

Abstract
Extensive technical advances in the past decade have substantially expanded quantitative proteomics in cardiovascular research. This has great promise for elucidating the mechanisms of cardiovascular diseases (CVD) and the discovery of cardiac biomarkers used for diagnosis and treatment evaluation. Global and targeted proteomics are the two major avenues of quantitative proteomics. While global approaches enable unbiased discovery of altered proteins via relative quantification at the proteome level, targeted techniques provide higher sensitivity and accuracy, and are capable of multiplexed absolute quantification in numerous clinical/biological samples. While promising, technical challenges need to be overcome to enable full utilization of these techniques in cardiovascular medicine. Here we discuss recent advances in quantitative proteomics and summarize applications in cardiovascular research with an emphasis on biomarker discovery and elucidating molecular mechanisms of disease. We propose the integration of global and targeted strategies as a high-throughput pipeline for cardiovascular proteomics. Targeted approaches enable rapid, extensive validation of biomarker candidates discovered by global proteomics. These approaches provide a promising alternative to immunoassays and other low-throughput means currently used for limited validation.
Keywords: Cardiovascular diseases, Proteomics, LC-MS, Biomarker, mechanism study, targeted quantification
1. Introduction: Proteomics and Cardiovascular Disease (CVD)
Cardiovascular disease (CVD) remains the leading cause of mortality worldwide, and represents a tremendous burden on global health and medical resources[1]. Investigation of the mechanisms of CVD, as well as the discovery of biomarkers for disease diagnosis, staging and therapy evaluation, will greatly improve therapeutic efforts, management, and risk stratification of CVD[2]. Conventionally, such studies often involve the examination of hypothesized or known targets in clinical or pre-clinical subjects using traditional methods such as ligand binding assays[3]. Despite considerable success over the past several years, these strategies remain suboptimal in that they are often laborious, time-consuming, and sometimes susceptible to bias[4]. By comparison, discovery-based investigations using “-omics” methods may provide systematic and unbiased insight into the molecular basis of cardiac physiological and pathological adaptations in health and disease, facilitating research on identifying novel disease mechanisms and biomarkers. Recently, genomic and transcriptomic approaches have been employed as high-throughput and powerful tools for CVD research[5]. One drawback for such strategies is that changes in messenger RNA expression may not quantitatively correlate well with the expression of proteins[6, 7]. For example, these strategies tend to correlate poorly with the expression of cell-surface proteins[6, 8]. An additional significant limitation of genomic and transcriptomic approaches arises from their inability to characterize post-translational modifications (PTM) which are prominently involved in modulating many biological processes.
Proteomic approaches enable accurate characterization of proteins and PTMs in highly complex biological systems, directly providing relevant information on altered biological cascades. The past decade has witnessed tremendous progress in cardiovascular proteomics research which is the result of rapid evolution of proteomics techniques; specifically, those based on liquid chromatography mass spectrometry (LC/MS). To date, cardiovascular proteomics remains a dynamic and rapidly developing field. It is dramatically advancing our knowledge of the complex myocardial physiology and pathophysiological states with an enormous potential to rapidly advance the identification of disease mechanisms. Quantitative proteomic techniques can be roughly divided into two categories: global approaches and targeted approaches[9], which are employed in discovery and hypothesis based investigations respectively. Global proteomics compares quantitative proteome expression under different conditions in a relative manner [10]. This strategy is usually employed as a hypothesis-free profiling tool to identify altered proteins in systems responding to perturbations or under distinct physiological or pathological states. These studies markedly contribute to the discovery of CVD biomarkers and provides the mechanisms underlying ischemia and myocardial infarction (MI). Targeted proteomics, on the other hand, specifically characterizes or quantifies a limited number of known target proteins in a complex system. One prominent paradigm for the application of targeted approach is the validation of biomarker candidates, often with superior specificity, accuracy and analytical throughput over traditional ligand binding assays[11]. The following sections will discuss both global and targeted proteomics techniques, and review their recent applications in cardiovascular research with an emphasis on pathophysiological mechanism and biomarker discovery.
2. Quantitative Global Proteomics Methods
2.1 Technical Overview of Quantitative Global Proteomics
Global proteomics compares two or more proteomes to identify differentially altered protein expression or PTM, for unbiased discovery of biomarkers or key players associated with specific physiological and pathological states, with little or no prior knowledge. A generic workflow of global quantitative proteomics using bottom-up strategy is shown in Figure. 1, which also lists some popular options for quantification approaches, and techniques for sample preparation , LC/MS analysis, protein identification and quantification A researcher should choose the optimal strategy by balancing the considerations based on the type of samples, the number of replicates per group, the complexity of the proteomes, the desired levels of quantitative accuracy, sensitivity and precision, and depth of analysis, as well as the budget and analytical expertise level.
Figure. 1. A generic workflow of global quantitative proteomics using a bottom-up (shotgun) approach.
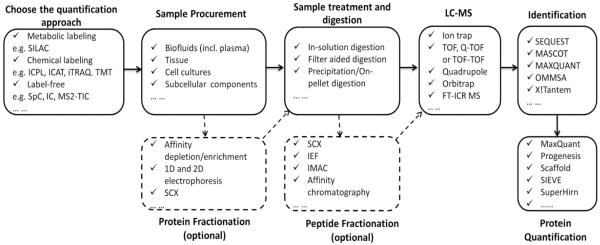
From a technical perspective, an ideal method for global proteomics should provide i) extensive proteome coverage, ii) high sensitivity, accuracy and precision for relative quantification, iii) low false-discovery of altered proteins (biomarkers) and iv) the ability to reliably compare multiple biological replicates in one set. This section discusses global proteomics techniques with the considerations of these desirable features.
Global proteomic approaches can be divided into two major categories: gel based (mainly the two-dimensional gel electrophoresis (2DE) and LC/MS-based. The latter can be further separated into isotope-labeling and label-free approaches. The vast majority of LC/MS-based methods employ a “shotgun” approach (i.e. samples are digested enzymatically before LC/MS analysis) which is effective for large-scale protein analysis [12].
2.1.1 2DE method
The 2DE method separates proteins by pI and molecular weight [13]. 2DE was the dominant method for cardiovascular proteomics research in the initial stage of proteomics (1990s-2000s), but has decreased in popularity in recent years, due to the rise of LC/MS-based approaches[14]. Compared with LC/MS, 2DE falls short in its low sensitivity, narrow dynamic range, low proteomic coverage and limited ability to analyze membrane proteins. Nevertheless, this low-cost, directly observable and robust technique has still contributed importantly to cardiovascular proteomic research [15]. Proteomics studies based on 2DE identified altered regulatory proteins associated with cardiomyopathy, characterized a number of sub-proteomes of the heart (e.g. mitochondrion), has been employed for biomarker discovery in animal models and has been used to characterize selected PTMs[16, 17].
2.1.2 Isotope labeling methods
Isotope labeling approaches play an important role in quantitative proteomics. These methods incorporate stable isotope coded and/or isobaric tags into proteins or peptides by either a chemical reaction, (e.g. Isotope-Coded Affinity Tag (ICAT)[18], Isobaric Tags for Relative and Absolute Quantification(iTRAQ)[19], Tandem Mass Tags(TMT)[20], and more recently, Neutron-encoded Mass Signatures(NeuCode)[21]) or metabolic process (e.g. Stable Isotope Labeling by Amino Acids in cell culture (SILAC)[22]). In the majority of these techniques, the different forms of labeled species exhibit almost identical physicochemical properties, allowing the incorporation of stable-isotope labels to correct for experimental bias and variation during the preparation step. Quantification of multiple conditions by LC/MS analysis can be achieved [9]. An extensive review of labeling strategies can be found in ref [23]. In cardiovascular research, chemical labeling methods are more prevalent due to their ability to study various types of proteomes (e.g. tissues and body fluids). As there are very few dividing cell culture systems for ventricular cardiomyocytes, metabolic methods such as SILAC have limited application in cardiovascular research [14](Supplementary Table I). Examples using SILAC for cardiovascular proteomics in animal models include cardiac morphogenesis of zebra fish[24] and profiling of mouse heart tissues[25].
2.1.3 Label-free methods: ion current and spectral counts
Label-free quantification does not employ any label, and samples are analyzed sequentially in individual LC/MS experiments. Quantitative features in each measurement are matched to individual peptides or proteins and then compared among samples to derive information of relative quantity. The basis of label-free approaches is the linear correlation between LC/MS abundance features and relative abundance of peptides [26]. Label-free strategies can be categorized by the abundance features utilized for quantification, including those based on the peptide precursor MS1 signals (ion current; IC) [27, 28], Spectral Counts(SpC) of protein obtained from MS2 product ion scans[29, 30], and a mixture of these features[31](a schematic representation of IC-based label-free quantification strategy is displayed in Figure.2A).
Figure. 2. Label-free quantification strategies.

(A) Illustration of the work flow of the ion current-based relative quantification; (B) comparison of the reproducibility of quantitative features by ion current-based vs. spectral count methods for the repetitive analysis of a tissue sample (reproduced from Ref[34]).
Until recently SpC has been the most prevalent label-free method for cardiovascular proteomics (Supplementary Table I), because the approach is easy to implement and more tolerant to imperfections in sample preparation and LC/MS analysis. Moreover, SpC does not require stringent matching of peptide IC peaks between runs, which is especially advantageous when low-resolution mass spectrometers are used. Because of this, a number of comparative studies based on low-mass-resolution LC-MS data concluded that spectral count is a more sensitive and precise way to identify protein changes in complex samples[32, 33]. However, the game has been dramatically changed by the recent rapidly-growing availability of high-resolution MS analyzers such as Fourier Transform-ion Cyclotron Resonance (FTICR) and Orbitrap[34]. The use of high-resolution analyzers permits extraction of peptide IC within a very narrow m/z range (e.g. <0.02 mass unit), substantially reducing chemical noise, to improve sensitivity and specificity of IC-based quantification[35]. Consequently, IC-based methods are markedly superior in terms of sensitivity and lack of of missing data when analyzing data from high-resolution LC/MS[36]. Our study showed that IC-based method (Figure. 2 B) achieved a better reproducibility and lower level of false-positive biomarker discovery than SpC [34]. Furthermore, we recently conducted a comprehensive comparison of IC-based method vs. SpC using high-resolution LC/MS data acquired from a wide variety of proteomes, and demonstrated that IC-based approaches achieved significantly better quantitative accuracy and precision, lower levels of missing data, and superior performance in biomarker discovery[37]. Such a sharp contrast can be attributed to two shortcomings of SpC: 1) MS2 acquisition of low abundant peptides is often suppressed by peptides of higher abundance and 2) Quantitative counting for lower-abundance proteins/peptides often results in “boundary” counts of "1" and "0", which precludes accurate quantification [32, 38]. In comparison, the IC-based quantification method relies on the measurement of peptide precursors (MS1), thus circumventing the above-mentioned problems associated with MS2.
