

Abstract
The metabolic syndrome (MS) is characterized by insulin resistance, dyslipidemia and hypertension. It is associated with increased risk of cardiovascular diseases and type-2 diabetes. Consumption of fructose is linked to increased prevalence of MS. Ursodeoxycholic acid (UDCA) is a steroid bile acid with antioxidant, anti-inflammatory activities and has been shown to improve insulin resistance. The current study aims to investigate the effect of UDCA (150 mg/kg) on MS induced in rats by fructose administration (10%) in drinking water for 12 weeks. The effects of UDCA were compared to fenofibrate (100 mg/kg), an agonist of PPAR-α receptors. Treatment with UDCA or fenofibrate started from the 6th week after fructose administration once daily. Fructose administration resulted in significant increase in body weight, elevations of blood glucose, serum insulin, cholesterol, triglycerides, advanced glycation end products (AGEs), uric acid levels, insulin resistance index and blood pressure compared to control rats. Moreover, fructose increased oxidative stress in aortic tissues indicated by significant increases of malondialdehyde (MDA), expression of iNOS and reduction of reduced glutathione (GSH) content. These disturbances were associated with decreased eNOS expression, increased infiltration of leukocytes and loss of aortic vascular elasticity. Treatment with UDCA successfully ameliorated the deleterious effects of fructose. The protective effect of UDCA could be attributed to its ability to decrease uric acid level, improve insulin resistance and diminish oxidative stress in vascular tissues. These results might support possible clinical application of UDCA in MS patients especially those present with liver diseases, taking into account its tolerability and safety. However, further investigations on human subjects are needed before the clinical application of UDCA for this indication.
Introduction
The metabolic syndrome (MS) is a pathological condition characterized by obesity, insulin resistance, dyslipidemia, atherosclerosis and hypertension [1], [2]. The metabolic disturbance associated with this syndrome has been shown to increase the risk of developing cardiovascular diseases and type-2 diabetes [3], [4]. These deleterious influences raised worldwide attention to the MS and to the possible strategies for its prevention [5].
The increased consumption of fructose, commonly used in processed food and soft drinks, is one of the most important factors contributing to the growing prevalence of the MS [6]. Experimentally, fructose overload in animals can result in insulin resistance, hyperinsulinemia, hypertriglyceridemia, impaired glucose tolerance and can raise blood pressure [7], [8]. These metabolic disturbances bear a resemblance to the human MS [9], [10]. Therefore, the model of fructose-drinking rats is being widely used to induce the MS independent of obesity or genetic contributions and to study the possible interventions [11].
Hyperuricemia and oxidative stress have been proposed as causative factors in the development of insulin resistance, hyperinsulinemia and the progression of MS in rats induced by fructose consumption [10], [12]. In addition, a causal link has been suggested to exist between insulin resistance/hyperinsulinemia and the development of hypertension in this model [10]. This link has been explained by different mechanisms such as increased activity of vasoconstrictors [13], [14], activation of the sympathetic nervous system [15] and impaired endothelium-dependent relaxation [16]. It has been reported that the development hypertension in fructose-drinking rats model can be prevented by inhibiting any one of these factors, with varied effects on insulin sensitivity [10].
Ursodeoxycholic acid (UDCA) is a steroid bile acid that has been widely used for the treatment of chronic cholestatic liver diseases [17]. Different mechanisms have been proposed for the actions of UDCA, including anti-inflammatory, anti-apoptotic, membrane stabilizing, antioxidant and immunomodulating effects [18], [19]. In addition, it has been reported that UDCA possesses a cardiovascular protective effect [20]. Recently, Ratziu and coworkers [21] reported that high-dose of UDCA improved glycemic parameters and insulin resistance in patients with nonalcoholic steatohepatitis. Furthermore, it has been shown that UDCA improved insulin resistance and hepatic steatosis in animals fed a high-fat diet [22]. In addition, Sinisalo and his colleagues showed that treatment of coronary heart disease patients with depressed endothelial function using UDCA could enhance NO-independent endothelial vasodilatation [23]. Taken together, these reports withdrew our attention for the probable effect of UDCA in preventing the MS and the associated vascular endothelial impairment induced by fructose in rats.
Therefore, the current study aims to investigate the effect of UDAC to prevent or ameliorate the MS in a model of fructose-drinking rats. In this attempt, we compared the effects of UDAC with fenofibrate, a peroxisome proliferator activated receptor (PPAR)-α ligand, which is used widely for the treatment of hypertriglyceridemia [24] and has been shown to exert a protective effect in MS patients [25].
Materials and Methods
Ethics Statement
Experimental design and animal handling procedures were approved by the local authorities, Ethical Committee for Animal Handling at Zagazig University (ECAHZU), at the Faculty of Pharmacy, Zagazig University, Egypt in accordance with the recommendations of the Weatherall report. Every effort was done to minimize the number of animals and their suffering.
Animals
Adult male Wistar rats (140–160 g) were obtained from the Faculty of Veterinary Medicine, Zagazig University, Egypt. Rats were acclimatized for one week before starting experiments. They were housed in stainless steel cages (three rats/cage) and kept at controlled temperature (23±2°C), humidity (60±10%) and light/dark (12/12 h) cycle. Rats were supplied with commercially available normal chow diet and water ad libitum.
Drugs
Fructose was purchased from El-Nasr Chemical Co. (Cairo, Egypt). Fenofibrate was supplied from Abbott (Egypt) and UDAC from Minapharm (Egypt). All other chemicals were of analytical grade. Drugs were suspended in 1% gum acacia in distilled water immediately before administration.
Experimental design
The animals were randomly divided into four experimental groups (n = 5–8). Group 1 (Control): rats received vehicle only (1% gum acacia); group 2 (FDR): rats received the vehicle and administered fructose (10%) in drinking water for 12 weeks; group 3 (UDCA): rats administered fructose (10%) in drinking water for 12 weeks plus UDCA (150 mg/kg) suspended in the vehicle; group 4 (FENO): rats administered fructose (10%) in drinking water for 12 weeks plus fenofibrate (100 mg/kg) suspended in the vehicle. Treatment with UDCA and fenofibrate started from the 6th week after fructose administration once daily every morning at 10:00 AM by oral gavage.
Determination of blood glucose
After the last dose of drugs, rats were fasted overnight. Fasting blood glucose was determined with an automatic blood glucose meter (Super Glucocard, Japan) using blood samples from the tail tip.
