

In recent years, cyanobacteria have emerged as attractive microbial hosts for hydrocarbon production.[1, [2] However, whilst appealing, the practicalities of producing biofuels in cyanobacteria remain challenging, requiring the identification and engineering of natural biocatalysts for alkane production and their integration into metabolic processes.[3] Cyanobacterial hydrocarbon biosynthesis is presumed to arise from fatty acid catabolism involving two main enzymatic reactions, an acyl carrier protein reductase (AAR; converting fatty acids to aldehydes) followed by loss of the carbonyl group to form alka(e)nes catalysed by an aldehyde-deformylating oxygenase (ADO), known formerly as aldehyde decarbonylase (AD; Scheme 1).[4–7] Cyanobacterial ADOs (cADOs) have been isolated and shown to decarbonylate long-chain aldehydes (C18 and above), which are the presumed physiological substrates, whilst also displaying in vitro activity with medium-chain aldehydes (e.g., heptanal).[8, [9]
Scheme 1.

Cyanobacterial hydrocarbon production. AAR: acyl carrier protein reductase; ACP: acyl carrier protein; ADO: aldehyde-deformylating oxygenase; HCO2−: formate.
There is intense interest in engineering cyanobacteria to accumulate short-chain alkanes and produce “drop in” fuels such as propane. This requires enzyme catalysts that can produce these hydrocarbons from precursors derived from central metabolism. Here, we set out to engineer improved variants of Procholorococcus marinus (strain MIT9313) cADO through structure-based engineering of the substrate-access tunnel. Our intention was to alter the natural specificity of cADO to favour reactivity against short-chain over long-chain aldehydes, and thereby produce new catalytic modules for metabolic engineering.
The availability of a crystal structure for P. marinus cADO (strain MIT9313; Joint Center for Structural Genomics; PDB ID: 20C5) enabled us to investigate the active site architecture, and identify residues that may influence substrate binding.[10] The structure reveals a mainly alpha helical architecture, with a ferritin-like four-helix bundle. The latter contains the di-iron centre, coordinated by two histidine residues and four carboxylates from glutamate side chains. Substrates access the active site through a tunnel-like hydrophobic pocket, as evidenced by the occupancy of an unknown ligand of extended chain length in the 20C5 crystal structure. One end of the unknown ligand is located close to the di-iron centre within the substrate-binding tunnel, where iron-catalysed decarbonylation occurs. The mechanism of the unusual iron-catalysed decarbonylation reaction has been studied recently. Two distinct mechanisms (oxygen dependent or hydrolytic) have been proposed.[11, [12] It was originally thought that decarbonylation proceeds hydrolytically in the absence of oxygen,[13] but more recent evidence suggests turnover requires it.[14, [15] Despite some progress on the mechanistic understanding of cADO it remains unclear why turnover with medium/long-chain aldehydes is slow (typically ∼3–5 turnovers per hour). With heptanal as substrate, an exponential burst phase with a kapp value of 0.27±0.03 s−1 has been reported.[16] However, kcat measurements under steady-state turnover conditions returned a value ∼1 min−1.
The unknown ligand observed crystallographically copurifies with cADO, and is presumably derived from the host strain, the most likely candidate being a natural fatty acid (e.g., palmitic acid). However, the identity of this ligand has not been reported previously, so we independently solved the structure in-house, and identified the ligand using GC-MS analysis (Figure S1). The bound ligand(s) extracted from cADO protein samples were identified as a mixture of long-chain fatty acids consisting of palmitic acid (molecular weight 256 Da), stearic acid (molecular weight 284 Da) and oleic acid (molecular weight 282 Da), see Figure S1 A.
Analysis of the fatty acid binding site highlighted two residues (V41 and A134) adjacent to the C9 position of the ligand that might influence fatty acid binding. Both residues present their respective side chains towards the cavity of the fatty acid binding pocket. We therefore hypothesised that mutating these residues to tyrosine and phenylalanine, respectively, may introduce a steric block in this position and thus impede the binding of fatty acid chains beyond the length of C9. Following site-directed mutagenesis, both V41Y and A134F variant proteins were overexpressed and purified to homogeneity. GC-MS analysis as performed for wild-type cADO (Figure S1 B) indicated host-derived fatty acid ligands were not bound to the purified variant proteins.