2.1.4 General comparison of labeling and label-free methods
Labeling methods provide universal quantitative references (i.e. internal standards) for all labeled and detectable species in proteomics samples, affording accurate quantification if the labeling process is complete and reproducible. With the use of an internal standard, the mixture of labeled proteins or peptides can be subjected to fractionation procedures prior to LC-MS analysis, reducing sample complexity and the dynamic range of the proteome [23]. Moreover, two or more labeled samples are combined and analyzed in the same LC/MS run, which reduces the required instrument time. Despite these advantages, labeling methods have several drawbacks. First, the labeling reagents are expensive rendering the method cost-prohibitive for relatively large scale studies. Second, for most methods, the labeling processes may be laborious and it is difficult to achieve complete labeling for all proteins [39]. As the quantification is only performed for the labeled species, this problem may introduce artificial bias and limit the number of quantifiable proteins. Third, while the ability to quantify a relatively large number of biological replicates is important to understand biological variability[40], the number of replicates that can be analyzed by labeling methods is significantly limited by the number of tag forms available[41, 42]. Although recently “high-plex” labeling methods have been developed, such as the 8-plex iTRAQ[43] and NeuCode[21], the number of replicates possible remains relatively low and the reagents are costly.
Since label-free methods do not employ any labeling reagent, higher proteome coverage can be achieved in a more cost-effective way. The elimination of the labeling procedure also allows simpler sample preparation procedures and more flexible method development [44, 45]. Most importantly, the label-free method has the capacity to quantify a large number of biological replicates in one set [31, 34]. Prominent challenges for label-free methods include the requirement of highly reproducible sample preparation and LC/MS analysis, which makes it challenging to employ fractionation techniques with high robustness. These issues are discussed in the following section.
2.1.5 Protein Identification in LC/MS based proteomics
Protein identification is a crucial step for in quantitative proteomics, where the fragmentation spectra are matched to peptides and then proteins. The most widely used approaches is database searching that compares MS2 spectra to theoretical spectra generated in silico based on theoretical protein and peptide sequences[46]. Many excellent search engines are available for this purpose, such as the Sequest, Mascot, OMSSA, X!Tandem and MaxQuant . When prior knowledge of peptide sequence is not available, de novo sequencing methods can be used. In order to achieve confident identification, estimation and control of the false-identification is indispensable in this process. Currently the most prevalent method for controlling false-identification is the target-decoy strategy, because of its limited assumptions. Other approaches include posterior probabilities and single spectrum confidence scores[47]. More details on protein identification can be found in recent review articles[46, 47].
2.1.6 Technical Challenges and strategies
Sample preparation
Global proteomic quantification requires efficient and quantitative extraction of all proteins from the proteome despite diverse physical and chemical properties. Quantitative preparation of cardiac tissue is particularly challenging since myocytes are composed of membranes (sarcolemma, mitochondrial membrane and sarcoplasmic reticulum), myofilaments and contain large numbers of mitochondria[14]. Even with targeted myocardial mitochondrial proteomics, approximately 80% of the inner mitochondrial membrane mass comprises membrane proteins [48]. Therefore a method to completely disrupt various compartments, effectively extract membrane-associated proteins, and adequately clean up the samples (i.e. the efficient removal of buffer compounds and non-protein matrix components) while maintaining high peptide recovery, is highly desirable.
A number of strategies were developed to address this need. For instance, we developed a straightforward and highly efficient work flow for sample preparation [28]. Protein extraction is sonicated in lysis buffer containing a high concentration of detergent cocktail, which effectively disrupts membrane components and achieves a complete and highly reproducible protein yield. Then, an optimized precipitation procedure removes detergents and non-protein matrix components to prevent them from compromising the protein preparation and LC/MS analysis procedure [49]. The precipitated pellet, containing isolated proteins, is then subjected to a 2-phase on-pellet-digestion procedure. This provides a clean digest with high and consistent peptide recovery across many biological replicates [50]. This method has been demonstrated to provide efficient quantitative recovery of proteins (including membrane-associated proteins) in tissue samples, such as heart tissues, heart mitochondria[28] andretina[34]. The same approach has also been employed to identify proteins in solution from bronchoalveolar lavage fluid[45] and swine plasma[51].
Dynamic protein concentration range
In order to extensively reveal proteome level changes, it is important to obtain high proteomic coverage for low-abundance proteins. These are frequently key regulators of cellular function but are difficult to characterize due to the wide dynamic range of protein concentrations in typical proteomes [35]. The problem is more pronounced for plasma proteomics, where protein concentrations span a dynamic range that is ~10 orders of magnitude as compared to ~6 orders of magnitude in typical tissues [52]. Unfortunately, the dynamic range achievable by current proteomic methods (e.g. ~3 orders of magnitude for LC/MS [51]) is quite limiting. To address this problem, immunodepletion methods that reduce the dynamic range of the plasma proteome by 1-2 orders of magnitude were employed prior to gel-based and/or LC/MS analysis of the human plasma proteome[53, 54]. Though widely practiced, these techniques may result in co-depletion of lower-abundance proteins and carry-over [55]; additionally, these methods are often not applicable to animal models because most antibodies are targeted to epitopes derived from human protein sequences and thus may not enable a complete depletion.
We reported a strategy for extensive analysis of animal plasma proteomes and applied it in the investigation of CVD-related proteins in swine plasma [51]. A Combinatorial Peptide Ligand Library technique that is not species-dependent, was utilized to markedly reduce the dynamic range of plasma. A mild denaturing condition was used with this method to significantly alleviate the co-depletion problem. Digestion was performed in-parallel respectively by two enzymes (trypsin and GluC) carrying orthogonal specificities. These digests were fractionated with high-resolution Strong Cation-Exchange (SCX) chromatography and then resolved on a long, heated nano-LC column. MS analysis was performed on an LTQ/Orbitrap respectively with two complementary activation methods (CID and ETD). By performing differential proteolysis and orthogonal tandem MS sequencing, proteome coverage was greatly enhanced. Using this strategy, a total of 3421 unique proteins were identified in swine plasma. Functional annotation analysis revealed that 657 proteins were significantly associated with CVD (p<0.05), from five major CVD-related pathways[51]. This method shows considerable potential for future biomarker discovery in animal models such as swine.
Even though depletion or dynamic range compression procedures mentioned above considerably enhance proteomic coverage of plasma, global quantitative proteome studies of plasma remain a daunting challenge because the resultant samples are still highly complex with a wide dynamic range[14]. The dynamic range in tissue proteomes, for which depletion methods are rarely available, are also much greater than the majority of quantitative proteomics methods[34]. Advances such as multi-dimensional chromatography can significantly enhance proteome coverage [56]. Although these methods can be employed with ease for labeling approaches, it is more challenging to apply them with label-free approaches owing to the requirement of highly precise fractionation of each of the samples in the quantification cohort.. One alternative to achieve high proteome coverage using label-free approaches is the use of a long reversed-phase column and a shallow gradient for extensive 1-D LC/MS analysis, which can greatly improve coverage of complex proteomes without fractionation[57]. However, most previous long-column methods lack the reproducibility necessary to quantify multiple biological replicates. Recently, we described a robust long-column LC/MS method that enables extensive, sensitive and reproducible IC-based quantification across many samples[34, 45, 50, 58]. Columns with lengths of 75 to 100-cm were tightly packed with a unique dual-direction protocol for constant performance. A low void volume setup, with dampened pump noise and homogenous heating in a tightly-fit thermo sheath, was employed to obtain high chromatographic resolution and reproducible separation among parallel long-gradient runs. Using this strategy, several thousand unique proteins were quantified across >20 biological replicates in tissue and cellular proteomics with highly stringent criteria, typically including low identification FDR. In addition, each protein was quantified with high-quality AUC data (e.g. S/N≥10) from ≥2 unique peptides and was free of missing data at the protein level[34, 50, 58].
False positives discovery of altered proteins
High false-positive discovery rates of differentially altered proteins is a common and significant problem for global quantitative proteomics[40]. This results in false biological leads that can significantly undermine the credibility of discovery-based proteomics research. These primarily arise from two sources; biological variability and technical variability[40, 59]. When the variations in protein levels among individuals are high or too few biological replicates are used, the intrinsic biological variability will frequently cause false biomarker discovery. This problem is amplified by the common practice of using pooled samples of biological replicates for relative quantification, often due to technical limitations such as the limited multiplex capacity of labeling methods or difficulty in running many replicates with high reproducibility when a label-free method is used[60, 61]. Technical variability can also cause false-positives arising from 1) The "multiple hypothesis testing" when significance testing is simultaneously performed on numerous proteins[62] and 2) Sample preparation and LC/MS analysis which are susceptible to analytical variations and systematic errors. This is especially problematic for lower-abundance proteins[59] where noisy signals may result in unrelaible relative quantification [63].
To reduce biological variability, use of multiple biological replicates is highly desirable. This can be accommodated by label-free methods as discussed previously. To address technical variability, reproducible and quantitative sample preparation and LC/MS procedures are essential. Furthermore, it is essential to accurately estimate and control the false altered protein discovery rate (FADR). Several parametric algorithms have been developed to adjust for the false discovery rate in “-omics” studies, e.g. Benjamini and Hochberg p-value Adjustment Method[64] and the q-value method [65]. However, these are rarely used in proteomics research [62] since parametric algorithms are based on several assumptions that do not work well with proteomics data[66, 67]. Additionally, these algorithms may not extensively examine other causes of false positives, such as variations introduced by the sample preparation and LC/MS analysis.
Evaluation of the FADR nonparametrically and experimentally enables a more extensive and accurate estimation of false discoveries, providing a promising alternative to the statistical approaches. Along these lines, our lab has developed an experimental null-based hypothesis strategy for evaluation and control of FADR in label-free proteomics experiments which is detailed in recent reports [34, 45, 50, 58]. This method provides a straightforward and reliable means to predict FADR by evaluating false positives arising from an experimentally-determined null distribution, and therefore greatly facilitates the optimization of proteomic methods for sensitive and specific biomarker discovery. Other methods using nonparametric strategies to control FADR include acquiring null distributions from statistical interference [68].
2.2 Application of Global Proteomics in Mechanistic Research
Global proteomics profiling can provide an unbiased systematic view of the cardiovascular system, which in turn may improve our understanding of cardiac pathology, diagnosis, disease staging and lead to the development of novel therapies. Global proteomics has been applied to study mechanisms of CVD such as hibernating myocardium [28], irreversible ischemia[69], diabetic cardiomyopathy[70, 71] and congestive heart failure[72]. Because of the wide protein dynamic range, most work has focused on subcellular proteomes and characterization of PTMs rather than protein changes in unfractionated cardiac tissue.
2.2.1 Subcellular proteomics
In the cardiovascular field, proteomics investigations are frequently focused on subcellular proteomes, such as mitochondrion, proteasome, extracellular matrix and myofilaments, owing to the important biological significance of subcellular compartments. Methods to isolate subcellular fractions include differential centrifugation, immune-based isolation and membrane protein enrichment [15]. For instance, Warren et al. employed differential centrifugation and organelle enrichment to isolate mitochondria, nuclei, cytoplasmic fractions, microsomes and sarcomere proteins, for quantitative analysis of these sub-proteomes in rat hearts subjected to regional ischemia. They concluded that fractionation improved the depth of proteomics analysis by nearly four-fold[73]. Below, we focus on two of the most-widely studied subcellular proteomes in cardiovascular research: mitochondria and extracellular matrix(ECM). Several other less extensively studied subcellular proteomes in CVD research including myofilaments[74], membrane proteins[75] , sacromere[76] and proteasomes[77] .