Blood pressure measurement
Rats were anaesthetized with urethane (120 mg/100 g, i.p.) given as 25% freshly prepared solution. A cut open was made in the skin of the neck region, and a slit incision was made in the platysma muscles. After the identification of trachea, a small incision was made on the cartilage, and tracheostomy was done using a rodent tracheal intubation tube. The carotid artery was exposed and cannulated using a fine polyethylene catheter filled with heparinised saline (100 IU/ml) and connected to a pressure transducer “PT400”. Systolic blood pressure (SBP) and diastolic blood pressure (DBP) were then recorded using Oscillograph 400 MD 4C-Palmer (Bioscience, Washington, USA). Mean arterial pressure (MAP) was calculated as follow: MAP = ⅓ SBP + ⅔ DBP.
Collection of blood samples and serum separation
After recording of blood pressure, blood samples weretaken out by cardiac puncture and then centrifuged for 20 min. at 3,000 g. Serum was stored at −20°C and thawed just before use for the determination of cholesterol, triglycerides, uric acid, advanced glycation end products (AGEs) and insulin levels.
Isolation of aortic sections
After blood collection, descending thoracic aorta was carefully excised after cleaning of excess connective tissues and fats. Isolated aorta was then cut into rings (2–3 mm length); some rings were rapidly stored in 10% phosphate-buffered formalin solution at room temperature for histopathological examination. The remaining aortic rings were immersed immediately in liquid nitrogen and kept at −80°C for the determination of GSH and MDA contents, as well as the expression of iNOS, eNOS and xanthine oxidase (XO).
Biochemical analysis
Determination of total cholesterol, triglycerides and uric acid levels
Serum total cholesterol, triglycerides and uric acid were determined using colorimetric kits (Biodignostic, Cairo, Egypt) using a UV-visible spectrophotometer (UV-1601PC, Shimadzu, Japan) following the manufacturer's instructions.
Determination of AGEs level
Serum level of AGEs was determined as described by Sampathkumar et al. [26]. In brief, serum aliquot was diluted in phosphate-buffered saline (1∶50) and the fluorescence was determined (λexcitation = 370 nm, λemission = 440 nm) using LS45 spectrofluorometer (PerkinElmer Inc., USA). The fluorescence intensity is directly proportional to the concentration of the AGEs in serum samples.
Determination of insulin level
Serum insulin level was measured by sandwich enzyme-linked immunosorbent assay (ELISA) using kits supplied by Millipore (Cairo, Egypt), following the manufacturer's instructions with microtiter plate coated with mouse monoclonal anti-rat insulin antibodies.
Calculation of insulin resistance
Insulin resistance was determined using the homeostasis model assessment index for insulin resistance (HOMA-IR) using the following formula: HOMA-IR index = [fasting glucose (mmol/L) × fasting insulin (µU/ml)]/22.5 as described by Matthews et al. [27].
Determination of oxidative stress markers in aortic sections
Aortic MDA content was determined colorimetrically as described by Satoh [28], using diagnostic kit supplied by Biodiagnostic, Egypt, following the manufacturer's instructions. The assay is based on the reaction between MDA in the homogenized tissue samples with thiobarbituric acid forming a pink adduct, whose absorbance can be measured at 534 nm. Aortic reduced GSH content was determined colorimetrically as described by Beutler et al. [29], using a diagnostic kit supplied by Biodiagnostic, Egypt, following the manufacturer's instructions. The assay is based on the reduction of 5,5′ dithiobis (2-nitrobenzoic acid) with GSH in the sample to produce a yellow product, whose absorbance can be measured at 405 nm.
Determination of iNOS, eNOS and xanthine oxidase gene expression using real time PCR
Total RNA was extracted from aorta using Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instruction. The RNA pellet was resuspended in DEPC-treated water. The quality and concentration of the RNA was assessed using the OD 260/280 ratio, and only samples with ratio above 1.5 were used in the experiments. Total RNA was reverse transcribed using RevertAid Premium Reverse Transcriptase-Kit (Fermentas International Inc., Burlington, Canada). Briefly, RevertAid H Minus M-MuLV Reverse Transcriptase was added to dNTP Mix (10 mM), 5× Reaction Buffer and random hexamer primers, the mixture was subjected to cDNA synthesis cycling conditions at 37°C for 30 min. and at 85°C for 5 min. Real-time quantitative polymerase chain reaction (RT-qPCR) was performed using ABI prism 7500 sequence detector system (Applied Biosystems, Foster City, CA), using the Maxima SYBR Green qPCR Kit (Fermentas International Inc., Burlington, Canada). Primer sequences were as follow: for iNOS gene, forward: 5′-CGGTTCACAGTCTTGGTGAAAG-3′, reverse: 5′-CAGGTGTTCCCCAGGTAGGTAG-3′; for eNOS gene, forward: 5′-CATACAGAACCCAGGATGGGCT-3′, reverse: 5′ TCCTCAGGAGGTCTTGCACATA-3′; for XO gene, forward: 5′-TGCCATTGATATTGGACAAGTAG-3′, reverse: 5′-TGCAGCTCCTCCATAGTGAA-3′ and for GAPDH gene, forward: 5′ TGCTGGTGCTGAGTATGTCG 3′, reverse: 5′ TTGAGAGCAATGCCAGCC 3′. Reaction mixtures contained 10 pmol/µL of each primer, 12.5 µl Maxima SYBR mix and 5.5 µl nuclease-free water. An amount of 5 µl of template cDNA was added to each reaction mix. The thermal cycling protocol consisted of 2 min. at 50°C and 10 min. at 95°C, followed by 40 cycles at 95°C for 30 s, 60°C for 30 s and 72°C for 30 s. Data from real-time assays were calculated using the v. 1·7 Sequence Detection Software from PE Biosystems (Foster City, CA). Relative expression of studied genes was calculated using the comparative Ct method. All values were normalized to the GAPDH gene expression and reported as fold change over background levels detected in diseased group [30].
Histopathological examination
The aorta from each rat was rapidly dissected out and tissue sections were fixed in 10% phosphate-buffered formalin solution at room temperature. After an overnight wash, specimens were dehydrated in graded ethanol, cleared in xylene and embedded in paraffin. Paraffin-embedded tissue sections of aorta (4–5 mm thick) were prepared, mounted on slides and kept at room temperature. Thereafter, slides were dewaxed in xylene, hydrated using graded ethanol, and stained for histopathological examination by hematoxylin and eosin (H&E) stain and Weigert's stain for elastic fibers. The sections were examined under light microscope and photographed with a digital camera (Canon, Japan).
