

Abstract
The endocannabinoid system, most popularly known as the target of the psychoactive component of marijuana, Δ9-tetrahydrocannabinol (THC), is a signaling network that modulates a diverse range of physiological processes including nociception, behavior, cognitive function, appetite, metabolism, motor control, memory formation, and inflammation. While THC and its derivatives have garnered notoriety in the eyes of the public, the endocannabinoid system is consists of two endogenous signaling lipids 2-arachidonoylglycerol (2-AG) and N-arachidonoylethanolamine (anandamide) that activate cannabinoid receptors CB1 and CB2 in the nervous system and peripheral tissues. This review will focus on recent efforts to chemically manipulate 2-AG signaling, through the development of inhibitors of the 2-AG-synthesizing enzyme diacylglycerol lipase (DAGL) or the 2-AG-degrading enzyme monoacylglycerol lipase (MAGL), and assessing the therapeutic potential of DAGL and MAGL inhibitors in pain, inflammation, degenerative diseases, tissue injury, and cancer.
1. Introduction
The endocannabinoid system is a neurotransmission pathway and the primary target of the psychoactive ligand in marijuana, Δ9-tetrahydrocannabinol (THC). Marijuana has been in use for centuries for both medicinal and recreational purposes and has profound effects on nociception, behavior, cognitive function, appetite, metabolism, motor control, memory formation, and immune suppression 1, 2. While THC has gained a certain notoriety in the public eye, the endogenous physiological function of the endocannabinoid system is to respond to the signaling lipids 2-arachidonoylglycerol (2-AG) and N-arachidonoylethanolamine (anandamide) 3, 4. THC and endocannabinoids act through the membrane-bound G-coupled protein receptors cannabinoid receptors type 1 and 2 (CB1 and CB2) to alter these varied aspects of mammalian physiology. CB1 is highly expressed in the central nervous system and to a lower extent in peripheral tissues. CB1 activation appears to control most of the neurogenic features associated with cannabinoid exposure, including hypothermia, hypomotility, anti-nociception, and catalepsy. In contrast, CB2 is expressed predominantly in immune cells such as monocytes, macrophages, CD4+ and CD8+ T cells, and B cells. Originally described as a peripheral cannabinoid receptor, mounting evidence shows that CB2 is also expressed in microglia, which are derived from macrophages, during neuroinflammatory and neurodegenerative disease states 4-10.
In recent decades, innovative chemical approaches and proteomic and metabolomic technologies have been applied to the endocannabinoid field towards understanding the roles of endocannabinoid signaling lipids in physiology and disease, through the development of inhibitors for endocannabinoid synthesis or degradation. 2-AG is synthesized by diacylglycerol lipase (DAGL) and is degraded by monoacylglycerol lipase (MAGL). Anandamide is synthesized by initial generation of N-arachidonoyl phosphatidylethanolamine followed by several postulated routes, and degraded by fatty acid amide hydrolase (FAAH).The (patho)physiological roles, biochemical regulation, and therapeutic potential of FAAH, FAAH inhibitors, and anandamide have been previously extensively studied and reviewed 4, 11. Here, we will focus this review on chemical approaches that have been applied to understanding 2-AG signaling and metabolism and its (patho)physiological roles in various disease states. We will also discuss the therapeutic potential of inhibitors for 2-AG degradation and synthesis.
2. Endocannabinoid signaling
Endocannabinoid signaling in neurons occurs by a non-vesicular calcium-dependent retrograde mechanism. Stimulation of the post-synapse triggers synthesis of endocannabinoids and their subsequent release, although the mechanism by which the endocannabinoid ligand travels to CB1 receptors at the presynaptic interneuron terminal is poorly understood 5. CB1 activation inhibits neurotransmitter release by activating Gi/o proteins, thereby inhibiting calcium and potassium channels 12. Originally discovered in 1995 as the second endocannabinoid signaling lipid, 2-AG has been shown to be the major mediator of CB1-dependent synaptic plasticity controlling retrograde neurotransmission through depolarization-induced suppression of inhibition (DSI) and excitation (DSE) 13-19. Endocannabinoids are lipid messengers rather than water-soluble metabolites, thus hydrophobic interactions make their storage in synaptic vesicles unlikely. Instead, endocannabinoids are likely mobilized “on demand” from membrane phospholipid precursors or potential storage sites such as lipid rafts 5, 20.
3. Generation of Inhibitors for 2-AG Degradation and Synthesis
Understanding the physiological roles of 2-AG signaling have been greatly accelerated in recent years through the development of enzymatic inhibitors for 2-AG metabolism.
3.1 Enzymes Controlling 2-AG Degradation and Synthesis
3.1.1 Monoacylglycerol Lipase (MAGL)
2-AG is degraded primarily by monoacylglycerol lipase (MAGL) to glycerol and arachidonic acid both in vitro and in vivo (Figure 1) 4, 11, 21. MAGL is a soluble serine hydrolase that peripherally associates with cell membranes and was originally isolated from adipose tissue as the enzyme responsible for the final lipolytic step in triacylglycerol catabolism. Immunodepletion of MAGL in rat brain reduced 2-AG hydrolytic activity by 50 % 22-25. Functional proteomic profiling of 2-AG hydrolytic activity in vitro showed that MAGL in the brain is responsible for 85 % of total 2-AG hydrolytic activity 26. MAGL-deficient mice show dramatically elevated levels of 2-AG levels in brain and peripheral tissues 27. Interestingly, these mice show partial desensitization of CB1 in the brain and blunted responses to exogenous CB1 agonists due to functional antagonism of the endocannabinoid system 27. Pan et al showed that MAGL -/- mice selectively enhanced theta burst stimulation-induced long-term potentiation in the CA1 region of hippocampal slices 13, 16.
Figure 1. Pathways that Control 2-AG Degradation and Synthesis.
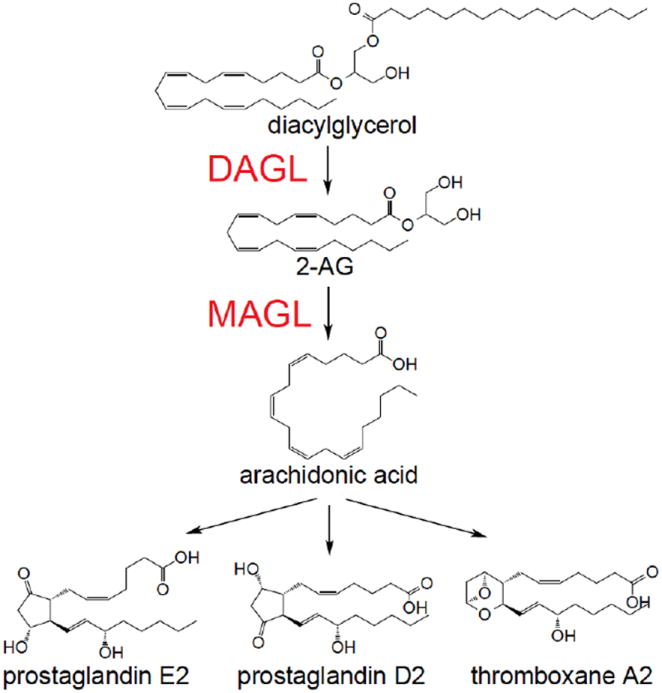
DAGL synthesizes 2-AG through hydrolysis of diacylglycerols and MAGL generates arachidonic acid for eicosanoid biosynthesis through the hydrolysis of 2-AG
There are also other serine hydrolases that have been implicated in 2-AG hydrolysis. Previous studies using inhibitors of MAGL in mice have found that approximately 15% of 2-AG hydrolytic activity persists after MAGL inhibition. Blankman et al. established that the serine hydrolases, α/β-hydrolase 6 and 12 (ABHD6 and 12), were responsible for the remaining 2-AG hydrolytic activity 26. While it is unclear what role ABHD6 and ABHD12 may play in 2-AG metabolism and signaling, recent studies indicate that these enzymes may have alternate physiological functions. Thomas et al. recently showed that genetic knockdown of ABHD6 protects mice against diet-induced obesity and acts as a general lysophospholipid hydrolase that turns over lysophosphatidylglycerol, lysophosphatidylethanolamine, lysophosphatidic acid, and lysophosphatidylserine 28. Blankman et al. recently discovered that ABHD12 hydrolyzes lysophosphatidylserine (LPS) and that ABHD12-deficient mice have elevated levels of brain LPS lipids, but not 2-AG, leading to increased Toll-like receptor activation and age-dependent microglial activation and auditory and motor deficits that resemble the behavioral phenotypes of the human polyneuropathy, hearing loss, ataxia, retinosis, and cataract (PHARC) disorder caused by ABHD12 loss-of-function 29.