Comparative study of the mitochondrial proteome represents one of the most popular areas in cardiovascular proteomics [78]. Mitochondria account for nearly 40% of the volume fraction of a healthy cardiomyocyte. They not only generate ATP for contractile function but also regulate programmed cell death. Accumulating evidence demonstrates a close association between CVD progression and changes in mitochondrial structure and function [79, 80]. Using an ion-current based label-free quantitative proteomics approach, we compared mitochondrial proteomes from myocardium of healthy swine versus those with hibernating myocardium [28], with a relatively large number biological replicates (n=10/group). Another early-stage label-free proteomic study was done by Zhang et al. to quantify changes in the mitochondrial proteome post ischemic stress using a modified SpC method[69]. Labeling methods have also been utilized. For example, Julling et al. used iTRAQ to characterize changes to the cardiac mitochondrial proteome in streptozotocin-diabetic rats[81] and aged spontaneously hypertensive rats[60]. Comparative mitochondrial proteomics has also been applied to study mechanisms of pressure overload-induced heart failure [72], atrial fibrillation[82], and type I [70] and type II diabetic heart[71]. Mitochondrial proteome changes elicited by system perturbations were also reported in estrogen deficiency[83], diabetic hearts with overexpression of mPHGPx[61] and GSK inhibition[84]. A study on mitochondrial proteome dynamics was recently conducted by Kim, et al., who used mitochondria from mice fed with 2H2O to examine the turnover of 458 proteins[85].
Another well-studied sub-cellular proteome is the extracellular matrix (ECM). ECM proteins anchor surrounding cardiac myocytes[86] and ECM remodeling is associated with heart failure and cardiac fibrosis [87, 88]. The ECM proteome was first characterized in 2010 using SpC in human aortic samples[89]. Similar approaches were applied to the characterization of ECM remodeling in abdominal aortic aneurysms[90] and myocardial ischemia/reperfusion injury[91]. Notably, in these three studies, the authors developed a reproducible procedure to improve ECM proteome analysis. Besides characterizing the ECM of tissues, evaluation of ECM proteins secreted from differentiating cells has also been carried out using iTRAQ[92].
2.2.2 Post translational modifications (PTMs)
PTMs of proteins regulate many key molecular pathways and quantification of PTMs provides highly valuable information for insights into disease-related signaling cascades. LC/MS-based technologies enable sensitive and high-throughput PTM analysis, which allow characterization of myocardial PTMs on a global proteome-level[93]. To date, >400 different PTMs were identified in higher organisms and the number is still increasing[94, 95]. To overcome the problem of low stoichiometry of PTMs, most global quantification methods involve a PTM-enrichment procedure, such as TiO2 enrichment and Immobilized Metal Affinity Chromatography(IMAC), or a fraction step (e.g. SCX) prior to LC/MS analysis. Subcellular fractions of cardiomyocytes were often used to achieve deeper PTM analysis[95].
Since phosphorylation plays a critical role in a broad range of signaling pathways associated with myocardial disease including contractile function, metabolism, and protein degradation[96], it is the most widely studied PTM in cardiovascular proteomics [93]. Initial work of quantitative phospho-proteomics was carried out by Boja et al. on porcine cardiac mitochondria[97]. They used TiO2 enrichment to capture phosphopeptides and iTRAQ/ HCD to analyze and quantify phosphorylation residues. In recent years, more extensive phosphoproteomic studies have been completed. Chang et al. used IMAC to enrich phosphorylated peptides followed by iTRAQ labeling to quantify the phosphoproteome of myocardium from pressure-overloaded mice in a time-profile manner[98]. Their study suggested Dynamin-related protein 1 may serve as a potential modulator of cardiac hypertrophy caused by pressure-overload. Scholten et al. used stable-isotope dimethyl labeling, SCX fractionation and CID/HCD fragmentation to identify putative downstream CaMKII targets in cardiac tissue from animals with cardiac-specific overexpression of a CaMKII inhibitor[99]. Another recent study designed to discover targeted residues phosphorylated by the beta-adrenergic receptor identified 670 altered phosphorylation sites [100]. Other PTMs studied in global cardiovascular proteomics include S-nitrosylation[101], glycosylation[102] and acetylation[103].
2.2.3 Myocardial tissue proteomics
Despite technical hurdles, it is extremely insightful to study the cardiac tissue proteome without fractionation so as to obtain a comprehensive and unbiased view of molecular mechanisms involved in CVD. One example is the use of a label-free method to identify altered proteins in heart tissue of mice which were subjected to cardiac-specific overexpression of activated calcineurin to cause cardiac hypertrophy[104]. Fractionation was employed to eliminate high-abundance contractile proteins from cardiac tissue lysate, and 290 proteins were found to be dysregulated out of 1918 cardiac proteins identified. For comparison, directly analyzing tissue lysates without fractionation results in fewer proteins identified, as shown in a study of radiation-induced changes in the cardiac tissues, where 662 proteins were identified[11]. By investigating cardiac tissue in murine animal models using time-series with label-free quantitative proteomics, a recent study discovered differentially expressed proteins following myocardial infarction[105].
2.3 Application of Global Proteomics in Biomarker Discovery
Global quantitative proteomics are widely used to discover biomarker candidates for disease diagnosis and evaluation of therapies, by comparing two or more groups of proteomes to identify differentially expressed proteins. There are five biomarkers widely employed in the evaluation of patients with coronary artery disease. These include cardiac troponin I(cTnI) and cardiac Troponin T(cTnT) employed in acute coronary syndromes and myocardial infarction, B-type natriuretic peptides and its N-terminal form to diagnose and manage congestive heart failure, and c-reactive protein to diagnose cardiac inflammation in atherosclerosis. Although these biomarkers greatly improve the diagnosis and therapy of CVD, limitations remain and new biomarkers and biomarker panels have emerged[3, 106]. Novel biomarkers are urgently needed to improve risk stratification and management of specific subsets of patients with CVD patients [106, 107].
Samples from well characterized human subjects are preferable for evaluation since it would yield the most clinically-relevant information. However, using human samples for biomarker discovery also carries several major disadvantages, such as high individual variability [108], and difficulty investigating certain cardiovascular events such as reversible ischemia and hibernating myocardium in patients[109]. Consequently, many biomarker studies are conducted with well-controlled animal disease models, such as these in mice[104], rats[105] and swine[91].
2.3.1 Tissue Biomarkers
As discovery of candidate circulating proteomic biomarkers in plasma can be technically difficult, an alternative is to analyze myocardial tissue samples for proteins that may be secreted in pathophysiological disease states [110]. In clinical proteomics, either myocardial biopsies or post-mortem tissues can be employed for discovery based investigation. For example, Hammer et al, used label-free quantitative proteomics to characterize inflammatory changes in myocardial biopsies from patients with dilated cardiomyopathy [111]. By comparing the proteome of 10 patients vs. 7 controls, they identified 174 differentially-expressed proteins that were involved in mitochondrial and cytoskeleton remodeling. More recently, a label-free analysis was conducted on myocardial tissue isolated from patients who succumbed to MI at postmortem to identify early stage biomarkers [112]. Sorbin and SH3 domain-containing protein 2 were reported as novel biomarker candidates. There have also been proteomic analyses of animal models to discover tissue biomarkers. For example, Holland et al. used label-free LC-MS/MS to quantify proteome changes of cardiomyopathic tissue in mouse models of Duchenne muscular dystrophy [113]. A total of 67 proteins showed altered expression while drastic changes were demonstrated for 17 proteins including Ig chains and transferrin, laminin, nidogen and annexin.
Since the ultimate objective is to discover circulating biomarkers, meaningful in-tissue biomarkers findings must in the end contribute to that goal. Therefore, in some studies, the authors validated in-tissue biomarkers in plasma samples. Chugh et al. used a global proteomics approach on myocardial tissue to analyze in-tissue biomarkers of heart failure on mouse models and validated 4 potential tissue biomarkers for heart failure in mouse and human plasma[114].
2.3.2 Circulating Biomarkers
Discovery of circulating plasma biomarkers is difficult due to the wide dynamic range[9] and the enormous biological variability among patients which necessitates the use of many samples for reliable discovery. As discussed above, depletion or fractionation techniques can be employed in circulating biomarker research to address the dynamic range problem. A study employing a multiple affinity removal spin column to deplete the three most abundant proteins identified several potential biomarker candidates for coronary artery disease from a transgenic mouse model[115]. To obtain extensive proteome coverage, the authors utilized 2-D LC separation. Both known biomarkers and novel candidates were discovered[115]. A similar high-abundance protein depletion strategy was used to discover biomarkers in patients diagnosed with an MI [116]. More recently, a more powerful depletion strategy, IgY14-supermix tandem depletion method, was utilized to seek predictive markers for near-term MI[117]. The workflow developed by the authors demonstrated good reproducibility of the measurement and the ability to quantify changes of low-abundance proteins. As reviewed in 2.1.5, chromatography can also be used to reduce the dynamic range of the plasma proteome. Mebazaa et al. used combined fractional diagonal chromatography to overcome technical difficulties posed by plasma samples. Their proteomics pipeline identified quiescin Q6 as a candidate biomarker for acute heart failure[118]. More recently, metabolomics-based biomarker discovery has gained increasing attention; it is worth noting that such strategies may mitigate some of the above-mentioned technical hurdles for plasma proteomics studies [119].
3. Quantitative Targeted Proteomics Methods
Due to the increasing utilization of Selected-Reaction Monitoring (SRM) operated on a triple-quadrupole MS, also known as Multiple-Reaction Monitoring(MRM), targeted proteomics is a rapidly growing area. Compared with global proteomics, targeted proteomics focuses on known targets to afford a much more robust analysis with significantly higher sensitivity, selectivity and accuracy. Absolute quantification can be achieved using peptide or protein calibrators[49, 120, 121]. The two targeted proteomic techniques employed in cardiovascular research are SRM and top-down methods. Here we provide a general review of the techniques followed by applications in cardiovascular fields.
3.1 Technical Overview
3.1.1 Top-down approaches
Unlike “bottom-up” strategies, top-down MS directly analyzes intact proteins in a high-resolution analyzer. This approach is capable of providing high sequence coverage and accurate PTM mapping of the target protein. This alleviates shortcomings of bottom-up methods such as the ambiguity in PTM localization, isoform identification and detection of degradation[122]. Nonetheless, performing top-down proteomics studies remains challenging due to technical difficulties associated with sensitivity, throughput and the requirement for protein enrichment and purification[123]. Despite the technical challenges, tremendous progress has been made in top-down proteomics for cardiovascular research. Top-down methods developed based on FTICR MS by Ge and co-workers have achieved comprehensive analysis of cTnI modifications, quantitative analysis of different phosphorylation forms of cTnI in heart failure[124, 125], sequencing and phosphorylation characterization of cTnT [126] and PTM analysis of integral membrane proteins[127].