To determine how the V41Y and A134F variants were able to discriminate against the binding of the native fatty acid ligands, we generated crystal structures of both V41Y and A134F in the presence of hexanoic acid. The structures were solved at 1.88 and 1.67 Å, respectively. A superimposition of V41Y and A134F reveals they have retained the same global architecture as wild-type cADO, with RMSDs of 0.262 and 0.177 Å, respectively, for all αC atoms. Previous attempts to crystallise wild-type cADO in the presence of shorter aldehydes had proved fruitless due to the presence of palmitic acid blocking the active site. However, clear electron density was observed for hexanoic acid (Figure 1) within the variant proteins, consistent with the lack of detectable palmitic acid in the purified enzyme forms.
Figure 1.
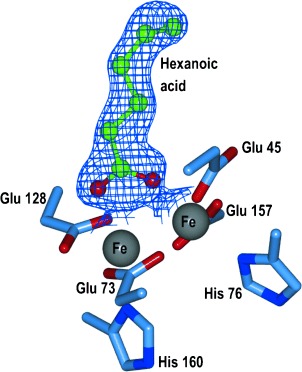
Ball and stick representation of hexanoic acid bound in the active site of A134F. Electron density shown as a blue mesh (2Fo−Fc, contoured at 1σ). Residues are shown as sticks with blue carbons, and iron ions are grey.
In both variant structures, V41Y and A134F, the mutated side chains encroach upon the palmitic acid binding site near the C11 position, effectively blocking the site and thus preventing palmitic acid binding. Importantly, neither mutation causes any occlusion of the active site channel that extends towards the di-iron centre, thus allowing the variants to retain binding and catalytic activity towards aldehydes shorter in length than approximately C10 (Figure 2).
Figure 2.
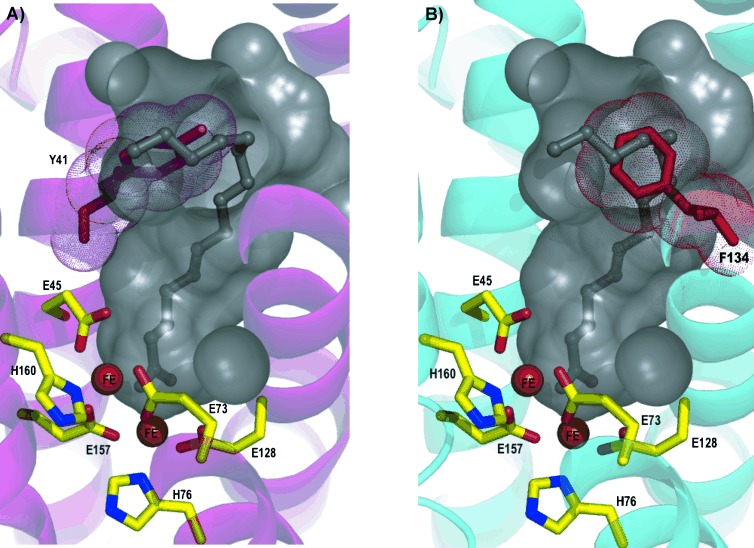
Cartoon representation of A) V41Y and B) A134F. Internal protein cavity of wild-type cADO shown as a grey surface. Palmitic acid is shown as a ball and stick representation (grey). Native residues are shown as sticks with yellow carbons, and iron ions are shown as orange spheres. Mutated residues Y41 (A) and F134 (B) are shown as pink and red sticks, respectively.
To evaluate how the engineered changes in the V41Y and A134F proteins altered the substrate selectivity of cADO, we measured the enzyme activity with a range of different aldehyde substrates. These experiments used phenazine methosulfate/reduced nicotinamide adenine dinucleotide (PMS/NADH) as the auxiliary reducing system.[17] The decarbonylation reaction was first monitored by following the conversion of butanal to propane by wild-type cADO enzyme using GC (Figure 3 A). Initial reaction velocities (Vo) displayed apparent Michaelis–Menten behaviour with respect to butanal concentration (Figure 3 B). Although butanal is decarbonylated to propane, the kcat (0.0031±0.0001 min−1) and Km (10.1±0.9 mm) values indicate that that wild-type cADO activity with this substrate is very low. In control studies, kinetic analyses performed with wild-type cADO and butanal showed that product accumulated linearly with respect to time and that reaction rates were, as expected, dependent on protein concentration (Figure 3 B, inset).
Figure 3.