Statistical analysis
All data were expressed as mean ± standard error of the mean (SEM). Statistical analysis was performed using Graphpad prism software v.5 (GraphPad Software Inc., La Jolla, CA, USA). The statistical significance of differences between groups was tested using one-way analysis of variance (ANOVA) followed by Tukey's Post-test. A significant difference was assumed for values of P<0.05.
Results
Effect on body weight, serum parameters and blood pressure
As shown in Table 1, fructose administration resulted in a significant increase in the percentage of body weight gain compared to control rats (27.8%±0.6 vs. 20.9%±1.0 increase from basal weight, P<0.001). After 12 weeks, FDR showed significant increases in fasting blood glucose (P<0.001), serum insulin (P<0.001), total cholesterol (P<0.001), triglycerides (P<0.001), AGEs (P<0.001) and uric acid (P<0.001) levels compared to the control group. In addition, significant increases in HOMA-IR (P<0.001), SBP (P<0.001), DBP (P<0.01) and MAP (P<0.001) were observed in FDR compared to the control rats. Treatment of FDR with either UDCA or fenofibrat significantly decreased the percentage increase in body weight compared to rats administered fructose alone (P<0.05, P<0.001, respectively). In addition, administration of UDCA or fenofibrat significantly reduced the elevated fasting blood glucose (P<0.001), serum insulin (P<0.001 and P<0.01, respectively), total cholesterol (P<0.05, P<0.001, respectively), triglycerides (P<0.001), AGEs (P<0.01) and uric acid levels (P<0.001) compared to rats administered fructose alone. Moreover, HOMA-IR index, SBP, DBP and MAP were significantly reduced (P<0.001) by treatment with UDCA or fenofibrat compared to rats administered fructose alone (Table 1).
Table 1. Effect of ursodeoxycholic acid (150 mg/kg, p.o.) and fenofibrate (100 mg/kg, p.o.) on body weight, serum parameters and blood pressure in rats administered fructose.
| Parameters | Groups | |||
| Control | FDR | UDCA | FENO | |
| 1. Δ Body weight (%)a | 20.9±1.0 | 27.8±0.6* | 24.3±0.8# | 22.3±0.8# |
| FBG (mmol/l) | 5.1±0.09 | 7.6±0.41* | 5.8±0.22# | 5.7±0.17# |
| Insulin (µU/ml) | 9.2±0.66 | 20.1±1.9* | 10.6±0.65# | 12.7±0.6# |
| HOMA-IR | 2.1±0.19 | 6.9±0.99* | 2.7±0.14# | 3.2±0.19*# |
| T. CH (mg/dl) | 43.5±3.9 | 78.6±5.6* | 56.6±4.5# | 43.5±4.8# |
| TG (mg/dl) | 57.7±4.3 | 117.9±3.9* | 55.5±4.3# | 52.12±4.7# |
| AGEs (pg/mg prot.) | 48.9±2.6 | 77.1±5.9* | 52.4±4.2# | 52.7±4.9# |
| Uric acid (mg/dl) | 0.37±0.07 | 2.42±0.1* | 1.1±0.12# | 0.82±0.09# |
| SBP (mmHg) | 105±2.4 | 140±5.9* | 103±4.2# | 86±4.3# |
| DBP (mmHg) | 72.3±1.6 | 100±5.7* | 70±3.3# | 53.7±5.7# |
| MAP (mmHg) | 86.5±1.3 | 113.3±5.6* | 81.0±3.4# | 64.6±5.1# |
Values are expressed as mean ± SEM (n = 5–8). aPercentage change in body weight from basal value before starting the experiment. *P<0.05 vs. control, #P<0.05 vs. FDR. FDR, fructose-drinking rats; UDCA, fructose-drinking rats treated with ursodeoxycholic acid (150 mg/kg, p.o.); FENO, fructose-drinking rats treated with fenofibrate (100 mg/kg, p.o.). FBG, fasting blood glucose; HOMA-IR, homeostasis model assessment index for insulin resistance; T. CH, total cholesterol; TG, triglycerides; AGEs, advanced glycation end products; prot., protein; SBP, systolic blood pressure; DBP, diastolic blood pressure; MAP, mean arterial pressure.
Oxidative stress markers
Aorta isolated from FDR showed a significantly elevated MDA content compared to control rats (38.46±1.68 vs. 8.05±0.78 nmol/g. tissue, P<0.001). This elevation was significantly diminished by treatment with either UDCA (14.95±0.74 vs. 38.46±1.68 nmol/g. tissue, P<0.001) or fenofibrate (15.58±1.39 vs. 38.46±1.68 nmol/g. tissue, P<0.001) as depicted in Fig. 1a. Another important marker of oxidative stress, reduced GSH, was significantly reduced in aortic sections isolated from rats administered fructose for 12 weeks compared to the control group (40.09±1.51 vs. 84.99±2.28 mg/g. tissue, P<0.001). Administration of UDCA or fenofibrate to FDR significantly increased the level of GSH (80.46±2.94 and 76.66±2.29 vs. 40.09±1.51 nmol/g. tissue, respectively, P<0.001) and restored it close to the control levels (P>0.05) as shown in Fig. 1b.
Figure 1. Effect of ursodeoxycholic acid (150 mg/kg, p.o.) and fenofibrate (100 mg/kg, p.o.) on aortic malondialdehyde (MDA) content (a) and aortic reduced glutathione (GSH) content (b) in fructose-drinking rats.

(n = 5–8). *P<0.05 vs. control, **P<0.05 vs. FDR. FDR, fructose-drinking rats; UDCA, fructose-drinking rats treated with ursodeoxycholic acid (150 mg/kg, p.o.); FENO, fructose-drinking rats treated with fenofibrate (100 mg/kg, p.o.).
Effect on aortic expression of iNOS, eNOS and XO
As depicted in Fig. 2, fructose consumption for 12 weeks induced significant reductions in the expression of eNOS (P<0.001), while it induced significant increases in the expression of iNOS (P<0.001) and XO (P<0.001) compared to the control group. Alterations of gene expression induced by fructose were ameliorated by treatment with UDCA or fenofibrate. Both drugs showed comparable effects resulting in a significant elevation of eNOS expression (P<0.05, P<0.01, respectively), significant reductions of iNOS expression (P<0.001, P<0.01, respectively) and XO expression (P<0.001, P<0.01, respectively) compared to FDR.