3.2.2 SRM
Compared with other tandem MS techniques, LC/SRM-MS exhibits higher sensitivity, better quantitative accuracy and wider dynamic range in targeted protein quantification, and can be easily multiplexed by quickly switching among different transitions[49]. SRM is most commonly utilized for the quantification of proteins in complex matrices. A typical procedure includes sample treatment, digestion using trypsin and quantification of the target proteins based on the selected Signature Peptides (SP) derived from the target. Stable isotope labeled (SIL) SP surrogate or SIL-full-length-protein is used as the internal standard. Extensive reviews can be found in references [128] and [129]. In recent years, SRM has emerged as a promising alternative to traditional ligand binding assays such as ELISA, in that this method is generally matrix independent, and method development is often faster at lower cost [130].
Enormous efforts have been devoted to advance LC/SRM-MS methods in terms of sample preparation, method development pipeline, instrumentation and data analysis [130, 131], which greatly improve sensitivity, precision and dynamic range[129, 132]. The rapidly evolving techniques have laid a solid foundation for the applications of SRM in future areas of cardiovascular research.
3.1.3 Challenges and strategies
Although SRM is a versatile tool for targeted proteomics investigation, several significant technical challenges remain. First, an optimal strategy for method development remains elusive [133]. One of the most important aspects for method development is the identification of an optimal SP that is sensitive, specific and stable for quantification. Currently, in silico prediction approaches are commonly used. This approach may not accurately predict the most sensitive, stable peptides[134] and matrix-dependent parameters such as chemical interferences [135, 136]. It is important to choose stable peptides as SP[135, 136]to avoid quantitative bias, but this issue has been often overlooked. Second, despite the high sensitivity of LC/SRM-MS, insufficient sensitivity continues to be a significant limitation due to the large molecular weight of the proteins and the need for multiple sample (blood and tissue) dilution (e.g. 10- to 50-fold) because of high protein content [135, 136]. Third, though SIL- internal standards (I.S.) are used, quantitative accuracy continues to be a major problem. Much of the current work employs synthetic peptides as the calibrator and SIL-peptides as I.S. These assume nearly 100% efficiency of the preparation and digestion procedures which may not be true[135]. Finally, most current methods use a lone SP for quantification, which carries a risk of error when proteins are truncated biologically outside the SP domain or certain residues within the SP domain are biologically modified[136, 137].
To address these difficulties methodologically, we devised a novel pipeline for developing SRM approaches, as illustrated in Figure 3. Instead of using an in silico method to predict the optimal SP, we employed an experimental strategy to discover and optimize many SP candidates, and then evaluate these candidates in target matrices prior to SP selection[135-137]. Briefly, the pool of SP candidates was generated by a data-dependent LC/MS experiment following a stringent filtering step. To evaluate these candidates, the target protein was spiked into the blank matrices (e.g. plasma or tissue extract) and then prepared and digested. The optimal LC/MS conditions of all SP candidates were accurately obtained by a high-throughput and on-the-fly orthogonal-array-optimization (OAO) procedure [135-137], which has the capacity to develop the SRM conditions for >100 candidates within one single LC/MS run. Using the developed LC/MS conditions, all candidates were thoroughly assessed for stability and signal-to-noise ratios (S/N) in the matrix digest. Among the stable peptides, two with the highest S/N were selected as the SP. The use of two SP from different domains of the same protein provides a versatile gauge for reliability of quantitative methods and results[135-137].
Figure. 3.

A high-throughput method development pipeline for sensitive, accurate and reproducible LC/SRM-MS quantification of target proteins in complex matrices, based on an on-the-fly orthogonal array optimization and experimental evaluation. Details can be found in Ref. [136, 137].
In order to improve sensitivity for targeted protein analysis, we’ve developed an optimized a robust nano-flow LC/MS strategy [135-137], which typically lowers the limit of quantification (LOQ) by ~30-50 fold compared to conventional-flow LC/MS. Another means to improve sensitivity is to enrich proteins or peptides before LC/SRM-MS analysis. For instance, the Stable Isotope Standards and Capture by Anti-peptide Antibodies (SISCAPA) technique was developed to enrich signature peptides using polyclonal antibodies[138]. More recently, Hendrik et al. developed a series of affinity-based methods for quantitative enrichment of target proteins and/or signature peptides in plasma, achieving ultra-sensitive quantification of circulating biomarkers in plasma[139]. Other approaches to improve the sensitivity of targeted quantification include SCX fractionation [140], high-pH fractionation before LC/MS analysis[130] and the use of long columns to obtain high S/N of target peptides[141].
Protein-level calibration methods (i.e. protein calibrator with SIL-protein I.S.) have recently been employed to overcome concerns about quantitative accuracy[142]. However, SIL-proteins are costly to produce and may be impractical for many classes of proteins. Our lab and others demonstrated accurate quantification of enzymes and protein drugs in plasma and tissues using “hybrid” calibration strategies (e.g. protein calibrator with SIL-peptide I.S.) [135-137, 143, 144], provided that technically reproducible sample preparation and digestion are achieved. Our recent investigation showed severe negative biases by the peptide and extended peptide calibration methods, while the hybrid approaches provide a cost-effective means for accurate quantification without expensive SIL-protein[145].
3.2 Application of LC/SRM-MS in CVD research
3.2.1 Biomarkers Quantification
An early cardiovascular study using LC/SRM-MS was conducted in 2009, when Kuhn successfully quantified the cardiac biomarker cTnI and interleukin 33 with high precision using SISCAPA coupled with SRM-MS [146]. Using this well-established approach, the same group measured CVD biomarkers concentrations in plasma with precision, reproducibility and sensitivity[140]. These two studies demonstrated the multiplex potential of SRM for CVD marker quantification. More recently, a large-scale biomarker validation study was carried out using LC/SRM-MS with 135 stable isotope-labeled peptides, for the quantification of 67 putative CVD biomarkers with good sensitivity, accuracy and quantitative linearity[147]. Shortly thereafter, the same group used a similar strategy for discovery of biomarkers characteristic of patients with or without coronary artery disease. SRM analysis was carried out for 44 proteins on plasma from 38 patients, and 5 of the proteins surfaced as potential biomarkers[148]. Other recent studies include using full-length stable-isotope-labeled proteins as I.S. in SRM to quantify 5 biomarkers of MI in prefractionated human plasma [142]. In agreement with what we mentioned in 3.1.3, this study confirmed introducing isotope-labeled proteins as internal standards before enzyme digestion, enabling more accurate protein quantification than peptide standards.
With SRM now recognized as an ideal validation tool for global quantitative proteomics biomarker discovery, some studies combined global proteomics and targeted proteomics to achieve a more extensive biomarker discovery pipeline. For instance, Cohen Freue et al. coupled multiplexed iTRAQ-LC-MALDI-TOF MS with SRM and immunoassays to discover/validate potential biomarkers on plasma of patients chosen for cardiac transplantation[149]. This pipeline provides a novel avenue for investigation of plasma biomarkers and is highly applicable to a wide range of biomarker studies. Another recent study used a similar strategy to discover circulating markers of atherosclerotic disease from 120 individual samples [150]. By combining results from global proteomics and SRM, the study revealed statistically significant upregulation of Vcl in acute coronary syndrome group.
3.2.2 PTM Quantification
Aside from applications in biomarker validation, SRM is also a promising tool to study PTMs in a relatively small subgroup of proteins. Lam et al. used SRM to quantify site-specific phosphorylation of several mitochondrial proteins of myocardium using 68 transitions [151]. Later, they expanded the SRM transitions to 176 to acquire better coverage of mitochondrial proteins [152]. Their research demonstrates the potential utility of SRM workflow to study the detailed function of mitochondrial phosphorylation. Some more focused PTM studies targeted well-established cardiac biomarkers such as cTnI and Troponin complex. For example, Zhang et al. used in silico prediction of phosphorylated sites followed by SRM based targeted proteomics to identify site-specific cTnI phosphorylated residues in failing human hearts (i.e. patients with ischemic heart disease or idiopathic dilated cardiomyopathy vs. healthy donors) [153]. Fourteen phosphorylation sites of cTnI were identified and quantified, while 6 new phosphorylated residues were identified. Among the 14 quantifiable phosphorylated residues of cTnI, 5 showed down-regulation and 8 showed up-regulation. They also employed a canine heart failure model with dysynchrony to demonstrate that 3 of the remodeled phosphorylated residues can be reversed after cardiac resynchronization with a biventricular pacing system. Another study used SRM to quantify changes of cTnI phosphorylated residues in human myocardium from clinical patients treated with PKCα and recombinant cTnI [154]. This study revealed several PKCα specific phosphorylated sites of the troponin complex.
4. Concluding Remarks
The techniques used for quantitative proteomics can be categorized into either global or targeted approaches. Generally, the global and targeted methods have distinct rationales and goals, e.g. hypothesis-free vs. hypothesis-driven or discovery-based vs. known-target-based, etc. Nonetheless, these two types of strategies are complimentary and can constitute an integral work flow for biomedical research which leads to a more comprehensive, versatile and high-throughput alternative than traditional methods such as ligand binding assays. That being said, proteomics studies still face considerable technical challenges. For global proteomics, more extensive coverage of the proteome, the ability to compare multiple biological replicates and sensitive biomarker discovery with low-false positive/negative rate are highly desirable but difficult to achieve owing to the high complexity of typical proteomes and the limited capacity of available methods[9, 14]. In comparison, the primary considerations for targeted methods are the determination of the levels of the target proteins/PTM with sufficient sensitivity, specificity, accuracy and analytical throughput [133]. These are challenging when target proteins are of low abundance and obscured by a background of high-abundance proteins. Some outstanding developments have been made in the last decade to alleviate many of these problems. To name a few: 1) High-plex approaches such as 8-plex iTRAQ[43] and 18-plex NeuCode [21] have substantially improved the number of biological replicates that can be analyzed by isotope-labeling methods, 2) A cohort of depletion and fractionation techniques markedly reduces the dynamic concentration ranges of typical proteomes, 3) Recent advances in high-resolution MS and reproducible long-column LC separation opens the possibility of extensive IC-based label-free proteomics profiling, 4) New enrichment methods have dramatically lowered the LOQ for LC/MS quantification of proteins in tissues and plasma and 5) Hybrid calibration approaches provide a cost-effective means for accurate quantification without expensive SIL-protein I.S.[135-137]. We have entered an exciting time where the proteomic techniques continue to evolve at an amazing pace, with many emerging new techniques applicable to high throughput analysis.
Although current proteomics techniques are not as heavily utilized in the cardiovascular field as in some other biomedical research areas such as cancer and immunological diseases, the application of proteomics in CVD research has been rapidly increasing[14]. As reviewed here, innovations in CVD proteomics permit the analysis of large numbers of plasma and tissue proteins, extensive analysis of PTM, and sensitive, accurate and robust quantification of target proteins of cardiovascular interest. However, some prominent factors still impede the progress of cardiovascular proteomics research. Two of these are highlighted below.