A) Decarbonylation of butanal to propane using wild-type cADO in the presence of a PMS/NADH chemical reducing system. Reactions contained wild-type cADO (10 μm), ferrous ammonium sulfate (20 μm), PMS (75 μm), NADH (1 mm) and C4 aldehyde (2 mm). Reactions were carried out micro-aerobically, incubated at 37 °C at 220 rpm for up to 3 h. Peaks A and B refer to propane and butanal, respectively. B) Michaelis–Menten plot of wild-type cADO activity versus butanal concentration, ranging from 0 to 40 mm. Inset: Wild-type cADO reaction kinetics at different enzyme concentrations. Circles, triangles and squares represent 10, 20 and 40 μm of wild-type cADO, respectively.
A range of aldehyde substrates from C4 to C18 was used to determine the specificity of wild-type cADO and variants V41Y and A134F. For each substrate, the apparent kcat (kapp) value was measured and relative activities determined (Figure 4). For clarity, the horizontal axis in Figure 4 A and B is broken to highlight differences in substrate concentration used. C4–10 aldehydes were screened individually at 2 mm; due to limited solubility, C11–18 aldehydes were screened at 300 μm.
Figure 4.
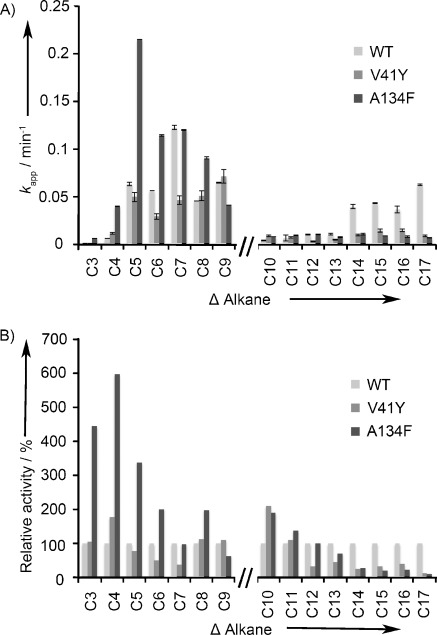
A) Substrate specificity of wild-type cADO, and variants V41Y and A134F were evaluated from initial steady-state velocity (Vo) measurements for different aldehyde chain lengths (C4–18). The wild-type cADO is indicated in light grey, the V41Y variant in grey and A134F variant in dark grey. B) Relative activity of wild-type cADO, V41Y and A134F variant for different alkyl chain lengths (C3–17). Wild-type cADO is set at 100 %. All reactions used a PMS/NADH reducing system with 10 μm cADO (see the Experimental Section for details).
Wild-type cADO has a broad substrate range catalysing C4–18 aldehyde decarbonylation. With C4–10 aldehydes, wild-type cADO exhibited maximal activity with the medium-chain aldehyde, octanal (kapp value of 0.122±0.002 min−1). With longer-chain aldehydes (C11–18), maximal activity was observed with octadecanal (kapp value of 0.06±0.001 min−1). In contrast, the V41Y variant cADO has maximal activity with decanal (kapp value of 0.07±0.001 min−1), and the relative activity with C4–10 aldehydes remains similar to that for wild-type cADO. Furthermore, V41Y shows an approximately sevenfold reduction in relative activity towards octadecanal when compared to wild-type cADO (kapp value of 0.01±0.001 min−1). The substrate specificity range of the V41Y variant is therefore narrower than that observed for wild-type cADO with a preference toward short-chain aldehydes.
The A134F variant shows a similar specificity to V41Y, being almost inactive with the majority of long-chain aldehyde substrates tested. For instance, A134F retains only 11 % relative activity for octadecanal (kapp value of 0.007±0.001 min−1). When compared to wild-type cADO, however, A134F exhibits enhanced general activity with shorter-chain aldehydes, with maximal activity for hexanal (kapp value increases to 0.215±0.0002 min−1). In addition, the A134F variant displayed an approximate fourfold increase in the rate of butanal consumption (kapp value of 0.003±0.0005 min−1) and approximately sixfold increase in pentanal consumption (kapp value of 0.023±0.001 min−1) compared to wild-type cADO. In an attempt to improve further the selectivity of cADO towards short-chain aldehydes, we combined the two beneficial aromatic residues (V41Y and A134F) into a single cADO variant and tested this new variant (V41Y/A134F) against C4–10 aldehydes (Table S3). However, no synergistic or additive effects were observed in reactions with short-chain aldehydes compared with reaction profiles with the single variants.