Figure 2. Effect of ursodeoxycholic acid (150 mg/kg, p.o.) and fenofibrate (100 mg/kg, p.o.) on gene expression of iNOS (a); eNOS (b) and XO (c) of aortic sections isolated from fructose-drinking rats.

(n = 5–8). *P<0.05 vs. control, **P<0.05 vs. FDR. FDR, fructose-drinking rats; UDCA, fructose-drinking rats treated with ursodeoxycholic acid; FENO, fructose-drinking rats treated with fenofibrate. iNOS, inducible nitric oxide synthase; eNOS, endothelial nitric oxide synthase; XO, xanthine oxidase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Histopathological examination
As depicted in Fig. 3a, staining aortic sections of control rats with H&E and Weigert's stain (elastic stain), respectively, revealed normal tunica adventitia with no apparent infiltration of leukocytes and wavy elastic fibers in tunica media. On the other hand, aortic sections from FDR showed multiple leukocytes infiltrating the tunica adventitia and elastic fibers that are relatively straighter (Fig. 3b) compared to the control rats. In rats administered UDCA (Fig. 3c) or fenofibrate (Fig. 3d), no infiltrating leukocytes were detected, while few straight and mostly wavy elastic fibers were observed.
Figure 3. Histological examination of aortic sections stained with H&E (×400) on the right panel and Weigert's elastic stain (×400) on the left panel.
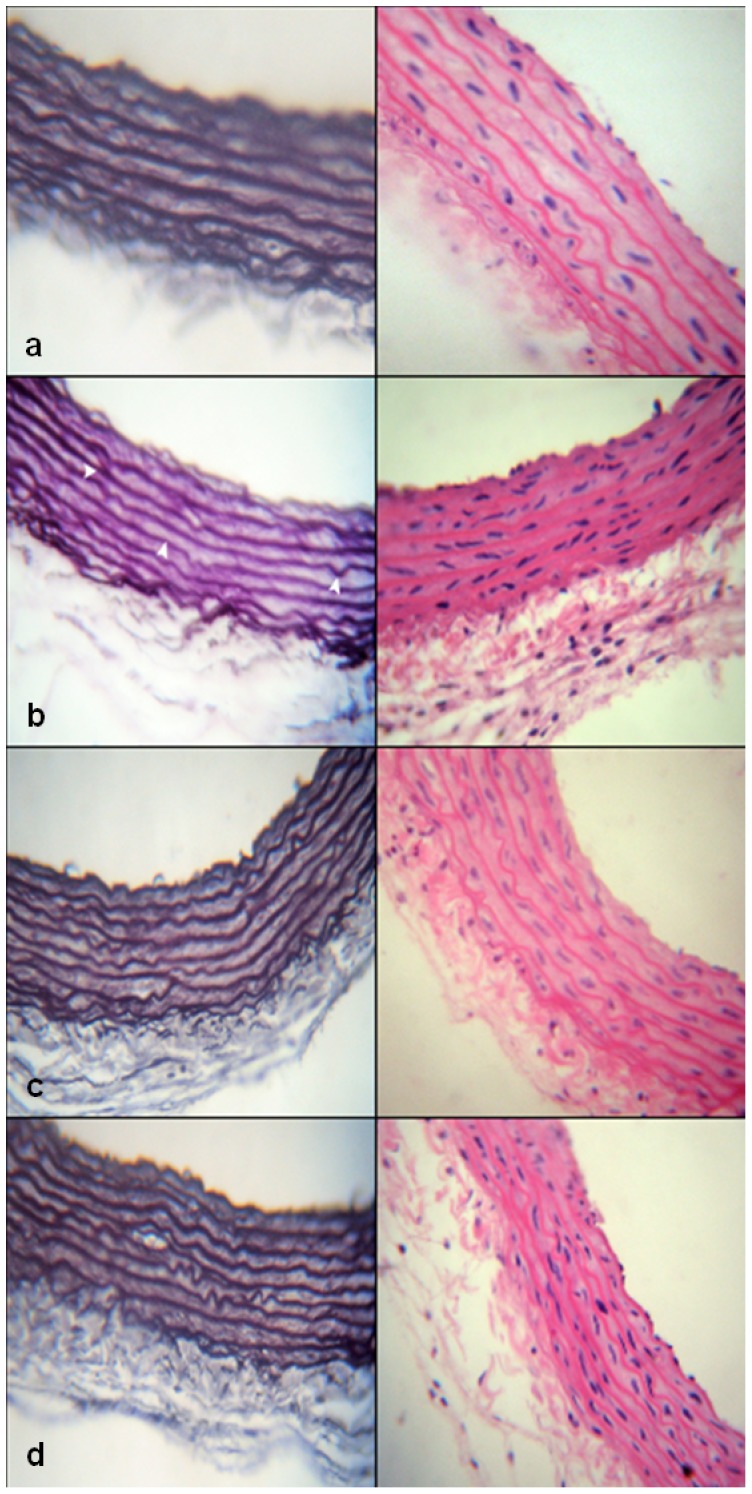
Representative aortic sections were obtained from control rats (a); FDR (b); FDR administered UDCA 150 mg/kg (c) and FDR administered fenofibrate 100 mg/kg (d). White arrowheads point to the relatively straight elastic laminae of aorta from FDR. FDR, fructose-drinking rats; UDCA, ursodeoxycholic acid.
Discussion
Metabolic syndrome (MS) comprises a constellation of disturbances such as abdominal obesity, insulin resistance and dyslipidemia, which eventually result in hypertension. These disturbances put the patient at a high risk of developing type-2 diabetes and cardiovascular morbidities [3], [31]. The consumption of fructose, among others dietary changes, has been linked to the escalated incidence of MS [32], [33]. This represents a major health risk because fructose intake has greatly increased over the last decades [34]. Experimentally, Fructose has been widely accepted as an animal model for MS that mimics the symptoms afflicting human subjects [9], [10].
