

Abstract
Identifying proteome changes of honey bee embryogenesis is of prime importance for unraveling the molecular mechanisms that they underlie. However, many proteomic changes during the embryonic period are not well characterized. We analyzed the proteomic alterations over the complete time course of honey bee worker embryogenesis at 24, 48, and 72 h of age, using mass spectrometry-based proteomics, label-free quantitation, and bioinformatics. Of the 1460 proteins identified the embryo of all three ages, the core proteome (proteins shared by the embryos of all three ages, accounting for 40%) was mainly involved in protein synthesis, metabolic energy, development, and molecular transporter, which indicates their centrality in driving embryogenesis. However, embryos at different developmental stages have their own specific proteome and pathway signatures to coordinate and modulate developmental events. The young embryos (<24 h) stronger expression of proteins related to nutrition storage and nucleic acid metabolism may correlate with the cell proliferation occurring at this stage. The middle aged embryos (24–48 h) enhanced expression of proteins associated with cell cycle control, transporters, antioxidant activity, and the cytoskeleton suggest their roles to support rudimentary organogenesis. Among these proteins, the biological pathways of aminoacyl-tRNA biosynthesis, β-alanine metabolism, and protein export are intensively activated in the embryos of middle age. The old embryos (48–72 h) elevated expression of proteins implicated in fatty acid metabolism and morphogenesis indicate their functionality for the formation and development of organs and dorsal closure, in which the biological pathways of fatty acid metabolism and RNA transport are highly activated. These findings add novel understanding to the molecular details of honey bee embryogenesis, in which the programmed activation of the proteome matches with the physiological transition observed during embryogenesis. The identified biological pathways and key node proteins allow for further functional analysis and genetic manipulation for both the honey bee embryos and other eusocial insects.
Embryogenesis is an important period during which the body plan of adult honey bees (Apis mellifera L.) is formed. This life stage, lasting 72 h, occurs during the egg laid by the queen before bees hatch as young larva. Worker bees are derived from fertilized eggs and develop through four distinct stages until the imago eventually emerges: egg, larva, pupa, and emerging adult (1–4). The worker is the dominate caste and engages in almost all aspects of social life: taking care of larvae, cleaning the hive, guarding the nest, and foraging for nectar and pollen for the colony. Understanding the developmental mechanism of embryogenesis of honey bee workers at the protein level is conducive to gaining a new insight into honey bee embryology, but information about the mechanisms of honey bee embryos at molecular level is still very limited.
The embryo is recognized as an ideal model for genetic modification as compared with larva, pupa, and emerged adults (5). The environment for embryonic development requires a constant temperature of 34 °C and 80% relative humidity, which can easily be simulated under laboratory conditions. In contrast, rearing larvae or pupae is more challenging because they demand a specific temperature, humidity, and nutrition in the colony environment (1, 5). Furthermore, the honey bee has adapted an evolutionary strategy for better colony survival that makes it difficult to rear experimentally modified larvae and pupae within the colony (6, 7), nurse bees use acute judgment to identify and remove abnormal eggs or larvae (8). This adaptation makes raising experimentally treated bees, such as genetically manipulated eggs and larvae, very difficult in the honey bee colony (9–11). Because of totipotency and multiple differentiation potential, modified eggs could be hatched out normally and eventually some of them could be induced to morphologically and physiologically normal adult queens (12), increasing their usefulness as a model system. Moreover, the chorion of honey bee egg is more suitable for puncturing a hole for microinjection as it is much thinner than that of the fruit fly (Drosophila melonogastero) or the silk worm (Bombyx mori) (0.1–0.25 μm for honey bees compared with ∼17 μm for silk worm) (13, 14). These superiorities are quite promising for in vivo transgenic research on honey bee embryos.
Until now, a number of genetic manipulations of the honey bee embryo have been developed. For example, embryonic cells in the pre-gastrula stage that have been transplanted with nuclear materials have developed into chimeric honey bee larvae (15). RNA interference (RNAi) has been used for honey bee embryos in vivo to characterize the functioning of specific genes (16) and for genetic effects on morphological differentiation (17, 18). Moreover, the cultivation of short-term (19–21), long-term (22), and immortalized cell lines (23), and the expression of non-Apis genes in cultured embryonic cells (24) have opened up a new era for genetic manipulation of honey bee embryos.
Like Drosophila, Apis is a long germ insect in which segmentation occurs across the whole body (25). To date, although several studies have examined morphological change (2, 26, 27) and gene expression (25, 28, 29) during the period of embryogenesis in the honey bee, only a few works report on the preliminary results of the unraveling molecular underpinnings of worker (30) and drone (31) embryogenesis at the proteomic level, identifying only 107 proteins. MS-based proteomics is the primary technology that enables a system-wide view of proteomes and their changes. The development of MS with high resolution, high mass accuracy, and high sequencing speed now allows routine identification and quantification of proteins in a comprehensive and unbiased manner in biological samples with high confidence (32). These technological advances in LC-MS now allow the study of protein expression on a system-wide level (33). Therefore, an in-depth characterization of the proteome changes during the honey bee embryogenesis will provide greater understanding of the molecular mechanisms that underlie the process of embryogenesis in honey bee workers, and offers new insights into the embryology of other social insects.
EXPERIMENTAL PROCEDURES
Embryo Sampling and Protein Extraction
Honey bee (A. m. lingustica) colonies were kept at the Institute of Apicultural Research, Chinese Academy of Agricultural Science, Beijing, China. A total of 1000 eggs were sampled from a worker comb 24, 48, and 72h after the queen had laid eggs. For each time point, we sampled eggs from five colonies, and pooled all eggs for further analysis (31). This procedure was repeated three times, so that we finally ended up with three independent biological replicates per time point, each consisting of 5000 eggs.
Protein extraction was performed as previously described (31). In short, lysis buffer (LB, 8 m urea, 2 m thiourea, 4% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), 20 mm Tris-base, 30 mm dithiothreitol (DTT), and 2% Bio-lyte pH 3–10, 1 mg/10 μl) was added to worker egg samples and centrifuged at 15,000 × g for 15 min at 4 °C. The supernatant was collected and proteins precipitated with ice-cold acetone at −20 °C for 30 min. The sample was centrifuged twice at 15,000 × g for 10 min at 4 °C. The supernatant was discarded, the pellet was dried by airing at room temperature for 10 min and dissolved in 40 mm (NH4)HCO3 finally. The final protein concentration was quantified using a Bradford assay.
Protein Digestion and MS Analysis
Denatured proteins were reduced with DTT (final concentration10 mm) and iodoacetamide (final concentration 50 mm) was added to samples to prevent reformation of disulfide bonds (34). The sample was digested in-solution according to previous methods (34). Finally, peptides were pooled and dried using a SpeedVac system (RVC 2–18, Marin Christ) for MS/MS analysis.