First, it should be well recognized that there is a wide gap between candidate proteomic biomarkers and clinically applicable biomarkers. Despite the numerous candidates reported in the proteomics literature, their subsequent contribution to diagnostics has been quite limited[106, 107]. This discrepancy stems, at least partially, from the lack of a robust pipeline to interface biomarker discovery to clinical validation[59]. One important reason for this absence is the long held belief that traditional methods based on ligand binding assays should be employed for validation of candidates[155]. For example, authors are often asked for immunoassays to validate in publications on proteomics-based tissue protein and biomarker discovery. Unfortunately, these traditional assays often can’t be multiplexed and may suffer from relatively poor quantitative accuracy and precision compared to LC-MS based methods. As an alternative, LC/SRM-MS provides multiplex capacity, high quantitative precision and selectivity, as reviewed above. In a recent article, Aebersold and colleagues argued that validation of quantitative MS data by Western blotting should no longer be required since the LC/SRM-MS method is a significantly better choice[155]. We expect the combination of global proteomics discovery and SRM based validation along with informatics innovations (e.g. a recent development of a discovery-validation pipeline[149]) will mitigate the gap between biomarker discovery and validation. Similar ideas have also been conveyed in other reviews[107, 156].
Secondly, there is an urgent need to link quantitative changes discovered in the proteome to biological function, in order to gain an in-depth understanding of the perplexing molecular basis underlying CVD and therapeutic effects. The development of function, annotation and visualization tools such as IPA(QIAGEN, Inc), STRING[157] and Metacore(Thomson Reuters, Inc) has greatly contributed in this regard, but more powerful functional analysis tools remain to be developed. Moreover, it is also very helpful to integrate proteomics with other high-throughput “-omics” tools in CVD research, such as transcriptomics[158] and metabolomics, which will enormously contribute to a systematic and comprehensive understanding of the cardiovascular system[159].
All in all, we have entered an exciting time where the proteomic techniques continue to evolve at an amazing pace, with many emerging new techniques applicable to high throughput analysis. For example, a recently released article demonstrated a comprehensive proteome analysis in only a 1.3-hour LC/MS run using an Orbitrap Fusion Tribrid MS analyzer [160]. These new techniques will soon change the face of cardiovascular research and enormously help to achieve a comprehensive understanding of CVD and optimal disease management and treatment.
Supplementary Material
5. Acknowledgement
This work was supported by NIH grants HL55324, HL61610 and HL75324 (JMC), and U54HD071594 (JQ), by the Center for Protein Therapeutics (JQ), the Department of Veterans Affairs (JMC), and by American Heart Association (AHA) award 12SDG9450036 (JQ).
Abbreviations:
- CVD
Cardiovascular Diseases
- cTnI
Cardiac Troponin I
- cTnT
Troponin T
- ECM
Extracellular Matrix
- FADR
False Altered Protein Discovery Rate
- FDR, for peptide protein identification
False Discovery Rates
- FTICR
Fourier Transform-ion Cyclotron Resonance
- I.S.
Internal Standards
- IC
Ion Currents
- IMAC
Immobilized Metal Affinity Chromatography
- LOD
Limit of Detection
- LOQ
Limit of Quantification
- MI
Myocardial Infarction
- SCX
Strong Cation-exchange
- SIL
Stable isotope labeled
- SISCAPA
Stable Isotope Standards and Capture by Anti-Peptide Antibodies
- SP
Signature Peptides
- SpC
Spectral Counts
Footnotes
The authors declared no conflicts of interests.
References
- [1].Yusuf S, Reddy S, Ounpuu S, Anand S. Global burden of cardiovascular diseases: Part II: variations in cardiovascular disease by specific ethnic groups and geographic regions and prevention strategies. Circulation. 2001;104:2855–2864. doi: 10.1161/hc4701.099488. [DOI] [PubMed] [Google Scholar]
- [2].Cui Z, Dewey S, Gomes AV. Cardioproteomics: advancing the discovery of signaling mechanisms involved in cardiovascular diseases. Am J Cardiovasc Dis. 2011;1:274–292. [PMC free article] [PubMed] [Google Scholar]
- [3].Arab S, Gramolini AO, Ping P, Kislinger T, et al. Cardiovascular proteomics: tools to develop novel biomarkers and potential applications. Journal of the American College of Cardiology. 2006;48:1733–1741. doi: 10.1016/j.jacc.2006.06.063. [DOI] [PubMed] [Google Scholar]
- [4].Hoofnagle AN, Wener MH. The fundamental flaws of immunoassays and potential solutions using tandem mass spectrometry. J Immunol Methods. 2009;347:3–11. doi: 10.1016/j.jim.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schnabel RB, Baccarelli A, Lin H, Ellinor PT, Benjamin EJ. Next steps in cardiovascular disease genomic research--sequencing, epigenetics, and transcriptomics. Clinical Chemistry. 2012;58:113–126. doi: 10.1373/clinchem.2011.170423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rugg-Gunn PJ, Cox BJ, Lanner F, Sharma P, et al. Cell-surface proteomics identifies lineage-specific markers of embryo-derived stem cells. Dev Cell. 2012;22:887–901. doi: 10.1016/j.devcel.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cox B, Kotlyar M, Evangelou AI, Ignatchenko V, et al. Comparative systems biology of human and mouse as a tool to guide the modeling of human placental pathology. Mol Syst Biol. 2009;5:279. doi: 10.1038/msb.2009.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lundberg E, Fagerberg L, Klevebring D, Matic I, et al. Defining the transcriptome and proteome in three functionally different human cell lines. Mol Syst Biol. 2010;6:450. doi: 10.1038/msb.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mallick P, Kuster B. Proteomics: a pragmatic perspective. Nat Biotechnol. 2010;28:695–709. doi: 10.1038/nbt.1658. [DOI] [PubMed] [Google Scholar]
- [10].de Godoy LM, Olsen JV, Cox J, Nielsen ML, et al. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature. 2008;455:1251–1254. doi: 10.1038/nature07341. [DOI] [PubMed] [Google Scholar]
- [11].Azimzadeh O, Sievert W, Sarioglu H, Yentrapalli R, et al. PPAR alpha: a novel radiation target in locally exposed Mus musculus heart revealed by quantitative proteomics. J Proteome Res. 2013;12:2700–2714. doi: 10.1021/pr400071g. [DOI] [PubMed] [Google Scholar]
- [12].Florens L, Washburn MP, Raine JD, Anthony RM, et al. A proteomic view of the Plasmodium falciparum life cycle. Nature. 2002;419:520–526. doi: 10.1038/nature01107. [DOI] [PubMed] [Google Scholar]
- [13].Rabilloud T, Chevallet M, Luche S, Lelong C. Two-dimensional gel electrophoresis in proteomics: Past, present and future. J Proteomics. 2010;73:2064–2077. doi: 10.1016/j.jprot.2010.05.016. [DOI] [PubMed] [Google Scholar]
- [14].Van Eyk JE. Overview: the maturing of proteomics in cardiovascular research. Circ Res. 2011;108:490–498. doi: 10.1161/CIRCRESAHA.110.226894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sharma P, Cosme J, Gramolini AO. Recent advances in cardiovascular proteomics. J Proteomics. 2013;81:3–14. doi: 10.1016/j.jprot.2012.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Page B, Young R, Iyer V, Suzuki G, et al. Persistent regional downregulation in mitochondrial enzymes and upregulation of stress proteins in swine with chronic hibernating myocardium. Circ Res. 2008;102:103–112. doi: 10.1161/CIRCRESAHA.107.155895. [DOI] [PubMed] [Google Scholar]
- [17].Banfi C, Brioschi M, Tremoli E. [Cardiovascular proteomics: challenges and opportunities for cardiologists of the future] G Ital Cardiol (Rome) 2013;14:495–503. doi: 10.1714/1308.14458. [DOI] [PubMed] [Google Scholar]
- [18].Gygi SP, Rist B, Gerber SA, Turecek F, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- [19].Dayon L, Hainard A, Licker V, Turck N, et al. Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal Chem. 2008;80:2921–2931. doi: 10.1021/ac702422x. [DOI] [PubMed] [Google Scholar]
- [20].Rauniyar N, Gao B, McClatchy DB, Yates JR., 3rd Comparison of protein expression ratios observed by sixplex and duplex TMT labeling method. J Proteome Res. 2013;12:1031–1039. doi: 10.1021/pr3008896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hebert AS, Merrill AE, Bailey DJ, Still AJ, et al. Neutron-encoded mass signatures for multiplexed proteome quantification. Nat Methods. 2013;10:332–334. doi: 10.1038/nmeth.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- [23].Lengqvist J, Sandberg A. Stable isotope labeling methods in protein profiling. Methods Mol Biol. 2013;1023:21–51. doi: 10.1007/978-1-4614-7209-4_3. [DOI] [PubMed] [Google Scholar]
- [24].Konzer A, Ruhs A, Braun H, Jungblut B, et al. Stable isotope labeling in zebrafish allows in vivo monitoring of cardiac morphogenesis. Mol Cell Proteomics. 2013;12:1502–1512. doi: 10.1074/mcp.M111.015594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Konzer A, Ruhs A, Braun T, Kruger M. Global protein quantification of mouse heart tissue based on the SILAC mouse. Methods Mol Biol. 2013;1005:39–52. doi: 10.1007/978-1-62703-386-2_4. [DOI] [PubMed] [Google Scholar]
- [26].Silva JC, Denny R, Dorschel CA, Gorenstein M, et al. Quantitative proteomic analysis by accurate mass retention time pairs. Anal Chem. 2005;77:2187–2200. doi: 10.1021/ac048455k. [DOI] [PubMed] [Google Scholar]
- [27].Gautier V, Mouton-Barbosa E, Bouyssie D, Delcourt N, et al. Label-free quantification and shotgun analysis of complex proteomes by one-dimensional SDS-PAGE/NanoLC-MS: evaluation for the large scale analysis of inflammatory human endothelial cells. Mol. Cell. Proteomics. 2012;11:527–539. doi: 10.1074/mcp.M111.015230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Duan X, Young R, Straubinger RM, Page B, et al. A straightforward and highly efficient precipitation/on-pellet digestion procedure coupled with a long gradient nano-LC separation and Orbitrap mass spectrometry for label-free expression profiling of the swine heart mitochondrial proteome. J Proteome Res. 2009;8:2838–2850. doi: 10.1021/pr900001t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gao J, Friedrichs MS, Dongre AR, Opiteck GJ. Guidelines for the routine application of the peptide hits technique. J Am Soc Mass Spectrom. 2005;16:1231–1238. doi: 10.1016/j.jasms.2004.12.002. [DOI] [PubMed] [Google Scholar]
- [30].Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- [31].Griffin NM, Yu J, Long F, Oh P, et al. Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis. Nat Biotechnol. 2010;28:83–89. doi: 10.1038/nbt.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Old WM, Meyer-Arendt K, Aveline-Wolf L, Pierce KG, et al. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics. 2005;4:1487–1502. doi: 10.1074/mcp.M500084-MCP200. [DOI] [PubMed] [Google Scholar]