Ursodeoxycholic acid (UDCA) is a hydrophilic bile acid used in the treatment of liver diseases such as primary biliary cirrhosis and cystic fibrosis-related cholestasis [35], [36]. Some reports described that UDCA can improve insulin resistance in patients with nonalcoholic steatohepatitis [21]. In addition, another report demonstrated that the taurine conjugate form of UDCA, tauroursodeoxycholic acid, could enhance insulin sensitivity in obese, insulin-resistant subjects [37]. Combined with its ability to reduce triglycerides, these data suggest a possible potential role of UDCA in ameliorating symptoms of the MS. Therefore, in the present study we aim to evaluate the effect of UDCA on fructose-induced MS and associated vascular endothelial impairment in rats. In this regard, the effects of UDCA were compared to fenofibrate. Our selection for fenofibrate as a reference drug is based on previous reports showing that PPAR-α agonists lowered triglycerides level and improved insulin sensitivity in different animal models including rats fed high-fat diet [38], rats fed high-fructose diet [39] and Zucker obese rats [40]. In addition, fenofibrate has been reported to inhibit vascular inflammation in experimental animals [41] and to improve endothelial function and arterial stiffness in obese glucose tolerant men [42].
Our results showed that fructose administration (10%) to rats in drinking water for 12 weeks induced the classic symptoms of the MS. Rats showed significant increase in weight gain compared to the control group. They also had significantly higher levels of blood glucose, serum insulin, total cholesterol, triglycerides, higher SBP, DBP and MAP compared the control rats that did not receive fructose. In addition, fructose induced insulin resistance as evidenced by the significant increase in HOMA-IR index. Together, these alterations confirm the proper induction of MS in our study, which is in agreement with previous reports [8], [43], [44].
Our results showed that fructose administration resulted in hyperuricemia, which is consistent with other previous reports [12], [45], [46]. Fructose-induced hyperuricemia might be attributed to the stimulation of AMP deaminase activity triggered by fructose metabolism resulting in increased formation of uric acid [47]. High level of uric acid during fructose consumption has been suggested as an important causative factor in the development of MS. This is supported by previous findings demonstrating that reducing uric acid level, using XO inhibitors or uricosuric agents, could prevent the development of MS signs in fructose-fed rats [46], [48].
Hyperuricemia plays a pivotal role in the development of insulin resistance and hypertension during MS through uric acid-induced endothelial dysfunction. Nakagawa and coworkers [48] showed that uric acid can block acetylcholine-mediated arterial dilation in a dose-dependent manner suggesting that uric acid can impair endothelial function. Furthermore, it has been shown that uric acid can reduce endothelial NO bioavailability in both cell culture and in experimental animal models [49]. In this context, our results show that fructose-induced hyperuricemia is associated with a significant reduction in the expression of eNOS in thoracic aorta sections. This reduction can contribute to insulin resistance through the inhibition of insulin-stimulated glucose uptake in skeletal muscle, which is actually mediated by increasing blood flow through a NO-dependent pathway [50]. Moreover, reduced production of endothelial NO cause the vascular smooth muscle cells to become more sensitive to the effects of vasoconstrictors, which can result in increased vascular tone and elevated blood pressure [10]. In this regard, our histopathological findings showed that the aortic sections from rats administered fructose lost some of its elasticity as shown by the relative straightening of elastic fibers in tunica media (Fig. 3b) compared to the control rats (Fig. 3a).
Another important causative factor that might be involved in the progression of fructose-induced impairment of vascular endothelium associated with insulin resistance is oxidative stress. The results of the present study demonstrate that fructose consumption increased oxidative stress in aortic tissues as evidenced by the increased production of MDA, GSH and increased expression of iNOS (Fig. 1a, 1b and 2a, respectively). Our results are in agreement with previous reports [51], [52]. Although increased expression of iNOS can result in increased production of NO in aortic tissues, however, under conditions of oxidative stress, superoxide radical is produced which then can interact with NO. This interaction can result in the degradation of NO and the formation of peroxynitrite, a highly toxic oxidant to the vascular tissues [53], [54]. Two main sources of ROS in vascular tissues have been described, namely NADPH oxidase and XO [55], [56]. Indeed, our results showed that the expression of XO in aortic rings from FDR was significantly higher compared to the control rats serving as a continued source of ROS production. In addition, we detected significant leukocyte infiltration in the tunica adventitia of aortic rings from FDR during the histopathological examination. These inflammatory cells play an important role in the consequent vascular dysfunction through increasing the production of ROS primarily by stimulating NADPH oxidase within the vasculature [57]. Our results are consistent with other reports showing also vascular inflammation induced by fructose [58], [59]. We found also that the vascular inflammation and endothelial impairment induced by fructose were not only confined to the aortic vasculature, but also were apparent in kidneys. Renal interstitium inflammation was manifested by the presence of lymphocytes and monocytes, as well as enhanced deposition of collagen fibers around venules in the renal cortex (data not published).
Moreover, we found that FDR had significantly higher levels of serum AGEs compared to the control rats (Table 1). Our results are in harmony with previous results showing that AGEs increase not only under hyperglycemic states, but also by fructose consumption [60], [61]. Fructose-induced elevation of AGEs level has many deleterious consequences on the vasculature including cross-linking of collagen, resulting in loss of elasticity and endothelial impairment. In addition, AGEs have been shown to quench NO in vitro and induce inflammation as well as oxidative stress that eventually can lead to vascular impairment and hypertension [62], [63].
Treatment of FDR with UDCA significantly diminished the percentage of body weight gain and ameliorated fructose-induced hyperglycemia, hyperinsulinemia, hypertriglyceridemia, hypercholesterolemia, hypertension, and improved insulin resistance as well (Table 1). The effects of UDCA were comparable to fenofibrate with relatively better effects regarding serum insulin and insulin resistance index. However, this difference does not reach statistical significance.
The protection exerted by UDCA might be attributed to several mechanisms including reduction of uric acid, antioxidant, anti-inflammatory effects and reduction of AGEs. On one hand, our results demonstrated that UDCA significantly reduced serum uric acid level compared to FDR. This reduction was associated with a significant improvement in the aortic expression of eNOS and aortic wall elasticity denoted by the presence of many wavy elastic fibers (Fig. 3c). UDCA-induced reduction of uric acid might be due reduced expression of XO, as we could find such an effect in aortic sections from rats treated with UDCA. In support to this postulation, Sokolovic et al., demonstrated also that UDCA can reduce XO activity in liver tissues [64]. On the other hand, UDCA exhibited remarkable antioxidant effect showed by significant reduction in aortic MDA content and expression of iNOS as well as significant increase in reduced GSH content (Fig. 1a, 1b and 2a, respectively). Moreover, UDCA reduced vascular endothelial inflammation as demonstrated by the absence of infiltrating leukocytes in the aortic tissues (Fig. 3c). UDCA also reduced the formation of AGEs, through improving insulin resistance, which may be involved also in reducing oxidative stress and vascular endothelial inflammation. All these effects help in ameliorating signs of MS and normalizing blood pressure.