3.1.2 Diacylglycerol Lipases (DAGL)
The biosynthetic pathway for 2-AG relies mainly on two enzymes, diacylglycerol lipase-α and -β (DAGLα and DAGLβ), to synthesize 2-AG from hydrolysis of arachidonoyl-containing diacylglycerols (DAGs) (Figure 1). DAGs are thought to be synthesized from membrane-bound phospholipids, primarily from sn-2 arachidonoyl phosphatidylinositol 4,5-biosphosphate by phospholipase Cβ. Two independent studies have confirmed the importance of the two DAGL isoforms in generating 2-AG in vivo. Interestingly, these studies have also demonstrated differential contributions of these two isoforms to 2-AG synthesis across various tissues. DAGLα primarily regulates 2-AG levels in the brain, with DAGLα and DAGLβ knockout mice showing ~80% and 50 % reduction in brain 2-AG levels, respectively 17, 19. Interestingly, DAGLα knockout mice show dramatic reduction of 2-AG in cortex, cerebellum, hypothalamus, and hippocampus while DAGLβ knockout mice showed lower 2-AG levels only in the hypothalamus, indicating differential contributions of these two isoforms even within different regions in the brain 17, 19. In contrast to the brain, DAGLβ is the dominant enzyme for 2-AG synthesis in the liver as evidenced by a ~90 % reduction in liver 2-AG levels in DAGLβ knockout mice, compared to ~50-60 % reduction in 2-AG levels in DAGLα-deficient livers. Studies using these knockout mice have shown an important role for the two isoforms of DAGL in retrograde endocannabinoid signaling and adult neurogenesis. The transient suppression of GABA-mediated transmission at inhibitory synapses induced by post-synaptic release of endocannabinoids is lost in DAGLα knockout, but not in DAGLβ knockout mice. Both DAGLα and DAGLβ knockout mice show compromised control of adult neurogenesis in the hippocampus or subventricular zone. These studies thus show that DAGL activity in the brain is essential for regulating retrograde synaptic plasticity and adult neurogenesis 17, 19.
3.2 First-generation MAGL and DAGL inhibitors
3.2.1 First-generation MAGL Inhibitors
First-generation MAGL inhibitors were non-selective or had modest in vivo activity (Figure 2a). Nonetheless, these inhibitors were initially used to indicate that MAGL was a 2-AG hydrolase and that MAGL blockade led to increased brain 2-AG levels in mice and rats. Both MAGL and FAAH activity can be attenuated with general serine hydrolase inhibitors such as methyl arachidonoylfluorophosphonate, phenylmethanesulfonyl fluoride, arachidonoyl trifluoromethylketone, and hexadecyl sulfonylfluoride 22, 30. MAGL, unlike FAAH and other serine hydrolases, is also sensitive to sulfhydryl-specific inhibitors, indicative of a free cysteine residue near the active site, such as mercury chloride, 4-chloromercuribenzoic acid, and N-ethylmaleimide. The first semi-selective MAGL inhibitors URB602, N-arachidonoyl maleimide (NAM), and OMDM169 exhibited modest increases in 2-AG concentration and proved to be effective against rodent models of pain. The carbamate compound URB602 showed an approximately two-fold increase in the concentration of 2-AG, but not anandamide, in rat central gray matter 31. URB602 has low potency in vivo and possible overlapping selectivity with FAAH in vitro 31-33, making it unsuitable for work distinguishing the functions of these two enzymes. NAM was found to nearly abolish 2-AG hydrolysis in vitro using rat cerebellar membranes and was found to have a permissive effect on exogenous 2-AG administration in mice 34. Though NAM is relatively selective for MAGL compared to FAAH and other serine hydrolases, NAM has limited utility since the maleimide group is a thiol-reactive electrophile likely to react with many cysteine-containing residues. Indeed, CB1-knockout mice treated with NAM plus 2-AG administration retained locomotor inhibition similar to wildtype mice, suggesting that NAM may have additional mechanisms of action. OMDM169, a derivative of tetrahydrolipostatin, was capable of a modest increase of 2-AG, but not anandamide, levels in neuroblastoma cells and in paws of formalin-treated mice. OMDM169 shared similar inhibitory effects for MAGL and pancreatic lipase while having an approximately 10-fold greater selectivity over FAAH and DAGLα 35.
Figure 2. First-generation MAGL and DAGL Inhibitors.

First-generation MAGL (a) and DAGL (b) inhibitors were non-selective, not potent, or not in vivo active.
The sarin analog isopropyl dodecylfluorophosphonate (IDFP) and, surprisingly, the insecticide chlorpyrifos were also used to study the in vivo effects of inhibiting MAGL 36. IDFP fully inhibited MAGL in vivo, but this inhibitor was non-selective, inhibiting MAGL, FAAH, and several other serine hydrolases. The insecticide chlorpyrifos completely blocked MAGL and partially blocked FAAH in vivo in the brain through bioactivation of this compound to the chlorpyrifos oxon. While this insecticide was more selective than IDFP, it also inhibited the lethal target acetylcholinesterase, Both IDFP and chlorpyrifos administration showed many cannabinoid-mediated behaviors including catalepsy, which was later found to be caused by dual blockade of MAGL and FAAH. IDFP and chlorpyrifos-treated mice showed >10-fold elevations in brain 2-AG and anandamide levels, and interestingly also showed a stoichiometric reduction in arachidonic acid levels, indicating that 2-AG and arachidonic acid levels may be linked in the brain through MAGL 36, 37. Nonetheless, these inhibitors were limited in their ability to specifically dissect the roles of MAGL in vivo due to their non-selectivity.