Peptide pellets were redissolved in 10 μl of 0.1% formic acid in distilled water. Then 8 μl of peptide sample was loaded onto a LTQ-Orbitrap Elite MS (Thermo Fisher Scientific) coupled to Easy-nLC 1000 (Thermo Fisher Scientific) using a nanoelectrospray ion source (spray voltage 2.3 kV, capillary temperature 275 °C and S-Lens RF 55%). Full MS scans were acquired (range from m/z 300–2000 with a resolution of 30,000 at m/z 400). The 20 most abundant ions were fragmented by higher energy collisional dissociation (HCD) with a normalized fragmentation energy of 35%. The HCD fragment ion spectra were acquired in the orbitrap analyzer with a resolution of 15,000 at m/z 400 and start from m/z 100. Ions with charge state of +1 were excluded. The reverse phase C18 was performed using an Easy-spray column packed with 2 μm C18 (100Å, 75 μm x 50 cm); The mobile phase buffer consisted of buffer A (0.1% formic acid, 2% acetonitrile in water) and buffer B (0.1% formic acid in acetonitrile). Peptides were separated at a flow rate of 250 nl/min in the EASY-nLC 1000 system using the following gradients: from 3 to 8% buffer B in 10 min, from 8 to 23% buffer B in 110 min, from 23 to 30% buffer B in 10 min, from 30 to 90% buffer B in 8 min, and 90% buffer B in 12 min.
Protein Identification
MS-MS spectra were retrieved using Xcalibur (version 2.2, Thermo Fisher Scientific). Using PEAKS software (version 6.0, Bioinformatics Solutions Inc.) the LTQ-Orbitrap raw files were searched against the sequence database generated from protein sequences of Apis mellifera (downloaded April, 2012) and augmented with sequences from Saccharomyces cerevisiae (downloaded April, 2012), totaling 61,380 entries (35). The search parameters were as follows: precursor and fragment mass tolerances were set to 50 ppm and 0.05 Da, respectively; tryptic cleavage specificity with up two missed cleavages; carbamidomethyl (C, +57.02) as a fixed modification; and oxidation (M, +15.99) as a variable modification. False discovery rate (FDR) was controlled using a target/decoy database approach for both protein identification and peptide identification applying the cut-off FDR of ≤1.0% (-10lgP ≥20.0). Protein identifications were only used if at least one unique peptide with at least one spectrum was identified.
Label-free Quantitation of Protein Abundance
For label-free quantitation, raw MS data was imported and processed in Progenesis LC-MS software (Version 4.1; Nonlinear Dynamics, Newcastle, UK) using a quantify-then-qualify strategy. One sample was automatically selected as reference and all other runs were aligned to it and further manual editing was done to correct the mismatched and unmatched features. MS1 spectra were subjected to peak modeling algorithm and quality control using default settings of the software. The result of peak detection is a set of features, and each feature represents a same peptide ion and its associated isotopes from all aligned samples. The abundance of discriminatory peptides was the sum of the peak areas within the isotope boundaries of the corresponding feature. The expression level of each protein was calculated in terms of its peptide ion abundance of three different replicate experiments. The differentially expressed proteins among the three time-points of embryos were considered to be statistically significant with p < 0.05 (using one-way ANOVA) and at least a twofold change. The q-value, an adjusted p value for multiple tests, was calculated to estimate false positive results. A merged peak list with differential abundance was generated by Progenesis LC-MS and then searched against the above search engine, protein identification database, and parameters. The search results of quantified protein were imported into Progenesis LC-MS software again to match each signal feature with the best peptide assignment. Similar proteins were grouped and only non-conflicting features were used for quantitation.
Bioinformatics Analysis
All identified proteins were assigned to gene ontology (GO)1 term using Blast2GO (36), using BLAST searches for each protein (BLASTp, NCBInr database, high scoring segment pair (HSP) cutoff length 33, report 20 hits, and maximum e-value 1e-10), followed by mapping and annotation (e-value hit filter 1e-10, annotation cutoff 55, GO weight 5, and HSP-hit coverage cutoff 20). Biological process terms were generated (sequence count 70, score alpha 0.6, and node score filter 0). The assigned GO-terms were manually checked. Because proteins usually perform multiple functions, it is therefore a protein may be classified into multiple categories.
To enrich the identified proteins into significant biological pathways, it was analyzed by KEGG Orthology-Based Annotation System (KOBAS, http://kobas.cbi.pku.edu.cn) (37). The protein sequences were blasted against the A.mellifera database, and pathway enrichment was then conducted by a hypergeometric statistic test. The Benjamini and Hochberg FDR correction was used to correct the probability values, and only the corrected p < 0.05 was considered as statistically significant enriched biological pathway.
To predict the protein–protein interaction (PPI) network, the identified proteins and the differentially expressed proteins were annotated by the Interologous Interaction Database (I2D) v1.9I2D (http://ophid.utoronto.ca/i2d) (38, 39), that integrated known and predicted PPI data sets from D. melanogaster and mapped them onto fly protein orthology. PPI networks were visualized using NAViGaTOR v2.2.1 (http://ophid.utoronto.ca/navigator/). Only proteins with more than 10 interaction degrees were considered.
To create an expressional profile of differentially expressed embryonic proteins, unsupervised hierarchical clustering (40) was analyzed (gene cluster 3.0) using uncentered Pearson correlation and average linkage, and visualized by Java Treeview software (41).
Quantitative Real-time PCR
Total RNA was extracted from 24, 48, and 72h old worker embryos (TRIzol reagent, Invitrogen, Carlsbad, CA) and used to generate cDNA using Reverse Transcriptase kit reagents (Transgen, Beijing, China), according to the manufacturer's instructions. Differentially expressed proteins from the PPI networks were selected for qRT-PCR analysis and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as an internal control to normalize the data. The primer pairs are provided in supplemental Table S1. Real-time PCR was conducted using an iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). PCR and the following data analysis were performed as in our previous reported protocol (31). The statistical analysis of gene expression was performed by one-way ANOVA (SPSS version 16.0, SPSS, Inc. IL) using Duncan's multiple-range test. An error probability p < 0.05 was considered statistically significant.
Western Blotting Analysis
Western blotting analysis of honey bee worker embryos were done as previously described (42) using the ECL (enhanced chemiluminescence) for Western blotting detection. All primary antibodies were from Abcam (Cambridge, MA, USA). The primary rabbit polyclonal antibodies were anti-nucleoside diphosphate kinase (NDPK), lysyl-tRNA synthetase (LysRS) elongation factor 1 alpha (eEF1A), eukaryotic translation initiation factor 5A (eIF-5A), and 40S ribosomal protein S11 (RPS11) at dilutions of 1:5000, 1:4000, 1:3000, 1:4000, and 1:5000, respectively; The secondary antibody was horseradish peroxidase-conjugated goat anti-rabbit at a dilution of 1:10000. Each lane (10 μg sample) was separated by stacking (4%) and separating (12%) SDS-PAGE gels with three replication runs of each sample. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as reference control. Immunoreactive protein bands were quantified by densitometry using Quantity One image analysis software (Bio-Rad, Hercules, CA, USA). The statistical analysis of protein abundance was done using the same software as the above gene expression.
RESULTS
MS-based proteomics with high resolution and high sensitivity (LTQ-Orbitrap Elite MS) identified 1460 individual proteins (FDR<1% at the peptide level) during the entire course of honey bee worker embryogenesis; 846, 1080, and 1118 proteins were discovered at 24h, 48h, and 72h, respectively (Fig. 1 and supplemental Table S2–4; the spectra of identified proteins with one unique peptide see supplemental Table S5–7).