These findings seem interesting talking into consideration the added therapeutic benefit of using UDCA in patients with non-alcoholic fatty liver disease (NAFLD), which is considered as a hepatic component of MS and diabetes mellitus type-2 and the most common cause of hepatic dysfunction [65]. Although the protective effects of UDCA observed in this study were almost comparable to fenofibrate, however, UDCA might be preferable considering safety. Fibrates, although generally well tolerated, may cause myopathy and rhabdomyolysis both in humans [66] and rats [67]. In addition, some case studies described hepatotoxicity associated with fenofibrate [68], [69]. On the other hand, UDCA has been shown to be well tolerated [70] and additionally proven to be safe even when administered during pregnancy [71].
In conclusion, the present study provides an evidence of the potential protective effect of UDCA against MS and associated hypertension induced by fructose in rats. These effects could be attributed to the ability of UDCA to decrease uric acid level and oxidative stress particularly in vascular tissues. In addition, these results may support future possible clinical application of UDCA in MS patients especially those present with NAFLD. However, this assumption needs further investigations on human subjects to be confirmed and clinically applied.
Acknowledgments
The authors acknowledge Dr. Ahmed Balah, Faculty of Veterinary Medicine, Zagazig University for the hispathological examination.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
The authors have no support or funding to report.
References
- 1. Reaven GM (1988) Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 37: 1595–1607. [DOI] [PubMed] [Google Scholar]
- 2. Isomaa B, Almgren P, Tuomi T, Forsen B, Lahti K, et al. (2001) Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes care 24: 683–689. [DOI] [PubMed] [Google Scholar]
- 3. Lorenzo C, Okoloise M, Williams K, Stern MP, Haffner SM (2003) The metabolic syndrome as predictor of type 2 diabetes: the San Antonio heart study. Diabetes care 26: 3153–3159. [DOI] [PubMed] [Google Scholar]
- 4. Malik S, Wong ND, Franklin SS, Kamath TV, L'Italien GJ, et al. (2004) Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease, and all causes in United States adults. Circulation 110: 1245–1250. [DOI] [PubMed] [Google Scholar]
- 5. Zimmet P, Alberti KG, Shaw J (2001) Global and societal implications of the diabetes epidemic. Nature 414: 782–787. [DOI] [PubMed] [Google Scholar]
- 6. Laville M, Nazare JA (2009) Diabetes, insulin resistance and sugars. Obesity reviews : an official journal of the International Association for the Study of Obesity 10 Suppl 1 24–33. [DOI] [PubMed] [Google Scholar]
- 7. Hulman S, Falkner B (1994) The effect of excess dietary sucrose on growth, blood pressure, and metabolism in developing Sprague-Dawley rats. Pediatric research 36: 95–101. [DOI] [PubMed] [Google Scholar]
- 8. Hwang IS, Ho H, Hoffman BB, Reaven GM (1987) Fructose-induced insulin resistance and hypertension in rats. Hypertension 10: 512–516. [DOI] [PubMed] [Google Scholar]
- 9. Tobey TA, Mondon CE, Zavaroni I, Reaven GM (1982) Mechanism of insulin resistance in fructose-fed rats. Metabolism: clinical and experimental 31: 608–612. [DOI] [PubMed] [Google Scholar]
- 10. Tran LT, Yuen VG, McNeill JH (2009) The fructose-fed rat: a review on the mechanisms of fructose-induced insulin resistance and hypertension. Molecular and cellular biochemistry 332: 145–159. [DOI] [PubMed] [Google Scholar]
- 11. Chou CL, Lai YH, Lin TY, Lee TJ, Fang TC (2011) Aliskiren prevents and ameliorates metabolic syndrome in fructose-fed rats. Archives of medical science : AMS 7: 882–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nakagawa T, Tuttle KR, Short RA, Johnson RJ (2005) Hypothesis: fructose-induced hyperuricemia as a causal mechanism for the epidemic of the metabolic syndrome. Nature clinical practice Nephrology 1: 80–86. [DOI] [PubMed] [Google Scholar]
- 13. Iyer SN, Katovich MJ (1994) Effect of chronic losartan potassium treatment on fructose-induced hypertension. Life sciences 55: PL139–144. [DOI] [PubMed] [Google Scholar]
- 14. Juan CC, Fang VS, Hsu YP, Huang YJ, Hsia DB, et al. (1998) Overexpression of vascular endothelin-1 and endothelin-A receptors in a fructose-induced hypertensive rat model. Journal of hypertension 16: 1775–1782. [DOI] [PubMed] [Google Scholar]
- 15. Verma S, Bhanot S, McNeill JH (1999) Sympathectomy prevents fructose-induced hyperinsulinemia and hypertension. European journal of pharmacology 373: R1–4. [DOI] [PubMed] [Google Scholar]
- 16. Miller AW, Katakam PV, Ujhelyi MR (1999) Impaired endothelium-mediated relaxation in coronary arteries from insulin-resistant rats. Journal of vascular research 36: 385–392. [DOI] [PubMed] [Google Scholar]
- 17. Luketic VA, Sanyal AJ (1994) The current status of ursodeoxycholate in the treatment of chronic cholestatic liver disease. The Gastroenterologist 2: 74–79. [PubMed] [Google Scholar]
- 18. Makino I, Tanaka H (1998) From a choleretic to an immunomodulator: historical review of ursodeoxycholic acid as a medicament. Journal of gastroenterology and hepatology 13: 659–664. [DOI] [PubMed] [Google Scholar]
- 19. Angulo P (2002) Use of ursodeoxycholic acid in patients with liver disease. Current gastroenterology reports 4: 37–44. [DOI] [PubMed] [Google Scholar]
- 20. Ma J, Nakajima T, Iida H, Iwasawa K, Terasawa K, et al. (2003) Inhibitory effects of ursodeoxycholic acid on the induction of nitric oxide synthase in vascular smooth muscle cells. European journal of pharmacology 464: 79–86. [DOI] [PubMed] [Google Scholar]