The decarbonylation reactions reported here were initially performed in vitro. To demonstrate that long-chain fatty acids are efficiently excluded from the A134F variant both in vitro and in vivo, whole-cell biotransformations were performed (Figures 5 and S2). From control experiments, small levels of propane were detected using an untransformed Escherichia coli strain (0.03±0.01 mg L−1), the origin of which is unclear as genome searches indicate this this organism does not contain cADO-related genes. In broad agreement with in vitro turnover data (Figure 4), the E. coli strain containing the A134F-variant cADO generated propane at a rate (0.46±0.04 mg L−1) approximately twofold greater than E. coli containing wild-type cADO (0.27±0.04 mg L−1). This elevation in activity is slightly less than that determined from the in vitro turnover measurements. We attribute this small difference to the approximately twofold lower expression of the A134F variant in E. coli compared with wild-type cADO (Figure S2).
Figure 5.
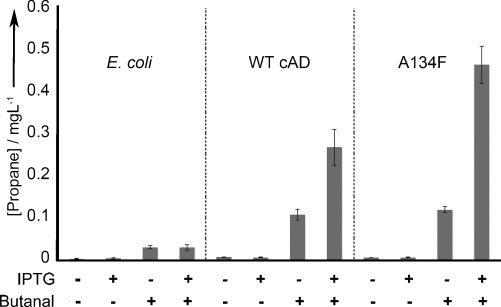
All strains (E. coli control lacking cADO, E. coli transformed with wild-type cADO or the A134F variant) were cultivated in lysogeny broth. All reactions contained butanal (10 mm) and propane concentration was quantified from whole-cell biotransformations. (see the Supporting Information for details).
In summary, based on crystallographic data we have isolated two variant forms of cADO that were strategically engineered to display improved specificity for short- to medium-chain aldehydes. The A134F cADO has also been shown to generate enhanced levels of propane production in whole-cell biotransformations compared to wild-type cADO. These studies define a region in the substrate channel that can be modified to exclude longer-chain aldehydes and improve reactivity with shorter-chain substrates. This simple switch in specificity releases cADO from potential complications arising from long-chain fatty acid binding in vivo. The A134F variant is an excellent catalytic module from which to now explore the role of second-shell residues to improve specificity and catalysis by cADO for production of drop-in hydrocarbon biofuels.
Experimental Section
Auxiliary PMS/NADH assay: All assays were performed under microaerobic conditions in an anaerobic glovebox (Belle Technology) under a nitrogen atmosphere (oxygen maintained at <2 ppm) unless otherwise stated. Enzyme assays were performed in potassium phosphate buffer (100 mm, pH 7.2) containing KCl (100 mm) and 10 % glycerol. Aldehyde substrates were made up as stock solutions in DMSO. Enzymatic reactions contained cAD (10 μm), ferrous ammonium sulfate (20 μm), PMS (75 μm) and NADH (1 mm), C4–10 (2 mm) or C11–18 (300 μm) aldehyde in a total volume of 0.5 mL, respectively.
Gas chromatography detection of volatile hydrocarbon product (C3–9 alkane) from enzyme reactions: Headspace analysis of volatile products (C3–9 alkane) was carried out using GC. The reaction mixtures were shaken continuously (190 rpm) at 37 °C. Headspace samples (1.0 mL) were manually drawn off, and injected into the GC with a syringe-lock needle at time intervals (0, 36, 72, 108, 144, 180 min for propane and 0, 5, 10, 20, 40, 60 min for n>3 alkanes). All kinetic assays were performed in duplicate.
A Varian 3800 GC equipped with a DB-WAX column (30 m×0.32 mm×0.25 μm film thickness, JW Scientific) was used to detect and quantify the hydrocarbon released from enzyme reactions. The column temperature was programmed as follows: 40 °C hold for 2 min, to 100 °C at 20 °C min−1 (for detection of propane) and 40 °C hold for 2 min, to 150 °C at 20 °C min−1 (for detection of n>3 alkanes). The injector temperature was 250 °C (10:1 split), and the FID temperature was set at 250 °C. The carrier gas was helium at a flow rate of 1 mL min−1. Peak identification of each alkane was achieved by comparison with pure alkane standards. Quantification of alkanes was achieved by comparison of integrated peak with calibration curves of standard pure alkanes.