Fig. 1.
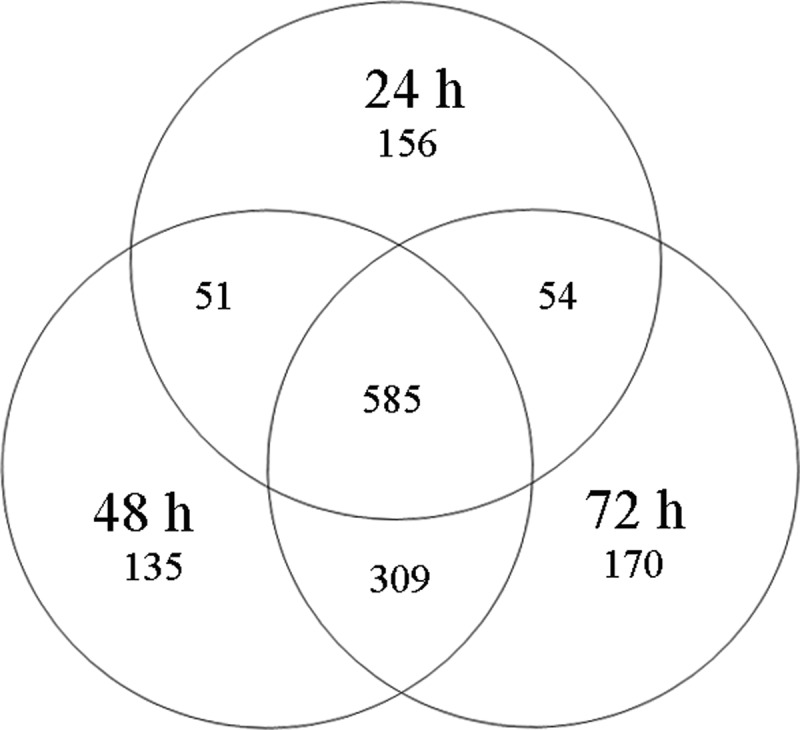
Global proteomics view across the three time points of the worker honey bee's embryo (A.m.ligustica). Venn diagram shows the numbers of shared and unique proteins identified in the worker honey bee's embryo at 24 h, 48 h, and 72 h.
In-depth Profiling of Time-resolved Proteomes
A function-level view of the proteome across the three time points in the honey bee embryo was generated by sorting protein groups into GO terms. All the 1460 identified proteins were classified into 17 functional GO terms on the basis of their biological processes (supplemental Fig. S1 and supplemental Table S8). The category containing the highest number of proteins was related to translation (12.1%), followed by proteins associated with folding/degradation (10.7%), development (10.1%), transporters (10.1%), carbohydrate metabolism/energy (9.4%), and transcription (9.2%). A similar coverage of functional categories was observed at the 24 h, 48 h, and 72 h as was identified in the total proteins (supplemental Fig. S2). Almost all of the GO terms in the embryo at age's 48 h and 72 h had higher representations of proteins than in the 24 h stage. Notably, the 72 h embryos had the highest representation of proteins related to carbohydrate metabolism/energy, folding/degradation, translation, transcription, signal transduction, and morphogenesis (Fig. 2).
Fig. 2.

Distribution of the identified proteins by their functional classification at three developmental stages of the worker bee embryo (A.m.ligustica). Color codes are different aged samples.
Noticeably, a “core” proteome of 585 proteins (accounting for 40% of total identified proteins) was shared across the three different aged embryos (Fig. 1). The major represented GO terms of the core proteome were proteins associated with (1) translation, (2) folding/degradation, (3) carbohydrate metabolism/energy, (4) development, and (5) transporters (Fig. 3). Also, we observed unique expression of 156, 135, and 170 proteins at 24 h, 48 h, and 72 h-aged embryos, respectively. Of these age-unique proteins, the GO term was totally different across three time points. Proteins involved in development, folding/degradation, and transporters were overrepresented at 24 h, whereas proteins associated with transporters, carbohydrate metabolism/energy and cell cycle control/apoptosis were predominant in 48 h embryos. Proteins related to folding/degradation, morphogenesis, and transcription were mainly expressed in 72 h embryos (supplemental Fig. S3).
Fig. 3.
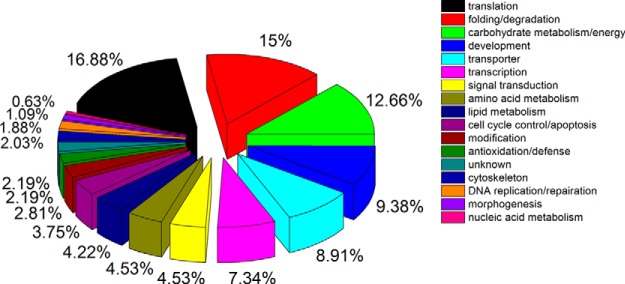
Functional category of the shared proteins identified (core proteome) across embryogenesis of honey bee workers (A.m.ligustica) at 24 h, 48 h, and 72 h. Proteins are classified based on the biological process of gene ontology (GO) using Blast2GO (36). Because proteins usually perform multiple fuctions, a protein may be classified into multiple categories. The percentage of each functional group is obtained on the basis of the number of proteins under each of the functional groups divided by the total number of shared proteins. Color codes represent different protein functional groups.
To better understand key metabolic pathways during the embryogenesis of honey bee workers, all of 845, 1078, and 1116 proteins were successfully annotated to the KEGG database at 24 h, 48 h, and 72 h, respectively. Six common metabolic pathways were significantly enriched in the embryos during the three stages: ribosome, proteasome, citrate cycle, protein processing in the endoplasmic reticulum, glycolysis/gluconeogenesis, and the pentose phosphate pathway (Fig. 4 and supplemental Table S9–11). In addition, protein export, β-alanine metabolism, and aminoacyl-tRNA biosynthesis pathways were significantly enriched in the 48 h embryo, whereas RNA transport and fatty acid metabolism were significantly enriched in 72 h embryo.
Fig. 4.

Biological pathway enrichment of identified proteins in the embryos of honey bee workers (A.m.ligustica). A, Comparison of enriched biological pathways of the identified proteins in the embryo of the honey bee workers (A.m.ligustica) aged 24h, 48h, and 72h. Significant enriched pathways are analyzed by KEGG Orthology-Based Annotation System (KOBAS, http://kobas.cbi.pku.edu.cn) (37). The pathway enrichment is conducted by a hypergeometric statistic test. The Benjamini and Hochberg FDR correction is used to correct the probability values, and only the corrected p < 0.05 is considered as statistically significant enriched pathways. B, Representative enriched pathway map from KOBAS. Green labeled boxes are the protein references of honey bee (A.m.ligustica) annotated to the KEGG PATHWAY database, highlighted red ones indicate the protein entries mapped to the significantly enriched pathway.