- 21. Ratziu V, de Ledinghen V, Oberti F, Mathurin P, Wartelle-Bladou C, et al. (2011) A randomized controlled trial of high-dose ursodesoxycholic acid for nonalcoholic steatohepatitis. Journal of hepatology 54: 1011–1019. [DOI] [PubMed] [Google Scholar]
- 22. Tsuchida T, Shiraishi M, Ohta T, Sakai K, Ishii S (2012) Ursodeoxycholic acid improves insulin sensitivity and hepatic steatosis by inducing the excretion of hepatic lipids in high-fat diet-fed KK-Ay mice. Metabolism: clinical and experimental 61: 944–953. [DOI] [PubMed] [Google Scholar]
- 23. Sinisalo J, Vanhanen H, Pajunen P, Vapaatalo H, Nieminen MS (1999) Ursodeoxycholic acid and endothelial-dependent, nitric oxide-independent vasodilatation of forearm resistance arteries in patients with coronary heart disease. Br J Clin Pharmacol 47: 661–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vamecq J, Latruffe N (1999) Medical significance of peroxisome proliferator-activated receptors. Lancet 354: 141–148. [DOI] [PubMed] [Google Scholar]
- 25. Koh KK, Han SH, Quon MJ, Yeal Ahn J, Shin EK (2005) Beneficial effects of fenofibrate to improve endothelial dysfunction and raise adiponectin levels in patients with primary hypertriglyceridemia. Diabetes care 28: 1419–1424. [DOI] [PubMed] [Google Scholar]
- 26. Sampathkumar R, Balasubramanyam M, Rema M, Premanand C, Mohan V (2005) A novel advanced glycation index and its association with diabetes and microangiopathy. Metabolism: clinical and experimental 54: 1002–1007. [DOI] [PubMed] [Google Scholar]
- 27. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, et al. (1985) Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28: 412–419. [DOI] [PubMed] [Google Scholar]
- 28. Satoh K (1978) Serum lipid peroxide in cerebrovascular disorders determined by a new colorimetric method. Clinica chimica acta; international journal of clinical chemistry 90: 37–43. [DOI] [PubMed] [Google Scholar]
- 29. Beutler E, Duron O, Kelly BM (1963) Improved method for the determination of blood glutathione. The Journal of laboratory and clinical medicine 61: 882–888. [PubMed] [Google Scholar]
- 30. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 31. Hunt KJ, Resendez RG, Williams K, Haffner SM, Stern MP (2004) National Cholesterol Education Program versus World Health Organization metabolic syndrome in relation to all-cause and cardiovascular mortality in the San Antonio Heart Study. Circulation 110: 1251–1257. [DOI] [PubMed] [Google Scholar]
- 32. Le KA, Tappy L (2006) Metabolic effects of fructose. Current opinion in clinical nutrition and metabolic care 9: 469–475. [DOI] [PubMed] [Google Scholar]
- 33. Elliott SS, Keim NL, Stern JS, Teff K, Havel PJ (2002) Fructose, weight gain, and the insulin resistance syndrome. The American journal of clinical nutrition 76: 911–922. [DOI] [PubMed] [Google Scholar]
- 34. Bray GA, Nielsen SJ, Popkin BM (2004) Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. The American journal of clinical nutrition 79: 537–543. [DOI] [PubMed] [Google Scholar]
- 35. de Caestecker JS, Jazrawi RP, Petroni ML, Northfield TC (1991) Ursodeoxycholic acid in chronic liver disease. Gut 32: 1061–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Comar KM, Sterling RK (2006) Review article: Drug therapy for non-alcoholic fatty liver disease. Aliment Pharmacol Ther 23: 207–215. [DOI] [PubMed] [Google Scholar]
- 37. Kars M, Yang L, Gregor MF, Mohammed BS, Pietka TA, et al. (2010) Tauroursodeoxycholic Acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 59: 1899–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ye JM, Doyle PJ, Iglesias MA, Watson DG, Cooney GJ, et al. (2001) Peroxisome proliferator-activated receptor (PPAR)-alpha activation lowers muscle lipids and improves insulin sensitivity in high fat-fed rats: comparison with PPAR-gamma activation. Diabetes 50: 411–417. [DOI] [PubMed] [Google Scholar]
- 39. Matsui H, Okumura K, Kawakami K, Hibino M, Toki Y, et al. (1997) Improved insulin sensitivity by bezafibrate in rats: relationship to fatty acid composition of skeletal-muscle triglycerides. Diabetes 46: 348–353. [DOI] [PubMed] [Google Scholar]
- 40. Guerre-Millo M, Gervois P, Raspe E, Madsen L, Poulain P, et al. (2000) Peroxisome proliferator-activated receptor alpha activators improve insulin sensitivity and reduce adiposity. The Journal of biological chemistry 275: 16638–16642. [DOI] [PubMed] [Google Scholar]
- 41. Tabernero A, Schoonjans K, Jesel L, Carpusca I, Auwerx J, et al. (2002) Activation of the peroxisome proliferator-activated receptor alpha protects against myocardial ischaemic injury and improves endothelial vasodilatation. BMC pharmacology 2: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ryan KE, McCance DR, Powell L, McMahon R, Trimble ER (2007) Fenofibrate and pioglitazone improve endothelial function and reduce arterial stiffness in obese glucose tolerant men. Atherosclerosis 194: e123–130. [DOI] [PubMed] [Google Scholar]
- 43. Sanchez-Lozada LG, Tapia E, Jimenez A, Bautista P, Cristobal M, et al. (2007) Fructose-induced metabolic syndrome is associated with glomerular hypertension and renal microvascular damage in rats. American journal of physiology Renal physiology 292: F423–429. [DOI] [PubMed] [Google Scholar]
- 45. Cavarape A, Feletto F, Mercuri F, Quagliaro L, Daman G, et al. (2001) High-fructose diet decreases catalase mRNA levels in rat tissues. Journal of endocrinological investigation 24: 838–845. [DOI] [PubMed] [Google Scholar]
- 46. Sanchez-Lozada LG, Tapia E, Bautista-Garcia P, Soto V, Avila-Casado C, et al. (2008) Effects of febuxostat on metabolic and renal alterations in rats with fructose-induced metabolic syndrome. American journal of physiology Renal physiology 294: F710–718. [DOI] [PubMed] [Google Scholar]