- [33].Asara JM, Christofk HR, Freimark LM, Cantley LC. A label-free quantification method by MS/MS TIC compared to SILAC and spectral counting in a proteomics screen. Proteomics. 2008;8:994–999. doi: 10.1002/pmic.200700426. [DOI] [PubMed] [Google Scholar]
- [34].Tu C, Li J, Jiang X, Sheflin L, et al. Ion current-based proteomic profiling of the retina in a rat model of Smith-Lemli-Opitz Syndrome. Mol Cell Proteomics. 2013 doi: 10.1074/mcp.M113.027847. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yates JR, Ruse CI, Nakorchevsky A. Proteomics by mass spectrometry: approaches, advances, and applications. Annu. Rev. Biomed. Eng. 2009;11:49–79. doi: 10.1146/annurev-bioeng-061008-124934. [DOI] [PubMed] [Google Scholar]
- [36].Matzke MM, Brown JN, Gritsenko MA, Metz TO, et al. A comparative analysis of computational approaches to relative protein quantification using peptide peak intensities in label-free LC-MS proteomics experiments. Proteomics. 2013;13:493–503. doi: 10.1002/pmic.201200269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tu C, Li J, Sheng Q, Zhang M, Qu J. Systematic Assessment of Survey Scan and MS2-based Abundance Strategies for Label-Free Quantitative Proteomics Using High-resolution MS Data. J Proteome Res. 2014 doi: 10.1021/pr401206m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lai X, Wang L, Tang H, Witzmann FA. A novel alignment method and multiple filters for exclusion of unqualified peptides to enhance label-free quantification using peptide intensity in LC-MS/MS. J. Proteome Res. 2011;10:4799–4812. doi: 10.1021/pr2005633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Filiou MD, Martins-de-Souza D, Guest PC, Bahn S, Turck CW. To label or not to label: applications of quantitative proteomics in neuroscience research. Proteomics. 2012;12:736–747. doi: 10.1002/pmic.201100350. [DOI] [PubMed] [Google Scholar]
- [40].Carr SA, Anderson L. Protein quantitation through targeted mass spectrometry: the way out of biomarker purgatory? Clin Chem. 2008;54:1749–1752. doi: 10.1373/clinchem.2008.114686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mueller LN, Brusniak MY, Mani DR, Aebersold R. An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J. Proteome Res. 2008;7:51–61. doi: 10.1021/pr700758r. [DOI] [PubMed] [Google Scholar]
- [42].Zhu W, Smith JW, Huang CM. Mass spectrometry-based label-free quantitative proteomics. J Biomed Biotechnol. 2010;2010:840518. doi: 10.1155/2010/840518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Karp NA, Huber W, Sadowski PG, Charles PD, et al. Addressing accuracy and precision issues in iTRAQ quantitation. Mol. Cell. Proteomics. 2010;9:1885–1897. doi: 10.1074/mcp.M900628-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Merl J, Ueffing M, Hauck SM, von Toerne C. Direct comparison of MS-based label-free and SILAC quantitative proteome profiling strategies in primary retinal Muller cells. Proteomics. 2012;12:1902–1911. doi: 10.1002/pmic.201100549. [DOI] [PubMed] [Google Scholar]
- [45].Tu C, Jacob Mammen M, Li J, Shen X, et al. Large-Scale, Ion-Current-Based Proteomics Investigation of Bronchoalveolar Lavage Fluid in Chronic Obstructive Pulmonary Disease Patients. J Proteome Res. 2013 doi: 10.1021/pr4007602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Vaudel M, Sickmann A, Martens L. Current methods for global proteome identification. Expert Rev Proteomics. 2012;9:519–532. doi: 10.1586/epr.12.51. [DOI] [PubMed] [Google Scholar]
- [47].Nesvizhskii AI. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J Proteomics. 2010;73:2092–2123. doi: 10.1016/j.jprot.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Taylor SW, Fahy E, Zhang B, Glenn GM, et al. Characterization of the human heart mitochondrial proteome. Nat Biotechnol. 2003;21:281–286. doi: 10.1038/nbt793. [DOI] [PubMed] [Google Scholar]
- [49].Qu J, Straubinger RM. Improved sensitivity for quantification of proteins using triply charged cleavable isotope-coded affinity tag peptides. Rapid Commun Mass Spectrom. 2005;19:2857–2864. doi: 10.1002/rcm.2138. [DOI] [PubMed] [Google Scholar]
- [50].Tu C, Li J, Bu Y, Hangauer D, Qu J. An ion-current-based, comprehensive and reproducible proteomic strategy for comparative characterization of the cellular responses to novel anti-cancer agents in a prostate cell model. J Proteomics. 2012;77:187–201. doi: 10.1016/j.jprot.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Tu C, Li J, Young R, Page BJ, et al. Combinatorial peptide ligand library treatment followed by a dual-enzyme, dual-activation approach on a nanoflow liquid chromatography/orbitrap/electron transfer dissociation system for comprehensive analysis of swine plasma proteome. Anal Chem. 2011;83:4802–4813. doi: 10.1021/ac200376m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics. 2002;1:845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- [53].Fu Q, Garnham CP, Elliott ST, Bovenkamp DE, Van Eyk JE. A robust, streamlined, and reproducible method for proteomic analysis of serum by delipidation, albumin and IgG depletion, and two-dimensional gel electrophoresis. Proteomics. 2005;5:2656–2664. doi: 10.1002/pmic.200402048. [DOI] [PubMed] [Google Scholar]
- [54].Liu T, Qian WJ, Mottaz HM, Gritsenko MA, et al. Evaluation of multiprotein immunoaffinity subtraction for plasma proteomics and candidate biomarker discovery using mass spectrometry. Mol Cell Proteomics. 2006;5:2167–2174. doi: 10.1074/mcp.T600039-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Gundry RL, White MY, Nogee J, Tchernyshyov I, Van Eyk JE. Assessment of albumin removal from an immunoaffinity spin column: critical implications for proteomic examination of the albuminome and albumin-depleted samples. Proteomics. 2009;9:2021–2028. doi: 10.1002/pmic.200800686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jaffe JD, Keshishian H, Chang B, Addona TA, et al. Accurate Inclusion Mass Screening: A Bridge from Unbiased Discovery to Targeted Assay Development for Biomarker Verification. Molecular & Cellular Proteomics. 2008;7:1952–1962. doi: 10.1074/mcp.M800218-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Nagaraj N, Alexander Kulak N, Cox J, Neuhauser N, et al. System-wide Perturbation Analysis with Nearly Complete Coverage of the Yeast Proteome by Single-shot Ultra HPLC Runs on a Bench Top Orbitrap. Molecular & Cellular Proteomics. 2012;11 doi: 10.1074/mcp.M111.013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Qu J, Lesse AJ, Brauer AL, Cao J, et al. Proteomic expression profiling of Haemophilus influenzae grown in pooled human sputum from adults with chronic obstructive pulmonary disease reveal antioxidant and stress responses. BMC Microbiol. 2010;10:162. doi: 10.1186/1471-2180-10-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol. 2006;24:971–983. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- [60].Jullig M, Hickey AJ, Chai CC, Skea GL, et al. Is the failing heart out of fuel or a worn engine running rich? A study of mitochondria in old spontaneously hypertensive rats. Proteomics. 2008;8:2556–2572. doi: 10.1002/pmic.200700977. [DOI] [PubMed] [Google Scholar]
- [61].Baseler WA, Dabkowski ER, Jagannathan R, Thapa D, et al. Reversal of mitochondrial proteomic loss in Type 1 diabetic heart with overexpression of phospholipid hydroperoxide glutathione peroxidase. Am J Physiol Regul Integr Comp Physiol. 2013;304:R553–565. doi: 10.1152/ajpregu.00249.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Diz AP, Carvajal-Rodriguez A, Skibinski DO. Multiple hypothesis testing in proteomics: a strategy for experimental work. Mol Cell Proteomics. 2011;10:M110. doi: 10.1074/mcp.M110.004374. 004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kim YN, Kim HK, Warda M, Kim N, et al. Toward a better understanding of preeclampsia: Comparative proteomic analysis of preeclamptic placentas. Proteomics Clin Appl. 2007;1:1625–1636. doi: 10.1002/prca.200700034. [DOI] [PubMed] [Google Scholar]
- [64].Benjamini Y. a. H., Yosef Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. 1995:289–300. Series B. [Google Scholar]
- [65].Storey JD. A direct approach to false discovery rates. Journal of the Royal Statistical Society: Series B. 2002;64:479–198. [Google Scholar]
- [66].Chang J, Van Remmen H, Ward WF, Regnier FE, et al. Processing of data generated by 2-dimensional gel electrophoresis for statistical analysis: missing data, normalization, and statistics. J Proteome Res. 2004;3:1210–1218. doi: 10.1021/pr049886m. [DOI] [PubMed] [Google Scholar]
- [67].Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- [68].Margolin AA, Ong SE, Schenone M, Gould R, et al. Empirical Bayes analysis of quantitative proteomics experiments. PLoS One. 2009;4:e7454. doi: 10.1371/journal.pone.0007454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zhang J, Liem DA, Mueller M, Wang Y, et al. Altered proteome biology of cardiac mitochondria under stress conditions. J Proteome Res. 2008;7:2204–2214. doi: 10.1021/pr070371f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Baseler WA, Dabkowski ER, Williamson CL, Croston TL, et al. Proteomic alterations of distinct mitochondrial subpopulations in the type 1 diabetic heart: contribution of protein import dysfunction. Am J Physiol Regul Integr Comp Physiol. 2011;300:R186–200. doi: 10.1152/ajpregu.00423.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Dabkowski ER, Baseler WA, Williamson CL, Powell M, et al. Mitochondrial dysfunction in the type 2 diabetic heart is associated with alterations in spatially distinct mitochondrial proteomes. Am J Physiol Heart Circ Physiol. 2010;299:H529–540. doi: 10.1152/ajpheart.00267.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Bugger H, Schwarzer M, Chen D, Schrepper A, et al. Proteomic remodelling of mitochondrial oxidative pathways in pressure overload-induced heart failure. Cardiovasc Res. 2010;85:376–384. doi: 10.1093/cvr/cvp344. [DOI] [PubMed] [Google Scholar]
- [73].Warren CM, Geenen DL, Helseth DL, Jr., Xu H, Solaro RJ. Sub-proteomic fractionation, iTRAQ, and OFFGEL-LC-MS/MS approaches to cardiac proteomics. J Proteomics. 2010;73:1551–1561. doi: 10.1016/j.jprot.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Yin X, Cuello F, Mayr U, Hao Z, et al. Proteomics analysis of the cardiac myofilament subproteome reveals dynamic alterations in phosphatase subunit distribution. Mol Cell Proteomics. 2010;9:497–509. doi: 10.1074/mcp.M900275-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Van Hoof D, Dormeyer W, Braam SR, Passier R, et al. Identification of cell surface proteins for antibody-based selection of human embryonic stem cell-derived cardiomyocytes. J Proteome Res. 2010;9:1610–1618. doi: 10.1021/pr901138a. [DOI] [PubMed] [Google Scholar]