For a network-wide overview of honey bee embryogenesis, proteins with high degrees of interaction in the PPI network were identified as key node proteins. Applying a cutoff of at least 10 interaction links, a total of 246, 283, and 308 proteins were identified in the PPI network of embryos at 24h, 48h, and 72h, respectively (Fig. 5, PPI networks of 48h and 72h aged embryos see supplemental Fig. S4, and supplemental Table S12–14). These key node proteins were mainly involved in translation, carbohydrate metabolism/energy, and folding/degradation (supplemental Fig. S5).
Fig. 5.

Protein-protein interaction (PPI) network of proteins expressed in the 24 h embryo of honey bee workers (A.m.ligustica). PPI is predicted using the Interologous Interaction Database (I2D) and visualized by NAViGaTOR software (38, 39). The color symbols represent identified proteins connected in the PPI network with more than ten interaction degrees (supplemental Table S9). The regular triangles stand for proteins up-regulated in the embryogenesis of honey bee worker at 24 h. Letters from “a” to “p” represent the categories of carbohydrate metabolism/energy, morphogenesis, nucleic acid metabolism, translation, modification, development, cytoskeleton, DNA replication/repairation, cell cycle control/apoptosis, transcription, lipid metabolism, antioxidation/defense, folding/degradation, signal transduction, amino acid metabolism, transporters, respectively. Blue lines indicate interactions between proteins. The intensity of the interaction degree is indicated by a color gradient as noted on the key bar on the bottom right side of the figure.
Quantitative Analysis of Honey bee Worker Embryogenesis
To quantify the proteomic changes during honey bee embryogenesis, a label-free LC-MS based approach was applied, using three replicates of each aged embryo sample. Overall, 4337 qualified peptides were screened for quantification and they were corresponded to 544 proteins. These differentially expressed proteins (fold change≥2 and p < 0.05, FDR<1%) represented about 37% of the total identified 1460 proteins (supplemental Table S15).
Proteins with significant differences in expression (544 proteins) were plotted against GO category. The four most abundant functional categories were proteins associated with (1) translation, (2) folding/degradation, (3) development, and (4) transporters (supplemental Fig. S6). This was generally consistent with the functional category coverage of all the 1460 identified proteins (supplemental Fig. S1).
Of the 544 proteins that had significant differentiations in expression, 543 were annotated into KEGG metabolic pathways. Biological pathways involved in ribosomes, fatty acid metabolism, fatty acid elongation, proteasome, and pentose phosphate were significantly enriched (supplemental Table S16). A total of 139 proteins were linked in the PPI network (supplemental Fig. S4C and supplemental Table S17), of which proteins implicated in translation, folding/degradation, and metabolism of carbohydrate or energy were overrepresented.
To better visualize protein expression during the embryogenesis of honey bee worker, the expression pattern of the 544 significant differential expressed proteins were analyzed (Fig. 6. Seven representative 3D montages were the abundance of representative peptides of a protein that uniquely or commonly expressed in the embryo of the honey bee workers aged 24 h, 48 h, and 72 h). Protein expression at 48 h and 72 h were clustered in one branch and protein expression at 24 h was clustered in another independent branch. Specifically, 97, 243, and 203 proteins were up-regulated in the three aged embryos, in which 56, 156, and 95 proteins were in the core proteome (supplemental Fig. S7). To further interpret the biological significance of cluster data, the up-regulated proteins at each time point were plotted against GO category (Fig. 7). The up-regulated proteins were mainly related to translation, folding/degradation, and carbohydrate metabolism/energy, which showed a similar distribution patterns as in the all identified and differential proteins (supplemental Figs. S1 and S6). Together with the above functional classification of the 544 differential proteins, the quantitative data could be more accurate representation of the qualitative results. Almost all the categories at 48 h and 72 h had more up-regulated proteins than at 24 h, except for those related to DNA replication/reparation. Additionally, proteins related to development and folding/degradation were overrepresented at the age of 24 h. Proteins involved in translation and folding/degradation were predominant at 48h, whereas proteins related to translation and development were overrepresented at 72 h. For the up-regulated proteins at each stage (including exclusively expressed proteins), no metabolic pathway was significantly enriched at 24h, but the ribosomal pathway was significantly enriched at both 48 h and 72 h. Biological pathways of protein export and fatty acid elongation were exclusively enriched at 48 h and 72 h, respectively (supplemental Table S18–20).
Fig. 6.

Unsupervised hierarchical clustering of the differentially expressed (fold change ≥ 2 and p < 0. 05) proteins in the embryo of honey bee workers (A.m.ligustica). The columns represent the embryonic age (24 h, 48 h, and 72 h), and the rows represent the individual proteins. The up- or down-regulated proteins are indicated by red and green color code, respectively. The color intensity changes with the protein expressional level as noted on the key bar on the top right. The histograms denote the expression trend of the representative proteins and the 3D montages are the abundance of representative peptides of a protein that uniquely (2, 3, 5, and 6) or commonly (1, 4, and 7) expressed in the embryo of the honey bee workers (A.m.ligustica) aged 24 h, 48 h, and 72 h, respectively.
Fig. 7.

Comparison of the up-regulated proteins by their functional classifications at three developmental stages of the honey bee worker (A. m.ligustica) embryo. Color codes denote the three aged samples.
Verification of Differentially Expressed Proteins at the Level of mRNA and Protein
To test the tendency of protein expression at the transcript level, twenty-nine key node proteins were selected for qRT-PCR analysis. The trends of mRNA expression showed that 16 genes were consistent with their protein expression, i.e. inorganic pyrophosphatase, importin subunit alpha-2, ubiquitin activating enzyme 1 isoform 1, 40S ribosomal protein S11, 60S ribosomal protein L11, isocitrate dehydrogenase [NAD] subunit beta, prefoldin subunit 5, nucleoside diphosphate kinase, peroxiredoxin 1, lysyl-tRNA synthetase, ATP synthase subunit beta, nucleolar complex protein 3 homolog, actin-related protein 2/3 complex subunit 2, eukaryotic translation initiation factor 5A, elongation factor 2 isoform 1, and elongation factor 1 alpha (Fig. 8). However, eight other gene expressions were not directly matched to the corresponding protein expression, which may be because the protein and its corresponding gene expression are not always synchronized (43).
Fig. 8.

Test of the 16 differentially expressed (fold change ≥2 and p < 0. 05) proteins at the mRNA level by quantitative real time PCR analysis. The mRNA expression is normalized with the reference gene (GAPDH). The color bars represent the relative expression values of mRNA and protein in different aged embryos. (a) is significantly higher than (b) and (c), (b) is significantly higher than (c). Error bar is standard deviation. Abbreviated protein names indicate different proteins as in supplemental Table S1.
Western blotting analysis was performed to verify the expression of key node proteins in PPI networks-NDPK, LysRS, eEF1A, eIF-5A, and RPS11, the results showed that they weree consistent with the proteomics data (Fig. 9).
Fig. 9.