3.2.2. First-generation DAGL Inhibitors
The synthesis of dual DAGL inhibitors or selective DAGLα or DAGLβ inhibitors has been hampered by a lack of resolved crystal structures to provide structural knowledge about the target and a dearth of functional assays to assess endogenous DAGL activity. In early studies, in vitro hydrolysis of exogenous sn-1-[14C]-oleoyl-2-arachidonoyl-glycerol was used as a readout of DAGL activity. The general lipase inhibitor tetrahydrolipostatin (THL, Orlistat) and the compound RHC-80267 inhibit DAGL-mediated synthesis of 2-AG, although at a higher concentration than needed to inhibit other lipases 15, 38 (Figure 2b). Both DAGL enzymes are also sensitive to treatment with the serine hydrolase inhibitors mercury chloride, 4-chloromercuribenzoic acid and methyl arachidonoylfluorophosphonate (MAFP) 39. Bisogno et al also developed MAFP organophosphorus analogs O-3640 and O-3841 that showed high selectivity for DAGLα over DAGLβ and other lipases but had poor potency, lack of stability, and poor cell penetrance 38. Further medicinal chemistry studies improved upon O-3841, yielding the similarly potent O-5596 but with better bioavailability and stability in physiological buffers 40. Interestingly, O-5596-treated mice displayed a significant decrease in ad libitum consumption of sweetened cereal, but not regular chow, compared to vehicle-treated mice. This is in agreement with other studies using CB1 antagonists showing that endocannabinoid biosynthesis might be upregulated in response to palatable food exposure 41, 42. Indeed, animal models of overnutrition have been linked to elevated 2-AG levels, suggesting that DAGL inhibitors may be of use as anti-obesity therapeutics 43.
3.3 Activity-Based Protein Profiling (ABPP) for the Development of DAGL and MAGL Inhibitors
The identification and molecular characterization of more potent, selective, and in vivo efficacious inhibitors of MAGL and DAGL have been greatly accelerated by the use of the chemoproteomic technology, activity-based protein profiling (ABPP) (Figure 3)11, 44, 45. ABPP uses active-site directed chemical probes to directly assess the functional state of large numbers of enzymes in complex biological systems. Activity-based probes consist of a reactive chemical moiety that reacts with the active-site of specific enzyme class(es) and is coupled to an analytical handle, such as a fluorophore or biotin, enabling the detection of enzyme activities by fluorescence or mass-spectrometry based proteomics 11, 44-46. Because these probes bind to the active sites of enzymes, small-molecule inhibitor libraries can be competed directly against probe labeling of either pure enzymes or enzymes in complex native proteomes, enabling an assay strategy for inhibitor development. Furthermore, because the probe not only binds to the enzyme of interest, but also the active-sites of other proteins in the enzyme class, ABPP facilitates the assessment of inhibitor selectivity on a proteome-wide scale. Additionally, with compounds that bind active sites irreversibly, target occupancy and selectivity of the inhibitors in vivo could be easily assessed ex vivo in any tissue of interest using ABPP platforms 47.
Figure 3. Activity-Based Protein Profiling (ABPP) for Developing MAGL and DAGL Inhibitors.
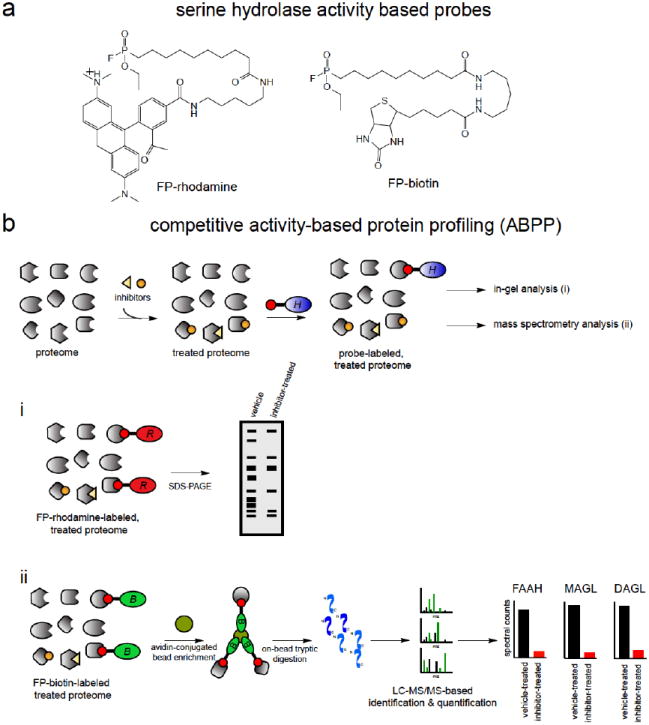
(a) Activity-based probes for serine hydrolases, fluorophosphonate (FP)-rhodamine and FP-biotin. (b) Competitive ABPP has been used to develop highly selective MAGL and DAGL inhibitors. ABPP uses active-site directed probes to assess the activities of large numbers of enzymes either by in-gel fluorescence (i) or by mass-spectrometry-based proteomics (ii). Because the probes bind to the active-sites, small-molecule inhibitor libraries can be competed against probe binding, facilitating an inhibitor-discovery platform. Selectivity can also be assessed across the entire enzyme class since probe-bound enzyme activities can be read-out in parallel.
Many of the enzymes involved in endocannabinoid metabolism, including MAGL, DAGL, and FAAH belong to the serine hydrolase superfamily of enzymes 11, 45. The fluorophosphonate (FP)-activity-based probes, FP-rhodamine and FP-biotin, were developed to assess the activities of serine hydrolases (Figure 3a) 48. These and other activity based probes have been used successfully in developing selective FAAH, MAGL, and DAGL inhibitors (Figure 3b). We will focus this review specifically on the development and effects of MAGL and DAGL inhibitors.
3.4 MAGL-selective inhibitors and their effects
Using ABPP platforms, Long et al. in 2009 put forth the first selective and in vivo active MAGL inhibitor JZL184, which contributed greatly in advancing our understanding of the physiological roles of MAGL (Figure 4a) 49, 50. JZL184 was developed through initial screening of a carbamate library of serine hydrolase inhibitors and subsequent optimization by traditional medicinal chemistry efforts. JZL184 is a piperidine carbamate that inhibits MAGL activity through irreversibly carbamylating the active-site catalytic serine nucleophile 49, 50. Competitive ABPP analysis using the FP-rhodamine probe revealed that JZL184 displayed 100-fold selectivity for MAGL over FAAH and was very selective against other mouse serine hydrolases expressed in the brain. Although highly selective in the brain, JZL184 had inhibitory effects on multiple carboxylesterase enzymes in peripheral tissues 49, 50. Inhibition of MAGL activity inhibited 2-AG hydrolysis by ~85 % in mouse brain membranes and led to dramatic elevations in bulk brain 2-AG levels and increases in depolarization-induced interstitial 2-AG levels in vivo. These results confirmed that MAGL is the primary enzyme involved in degrading 2-AG in vivo. A single dose of JZL184 at 16 mg/kg was capable of inhibiting MAGL for up to 24hr, with maximal 8-fold elevation of brain 2-AG levels for at least 8 hours 49, 50. Acute MAGL blockade with JZL184 has been shown to exhibit a wide range of beneficial effects including alleviation of pain, inflammation, emesis, anxiety, opiate-induced withdrawal symptoms, colitis, neurodegeneration, inflammation-induced lung and liver injury, and cancer pathogenicity 21, 51-55. These effects are discussed further below. Interestingly, MAGL inhibitors do not cause full-blown cannabinoid behaviors such as hypothermia and catalepsy, although they lower motility in open-field tests in mice despite apparently normal cage behavior 49, 50. Chronic and complete pharmacological blockade of MAGL, as observed in MAGL -/- mice, leads to functional antagonism of the cannabinoid system, leading to loss of cannabinoid-mediated effects, physical dependency, and desensitization of CB1 receptors in the brain. Thus, the MAGL inhibitor has been especially useful compared to full genetic knockout mouse models, since the cannabinoid effects are ablated upon chronic and complete inactivation of MAGL in MAGL -/- mice 27. Subsequent studies have shown that partial and chronic blockade of MAGL avoids this functional antagonism of CB1 and thus maintains the cannabinoid-mediated effects 56.
