ligand and protein interface exhibit significant conformational freedom, and experimental data may well represent an average over several sampled structures. Nevertheless, the docked conformation shown must be a major contributor to this average. It has reasonable molecular interactions and presents a best fit to available data.
An important aspect of the structure is the direction suggested for linkage to additional sugars found in potential native ligands, in other words, the direction in which the reducing end GlcNAc leaves the CRD. In our structure, the GlcNAc residue is constrained in orientation by both its own interactions with the protein and the bidentate interaction of the Gal and Fuc residues with the protein. The orientation of the reducing end GlcNAc anomeric oxygen is out and slightly to the right, as depicted in Figure 5. In contrast, while the 3,4 hydroxyl oxygens of the bound mannose of Man4 in structure 1SL4 are involved in Ca2+ coordination,24 much like fucose, the reducing end of the ligand, when the protein is similarly oriented, extends to the left. These differences in orientation are depicted in Figure 5. In a more biological context, the multiple CRDs of the membrane-bound DC-SIGN tetramer must interact with multiple glycans displayed on a membrane surface. Here, relative directions in which CRD binding sites are oriented must mesh with cell-surface glycan density and conformational distributions. Differences in reducing end directions could, therefore, have implications for how mannoside vs fucoside ligands on natural targets such as bacteria, viruses, and immune cells are differentiated.
Figure 5.
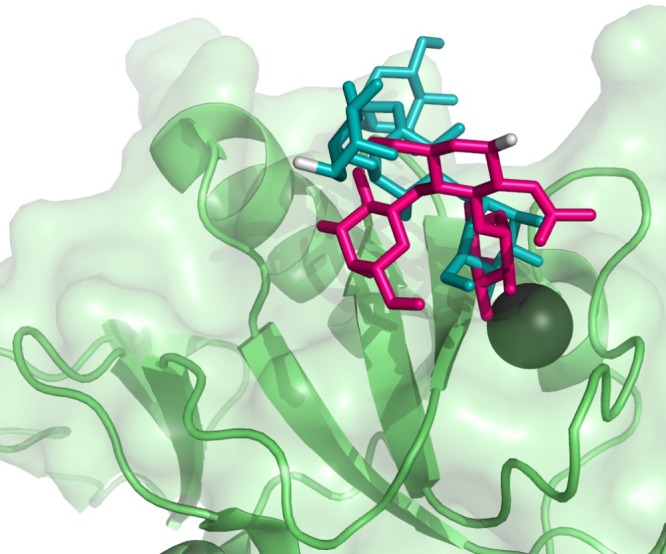
Crystal structure of the DC-SIGN CRD (green) with Man4 (teal) bound (PDB ID: 1SL4). The conformation of the bound LewisX in the best HADDOCK structure (pink) is overlaid into the binding site. The reducing ends of the ligands (highlighted in gray) are oriented in different directions, which could affect the binding of larger glycosylated targets containing these terminal ligands, especially to full DC-SIGN tetrameric receptors as presented on cell membranes.
Conclusions
Solution-based NMR data combined with molecular docking has produced an improved picture of how the LewisX trisaccharide binds to the CRD of DC-SIGN. In addition to commonly used restraints based on STD identification of ligand epitopes and chemical shift perturbation identification of residues in the binding pocket, simulation of STD build-up curves proved to be a useful way of scoring docked poses. The selected structure satisfies experimental data and produces a physically reasonable set of molecular interactions. The structure does depart significantly from that shown in a previously determined crystal structure; however, that structure had additional interaction between the ligand and a protein molecule in an adjacent unit cell that may explain the departure. The changes in the way the reducing end of the trisaccharide emerges from the binding site could have implications for target recognition in the larger context of tetrameric DC-SIGN interacting with glycans displayed on a membrane surface.
Acknowledgments
Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
Supporting Information Available
Surface plasmon resonance experimental methods and results. This material is available free of charge via the Internet at http://pubs.acs.org.