Western blotting analysis of nucleoside diphosphate kinase (NDPK), lysyl-tRNA synthetase (LysRS), elongation factor 1 alpha (eEF1A), eukaryotic translation initiation factor 5A (eIF-5A), 40S ribosomal protein S11 (RPS11). The protein samples from worker embryos (A.m.ligustica) are subjected to SDS-PAGE followed by Western blotting analysis. NDPK, LysRS, eEF1A, eIF-5A, and RPS11 are detected using the corresponding polyclonal antibody. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) is used as reference control. A, The Western blotting bands of NDPK, LysRS, eEF1A, eIF-5A, RPS11, and GAPDH. B, The relative expression values of NDPK, LysRS, eEF1A, eIF-5A, and RPS11 in the three aged honey bee (A.m.ligustica) embryos (normalized by GAPDH). (a) is significantly higher than (b) and (c), (b) is significantly higher than (c). Error bar is standard deviation.
DISCUSSION
Here we provide an in-depth comparison of the embryonic proteomes of workers of different ages and identify a total of 1460 individual proteins across the entire embryogenesis process, thereby increasing the proteome coverage of the honey bee embryo by about 15-fold (31, 44). During the whole course of development, the honey bee embryos require a core proteome mainly involved in protein synthesis, metabolic energy, development, and molecular transporter as driving forces for embryogenesis. Noticeably, the specified protein signatures of each embryonic stage match with the known physiological status of honey bee embryogenesis, that is, younger embryos (24 h) express high levels of proteins related to the initiation of cell proliferation to prepare for the subsequent organogenesis; whereas older embryos of 48 and 72 h of age up-regulated more proteins to support organogenesis, that is, the formation of rudimentary organs of the reproductive system, nervous system, tracheal respiratory system, digestive system, and circulatory system (Figs. 6, 7). The activated biological pathways in 48 h and 72 h old embryos (protein export, β-alanine metabolism, aminoacyl-tRNA biosynthesis, RNA transport, and fatty acid metabolism) are believed to modulate honey bee embryogenesis in a progressive activation manner coinciding with the development of the organ establishment (Fig. 4 and supplemental Table S9–11). Although the lineages of Drosophila and Apis mellifera have separated ∼300 million years ago, the vast majority of developmental genes are evolutionarily conserved (45). Therefore, our data add vital new knowledge not only for the further embryological research on the honey bee but also for holometabolous insects as well.
Although embryos at all three life stages have their own proteomic characteristics, about 40% of the identified proteins were present in all developmental stages of the embryos. This identified “core” proteome suggests its essential role to support embryogenesis of honey bee workers. In the core proteome, the high representation of proteins related to translation, folding/degradation, and carbohydrate metabolism/energy suggests their importance in driving embryogenesis by providing newly synthesized proteins and metabolic energy (Fig. 3). This major driving force is also reflected in the quantitative analysis that about 50% of the up-regulated proteins were in core proteome (supplemental Tables S2–4, S15), and they were mainly implicated in translation, folding/degradation, and carbohydrate metabolism/energy during the embryogenesis (supplemental Fig. S7).
Translational machinery is the important process of encoding new proteins in accordance with mRNA. In honey bee embryo, about one-fifth of the core proteome were proteins related to translation, such as ribosomal proteins (accounting for >50% of the translation related proteins in core proteome), eukaryotic translation initiation factor (eIF), and elongation factor (supplemental Table S8). These proteins are supposed to be working together for the synthesis of new peptides in accordance with the mRNA to provide the building blocks for the formation of new organs.
The normal functionality of proteins in a living cell requires the involvement of new protein synthesis and degradation activities, in which molecular chaperones and proteases are involved (46). These proteins control the balance between native folded functional proteins and aggregation-prone misfolded or invalidated proteins (47). To this effect, proteins involved in configuration of nascent peptide chains are expressed as heat shock proteins (Hsps), T-complex protein 1 (TCP-1), Dna J, and protein disulfide-isomerase (PDI). On the other hand, proteins with degradative function are also required in honey bee embryogenesis in parallel with protein synthesis as in human cells (48), which is achieved by the expression of proteasome related proteins, such as protein DJ-1, cathepsin L, lysosomal Pro-X carboxypeptidase, and xaa-Pro aminopeptidase. Overall, the expression of proteins related to synthesis/degradation activities suggests their pivotal roles in the normal functionality of honey bee embryonic organogenesis by their correct configuration of new proteins and cyclic utilization of discarded proteins (49).
Of the overrepresented proteins related to carbohydrate metabolism/energy, 30% were implicated in the oxidative phosphorylation pathway (supplemental Table S8), such as ATP synthases and cytochromes, which produce high energetic molecular ATP that powers most cellular reactions (50, 51). The higher number of proteins involved in glycolysis, the citric acid cycle, and the pentose phosphate pathways such as enolase, glucose-6-phosphate dehydrogenase, malate dehydrogenase, succinate dehydrogenase, transaldolase, and 6-phosphogluconate dehydrogenase indicates that embryogenesis requires intensive metabolic energy to sustain normal embryonic development (Fig. 4 and supplemental Table S9–11).
Similar to Drosophila, honey bee embryogenesis is a crucial stage in which the rudiments of organs or imaginal discs of adult bees' specific structures begin to form, such as compound eyes, head appendages, legs, genitalia, and wings of the adult appendages (52). To achieve this goal, a wide array of proteins related to the development in the core proteome suggests the importance as the driving force for embryonic organogenesis to modulate cell divisions, and tissue differentiations (31, 53) (supplemental Table S8). For instance, 14–3-3 protein family is the key regulator of cell division, signaling, and apoptosis during the embryogenesis of the honey bee (53). Restin homolog has a role in cellularization to regulate the Microtubule-dependent, Golgi-derived membrane vesicle export mechanism (55). Protein tumorous imaginal disc is a promoter in the formation of imaginal discs (56). As a multifunctional regulator, vitellogenin has a functionality of stimulating embryogenesis in the honey bee drone (31). In all, a large number of developmental proteins expressed by the embryo suggest they are vital for the enhancement of cellular activities during organogenesis, tissue elongation, and body segmentation (30, 31, 57).
For normal cell functionality, proteins acting as molecular transporters are used to deliver macromolecules (e.g. proteins and polynucleotides) to a desirable subcellular location (58), which is important for proper functioning in the developing honey bee embryos (57). Transporters involved in protein delivery represented the major category of the transporters in the core proteome, such as family members of coatomers, exportins, and importins, indicating the significance of new protein distribution in embryo tissue establishment and organogenesis (supplemental Table S5). Besides, other macromolecule transporters associated with polynucleotides (hrp65 protein), sugars (sugar transporter), and monomeric lipids (phosphatidylinositol transfer protein) are supposed to maintain coherence of the material supply and homeostasis. Moreover, as ion transporter, sodium/potassium-transporting ATPase is to create the electrochemical gradients of sodium and potassium ions in plasma membranes to provide the energy via active transport of various nutrients (59).
In general, the identified high number of proteins implicated in translation, folding/degradation, metabolism of carbohydrates/energy, development, and transporters at different aged embryos signifies their importance in providing sufficient metabolic fuel and protein building blocks to ensure normal embryonic organogenesis. The centrality of these five protein families are also consistent with the high representation in the post-embryonic development of other honey bee organs, such as the pupal head (60), brain (61), hemolymph (62), hypopharyngeal gland (63, 64), and salivary gland (65).