Figure 4. Selective MAGL and DAGL Inhibitors and Probes.
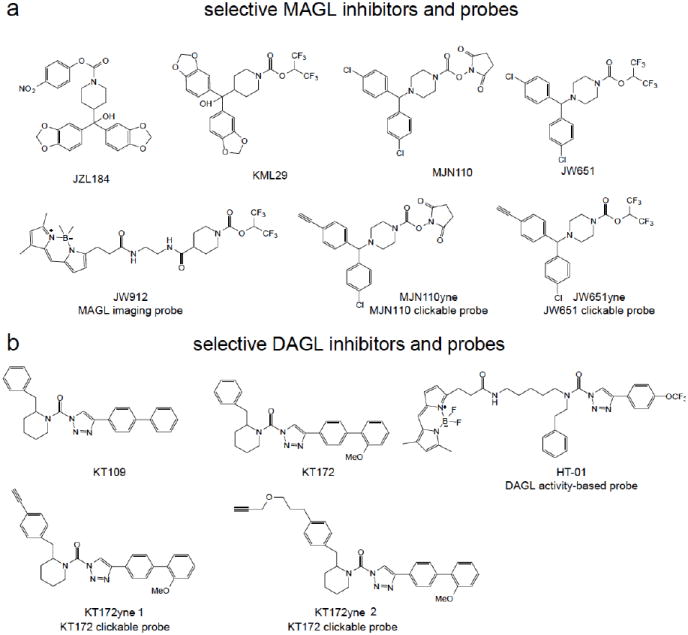
Selective MAGL (a, JZL184, KML29, MJN110, JW651) and DAGL (b, KT109, KT172) inhibitors, MAGL imaging probe (JW912), MAGL and DAGL clickable probes (MJN110yne, JW651yne, KT172yne 1 and 2), and the DAGL activity-based probe (HT-01).
Recent studies have yielded next-generation MAGL inhibitors with improved selectivity and cross-species activity compared to JZL184. These include the O-hexafluoroisopropyl carbamates and the N-hydroxysuccinimidyl (NHS) carbamates. The O-hexafluoroisopropyl leaving group on the newer MAGL inhibitors displayed greater selectivity towards MAGL over FAAH and, importantly, carboxylesterase enzymes both in vitro and in vivo. KML29, an O-hexafluoroisopropyl analog of JZL184, was completely selective for MAGL over FAAH even in chronically dosed mice using ABPP. This hexafluoroisopropyl leaving group of KML29 was found to be bioisoteric with the 2-AG substrate, indicating that serine hydrolase inhibitor selectivity may be better achieved my developing inhibitors bearing reactive groups resembling the structures of endogenous substrates 57. JZL184 had limited efficacy toward rat MAGL both in vitro and in vivo. In contrast, KML29 treatment showed near complete MAGL blockade and increased brain 2-AG levels in rats 57. A subsequent report also showed that a close analog of KML29, JW651, also selectively inhibited MAGL in vitro and in vivo 58. Recent studies have also uncovered the N-hydroxysuccinimidyl (NHS) carbamates as a promising class of MAGL inhibitors. Niphakis et al. reported MNJ110, as a highly potent, selective, and in vivo active NHS carbamate inhibitor of MAGL (Figure 4a)58.
Both Niphakis et al. and Chang et al. recently also used click-chemistry-ABPP using alkyne-bearing “clickable” analogs of highly selective MAGL inhibitors to confirm the selectivity of these compounds across the entire proteome by comprehensively mining all covalent probe-protein interactions (Figure 4a) 58, 59. The alkyne-bearing inhibitor protein targets are detected by conjugation with rhodamine-azide reporter tag using copper-catalyzed azide-alkyne cycloaddition chemistry. The click-chemistry carbamate probes, such as JW651yne and MJN110yne showed selective labeling of MAGL at low concentrations with FAAH, ABHD6, as well as other enzymes not detected by ABPP, as off-targets at higher concentrations 59.
Chang et al. also developed a fluorescent imaging probe for MAGL and ABHD6, based on the O-hexafluoroisopropyl carbamates that showed highly specificity for these two enzymes, resulting in the BODIPY-containing O-hexafluoroisopropyl carbamate JW912 (Figure 4a). This probe selectively labeled MAGL and ABHD6 as determined by in-gel fluorescence and the authors subsequently used this probe to visualize MAGL and ABHD6 localization in various cancer cells. To distinguish cellular localization of MAGL versus ABHD6, the authors used MAGL and ABHD6-selective inhibitors. They were able to show that MAGL in certain cancer cells display fluorescent signal on intracellular membranes and punctate staining patterns indicating localization to endosomes and other organelles 59.
These selective MAGL inhibitors have been invaluable in elucidating the biochemical and physiological roles of MAGL in vivo as well as establishing the therapeutic potential of MAGL inhibitors in various pathological states. The insights attained from these inhibitors are described in more detail below.
3.4.1. MAGL Also Controls Arachidonic Acid Pools that Generate Pro-Inflammatory Eicosanoids in Select Tissues
In addition to the role of MAGL in terminating 2-AG signaling, studies using both non-selective and highly selective MAGL inhibitors and MAGL knockout mice have found that MAGL is the primary source of arachidonic acid for the generation of pro-inflammatory eicosanoids in certain tissues, including the brain, liver, and the lung 51. Both pharmacological blockade with JZL184 and genetic ablation of MAGL lowers basal and lipopolysaccharide-induced arachidonic acid and eicosanoid levels in these tissues. These results were surprising, since cytosolic phospholipase A2 (cPLA2) has been historically considered to be the primary source of arachidonic acid for eicosanoid synthesis 51. Using MAGL inhibitors and MAGL -/- and cPLA2 -/- mice, Nomura et al found that MAGL contributes ~80 % of LPS-stimulated eicosanoids in mouse brain while cPLA2 contributes ~20 %. However, in spleen and the gastrointestinal tract, the authors showed that cPLA2 is the dominant enzyme that controls arachidonic acid release for prostaglandin production. Thus, MAGL, cPLA2, and potentially other enzymes differentially control arachidonic acid release in a tissue-specific manner 51.
3.4.2 The Effect of MAGL Inhibitors in Pain, Inflammation, and Mood
MAGL blockade with JZL184 has been shown by many studies to elicit CB1-dependent antinociceptive effects in various mouse models of pain, including noxious chemical, inflammatory, thermal, and neuropathic pain 49, 60, 61. MAGL blockade reduces mechanical and acetone-induced cold allodynia in mice with sciatic nerves that had previously undergone chronic constriction injury 60. MAGL blockade is also protective in mouse models of inflammatory bowel disease, in which MAGL blockade by JZL184 reduces colon cytopathology, inflammatory cytokine levels, and restores intestinal barrier function in a trinitrobenzene sulfonic acid-induced colitis model, thereby reducing endotoxemia and systemic inflammation in a CB1 or CB2-dependent manner 62.
Multiple studies have shown that MAGL blockade by JZL184 also exerts effects upon mood and reward behavior. In a marble burying model of repetitive and compulsive behavior inherent to anxiety disorders, MAGL blockade reduced marble burying 63. MAGL blockade also exerts anxiolytic effects in an elevated plus-maze paradigm for anxiety 64. Chronic MAGL blockade with JZL184 also prevented chronic stress-induced anxiety-like behavior and long-term depression of GABAergic transmission, indicating that MAGL inhibition prevents behavioral and synaptic adaptations to chronic stress that may lead to the worsening of affective disorders 65. MAGL inhibitors also improve withdrawal symptoms from naloxone-preciptated morphine withdrawal in a CB1-dependent manner 66.