The research reported was initiated with support of a bridging grant from the Consortium for Functional Glycomics. Continuing support was provided by the National Institute of General Medical Sciences of the National Institutes of Health under award nos. R01GM33225 and P41GM103390. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Banchereau J.; Steinman R. M. (1998) Dendritic cells and the control of immunity. Nature 392, 245–252. [DOI] [PubMed] [Google Scholar]
- Bell D.; Young J. W.; Banchereau J. (1999) Dendritic cells. Adv. Immunol. 72, 255–324. [DOI] [PubMed] [Google Scholar]
- Geijtenbeek T. B. H.; Kwon D. S.; Torensma R.; van Vliet S. J.; van Duijnhoven G. C. F.; Middel J.; Cornelissen I. L. M. H. A.; Nottet H. S. L. M.; KewalRamani V. N.; Littman D. R.; Figdor C. G.; van Kooyk Y. (2000) DC-SIGN, a dendritic cell–specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 100, 587–597. [DOI] [PubMed] [Google Scholar]
- Alvarez C. P.; Lasala F.; Carrillo J.; Muñiz O.; Corbí A. L.; Delgado R. (2002) C-type lectins DC-SIGN and L-SIGN mediate cellular entry by ebola virus in cis and in trans. J. Virol. 76, 6841–6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig I. S.; Lekkerkerker A. N.; Depla E.; Bosman F.; Musters R. J. P.; Depraetere S.; van Kooyk Y.; Geijtenbeek T. B. H. (2004) Hepatitis C virus targets DC-SIGN and L-SIGN to escape lysosomal degradation. J. Virol. 78, 8322–8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambi A.; Gijzen K.; de Vries I. J. M.; Torensma R.; Joosten B.; Adema G. J.; Netea M. G.; Kullberg B.-J.; Romani L.; Figdor C. G. (2003) The C-type lectin DC-SIGN (CD209) is an antigen-uptake receptor for Candida albicans on dendritic cells. Eur. J. Immunol. 33, 532–538. [DOI] [PubMed] [Google Scholar]
- Geijtenbeek T. B. H.; van Vliet S. J.; Koppel E. A.; Sanchez-Hernandez M.; Vandenbroucke-Grauls C. M. J. E.; Appelmelk B.; van Kooyk Y. (2003) Mycobacteria target DC-SIGN to suppress dendritic cell function. J. Exp. Med. 197, 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gringhuis S. I.; den Dunnen J.; Litjens M.; van der Vlist M.; Geijtenbeek T. B. H. (2009) Carbohydrate-specific signaling through the DC-SIGN signalosome tailors immunity to Mycobacterium tuberculosis, HIV-1 and Helicobacter pylori. Nat. Immunol. 10, 1081–1088. [DOI] [PubMed] [Google Scholar]
- van Kooyk Y.; Geijtenbeek T. B. H. (2003) DC-SIGN: escape mechanism for pathogens. Nat. Rev. Immunol. 3, 697–709. [DOI] [PubMed] [Google Scholar]
- Tabarani G.; Reina J. J.; Ebel C.; Vivès C.; Lortat-Jacob H.; Rojo J.; Fieschi F. (2006) Mannose hyperbranched dendritic polymers interact with clustered organization of DC-SIGN and inhibit gp120 binding. FEBS Lett. 580, 2402–2408. [DOI] [PubMed] [Google Scholar]
- Martinez-Avila O.; Hijazi K.; Marradi M.; Clavel C.; Campion C.; Kelly C.; Penades S. (2009) Gold manno-glyconanoparticles: multivalent systems to block HIV-1 gp120 binding to the lectin DC-SIGN. Chem.—Eur. J. 15, 9874–9888. [DOI] [PubMed] [Google Scholar]
- Becer C. R.; Gibson M. I.; Geng J.; Ilyas R.; Wallis R.; Mitchell D. A.; Haddleton D. M. (2010) High-affinity glycopolymer binding to human DC-SIGN and disruption of DC-SIGN interactions with HIV envelope glycoprotein. J. Am. Chem. Soc. 132, 15130–15132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciobanu M.; Huang K.-T.; Daguer J.-P.; Barluenga S.; Chaloin O.; Schaeffer E.; Mueller C. G.; Mitchell D. A.; Winssinger N. (2011) Selection of a synthetic glycan oligomer from a library of DNA-templated fragments against DC-SIGN and inhibition of HIV gp120 binding to dendritic cells. Chem. Commun. 47, 9321–9323. [DOI] [PubMed] [Google Scholar]
- Garcia-Vallejo J. J.; Koning N.; Ambrosini M.; Kalay H.; Vuist I.; Sarrami-Forooshani R.; Geijtenbeek T. B. H.; van Kooyk Y. (2013) Glycodendrimers prevent HIV transmission via DC-SIGN on dendritic cells. Int. Immunol. 25, 221–233. [DOI] [PubMed] [Google Scholar]