Proteins Expressed by 24h Embryos Play Key Roles in Nutrition and Cell Proliferation to Form Blastoderm
Embryos of honey bee worker aged 24 h cleave the zygote and form the early blastoderm through an accumulation of embryonic cell numbers (4, 66). However, in the initial stage of embryogenesis, the young embryo still requires high levels of nutrition for survival. In this reality, proteins that serve as nutritive sources must be expressed at high levels in the young embryos as in the silkworm (67, 68) and fruit fly (69). This is reflected in our data that the young embryos uniquely expressed proteins associated with major royal jelly proteins (MRJPs), vitellogenin, and ferritin are likely to serve their roles as nutritive sources to support the developing young embryos (Fig. 2 and supplemental Table S2). Despite the role as nutrition for the honey bee (70), MRJPs are pluripotent proteins that carry out biological missions in facilitating honey bee caste differentiation (71) and brain development (72). Thus it is believed that MRJPs may act both as a nutritive source to nourish the young embryos and an enhancer of cell proliferation for them. Vitellogenin, also a pluripotent protein, is the precursor of the lipoproteins and phosphoproteins that make up yolk protein, which may also provide more efficient nutrition for the embryos (73). Ferritin, the iron storage protein, has a similar nutritive role as yolk protein (74). Therefore, these proteins may work together as either a nutrition reservoir or as development promoters that ensure the growth of young embryos.
Embryonic growth depends on rapid cell division (75) and thus requires the intensive involvement of proteins associated with nucleic acid metabolism. For instance, the stronger expressed 10-formyltetrahydrofolate dehydrogenase and UMP-CMP kinase at this stage, suggest their involvement in the biosynthesis of purine (76) and pyrimidine (77), both are essential nucleotide precursors needed to form novel cell nuclei in the first 10 h post egg laying (66). The vital role of proteins related to nucleic acid metabolism is also corroborated by their higher representation in the PPI network (supplemental Fig. S5). Additionally, the highly expressed cytoskeletal proteins (microtubule-associated proteins and actin-related proteins) may be associated with the promotion of chromosome movement in mitosis (78), and with the migration of cleavage cells into the cortical cytoplasm to form the blastoderm, a layer of cells occurring after the tenth mitotic cycle (66). Interestingly, although the rudimentary organs of the embryo have not formed at this age, a high expression of proteins related to morphogenesis (BAG family molecular chaperone regulator, protein sly1 homolog, serine/threonine-protein kinase polo), and modification (oligosaccharyltransferase complex, dolichol-phosphate mannosyltransferase, protein arginine N-methyltransferase) (Fig. 2, supplemental Table S2, S8) indicates that the molecular event of organogenesis starts earlier than the morphological change by preparing a wide spectrum of molecular material for the upcoming organ creation.
The true functional units of metabolic systems are the biological pathways, so their identification in embryogenesis is vitally important in understanding the molecular mechanisms. The different biological pathways mapped at the three time point of the embryos suggest that embryos at each developmental stage have their own metabolic signature coinciding with their physiological change (Fig. 4 and supplemental Tables S9–11). For the proteins enhanced expression of 24h aged embryos, there were no significant enriched biological pathways (supplemental Table S18), indicating the metabolic activities of young embryos are still not fully activated since forming the rudimentary organogenesis are still at the beginning stage of embryogenesis (66).
Proteins Expressed by 48h Embryos are Mainly to Construct the Rudiments of Organs
Embryos at an age of 48h undertake segmentation, germ band formation, and the onset of rudimentary organogenesis, such as brain, nerve, stomodeum, trachea, and malpighiam tubules (5, 66). This is a critical period for the formation of the basic embryo configuration, which is achieved by the up-regulated 243 proteins that were mainly related to translation, folding/degradation, and carbohydrate metabolism/energy (104 proteins) (Figs. 6, 7, and supplemental Table S15), and biological pathways (65 proteins), such as ribosome, protein processing in endoplasmic reticulum (supplemental Table S19). These proteins are used either as metabolic fuel or as protein building blocks to shape the newly emerged organs. To ensure normal development of the newly established organs, the strongly activated biological pathways of aminoacyl-tRNA biosynthesis and β-alanine metabolism indicate that they are involved in processing materials of nascent peptides for protein synthesis of 48h embryos (Fig. 4 and supplemental Table S10).
Cell cycle control/apoptosis related proteins play major roles in providing a homeostatic balance of cell population during the development of the honey bee embryo (79). The higher expression of proteins associated with cell cycle control/apoptosis indicates the occurrence of mitotic divisions in this embryonic stage (supplemental Table S3). This is in line with the fact that mitosis starts in cells located in the ectodermal layer and in a stretch of mesoderm after a division break of about 25 h since the 14th mitotic cycle (66). The importance of cell cycle control/apoptosis is also emphasized by their high involvement in the PPI network of proteins related to DNA replication/reparation and cell cycle control/apoptosis. Again, the stronger expression of proteins associated with purine/pyrimidine biosynthesis (bifunctional purine biosynthesis protein PURH, and dihydroorotate dehydrogenase) indicates that the mitosis still remains at high level in the middle aged embryos. The enhanced expression of proteins involved in apoptosis at this stage, such as peptidyl-prolyl cis-trans isomerase (FKBP8), and immediate early response 3-interacting protein (IER3IP1), are likely related to programmed cell death of the dorsal blastoderm, in which the dorsal blastoderm contracts, degenerates, and finally disappears at the late blastoderm stage (5).
As aforesaid the roles played in core proteome, the transporters with elevated expression in the middle aged embryos specifically function in protein transport, which coincides with the initiation of rudimentary embryonic organs. The significantly enriched biological pathways of protein export (Fig. 4 and supplemental Table S10) and protein transport (supplemental Table S19) indicate their vital roles of protein sorting (targeting proteins to specific intracellular locations such as organelles) in the embryo at this stage. As of sorting transporters, AP-1 (adapter-related protein), and AP-2 are two complex subunits of AP that mediate both the recruitment of clathrin to membranes and the recognition of sorting signals within the cytosolic tails of transmembrane cargo molecules (80). Moreover, the enhanced expression of proteins associated with protein sorting (metaxin-1 (MTX1), sorting nexin-12 (SNX12), coatomer subunit, and sorting and assembly machinery component 50 homolog), reflects their roles in locating proteins to the appropriate destinations to form the blastoderm, early germ band, and the rudiment of embryonic organs (66). Overall, the highly expressed transporter proteins at 48 h indicate that middle-aged embryos employ robust transportation activities to deliver the highly needed biomaterial to boost the establishment of rudimentary organs. In addition, the stronger expression of proteins implicated in signal transduction at this stage is thought to cooperate with transporter to deliver the newly synthesized proteins to their destination in a more efficient manner. For instance, signal recognition particle (SRP) and SRP receptor subunits are involved in targeting secretory proteins to the rough endoplasmic reticulum (RER) membrane (81) to activate cellular functions, cell differentiation, and cell proliferation (82).