Gas chromatography-mass spectrometry (GC-MS) detection of less volatile hydrocarbon products (C10–17): Liquid-phase analysis of nonvolatile products (C10–17 alkanes) was carried out using GC-MS. At time intervals (0, 5, 10, 20, 40, 60, 80, 100 and 120 min), of the reaction (1 mL) was terminated by extraction with ethyl acetate (900 μL), containing an internal standard (0.005 % limonene), and dried over MgSO4. A 1 μL sample was then analysed using GC-MS on a Varian 3800 GC instrument equipped with a Saturn 2000 ion trap MS and a CP-8400 autosampler. A DB-WAX column (30 m×0.25 mm×0.25 μm film thickness, JW Scientific) was used with the following temperature programme: 70 °C hold for 2 min, to 250 °C at 20 °C min−1, hold for 2 min. The injector temperature was 250 °C (10:1 split), and the carrier gas was helium at a flow rate of 1 mL min−1. The transfer line, manifold and ion trap temperatures were set to 250, 35 and 150 °C, respectively. All kinetic assays were performed in duplicate.
Structural data have been deposited at RCSB Protein Data Bank (IDs: 4KVQ, 4KVR and 4KVS)
Acknowledgments
This work was supported by grants from the European Union FP-7 256808 to E.N.G.M. and N.S.S., and from NIH GM 093088 and NSF CHE 1152055 to E.N.G.M. N.S.S. is a Royal Society Wolfson Merit Awardee and holds an Engineering and Physical Sciences Research Council (EPSRC) Established Career Fellowship. We thank Diamond Light Source for access to beamlines I03 (MX1224-22) and I02 (MX7146-13), which contributed to the results presented here.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1.Lu X. Biotechnol. Adv. 2010;28:742–746. doi: 10.1016/j.biotechadv.2010.05.021. [DOI] [PubMed] [Google Scholar]
- 2.Machado IM, Atsumi S. J. Biotechnol. 2012;162:50–56. doi: 10.1016/j.jbiotec.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 3.McEwen JT, Atsumi S. Curr. Opin. Biotechnol. 2012;23:744–750. doi: 10.1016/j.copbio.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 4.Cheesbrough TM, Kolattukudy PE. Proc. Natl. Acad. Sci. USA. 1984;81:6613–6617. doi: 10.1073/pnas.81.21.6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dennis M, Kolattukudy PE. Proc. Natl. Acad. Sci. USA. 1992;89:5306–5310. doi: 10.1073/pnas.89.12.5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vioque J, Kolattukudy PE. Arch. Biochem. Biophys. 1997;340:64–72. doi: 10.1006/abbi.1997.9932. [DOI] [PubMed] [Google Scholar]
- 7.Warui DM, Li N, Norgaard H, Krebs C, Bollinger JM, Jr, Booker SJ. J. Am. Chem. Soc. 2011;133:3316–3319. doi: 10.1021/ja111607x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Das D, Eser BE, Han J, Sciore A, Marsh EN. Angew. Chem. 2011;123:7286–7290. doi: 10.1002/anie.201101552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2011;50 [Google Scholar]
- 9.Schirmer A, Rude MA, Li X, Popova E, del Cardayre SB. Science. 2010;329:559–562. doi: 10.1126/science.1187936. [DOI] [PubMed] [Google Scholar]
- 10.Axelrod HL, Paddock ML, Chiu H-J, Miller MM, Deacon AM, Wilson IA. Biophys. J. 2012;102:271a. [Google Scholar]
- 11.Li N, Norgaard H, Warui DM, Booker SJ, Krebs C, Bollinger JM., Jr J. Am. Chem. Soc. 2011;133:6158–6161. doi: 10.1021/ja2013517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krebs C, Bollinger JM, Jr, Booker SJ. Curr. Opin. Chem. Biol. 2011;15:291–303. doi: 10.1016/j.cbpa.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eser BE, Das D, Han J, Jones PR, Marsh EN. Biochemistry. 2012;51:5703. doi: 10.1021/bi300837j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paul B, Das D, Ellington B, Marsh EN. J. Am. Chem. Soc. 2013;135:5234–5237. doi: 10.1021/ja3115949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andre C, Kim SW, Yu XH, Shanklin J. Proc. Natl. Acad. Sci. USA. 2013;110:3191–3196. doi: 10.1073/pnas.1218769110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li N, Chang WC, Warui DM, Booker SJ, Krebs C, Bollinger JM., Jr Biochemistry. 2012;51:7908–7916. doi: 10.1021/bi300912n. [DOI] [PubMed] [Google Scholar]
- 17.Zaugg WS. J. Biol. Chem. 1964;239:3964–3970. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information