- Tabarani G.; Thépaut M.; Stroebel D.; Ebel C.; Vivès C.; Vachette P.; Durand D.; Fieschi F. (2009) DC-SIGN neck domain is a pH-sensor controlling oligomerization: SAXS and hydrodynamic studies of extracellular domain. J. Biol. Chem. 384, 21229–21240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg H.; Mitchell D. A.; Drickamer K.; Weis W. I. (2001) Structural basis for selective recognition of oligosaccharides by DC-SIGN and DC-SIGNR. Science 294, 2163–2166. [DOI] [PubMed] [Google Scholar]
- Mitchell D. A.; Fadden A. J.; Drickamer K. (2001) A novel mechanism of carbohydrate recognition by the C-type lectins DC-SIGN and DC-SIGNR. J. Biol. Chem. 276, 28939–28945. [DOI] [PubMed] [Google Scholar]
- Feinberg H.; Guo Y.; Mitchell D. A.; Drickamer K.; Weis W. I. (2005) Extended neck regions stabilize tetramers of the receptors DC-SIGN and DC-SIGNR. J. Biol. Chem. 280, 1327–1335. [DOI] [PubMed] [Google Scholar]
- Yu Q. D.; Oldring A. P.; Powlesland A. S.; Tso C. K. W.; Yang C.; Drickamer K.; Taylor M. E. (2009) Autonomous tetramerization domains in the glycan-binding receptors DC-SIGN and DC-SIGNR. J. Mol. Biol. 387, 1075–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon S.; Rosenberg K.; Graham S. A.; Ward E. M.; Taylor M. E.; Drickamer K.; Leckband D. E. (2009) Binding-site geometry and flexibility in DC-SIGN demonstrated with surface force measurements. Proc. Natl. Acad. Sci. U.S.A. 106, 11524–11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambi A.; de Lange F.; van Maarseveen N. M.; Nijhuis M.; Joosten B.; van Dijk E. M. H. P.; de Bakker B. I.; Fransen J. A. M.; Bovee-Geurts P. H. M.; van Leeuwen F. N.; van Hulst N. F.; Figdor C. G. (2004) Microdomains of the C-type lectin DC-SIGN are portals for virus entry into dendritic cells. J. Cell Biol. 164, 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bakker B. I.; de Lange F.; Cambi A.; Korterik J. P.; van Dijk E. M. H. P.; van Hulst N. F.; Figdor C. G.; Garcia-Parajo M. F. (2007) Nanoscale organization of the pathogen receptor DC-SIGN mapped by single-molecule high-resolution fluorescence microscopy. ChemPhysChem 8, 1473–1480. [DOI] [PubMed] [Google Scholar]
- van Liempt E.; Bank C. M. C.; Mehta P.; Garcıá-Vallejo J. J.; Kawar Z. S.; Geyer R.; Alvarez R. A.; Cummings R. D.; van Kooyk Y.; van Die I. (2006) Specificity of DC-SIGN for mannose- and fucose-containing glycans. FEBS Lett. 580, 6123–6131. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Feinberg H.; Conroy E.; Mitchell D. A.; Alvarez R.; Blixt O.; Taylor M. E.; Weis W. I.; Drickamer K. (2004) Structural basis for distinct ligand-binding and targeting properties of the receptors DC-SIGN and DC-SIGNR. Nat. Struct. Mol. Biol. 11, 591–598. [DOI] [PubMed] [Google Scholar]
- Appelmelk B. J.; van Die I.; van Vliet S. J.; Vandenbroucke-Grauls C. M. J. E.; Geijtenbeek T. B. H.; van Kooyk Y. (2003) Cutting edge: carbohydrate profiling identifies new pathogens that interact with dendritic cell-specific ICAM-3-grabbing nonintegrin on dendritic cells. J. Immunol. 170, 1635–1639. [DOI] [PubMed] [Google Scholar]
- Feinberg H.; Castelli R.; Drickamer K.; Seeberger P. H.; Weis W. I. (2007) Multiple modes of binding enhance the affinity of DC-SIGN for high mannose N-linked glycans found on viral glycoproteins. J. Biol. Chem. 282, 4202–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuriev E.; Farrugia W.; Scott A. M.; Ramsland P. A. (2005) Three-dimensional structures of carbohydrate determinants of Lewis system antigens: implications for effective antibody targeting of cancer. Immunol. Cell Biol. 83, 709–717. [DOI] [PubMed] [Google Scholar]
- Dominguez C.; Boelens R.; Bonvin A. M. J. J. (2003) HADDOCK: a protein–protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737. [DOI] [PubMed] [Google Scholar]
- Jayalakshmi V.; Krishna N. R. (2002) Complete relaxation and conformational exchange matrix (CORCEMA) analysis of intermolecular saturation transfer effects in reversibly forming ligand–receptor complexes. J. Magn. Reson. 155, 106–118. [DOI] [PubMed] [Google Scholar]
- Zierke M.; Smiesko M.; Rabbani S.; Aeschbacher T.; Cutting B.; Allain F. H.-T.; Schubert M.; Ernst B. (2013) Stabilization of branched oligosaccharides: LewisX benefits from a nonconventional C–H···O hydrogen bond. J. Am. Chem. Soc. 135, 13464–13472. [DOI] [PubMed] [Google Scholar]