- [76].Scruggs SB, Reisdorph R, Armstrong ML, Warren CM, et al. A novel, in-solution separation of endogenous cardiac sarcomeric proteins and identification of distinct charged variants of regulatory light chain. Mol Cell Proteomics. 2010;9:1804–1818. doi: 10.1074/mcp.M110.000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Fessart D, Martin-Negrier ML, Claverol S, Thiolat ML, et al. Proteomic remodeling of proteasome in right heart failure. J Mol Cell Cardiol. 2013;66C:41–52. doi: 10.1016/j.yjmcc.2013.10.015. [DOI] [PubMed] [Google Scholar]
- [78].Gregersen N, Hansen J, Palmfeldt J. Mitochondrial proteomics--a tool for the study of metabolic disorders. J Inherit Metab Dis. 2012;35:715–726. doi: 10.1007/s10545-012-9480-3. [DOI] [PubMed] [Google Scholar]
- [79].McDonald TG, Van Eyk JE. Mitochondrial proteomics. Undercover in the lipid bilayer. Basic Res Cardiol. 2003;98:219–227. doi: 10.1007/s00395-003-0417-8. [DOI] [PubMed] [Google Scholar]
- [80].Edwards AV, White MY, Cordwell SJ. The role of proteomics in clinical cardiovascular biomarker discovery. Mol Cell Proteomics. 2008;7:1824–1837. doi: 10.1074/mcp.R800007-MCP200. [DOI] [PubMed] [Google Scholar]
- [81].Jullig M, Hickey AJ, Middleditch MJ, Crossman DJ, et al. Characterization of proteomic changes in cardiac mitochondria in streptozotocin-diabetic rats using iTRAQ isobaric tags. Proteomics Clin Appl. 2007;1:565–576. doi: 10.1002/prca.200600831. [DOI] [PubMed] [Google Scholar]
- [82].Goudarzi M, Ross MM, Zhou W, Van Meter A, et al. Development of a novel proteomic approach for mitochondrial proteomics from cardiac tissue from patients with atrial fibrillation. J Proteome Res. 2011;10:3484–3492. doi: 10.1021/pr200108m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Lancaster TS, Jefferson SJ, Hunter JC, Lopez V, et al. Quantitative proteomic analysis reveals novel mitochondrial targets of estrogen deficiency in the aged female rat heart. Physiol Genomics. 2012;44:957–969. doi: 10.1152/physiolgenomics.00184.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Nguyen T, Wong R, Wang G, Gucek M, et al. Acute inhibition of GSK causes mitochondrial remodeling. Am J Physiol Heart Circ Physiol. 2012;302:H2439–2445. doi: 10.1152/ajpheart.00033.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Kim TY, Wang D, Kim AK, Lau E, et al. Metabolic labeling reveals proteome dynamics of mouse mitochondria. Mol Cell Proteomics. 2012;11:1586–1594. doi: 10.1074/mcp.M112.021162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Weber KT. Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol. 1989;13:1637–1652. doi: 10.1016/0735-1097(89)90360-4. [DOI] [PubMed] [Google Scholar]
- [87].Spinale FG, Zile MR. Integrating the myocardial matrix into heart failure recognition and management. Circ Res. 2013;113:725–738. doi: 10.1161/CIRCRESAHA.113.300309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Ghosh AK, Vaughan DE. Fibrosis: is it a coactivator disease? Front Biosci (Elite Ed) 2012;4:1556–1570. doi: 10.2741/e480. [DOI] [PubMed] [Google Scholar]
- [89].Didangelos A, Yin X, Mandal K, Baumert M, et al. Proteomics characterization of extracellular space components in the human aorta. Mol Cell Proteomics. 2010;9:2048–2062. doi: 10.1074/mcp.M110.001693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Didangelos A, Yin X, Mandal K, Saje A, et al. Extracellular matrix composition and remodeling in human abdominal aortic aneurysms: a proteomics approach. Mol Cell Proteomics. 2011;10:M111. doi: 10.1074/mcp.M111.008128. 008128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Barallobre-Barreiro J, Didangelos A, Schoendube FA, Drozdov I, et al. Proteomics analysis of cardiac extracellular matrix remodeling in a porcine model of ischemia/reperfusion injury. Circulation. 2012;125:789–802. doi: 10.1161/CIRCULATIONAHA.111.056952. [DOI] [PubMed] [Google Scholar]
- [92].Higuchi S, Lin Q, Wang J, Lim TK, et al. Heart extracellular matrix supports cardiomyocyte differentiation of mouse embryonic stem cells. J Biosci Bioeng. 2013;115:320–325. doi: 10.1016/j.jbiosc.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Edwards AV, Cordwell SJ, White MY. Phosphoproteomic profiling of the myocyte. Circ Cardiovasc Genet. 2011;4:575. doi: 10.1161/CIRCGENETICS.110.957787. [DOI] [PubMed] [Google Scholar]
- [94].Hart GW, Ball LE. Post-translational modifications: a major focus for the future of proteomics. Mol Cell Proteomics. 2013;12:3443. doi: 10.1074/mcp.E113.036491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Agnetti G, Husberg C, Van Eyk JE. Divide and conquer: the application of organelle proteomics to heart failure. Circ Res. 2011;108:512–526. doi: 10.1161/CIRCRESAHA.110.226910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Van Eyk JE. Lessons from old and new kinases. Circ Res. 2004;94:135–137. doi: 10.1161/01.RES.0000117526.17737.40. [DOI] [PubMed] [Google Scholar]
- [97].Boja ES, Phillips D, French SA, Harris RA, Balaban RS. Quantitative mitochondrial phosphoproteomics using iTRAQ on an LTQ-Orbitrap with high energy collision dissociation. J. Proteome Res. 2009;8:4665–4675. doi: 10.1021/pr900387b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Chang YW, Chang YT, Wang Q, Lin JJ, et al. Quantitative phosphoproteomic study of pressure-overloaded mouse heart reveals dynamin-related protein 1 as a modulator of cardiac hypertrophy. Mol Cell Proteomics. 2013;12:3094–3107. doi: 10.1074/mcp.M113.027649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Scholten A, Preisinger C, Corradini E, Bourgonje VJ, et al. Phosphoproteomics study based on in vivo inhibition reveals sites of calmodulin-dependent protein kinase II regulation in the heart. J Am Heart Assoc. 2013;2:e000318. doi: 10.1161/JAHA.113.000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Lundby A, Andersen MN, Steffensen AB, Horn H, et al. In vivo phosphoproteomics analysis reveals the cardiac targets of beta-adrenergic receptor signaling. Sci Signal. 2013;6:rs11. doi: 10.1126/scisignal.2003506. [DOI] [PubMed] [Google Scholar]
- [101].Murray CI, Uhrigshardt H, O'Meally RN, Cole RN, Van Eyk JE. Identification and quantification of S-nitrosylation by cysteine reactive tandem mass tag switch assay. Mol Cell Proteomics. 2012;11:M111. doi: 10.1074/mcp.M111.013441. 013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Parker BL, Palmisano G, Edwards AV, White MY, et al. Quantitative N-linked glycoproteomics of myocardial ischemia and reperfusion injury reveals early remodeling in the extracellular environment. Mol Cell Proteomics. 2011;10:M110. doi: 10.1074/mcp.M110.006833. 006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Lundby A, Lage K, Weinert BT, Bekker-Jensen DB, et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012;2:419–431. doi: 10.1016/j.celrep.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Bousette N, Chugh S, Fong V, Isserlin R, et al. Constitutively active calcineurin induces cardiac endoplasmic reticulum stress and protects against apoptosis that is mediated by alpha-crystallin-B. Proc Natl Acad Sci U S A. 2010;107:18481–18486. doi: 10.1073/pnas.1013555107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Li C, Qiu Q, Wang Y, Li P, et al. Time course label-free quantitative analysis of cardiac muscles of rats after myocardial infarction. Mol Biosyst. 2014 doi: 10.1039/c3mb70422j. [DOI] [PubMed] [Google Scholar]
- [106].Dubois E, Fertin M, Burdese J, Amouyel P, et al. Cardiovascular proteomics: translational studies to develop novel biomarkers in heart failure and left ventricular remodeling. Proteomics Clin Appl. 2011;5:57–66. doi: 10.1002/prca.201000056. [DOI] [PubMed] [Google Scholar]
- [107].Gerszten RE, Carr SA, Sabatine M. Integration of proteomic-based tools for improved biomarkers of myocardial injury. Clin Chem. 2010;56:194–201. doi: 10.1373/clinchem.2009.127878. [DOI] [PubMed] [Google Scholar]
- [108].McGregor E, Dunn MJ. Proteomics of the heart: unraveling disease. Circ Res. 2006;98:309–321. doi: 10.1161/01.RES.0000201280.20709.26. [DOI] [PubMed] [Google Scholar]
- [109].Canty JM, Jr., Suzuki G. Myocardial perfusion and contraction in acute ischemia and chronic ischemic heart disease. J. Mol. Cell. Cardiol. 2012;52:822–831. doi: 10.1016/j.yjmcc.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Stastna M, Van Eyk JE. Secreted proteins as a fundamental source for biomarker discovery. Proteomics. 2012;12:722–735. doi: 10.1002/pmic.201100346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Hammer E, Goritzka M, Ameling S, Darm K, et al. Characterization of the human myocardial proteome in inflammatory dilated cardiomyopathy by label-free quantitative shotgun proteomics of heart biopsies. J Proteome Res. 2011;10:2161–2171. doi: 10.1021/pr1008042. [DOI] [PubMed] [Google Scholar]
- [112].Kakimoto Y, Ito S, Abiru H, Kotani H, et al. Sorbin and SH3 domain-containing protein 2 is released from infarcted heart in the very early phase: proteomic analysis of cardiac tissues from patients. J Am Heart Assoc. 2013;2:e000565. doi: 10.1161/JAHA.113.000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Holland A, Dowling P, Zweyer M, Swandulla D, et al. Proteomic profiling of cardiomyopathic tissue from the aged mdx model of Duchenne muscular dystrophy reveals a drastic decrease in laminin, nidogen and annexin. Proteomics. 2013;13:2312–2323. doi: 10.1002/pmic.201200578. [DOI] [PubMed] [Google Scholar]
- [114].Chugh S, Ouzounian M, Lu Z, Mohamed S, et al. Pilot study identifying myosin heavy chain 7, desmin, insulin-like growth factor 7, and annexin A2 as circulating biomarkers of human heart failure. Proteomics. 2013;13:2324–2334. doi: 10.1002/pmic.201200455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Jing L, Parker CE, Seo D, Hines MW, et al. Discovery of biomarker candidates for coronary artery disease from an APOE-knock out mouse model using iTRAQ-based multiplex quantitative proteomics. Proteomics. 2011;11:2763–2776. doi: 10.1002/pmic.201000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Silbiger VN, Luchessi AD, Hirata RD, Neto LG, et al. Time course proteomic profiling of human myocardial infarction plasma samples: an approach to new biomarker discovery. Clin Chim Acta. 2011;412:1086–1093. doi: 10.1016/j.cca.2011.02.030. [DOI] [PubMed] [Google Scholar]
- [117].Juhasz P, Lynch M, Sethuraman M, Campbell J, et al. Semi-targeted plasma proteomics discovery workflow utilizing two-stage protein depletion and off-line LC-MALDI MS/MS. J Proteome Res. 2011;10:34–45. doi: 10.1021/pr100659e. [DOI] [PubMed] [Google Scholar]
- [118].Mebazaa A, Vanpoucke G, Thomas G, Verleysen K, et al. Unbiased plasma proteomics for novel diagnostic biomarkers in cardiovascular disease: identification of quiescin Q6 as a candidate biomarker of acutely decompensated heart failure. Eur Heart J. 2012;33:2317–2324. doi: 10.1093/eurheartj/ehs162. [DOI] [PubMed] [Google Scholar]