3.4.3 The Effect of MAGL Inhibitors in Neuroinflammation and Neurodegenerative Diseases
Both pharmacological and genetic ablation of MAGL show anti-inflammatory effects in the brain and neuroprotective effects in mouse models of Parkinson’s and Alzheimer’s disease 51, 53, 54. MAGL inhibition lowers LPS-stimulated pro-inflammatory cytokine levels in the brain through lowering neuroinflammatory eicosanoids, in a CB1 and CB2-independent manner 51. MAGL blockade with JZL184 or MAGL deficiency also protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurodegeneration and dopamine loss through lowering pro-inflammatory eicosanoids and suppressing neuroinflammation 51.
Two studies recently showed that MAGL inhibition with JZL184 or MAGL deficiency both lower amyloid-β plaque levels in Alzheimer’s disease mouse models in-part through lowering eicosanoids and suppressing microglial and astrocyte activation 53, 54. Piro et al. showed that MAGL -/- mice crossed with the presinilin/amyloid precursor peptide (PS/APP) transgenic Alzheimer’s disease mouse model possessed significantly lower amyloid-β peptide and plaque levels concomitant with reduced neuroinflammation. JZL184 administration recapitulated the lowering of inflammatory cytokines in PS/APP-transgenic mice and CB1 or CB2 receptor antagonists did not attenuate this reduction. Chen et al. also showed that JZL184 robustly suppressed the production and accumulation of amyloid-β through downregulating β–site amyloid precursor protein cleaving enzyme 1, concomitant with a suppression of neuroinflammation, in the 5X FAD APP transgenic mice. They also confirmed that this phenomenon was CB1 and CB2-independent. Quite provocatively, Chen et al. also showed that JZL184 also reduced neurodegeneration, maintained integrity of hippocampal synaptic structure and function, and improved long-term synaptic plasticity, spatial learning, and memory in this Alzheimer’s disease mouse model.
Thus, several studies have shown the potential therapeutic utility of MAGL inhibitors in attenuating neuroinflammation and protecting against neurodegeneration in both Parkinson’s and Alzheimer’s disease mouse models and indicates that MAGL inhibitors may even improve memory and learning function, likely through lowering the arachidonic acid and pro-inflammatory eicosanoid levels in the brain, and not through enhancing endocannabinoid signaling.
3.4.4. The Effect of MAGL Inhibitors on Inflammatory Tissue Injury
Recent studies have also shown that MAGL inhibitors may have therapeutic windows not only in neuroinflammatory or neurodegenerative diseases, but also in peripheral inflammatory tissue injury as well. Cao et al. showed that the endocannabinoid and eicosanoid levels in liver are elevated upon ischemia-reperfusion injury in mice and that pharmacological or genetic MAGL inactivation significantly protects against hepatocellular cell death as evidenced by lower hepatic necrosis, reduction in liver-damage blood serum markers ALT and AST, and lower levels of liver cell death markers. MAGL inactivation also lowered hepatic inflammation caused by ischemia-reperfusion injury through lowering neutrophil infiltration, inflammatory cytokines, and reactive oxygen stress. Quite intriguingly, in contrast to the previously described models where the phenotypes observed were either due to enhanced CB1/CB2 signaling or lower eicosanoid levels, this hepatoprotective phenotype appeared to be due to a combination of enhanced CB2 signaling and lower eicosanoid levels. The authors also provocatively demonstrated that JZL184 could even protect against liver injury when provided 3 h after reperfusion. Cao et al. also showed that JZL184 was protective in the carbon tetrachloride and galactosamine/LPS models of liver injury in mice 52.
Costola-de-Souza et al. recently showed that JZL184 protected against lung injury in a LPS-induced acute lung injury model. The authors showed that an acute treatment with JZL184 reduced leukocyte migration into the lungs, vascular permeability, and inflammatory cytokine and chemokine levels in bronchoalveolar lavage fluid. These protective effects appeared to be mediated through CB1 and CB2 receptors, as the effects were attenuated with CB1 and CB2 selective antagonists 55.
3.4.5. The Effect of MAGL inhibitors on Cancer
Using ABPP platforms, Nomura et al. showed that MAGL is highly upregulated across multiple types of aggressive human cancer cells and primary high-grade tumors 67, 68. Both genetic and pharmacological ablation of MAGL in aggressive cancer cells impaired cellular migration, invasiveness, serum-free cell survival, and in vivo tumor growth. Metabolomic analysis showed that MAGL in aggressive cancer cells controls the lipolytic release of free fatty acids which are in turn remodeled into various lysophosholipids and eicosanoids. MAGL blockade lowered cellular fatty acid levels and downstream tumor-promoting lipid signaling molecules such as eicosanoids and lysophosphatidic acid, leading to impairments in cancer pathogenicity67, 68. In ovarian, breast, and melanoma cancer cells, the anti-cancer effects of MAGL were through reducing the fatty acid network of oncogenic signaling lipids, but not through CB1 or CB2-dependent mechanisms. In contrast, in aggressive prostate cancer cells, the anti-tumorigenic phenotypes associated with MAGL blockade were due to a combination of heightened CB1 signaling and reduced fatty acid and fatty acid-derived lipid signaling 67, 68. Ye et al. also showed that MAGL blockade impairs colorectal cancer cell pathogenicity and tumor growth through downregulation of cyclin D1 and Bcl-2 69.
MAGL blockade has also been shown to alleviate pain associated with cancer through heightened CB2 signaling in a mouse model of mechanical hyperalgesia evoked by the growth of a fibrosarcoma tumor in the calcaneus bone 70. MAGL blockade also shows anti-emetic effects in a lithium chloride model of vomiting 71.
3.5 DAGL-selective inhibitors and their effects
Recently, the first specific and in vivo active DAGLβ inhibitors were reported, based on the triazole urea scaffold (Figure 4b) 72, 73. Using competitive ABPP platforms, Hsu et al. screened recombinantly expressed DAGL enzymes against a synthetic library of 1,2,3-triazole ureas, a chemotype that was previously shown to possess well-suited features for serine hydrolase inhibitor development. Two compounds from this screen, KT109 and KT172, potently inhibited DAGLβ with a ~60-fold selectivity over DAGLα. These inhibitors showed high selectivity for DAGLβ over other serine hydrolases, but both inhibitors showed ABHD6 as an off-target. KT109 and KT172 inhibit DAGLβ with an IC50 of 82 and 71 nM, respectively. At higher concentrations, KT109 and KT172 showed some inhibitory activity against PLA2G7 (IC50 1000 nM) and MAGL (IC50 5000 nM), respectively. To exclude the effects of ABHD6 off-target effects from their studies, Hsu et al. also developed a control inhibitor KT195 that was a close structural analog of KT109 and KT172 that did not inhibit DAGLβ but inhibited ABHD6 72, 73.
While the serine hydrolase activity-based FP-rhodamine probes were able to easily assess recombinantly overexpressed DAGL activity, the low endogenous expression level of DAGLβ in cells and tissues prohibited its detection by broad-based probes such as FP-rhodamine. To easily confirm target-engagement of KT109 and KT172 in vitro, in situ, and in vivo, Hsu et al. also developed a tailored activity-based probe for DAGLβ based on the 1,2,3-triazole urea scaffold, HT-01, a BODIPY-conjugated 1,2,3-triazole urea probe that selectively labeled endogenous DAGLβ in complex proteomes (Figure 4b). Using this HT-01 probe, Hsu et al. showed that KT109 and KT172 inhibited DAGLβ in situ in Neuro2A cells and in vivo in mouse macrophages. The authors also used ABPP using the FP-biotin probe coupled to proteomic-based methods to confirm target occupancy and selectivity of KT109 and KT172 in situ and in vivo. Hsu et al. in a subsequent study also developed “clickable” analogs of KT172, confirming the selectivity of this inhibitor for DAGLβ and ABHD6 using click-chemistry-ABPP (Figure 4b) 72, 73.