The antioxidant system protects the tissues and organs from the oxidative damage of reactive oxygen species (ROS) (83). As organs begin to establish, a wide range of metabolic activities in middle aged embryos have already begun, this in turn increases ROS production that causes oxidative damage to the cell. As an ROS scavenger, the antioxidant system prevents cellular components from oxidative damage by removing free radicals and subsequently inhibiting other oxidative reactions (84). The up-regulation of glutathione S-transferase S1, peroxidase, and peroxiredoxin (Figs. 6, 7, and supplemental Table S15) signifies their roles in protecting the embryo from ROS-mediated organ damage, rescuing the cellular components from oxidative damage during strengthened metabolic activities of embryogenesis at this age.
A cytoskeletal scaffold is required to maintain their shape in the formation of organs (85). The up-regulation of cytoskeletal proteins (Fig. 7), atlastin, actin-interacting protein, myosin heavy chain, and kinesin, suggests their key roles in cellular mitosis and segmentation of the middle-aged embryo. This is consistent with the high levels of mitotic activity maintained in the mesoderm during segmentation (66).
After ∼31 h of development, the cells of intact embryos begin to exhibit marked morphogenesis (5). The up-regulation of proteins implicated in morphogenesis is believed to boost the onset of organogenesis (supplemental Table S15). For instance, lachesin (Lac), serine/threonine-protein kinase polo, and Ras-related protein (Rac1) are related to the generation of the tracheal system (86) and heart (87), rudimentary formation of heart tubes (88), and regulation of dendritic neurons morphogenesis (89), respectively, as in Drosophila embryos. In all, the enhanced expression of proteins associated with morphogenesis suggests their importance in promoting the establishment of rudimentary organs for middle aged embryos (66).
Protein Expressed by 72h Embryos Further Complete the Rudiments of Organs and the Dorsal Closure
The embryo at 72 h is at the last stage of honey bee embryogenesis, during which the rudimentary organs and the fine structures of the hindgut, mouthparts, and larval cuticle, are completed, and finally the larval body is shaped (5, 66). These biological changes are achievable by stronger expression of proteins involved in translational activities and protein folding, in particular the transcriptional proteins (Fig. 2 and supplemental Table S4). The escalated expression of proteins associated with transcription, translation, and folding/degradation indicates the new proteins are provided as biomaterials to sustain the developing embryos. This is in line with 40% of up-regulated proteins were associated with transcription, translation, and folding/degradation (Fig. 7 and supplemental Table S15), and 50 of them were enriched into biological pathways, such as ribosome, protein processing in endoplasmic reticulum (supplemental Table S20). The importance of proteins related to novel protein synthesis for the old embryos is further emphasized by the highly interacted proteins associated with translational activities and protein folding in the PPI network (supplemental Fig. S4 and supplemental Table S14). Moreover, the old embryos' strongly activated biological pathway of RNA transport (Fig. 4 and supplemental Table S11) substantiate protein synthesis is still significant via transcriptional and translational machinery.
Metabolic balances of fatty acids between triacylglycerol deposition and mobilization are to control switch of energy resources and storage compartments (90). The strongly activated biological pathways of fatty acid metabolism (Fig. 4 and Supplemental Table S11) and increased expression of proteins involved in lipid metabolism (trans-2-enoyl-CoA reductase, trans-2, 3-enoyl-CoA reductase, and long-chain-fatty-acid-CoA ligase 3) indicate the energy demand still remain high of embryos at 72h. The metabolic energy requirement of the old embryos could be met by the decomposition of yolk to supply cell proliferation and development by vitellophag (91) as the yolk still presents in the 72h embryo (66), which is consistent with the significant weight loss of embryos at this age because of the depletion of yolk (27). Moreover, depletion of the enclosed yolk would fill in the vesicular invagination necessary to form organs such as the midgut and ventriculus (66). The activated fatty acid elongation pathways (supplemental Table S20) indicate fatty acid synthesis (fatty acid synthase and protein NPC2 homolog) still occurs at high levels in the old embryos. The stronger synthesis of fatty acids is likely involved in neurogenesis of the old embryos because of how the nervous system has shaped 44h after oviposition (66), which is vital for synthesis of lipoprotein for neuro-activity (92).
Apart from the role in the maintenance of cell shape and structure, the cytoskeletal proteins also have key roles in intracellular transport and cellular division (85). The up-regulation of cytoskeletal proteins in the 72 h embryo (kinectin, vinculin, and microtubule-associated protein Jupiter; Fig. 7 and supplemental Table S15) implies their critical roles in germinal layer movement and organ structure (93). The cytoskeleton is also important in driving blastokinesis in honey bee embryos, after which the orientation of the embryo may turn 180 degrees along the axis of the egg with the aim of taking full advantage of the surrounding nutrition (94). Additionally, the process of dorsal closure, a symbol of completed embryo formation involved in the joining of opposing epithelial cell sheets, also needs the assistance of cytoskeletal proteins (95).
Through programmed development, embryogenesis is completed and the most rudimentary organs are formed by 72 h (91). Accordingly, morphogenesis proteins expressed at high level in older embryos suggest that organogenesis has reached the peak time (supplemental Fig. S3). At this age, neurogenesis is a must, which requires enhanced expression of proteins involved in RNA-binding protein pno1, small minded, nucleolar protein 56, tropomyosin-1, neuroglian, ELAV protein 2, and lamin-B receptor to match the physiological changes. In addition, to complete the brain morphogenesis and heart development, laminin family members such as subunit beta-1 and gamma-1 were also increased expression. Generally, temporal and spatial progressive activation of the proteome and pathway trigger the complete embryonic process from organogenesis (inner embryo) to dorsal closure (outer embryo), and eventually an intact egg hatches out as a larva.
CONCLUSIONS
Our in-depth proteomic data substantially expand the proteome coverage of honey bee worker embryos and therefore new insight can be gained into the honey bee embryonic development. The embryogenesis requires a core proteome and common biological pathways to drive the development. However, embryos at different developmental stages have their own specific proteome and pathway signatures to coordinate and modulate developmental events. The young embryos (<24 h) stronger expression of proteins related to nutrition storage and nucleic acid metabolism are believed to correlate well with the cell proliferation occurring at this stage. The middle aged embryos (24–48 h) enhanced express proteins associated with cell cycle control, transporters, antioxidant activity, and the cytoskeleton to support rudimentary organogenesis. Of which, the function of biological pathways of aminoacyl-tRNA biosynthesis, β-alanine metabolism, and protein export are strongly activated. The old embryos (48–72 h) elevated express proteins implicated in fatty acid metabolism and morphogenesis indicates their functionality for the formation and development of organs and dorsal closure, in which the biological pathways of fatty acid metabolism and RNA transport are highly activated. Our deepen proteomics data constitute a proof-of-concept of molecular details during the honey bee embryogenesis, in which the programmed activation of the proteome matches with the physiological transition observed during embryogenesis. The identified biological pathways and key node proteins allow for further functional analysis and genetic manipulation for both the honey bee embryos and other eusocial insects.
Supplementary Material
Acknowledgments
We thank Meghan Milbrath from Michigan State University, USA, for her help with the language of the manuscript.
Footnotes
Author contributions: Y.F. and J.L. designed research; Y.F. and M.F. performed research; J.L. contributed new reagents or analytic tools; Y.F., B.H., X.L., and H.R. analyzed data; Y.F., M.F., and J.L. wrote the paper.