- 47. Hallfrisch J (1990) Metabolic effects of dietary fructose. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 4: 2652–2660. [DOI] [PubMed] [Google Scholar]
- 48. Nakagawa T, Hu H, Zharikov S, Tuttle KR, Short RA, et al. (2006) A causal role for uric acid in fructose-induced metabolic syndrome. American journal of physiology Renal physiology 290: F625–631. [DOI] [PubMed] [Google Scholar]
- 49. Khosla UM, Zharikov S, Finch JL, Nakagawa T, Roncal C, et al. (2005) Hyperuricemia induces endothelial dysfunction. Kidney international 67: 1739–1742. [DOI] [PubMed] [Google Scholar]
- 50. Roy D, Perreault M, Marette A (1998) Insulin stimulation of glucose uptake in skeletal muscles and adipose tissues in vivo is NO dependent. The American journal of physiology 274: E692–699. [DOI] [PubMed] [Google Scholar]
- 51. Delbosc S, Paizanis E, Magous R, Araiz C, Dimo T, et al. (2005) Involvement of oxidative stress and NADPH oxidase activation in the development of cardiovascular complications in a model of insulin resistance, the fructose-fed rat. Atherosclerosis 179: 43–49. [DOI] [PubMed] [Google Scholar]
- 52. Polizio AH, Gonzales S, Munoz MC, Pena C, Tomaro ML (2006) Behaviour of the anti-oxidant defence system and heme oxygenase-1 protein expression in fructose-hypertensive rats. Clinical and experimental pharmacology & physiology 33: 734–739. [DOI] [PubMed] [Google Scholar]
- 53. Beckman JS, Koppenol WH (1996) Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. The American journal of physiology 271: C1424–1437. [DOI] [PubMed] [Google Scholar]
- 54. Shinozaki K, Kashiwagi A, Nishio Y, Okamura T, Yoshida Y, et al. (1999) Abnormal biopterin metabolism is a major cause of impaired endothelium-dependent relaxation through nitric oxide/O2- imbalance in insulin-resistant rat aorta. Diabetes 48: 2437–2445. [DOI] [PubMed] [Google Scholar]
- 55. Spiekermann S, Landmesser U, Dikalov S, Bredt M, Gamez G, et al. (2003) Electron spin resonance characterization of vascular xanthine and NAD(P)H oxidase activity in patients with coronary artery disease: relation to endothelium-dependent vasodilation. Circulation 107: 1383–1389. [DOI] [PubMed] [Google Scholar]
- 56. Ulker S, McMaster D, McKeown PP, Bayraktutan U (2003) Impaired activities of antioxidant enzymes elicit endothelial dysfunction in spontaneous hypertensive rats despite enhanced vascular nitric oxide generation. Cardiovascular research 59: 488–500. [DOI] [PubMed] [Google Scholar]
- 57. Hink U, Li H, Mollnau H, Oelze M, Matheis E, et al. (2001) Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circulation research 88: E14–22. [DOI] [PubMed] [Google Scholar]
- 58. Mahmoud MF, Hassan NA, El Bassossy HM, Fahmy A (2013) Quercetin protects against diabetes-induced exaggerated vasoconstriction in rats: effect on low grade inflammation. PloS one 8: e63784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mahmoud MF, El-Nagar M, El-Bassossy HM (2012) Anti-inflammatory effect of atorvastatin on vascular reactivity and insulin resistance in fructose fed rats. Archives of pharmacal research 35: 155–162. [DOI] [PubMed] [Google Scholar]
- 60. Goldin A, Beckman JA, Schmidt AM, Creager MA (2006) Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 114: 597–605. [DOI] [PubMed] [Google Scholar]
- 61. Ardestani A, Yazdanparast R (2007) Cyperus rotundus suppresses AGE formation and protein oxidation in a model of fructose-mediated protein glycoxidation. International journal of biological macromolecules 41: 572–578. [DOI] [PubMed] [Google Scholar]
- 62. Vlassara H (2001) The AGE-receptor in the pathogenesis of diabetic complications. Diabetes/metabolism research and reviews 17: 436–443. [DOI] [PubMed] [Google Scholar]
- 63. Miller A, Adeli K (2008) Dietary fructose and the metabolic syndrome. Current opinion in gastroenterology 24: 204–209. [DOI] [PubMed] [Google Scholar]
- 64. Sokolovic D, Nikolic J, Kocic G, Jevtovic-Stoimenov T, Veljkovic A, et al. (2013) The effect of ursodeoxycholic acid on oxidative stress level and DNase activity in rat liver after bile duct ligation. Drug Chem Toxicol 36: 141–148. [DOI] [PubMed] [Google Scholar]
- 65. Tessari P, Coracina A, Cosma A, Tiengo A (2009) Hepatic lipid metabolism and non-alcoholic fatty liver disease. Nutrition, metabolism, and cardiovascular diseases : NMCD 19: 291–302. [DOI] [PubMed] [Google Scholar]
- 66. Hodel C (2002) Myopathy and rhabdomyolysis with lipid-lowering drugs. Toxicology letters 128: 159–168. [DOI] [PubMed] [Google Scholar]
- 67. Okada M, Sano F, Ikeda I, Sugimoto J, Takagi S, et al. (2009) Fenofibrate-induced muscular toxicity is associated with a metabolic shift limited to type-1 muscles in rats. Toxicologic pathology 37: 517–520. [DOI] [PubMed] [Google Scholar]
- 68. Ho CY, Kuo TH, Chen TS, Tsay SH, Chang FY, et al. (2004) Fenofibrate-induced acute cholestatic hepatitis. Journal of the Chinese Medical Association : JCMA 67: 245–247. [PubMed] [Google Scholar]
- 69. Rigal J, Furet Y, Autret E, Breteau M (1989) [Severe mixed hepatitis caused by fenofibrate? A review of the literature apropos of a case]. La Revue de medecine interne/fondee par la Societe nationale francaise de medecine interne 10: 65–67. [DOI] [PubMed] [Google Scholar]
- 70. Hempfling W, Dilger K, Beuers U (2003) Systematic review: ursodeoxycholic acid—adverse effects and drug interactions. Alimentary pharmacology & therapeutics 18: 963–972. [DOI] [PubMed] [Google Scholar]
- 71. Kondrackiene J, Beuers U, Kupcinskas L (2005) Efficacy and safety of ursodeoxycholic acid versus cholestyramine in intrahepatic cholestasis of pregnancy. Gastroenterology 129: 894–901. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.