3.5.1. Effects of DAGL Inhibitors on Endocannabinoids, Eicosanoids, and Inflammation
KT109 and KT172 were used to show that DAGLβ blockade lowers the levels of 2-AG, arachidonic acid, and prostaglandins in Neuro2A cells, mouse peritoneal macrophages, mouse liver, and human prostate cancer cells (Hsu 2013), indicating that the DAGL/MAGL and cPLA2 pathways both play complementary roles in arachidonic acid release for eicosanoid biosynthesis. While DAGLβ blockade alone lowers LPS-stimulated TNF-α release, which was also recapitulated in DAGLβ-deficient mice, DAGLβ and cPLA2 dual inactivation leads to an increase in TNF-α release 72.
Until now, understanding the role of DAGLs in mammalian physiology and pathophysiology has been hindered by a lack of inhibitors that are not just specific for DAGLs over other serine hydrolases, but are specific to only one isoform. These newest DAGL inhibitors will be immensely useful in future studies on understanding the nodal role of DAGL in regulating diacylglycerol, endocannabinoid, and eicosanoid signaling networks.
4. Potential Liabilities of DAGL and MAGL Inhibitors
With the many beneficial effects associated with DAGL and MAGL blockade, a key question is whether there may be any adverse effects associated with DAGL and MAGL inhibitors. One potential liability that may be associated with DAGL inhibitors is a potential impairment in adult neurogenesis as has been shown in DAGL knockout mice 17. Another potential adverse effect that may arise from DAGL blockade may be those phenotypes associated with functional antagonism of CB1. Previous studies have shown that CB1 antagonists show beneficial effects towards weight loss and improved serum lipid profiles, insulin sensitivity, and cardiometabolic parameters 74. However, CB1 antagonists were discontinued and clinical trials were terminated due to increased anxiety and depression75. One can conceivably avoid these potential adverse effects by developing either reversible DAGL inhibitors or small-molecules that do not cross the blood-brain barrier.
Liabilities that may be associated with MAGL inhibitors also include potential psychiatric effects that may arise from functional antagonism of CB1 in the brain. Previous studies have demonstrated that complete and chronic blockade of MAGL leads to a functional antagonism of CB1 in the brain, leading to reduced sensitivity to exogenous CB1 agonists and physical dependence as well as a loss of CB1-mediated phenotypes associated with acute MAGL blockade 27. This occurs due to prolonged heightening of 2-AG levels and CB1 stimulation, leading to desensitized brain CB1. These potential adverse effects may be avoided by either lowering the dose of currently available irreversible inhibitors to ensure that MAGL is not completely inactivated or by developing potent and selective reversible MAGL inhibitors. Kinsey et al. have already shown that repeated low-dose administration of JZL184 retains CB1-mediated antinociceptive and gastroprotective effects in mice. Cisneros et al. recently described the structure-activity relationships of a new series of reversible dual MAGL and FAAH inhibitors (±)-oxiran-2ylmethyl 6-(1,1’-biphenyl-4-yl)hexanoate and (2R)-(-)-oxiran-2-ylmethyl(4-benzylphenyl)acetate. While the selectivity of these inhibitors across other proteins is not known, developing reversible and selective MAGL inhibitors will be of future importance in understanding the therapeutic potential of MAGL inhibitors in relation to the currently available selective irreversible inhibitors.
Furthermore, it is important that any future MAGL inhibitor therapy that crosses the blood brain barrier be selective for MAGL over FAAH, since studies have shown that dual MAGL and FAAH blockade results in effects reminiscent of THC, such as catalepsy, not observed with either MAGL or FAAH inhibition alone76.
5. Future Therapeutic Potential of MAGL and DAGL Inhibitors
There are still many unanswered questions in the endocannabinoid field that will hopefully be addressed with the development and utilization of even more advanced MAGL and DAGL inhibitors. While Hsu et al. developed the first selective and in vivo active DAGLβ inhibitors (that also show DAGLα inhibition at higher concentrations), KT172 does not appear to cross the blood-brain barrier72. Thus, it will be of future interest to develop highly selective in vivo active and brain penetrant DAGLα inhibitors and to test their effects upon memory, synaptic plasticity, neuroinflammation, and in neurodegenerative disease models.With previous studies showing the importance of the endocannabinoid system in satiety, lipid metabolism, obesity, diabetes, and cardiovascular disease, it will also be of future interest to understand the effects of the newer and selective MAGL and DAGL inhibitors obesity and diabetes paradigms.
With the diverse roles of diacylglycerol, 2-AG, and eicosanoid signaling pathways, DAGL and MAGL inhibitors are likely to be critical in future investigations into dissecting the individual roles of these lipid signaling pathways as well as the complementary roles of cPLA2 and other phospholipases in eicosanoid metabolism, signaling, and associated (patho)physiological effects.
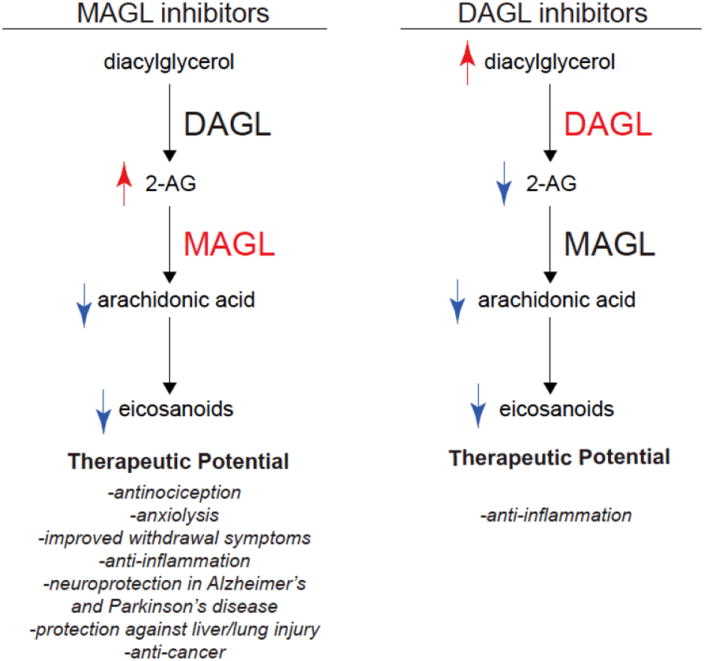
We have reviewed here the utility of modern chemical proteomic technologies such as ABPP in developing highly selective inhibitors for 2-AG degradation and synthesis. Studies using highly selective and in vivo active MAGL inhibitors have shown that these inhibitors may have potential therapeutic utility towards attenuating pain, inflammation, drug-withdrawal symptoms, anxiety, neurodegenerative diseases, ischemia-reperfusion tissue injuries, inflammation-induced injuries in liver and lung, and cancer and cancer-associated symptoms. DAGLβ inhibitors have been shown to elicit anti-TNF-α effects, which may have therapeutic utility in inflammatory disease such as rheumatoid arthritis where anti-TNF-α antibodies have shown favorable effects (Figure 5). The next steps in the clinical development of MAGL and DAGL inhibitors will be to test their toxicological properties, optimize pharmacokinetic parameters, and further show their efficacy in pre-clinical models towards advancing these inhibitors into the clinic to treat various human diseases that show dysregulated diacylglycerol, endocannabinoid, or eicosanoid signaling pathways.