- Veldkamp C. T.; Peterson F. C.; Hayes P. L.; Mattmiller J. E.; Haugner J. C. III; de la Cruz N.; Volkman B. F. (2007) On-column refolding of recombinant chemokines for NMR studies and biological assays. Protein Expression Purif. 52, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzesiek S.; Bax A. (1992) Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J. Am. Chem. Soc. 114, 6291–6293. [Google Scholar]
- Wittekind M.; Mueller L. (1993) HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the alpha- and beta-carbon resonances in proteins. J. Magn. Reson., Ser. B 101, 201–205. [Google Scholar]
- Kay L. E.; Ikura M.; Tschudin R.; Bax A. (1990) Three-dimensional triple-resonance NMR spectroscopy of isotopically enriched proteins. J. Magn. Reson. 89, 496–514. [DOI] [PubMed] [Google Scholar]
- Williamson M. P. (2013) Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 73, 1–16. [DOI] [PubMed] [Google Scholar]
- Barb A. W.; Wang X.; Prestegard J. H. (2013) Refolded recombinant Siglec5 for NMR investigation of complex carbohydrate binding. Protein Expression Purif. 88, 183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris R. K. (1986) Chemical exchange and quadrupolar effects, In Nuclear Magnetic Resonance Spectroscopy, pp 119–130, Longman Scientific & Technical, Essex, England. [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Goddard T. D., and Keneller D. G. (2006) Sparky 3, University of California, San Francisco, CA. [Google Scholar]
- Johnson B. A. (2004) Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 278, 313–352. [DOI] [PubMed] [Google Scholar]
- Johnson B. A.; Blevins R. A. (1994) NMRView: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614. [DOI] [PubMed] [Google Scholar]
- Moseley H. N. B.; Curto E. V.; Krishna N. R. (1995) Complete relaxation and conformational exchange matrix (CORCEMA) analysis of NOESY spectra of interacting systems; two-dimensional transferred NOESY. J. Magn. Reson., Ser. B 108, 243–261. [DOI] [PubMed] [Google Scholar]
- Han B.; Liu Y.; Ginzinger S.; Wishart D. (2011) SHIFTX2: significantly improved protein chemical shift prediction. J. Biomol. NMR 50, 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thépaut M.; Guzzi C.; Sutkeviciute I.; Sattin S.; Ribeiro-Viana R.; Varga N.; Chabrol E.; Rojo J.; Bernardi A.; Angulo J.; Nieto P. M.; Fieschi F. (2013) Structure of a glycomimetic ligand in the carbohydrate recognition domain of C-type lectin DC-SIGN. Structural requirements for selectivity and ligand design. J. Am. Chem. Soc. 135, 2518–2529. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Lee R. T.; Ichikawa Y.; Fay M.; Drickamer K.; Shao M. C.; Lee Y. C. (1991) Ligand-binding characteristics of rat serum-type mannose-binding protein (MBP-A). Homology of binding site architecture with mammalian and chicken hepatic lectins. J. Biol. Chem. 266, 4810–4815. [PubMed] [Google Scholar]
- Engering A. J.; Cella M.; Fluitsma D.; Brockhaus M.; Hoefsmit E. C. M.; Lanzavecchia A.; Pieters J. (1997) The mannose receptor functions as a high capacity and broad specificity antigen receptor in human dendritic cells. Eur. J. Immunol. 27, 2417–2425. [DOI] [PubMed] [Google Scholar]
- Drickamer K. (1992) Engineering galactose-binding activity into a C-type mannose-binding protein. Nature 360, 183–186. [DOI] [PubMed] [Google Scholar]
- Lee R. T.; Hsu T.-L.; Huang S. K.; Hsieh S.-L.; Wong C.-H.; Lee Y. C. (2011) Survey of immune-related, mannose/fucose-binding C-type lectin receptors reveals widely divergent sugar-binding specificities. Glycobiology 21, 512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodet G.; Poggi L.; Abergel D.; Gourmala C.; Dong D.; Zhang Y.; Mallet J.-M.; Bodenhausen G. (2007) Weak calcium-mediated interactions between Lewis X-related trisaccharides studied by NMR measurements of residual dipolar couplings. J. Am. Chem. Soc. 129, 9080–9085. [DOI] [PubMed] [Google Scholar]
- Poppe L.; Brown G. S.; Philo J. S.; Nikrad P. V.; Shah B. H. (1997) Conformation of sLex tetrasaccharide, free in solution and bound to E-, P-, and L-selectin. J. Am. Chem. Soc. 119, 1727–1736. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.