- [119].Lewis GD, Gerszten RE. Toward metabolomic signatures of cardiovascular disease. Circ Cardiovasc Genet. 2010;3:119–121. doi: 10.1161/CIRCGENETICS.110.954941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Li M, Gray W, Zhang H, Chung CH, et al. Comparative shotgun proteomics using spectral count data and quasi-likelihood modeling. J Proteome Res. 2010;9:4295–4305. doi: 10.1021/pr100527g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Qu J, Jusko WJ, Straubinger RM. Utility of cleavable isotope-coded affinity-tagged reagents for quantification of low-copy proteins induced by methylprednisolone using liquid chromatography/tandem mass spectrometry. Anal Chem. 2006;78:4543–4552. doi: 10.1021/ac0521697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Schluter H, Apweiler R, Holzhutter HG, Jungblut PR. Finding one's way in proteomics: a protein species nomenclature. Chem Cent J. 2009;3:11. doi: 10.1186/1752-153X-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Chait BT. Chemistry. Mass spectrometry: bottom-up or top-down? Science. 2006;314:65–66. doi: 10.1126/science.1133987. [DOI] [PubMed] [Google Scholar]
- [124].Zhang J, Guy MJ, Norman HS, Chen YC, et al. Top-down quantitative proteomics identified phosphorylation of cardiac troponin I as a candidate biomarker for chronic heart failure. J Proteome Res. 2011;10:4054–4065. doi: 10.1021/pr200258m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Dong X, Sumandea CA, Chen YC, Garcia-Cazarin ML, et al. Augmented phosphorylation of cardiac troponin I in hypertensive heart failure. J Biol Chem. 2012;287:848–857. doi: 10.1074/jbc.M111.293258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Zhang J, Zhang H, Ayaz-Guner S, Chen YC, et al. Phosphorylation, but not alternative splicing or proteolytic degradation, is conserved in human and mouse cardiac troponin T. Biochemistry (Mosc) 2011;50:6081–6092. doi: 10.1021/bi2006256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Ryan CM, Souda P, Bassilian S, Ujwal R, et al. Post-translational modifications of integral membrane proteins resolved by top-down Fourier transform mass spectrometry with collisionally activated dissociation. Mol Cell Proteomics. 2010;9:791–803. doi: 10.1074/mcp.M900516-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Lange V, Picotti P, Domon B, Aebersold R. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol Syst Biol. 2008;4:222. doi: 10.1038/msb.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Liebler DC, Zimmerman LJ. Targeted quantitation of proteins by mass spectrometry. Biochemistry (Mosc) 2013;52:3797–3806. doi: 10.1021/bi400110b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Shi T, Fillmore TL, Sun X, Zhao R, et al. Antibody-free, targeted mass-spectrometric approach for quantification of proteins at low picogram per milliliter levels in human plasma/serum. Proc Natl Acad Sci U S A. 2012;109:15395–15400. doi: 10.1073/pnas.1204366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].MacLean B, Tomazela DM, Shulman N, Chambers M, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Shi T, Su D, Liu T, Tang K, et al. Advancing the sensitivity of selected reaction monitoring-based targeted quantitative proteomics. Proteomics. 2012;12:1074–1092. doi: 10.1002/pmic.201100436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Pan S, Aebersold R, Chen R, Rush J, et al. Mass spectrometry based targeted protein quantification: methods and applications. J Proteome Res. 2009;8:787–797. doi: 10.1021/pr800538n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Stergachis AB, MacLean B, Lee K, Stamatoyannopoulos JA, MacCoss MJ. Rapid empirical discovery of optimal peptides for targeted proteomics. Nat Methods. 2011;8:1041–1043. doi: 10.1038/nmeth.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Cao J, Gonzalez-Covarrubias V, Straubinger RM, Wang H, et al. A rapid, reproducible, on-the-fly orthogonal array optimization method for targeted protein quantification by LC/MS and its application for accurate and sensitive quantification of carbonyl reductases in human liver. Anal Chem. 2010;82:2680–2689. doi: 10.1021/ac902314m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Duan X, Abuqayyas L, Dai L, Balthasar JP, Qu J. High-throughput method development for sensitive, accurate, and reproducible quantification of therapeutic monoclonal antibodies in tissues using orthogonal array optimization and nano liquid chromatography/selected reaction monitoring mass spectrometry. Anal Chem. 2012;84:4373–4382. doi: 10.1021/ac2034166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Duan X, Dai L, Chen SC, Balthasar JP, Qu J. Nano-scale liquid chromatography/mass spectrometry and on-the-fly orthogonal array optimization for quantification of therapeutic monoclonal antibodies and the application in preclinical analysis. J Chromatogr A. 2012;1251:63–73. doi: 10.1016/j.chroma.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Anderson NL, Anderson NG, Haines LR, Hardie DB, et al. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA) J Proteome Res. 2004;3:235–244. doi: 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- [139].Neubert H, Muirhead D, Kabir M, Grace C, et al. Sequential protein and peptide immunoaffinity capture for mass spectrometry-based quantification of total human beta-nerve growth factor. Anal Chem. 2013;85:1719–1726. doi: 10.1021/ac303031q. [DOI] [PubMed] [Google Scholar]
- [140].Keshishian H, Addona T, Burgess M, Mani DR, et al. Quantification of cardiovascular biomarkers in patient plasma by targeted mass spectrometry and stable isotope dilution. Mol Cell Proteomics. 2009;8:2339–2349. doi: 10.1074/mcp.M900140-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Shi T, Fillmore TL, Gao Y, Zhao R, et al. Long-gradient separations coupled with selected reaction monitoring for highly sensitive, large scale targeted protein quantification in a single analysis. Anal Chem. 2013 doi: 10.1021/ac402105s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Huillet C, Adrait A, Lebert D, Picard G, et al. Accurate quantification of cardiovascular biomarkers in serum using Protein Standard Absolute Quantification (PSAQ) and selected reaction monitoring. Mol Cell Proteomics. 2012;11:M111. doi: 10.1074/mcp.M111.008235. 008235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Jiang H, Zeng J, Titsch C, Voronin K, et al. Fully validated LC-MS/MS assay for the simultaneous quantitation of coadministered therapeutic antibodies in cynomolgus monkey serum. Anal. Chem. 2013;85:9859–9867. doi: 10.1021/ac402420v. [DOI] [PubMed] [Google Scholar]
- [144].Ocaña MF, Neubert H. An immunoaffinity liquid chromatography–tandem mass spectrometry assay for the quantitation of matrix metalloproteinase 9 in mouse serum. Anal. Biochem. 2010;399:202–210. doi: 10.1016/j.ab.2010.01.002. [DOI] [PubMed] [Google Scholar]
- [145].Nouri-Nigjeh E, Zhang M, Ji T, Yu H, et al. Effects of Calibration Approaches on the Accuracy for LC/MS Targeted Quantification of Therapeutic Protein. Anal Chem. 2014 doi: 10.1021/ac5001477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Kuhn E, Addona T, Keshishian H, Burgess M, et al. Developing multiplexed assays for troponin I and interleukin-33 in plasma by peptide immunoaffinity enrichment and targeted mass spectrometry. Clin Chem. 2009;55:1108–1117. doi: 10.1373/clinchem.2009.123935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Domanski D, Percy AJ, Yang J, Chambers AG, et al. MRM-based multiplexed quantitation of 67 putative cardiovascular disease biomarkers in human plasma. Proteomics. 2012;12:1222–1243. doi: 10.1002/pmic.201100568. [DOI] [PubMed] [Google Scholar]
- [148].Cohen Freue GV, Borchers CH. Multiple reaction monitoring (MRM): principles and application to coronary artery disease. Circ Cardiovasc Genet. 2012;5:378. doi: 10.1161/CIRCGENETICS.111.959528. [DOI] [PubMed] [Google Scholar]
- [149].Cohen Freue GV, Meredith A, Smith D, Bergman A, et al. Computational biomarker pipeline from discovery to clinical implementation: plasma proteomic biomarkers for cardiac transplantation. PLoS Comput Biol. 2013;9:e1002963. doi: 10.1371/journal.pcbi.1002963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Kristensen LP, Larsen MR, Mickley H, Saaby L, et al. Plasma proteome profiling of atherosclerotic disease manifestations reveals elevated levels of the cytoskeletal protein vinculin. J Proteomics. 2013 doi: 10.1016/j.jprot.2013.12.011. [DOI] [PubMed] [Google Scholar]
- [151].Lam MP, Scruggs SB, Kim TY, Zong C, et al. An MRM-based workflow for quantifying cardiac mitochondrial protein phosphorylation in murine and human tissue. J Proteomics. 2012;75:4602–4609. doi: 10.1016/j.jprot.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Lam MP, Lau E, Scruggs SB, Wang D, et al. Site-specific quantitative analysis of cardiac mitochondrial protein phosphorylation. J Proteomics. 2013;81:15–23. doi: 10.1016/j.jprot.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Zhang P, Kirk JA, Ji W, dos Remedios CG, et al. Multiple reaction monitoring to identify site-specific troponin I phosphorylated residues in the failing human heart. Circulation. 2012;126:1828–1837. doi: 10.1161/CIRCULATIONAHA.112.096388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Kooij V, Zhang P, Piersma SR, Sequeira V, et al. PKCalpha-Specific Phosphorylation of the Troponin Complex in Human Myocardium: A Functional and Proteomics Analysis. PLoS One. 2013;8:e74847. doi: 10.1371/journal.pone.0074847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Aebersold R, Burlingame AL, Bradshaw RA. Western blots versus selected reaction monitoring assays: time to turn the tables? Mol Cell Proteomics. 2013;12:2381–2382. doi: 10.1074/mcp.E113.031658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Fu Q, Schoenhoff FS, Savage WJ, Zhang P, Van Eyk JE. Multiplex assays for biomarker research and clinical application: translational science coming of age. Proteomics Clin Appl. 2010;4:271–284. doi: 10.1002/prca.200900217. [DOI] [PubMed] [Google Scholar]
- [157].Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Black KM, Barnett RJ, Bhasin MK, Daly C, et al. Microarray and proteomic analysis of the cardioprotective effects of cold blood cardioplegia in the mature and aged male and female. Physiol Genomics. 2012;44:1027–1041. doi: 10.1152/physiolgenomics.00011.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Langley SR, Dwyer J, Drozdov I, Yin X, Mayr M. Proteomics: from single molecules to biological pathways. Cardiovasc Res. 2013;97:612–622. doi: 10.1093/cvr/cvs346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Hebert AS, Richards AL, Bailey DJ, Ulbrich A, et al. The one hour yeast proteome. Mol Cell Proteomics. 2014;13:339–347. doi: 10.1074/mcp.M113.034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.