Figure 5. Metabolic and Biological Effects of MAGL and DAGL Inhibitors.
Acknowledgments
We thank the members of the Nomura Research Group for critical reading of the manuscript. This work was supported by grants from the National Institutes of Health (R00DA030908), the Searle Scholar Foundation, and Michael J. Fox Target Validation Award.
References
- 1.Adams IB, Martin BR. Addiction. 1996;91:1585–1614. [PubMed] [Google Scholar]
- 2.Di Marzo V, Bisogno T, De Petrocellis L. Chemistry & biology. 2007;14:741–756. doi: 10.1016/j.chembiol.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 3.Di Marzo V, Petrosino S. Curr Opin Lipidol. 2007;18:129–140. doi: 10.1097/MOL.0b013e32803dbdec. [DOI] [PubMed] [Google Scholar]
- 4.Ahn K, McKinney MK, Cravatt BF. Chemical reviews. 2008;108:1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alger BE, Kim J. Trends in neurosciences. 2011;34:304–315. doi: 10.1016/j.tins.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ashton JC, Glass M. Curr Neuropharmacol. 2007;5:73–80. doi: 10.2174/157015907780866884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berdyshev EV. Chemistry and physics of lipids. 2000;108:169–190. doi: 10.1016/s0009-3084(00)00195-x. [DOI] [PubMed] [Google Scholar]
- 8.Derocq JM, Segui M, Marchand J, Lefur G, Casellas P. Febs Lett. 1995;369:177–182. doi: 10.1016/0014-5793(95)00746-v. [DOI] [PubMed] [Google Scholar]
- 9.Galiegue S, Mary S, Marchand J, Dussossoy D, Carriere D, Carayon P, Bouaboula M, Shire D, Le Fur G, Casellas P. European journal of biochemistry / FEBS. 1995;232:54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- 10.Miller AM, Stella N. British journal of pharmacology. 2008;153:299–308. doi: 10.1038/sj.bjp.0707523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blankman JL, Cravatt BF. Pharmacological reviews. 2013;65:849–871. doi: 10.1124/pr.112.006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Howlett AC, Blume LC, Dalton GD. Curr Med Chem. 2010;17:1382–1393. doi: 10.2174/092986710790980023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan B, Wang W, Zhong P, Blankman JL, Cravatt BF, Liu QS. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:13420–13430. doi: 10.1523/JNEUROSCI.2075-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. Biochemical and biophysical research communications. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- 15.Stella N, Schweitzer P, Piomelli D. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- 16.Pan B, Wang W, Long JZ, Sun D, Hillard CJ, Cravatt BF, Liu QS. The Journal of pharmacology and experimental therapeutics. 2009;331:591–597. doi: 10.1124/jpet.109.158162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, Shen R, Zhang MY, Strassle BW, Lu P, Mark L, Piesla MJ, Deng K, Kouranova EV, Ring RH, Whiteside GT, Bates B, Walsh FS, Williams G, Pangalos MN, Samad TA, Doherty P. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:2017–2024. doi: 10.1523/JNEUROSCI.5693-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshino H, Miyamae T, Hansen G, Zambrowicz B, Flynn M, Pedicord D, Blat Y, Westphal RS, Zaczek R, Lewis DA, Gonzalez-Burgos G. J Physiol-London. 2011;589:4857–4884. doi: 10.1113/jphysiol.2011.212225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, Kita Y, Hashimoto K, Shimizu T, Watanabe M, Sakimura K, Kano M. Neuron. 2010;65:320–327. doi: 10.1016/j.neuron.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 20.Min R, Di Marzo V, Mansvelder HD. Neuroscientist. 2010;16:608–613. doi: 10.1177/1073858410373281. [DOI] [PubMed] [Google Scholar]
- 21.Mulvihill MM, Nomura DK. Life sciences. 2013;92:492–497. doi: 10.1016/j.lfs.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dinh TP, Kathuria S, Piomelli D. Molecular pharmacology. 2004;66:1260–1264. doi: 10.1124/mol.104.002071. [DOI] [PubMed] [Google Scholar]
- 24.Karlsson M, Contreras JA, Hellman U, Tornqvist H, Holm C. The Journal of biological chemistry. 1997;272:27218–27223. doi: 10.1074/jbc.272.43.27218. [DOI] [PubMed] [Google Scholar]
- 25.Tornqvist H, Belfrage P. Journal of Biological Chemistry. 1976;251:813–819. [PubMed] [Google Scholar]
- 26.Blankman JL, Simon GM, Cravatt BF. Chemistry & biology. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, Nguyen PT, Ramesh D, Booker L, Burston JJ, Thomas EA, Selley DE, Sim-Selley LJ, Liu QS, Lichtman AH, Cravatt BF. Nature neuroscience. 2010;13:1113–1119. doi: 10.1038/nn.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas G, Betters JL, Lord CC, Brown AL, Marshall S, Ferguson D, Sawyer J, Davis MA, Melchior JT, Blume LC, Howlett AC, Ivanova PT, Milne SB, Myers DS, Mrak I, Leber V, Heier C, Taschler U, Blankman JL, Cravatt BF, Lee RG, Crooke RM, Graham MJ, Zimmermann R, Brown HA, Brown JM. Cell reports. 2013;5:508–520. doi: 10.1016/j.celrep.2013.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blankman JL, Long JZ, Trauger SA, Siuzdak G, Cravatt BF. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:1500–1505. doi: 10.1073/pnas.1217121110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saario SM, Savinainen JR, Laitinen JT, Jarvinen T, Niemi R. Biochem Pharmacol. 2004;67:1381–1387. doi: 10.1016/j.bcp.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 31.Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- 32.Muccioli GG, Xu C, Odah E, Cudaback E, Cisneros JA, Lambert DM, Lopez Rodriguez ML, Bajjalieh S, Stella N. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:2883–2889. doi: 10.1523/JNEUROSCI.4830-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vandevoorde S, Jonsson KO, Labar G, Persson E, Lambert DM, Fowler CJ. British journal of pharmacology. 2007;150:186–191. doi: 10.1038/sj.bjp.0706971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burston JJ, Sim-Selley LJ, Harloe JP, Mahadevan A, Razdan RK, Selley DE, Wiley JL. Journal of Pharmacology and Experimental Therapeutics. 2008;327:546–553. doi: 10.1124/jpet.108.141382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bisogno T, Ortar G, Petrosino S, Morera E, Palazzo E, Nalli M, Maione S, Di Marzo V, Grp ER. Bba-Mol Cell Biol L. 2009;1791:53–60. doi: 10.1016/j.bbalip.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 36.Nomura DK, Blankman JL, Simon GM, Fujioka K, Issa RS, Ward AM, Cravatt BF, Casida JE. Nature chemical biology. 2008;4:373–378. doi: 10.1038/nchembio.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nomura DK, Hudak CS, Ward AM, Burston JJ, Issa RS, Fisher KJ, Abood ME, Wiley JL, Lichtman AH, Casida JE. Bioorganic & medicinal chemistry letters. 2008;18:5875–5878. doi: 10.1016/j.bmcl.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bisogno T, Cascio MG, Saha B, Mahadevan A, Urbani P, Minassi A, Appendino G, Saturnino C, Martin B, Razdan R, Di Marzo V. Biochimica et biophysica acta. 2006;1761:205–212. doi: 10.1016/j.bbalip.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 39.Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, Gangadharan U, Hobbs C, Di Marzo V, Doherty P. The Journal of cell biology. 2003;163:463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bisogno T, Burston JJ, Rai R, Allara M, Saha B, Mahadevan A, Razdan RK, Wiley JL, Di Marzo V. ChemMedChem. 2009;4:946–950. doi: 10.1002/cmdc.200800442. [DOI] [PubMed] [Google Scholar]
- 41.Di Marzo V, Matias I. Nature neuroscience. 2005;8:585–589. doi: 10.1038/nn1457. [DOI] [PubMed] [Google Scholar]
- 42.Kirkham TC, Williams CM, Fezza F, Di Marzo V. British journal of pharmacology. 2002;136:550–557. doi: 10.1038/sj.bjp.0704767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Di Marzo V, Goparaju SK, Wang L, Liu J, Batkai S, Jarai Z, Fezza F, Miura GI, Palmiter RD, Sugiura T, Kunos G. Nature. 2001;410:822–825. doi: 10.1038/35071088. [DOI] [PubMed] [Google Scholar]
- 44.Bachovchin DA, Cravatt BF. Nature reviews. Drug discovery. 2012;11:52–68. doi: 10.1038/nrd3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simon GM, Cravatt BF. Journal of Biological Chemistry. 2010;285:11051–11055. doi: 10.1074/jbc.R109.097600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nomura DK, Dix MM, Cravatt BF. Nature reviews. Cancer. 2010;10:630–638. doi: 10.1038/nrc2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Medina-Cleghorn D, Nomura DK. Pflugers Archiv : European journal of physiology. 2013;465:427–440. doi: 10.1007/s00424-012-1201-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Y, Patricelli MP, Cravatt BF. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavon FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF. Nature chemical biology. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Long JZ, Nomura DK, Cravatt BF. Chemistry & biology. 2009;16:744–753. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, Ward AM, Hahn YK, Lichtman AH, Conti B, Cravatt BF. Science. 2011;334:809–813. doi: 10.1126/science.1209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cao Z, Mulvihill MM, Mukhopadhyay P, Xu H, Erdelyi K, Hao E, Holovac E, Hasko G, Cravatt BF, Nomura DK, Pacher P. Gastroenterology. 2013;144:808–817. e815. doi: 10.1053/j.gastro.2012.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Piro JR, Benjamin DI, Duerr JM, Pi Y, Gonzales C, Wood KM, Schwartz JW, Nomura DK, Samad TA. Cell reports. 2012;1:617–623. doi: 10.1016/j.celrep.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen RQ, Zhang J, Wu Y, Wang DQ, Feng GP, Tang YP, Teng ZQ, Chen C. Cell reports. 2012;2:1329–1339. doi: 10.1016/j.celrep.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Costola-de-Souza C, Ribeiro A, Ferraz-de-Paula V, Calefi AS, Aloia TP, Gimenes-Junior JA, de Almeida VI, Pinheiro ML, Palermo-Neto J. PloS one. 2013;8:e77706. doi: 10.1371/journal.pone.0077706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kinsey SG, Wise LE, Ramesh D, Abdullah R, Selley DE, Cravatt BF, Lichtman AH. The Journal of pharmacology and experimental therapeutics. 2013;345:492–501. doi: 10.1124/jpet.112.201426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang JW, Niphakis MJ, Lum KM, Cognetta AB, 3, Wang C, Matthews ML, Niessen S, Buczynski MW, Parsons LH, Cravatt BF. Chemistry & biology. 2012;19:579–588. doi: 10.1016/j.chembiol.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Niphakis MJ, Cognetta AB, 3, Chang JW, Buczynski MW, Parsons LH, Byrne F, Burston JJ, Chapman V, Cravatt BF. ACS chemical neuroscience. 2013;4:1322–1332. doi: 10.1021/cn400116z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chang JW, Cognetta AB, 3, Niphakis MJ, Cravatt BF. ACS chemical biology. 2013 doi: 10.1021/cb400261h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kinsey SG, Long JZ, O’Neal ST, Abdullah RA, Poklis JL, Boger DL, Cravatt BF, Lichtman AH. The Journal of pharmacology and experimental therapeutics. 2009;330:902–910. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guindon J, Guijarro A, Piomelli D, Hohmann AG. British journal of pharmacology. 2011;163:1464–1478. doi: 10.1111/j.1476-5381.2010.01192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alhouayek M, Lambert DM, Delzenne NM, Cani PD, Muccioli GG. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2011;25:2711–2721. doi: 10.1096/fj.10-176602. [DOI] [PubMed] [Google Scholar]
- 63.Kinsey SG, O’Neal ST, Long JZ, Cravatt BF, Lichtman AH. Pharmacology, biochemistry, and behavior. 2011;98:21–27. doi: 10.1016/j.pbb.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sciolino NR, Zhou W, Hohmann AG. Pharmacological research : the official journal of the Italian Pharmacological Society. 2011;64:226–234. doi: 10.1016/j.phrs.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sumislawski JJ, Ramikie TS, Patel S. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2011;36:2750–2761. doi: 10.1038/npp.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ramesh D, Ross GR, Schlosburg JE, Owens RA, Abdullah RA, Kinsey SG, Long JZ, Nomura DK, Sim-Selley LJ, Cravatt BF, Akbarali HI, Lichtman AH. The Journal of pharmacology and experimental therapeutics. 2011;339:173–185. doi: 10.1124/jpet.111.181370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nomura DK, Lombardi DP, Chang JW, Niessen S, Ward AM, Long JZ, Hoover HH, Cravatt BF. Chemistry & biology. 2011;18:846–856. doi: 10.1016/j.chembiol.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Cell. 2010;140:49–61. doi: 10.1016/j.cell.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ye L, Zhang B, Seviour EG, Tao KX, Liu XH, Ling Y, Chen JY, Wang GB. Cancer letters. 2011;307:6–17. doi: 10.1016/j.canlet.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 70.Khasabova IA, Chandiramani A, Harding-Rose C, Simone DA, Seybold VS. Pharmacological research : the official journal of the Italian Pharmacological Society. 2011;64:60–67. doi: 10.1016/j.phrs.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sticht MA, Long JZ, Rock EM, Limebeer CL, Mechoulam R, Cravatt BF, Parker LA. British journal of pharmacology. 2012;165:2425–2435. doi: 10.1111/j.1476-5381.2011.01407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hsu KL, Tsuboi K, Adibekian A, Pugh H, Masuda K, Cravatt BF. Nature chemical biology. 2012;8:999–1007. doi: 10.1038/nchembio.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hsu KL, Tsuboi K, Chang JW, Whitby LR, Speers AE, Pugh H, Cravatt BF. Journal of medicinal chemistry. 2013;56:8270–8279. doi: 10.1021/jm400899c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Di Marzo V. Drug Discov Today. 2008;13:1026–1041. doi: 10.1016/j.drudis.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 75.Christensen R, Kristensen PK, Bartels EM, Bliddal H, Astrup A. Lancet. 2007;370:1706–1713. doi: 10.1016/S0140-6736(07)61721-8. [DOI] [PubMed] [Google Scholar]
- 76.Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, Burston JJ, Sim-Selley LJ, Lichtman AH, Wiley JL, Cravatt BF. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20270–20275. doi: 10.1073/pnas.0909411106. [DOI] [PMC free article] [PubMed] [Google Scholar]