* This work is supported by the earmarked fund for Modern Agro-industry Technology Research System (CARS-45), Key Projects of the National Scientific Supporting Plan of the 12th Five-Year Development (2011–2015) (2011BAD33B04).
 This article contains supplemental Figs. S1 to S7 and Tables S1 to S20.
This article contains supplemental Figs. S1 to S7 and Tables S1 to S20.
1 The abbreviations used are:
- GO
- gene ontology
- HSP
- high scoring segment pair
- KOBAS
- KEGG Orthology-Based Annotation System
- PPI
- protein–protein interaction
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- NDPK
- nucleoside diphosphate kinase
- LysRS
- lysyl-tRNA synthetase
- eEF1A
- elongation factor 1 alpha
- eIF-5A
- eukaryotic translation initiation factor 5A
- RPS11
- 40S ribosomal protein S11
- eIF
- eukaryotic translation initiation factor
- MRJPs
- major royal jelly proteins
- FKBP8
- peptidyl-prolyl cis-trans isomerase
- IER3IP1
- immediate early response 3-interacting protein
- AP
- adapter-related protein
- MTX1
- metaxin-1
- SNX12
- sorting nexin-12
- SRP
- signal recognition particle
- RER
- rough endoplasmic reticulum
- ROS
- reactive oxygen species
- Lac
- lachesin
- Rac1
- Ras-related protein.
REFERENCES
- 1. Winston M. L. (1987) The Biology of the Honey Bee, Harvard University Press, Cambridge, London [Google Scholar]
- 2. Fleig R., Sander K. (1986) Embryogenesis of the honeybee Apis mellifera L. (hymenoptera : apidae): An sem study. Int. J. Insect Morphol. 15, 449–462 [Google Scholar]
- 3. Schnetter M. (1934) Morphologische Untersuchungen uber das Differenzierungszentrum in der Embryonalentwicklung der Honigbien. Zeitschr Wiss Biol Abt A Zeitschr Morph U Okol Tiere 29, 114–195 [Google Scholar]
- 4. Nelson J. A. (1915) The embryology of the honey bee, Princeton University Press [Google Scholar]
- 5. DuPraw E. (1967) The honeybee embryo. Methods in Developmental Biology, pp. 183–217, Crowell, New York [Google Scholar]
- 6. Aase A. L. T. O., Amdam G. V., Hagen A., Omholt S. W. (2005) A new method for rearing genetically manipulated honey bee workers. Apidologie 36, 293–299 [Google Scholar]
- 7. Amdam G., Simoes Z., Guidugli K., Norberg K., Omholt S. (2003) Disruption of vitellogenin gene function in adult honeybees by intra-abdominal injection of double-stranded RNA. BMC Biotechnol. 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gary N. E. (1993) Activities and behavior of honey bees. The hive and the honey bee, pp. 318–319, Dadan & Son, Hamilton [Google Scholar]
- 9. Went D. F. (1982) Egg activation and parthenogenetic reproduction in insects. Biol. Rev. 57, 319–344 [Google Scholar]
- 10. Sasaki K., Obara Y. (2002) Egg activation and timing of sperm acceptance by an egg in honey bees (Apis mellifera L.). Insect. Soc. 49, 234–240 [Google Scholar]
- 11. Osborne P., Dearden P. K. (2005) Non-radioactive in-situ hybridisation to honey bee embryos and ovaries. Apidologie 36, 113–118 [Google Scholar]
- 12. Yu R., Hagen A., Omholt S. W. (1998) Biopsied preblastoderm honey bee embryos develop into normal honey bee queens. Apidologie 29, 547–554 [Google Scholar]
- 13. Omholt S. W., Rishovd S., Hagen A., Elmholdt O., Dalsgard B., Fromm S. (1995) Successful production of chimeric honey bee larvae. J. Exp. Zool. 272, 410–412 [Google Scholar]
- 14. Milne C. P. J., Phillips J. P., Krell P. J. (1988) Microinjection of early honey bee embryos. J. Apicult. Res. 27, 84–89 [Google Scholar]
- 15. Bergem M., Norberg K., Roseth A., Meuwissen T., Lien S., Aamodt R. H. (2006) Chimeric honey bees (Apis mellifera) produced by transplantation of embryonic cells into pre-gastrula stage embryos and detection of chimerism by use of microsatellite markers. Mol. Reprod. Dev. 73, 475–481 [DOI] [PubMed] [Google Scholar]
- 16. Maleszka J., Foret S., Saint R., Maleszka R. (2007) RNAi-induced phenotypes suggest a novel role for a chemosensory protein CSP5 in the development of embryonic integument in the honey bee (Apis mellifera). Dev. Genes Evol. 217, 189–196 [DOI] [PubMed] [Google Scholar]
- 17. Patel A., Fondrk M. K., Kaftanoglu O., Emore C., Hunt G., Frederick K., Amdam G. V. (2007) The making of a queen: TOR pathway is a key player in diphenic caste development. PLoS One 2, e509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kucharski R., Maleszka J., Foret S., Maleszka R. (2008) Nutritional control of reproductive status in honey bees via DNA methylation. Science 319, 1827–1830 [DOI] [PubMed] [Google Scholar]
- 19. Beisser K., Munz E., Reimann M., Renner-Müller I. C. E. (1990) Experimentelle Untersuchungen zur in vitro-Kultivierung von Zellen der Kärntner Honigbiene (Apis mellifera carnica Pollmann, 1879). J. Vet. Med. B 37, 509–519 [DOI] [PubMed] [Google Scholar]
- 20. Kreissl S., Bicker G. (1992) Dissociated neurons of the pupal honey bee brain in cell culture. J. Neurocytol. 21, 545–556 [DOI] [PubMed] [Google Scholar]
- 21. Gascuel J., Masson C., Bermudez I., Beadle D. J. (1994) Morphological analysis of honey bee antennal cells growing in primary cultures. Tissue Cell 26, 551–558 [DOI] [PubMed] [Google Scholar]
- 22. Bergem M., Norberg K., Aamodt R. M. (2006) Long-term maintenance of in vitro cultured honey bee (Apis mellifera) embryonic cells. BMC Dev. Biol. 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kitagishi Y., Okumura N., Yoshida H., Nishimura Y., Takahashi J., Matsuda S. (2011) Long-term cultivation of in vitro Apis mellifera cells by gene transfer of human c-myc proto-oncogene. In Vitro Cell Dev. An. 47, 451–453 [DOI] [PubMed] [Google Scholar]
- 24. Chan M. M., Choi S. Y., Chan Q. W., Li P., Guarna M. M., Foster L. J. (2010) Proteome profile and lentiviral transduction of cultured honey bee (Apis mellifera L.) cells. Insect Mol. Biol. 19, 653–658 [DOI] [PubMed] [Google Scholar]
- 25. Osborne P. W., Dearden P. K. (2005) Expression of Pax group III genes in the honey bee (Apis mellifera). Dev. Genes Evol. 215, 499–508 [DOI] [PubMed] [Google Scholar]
- 26. Katzav-Gozansky T., Soroker V., Kamer J., Schulz C. M., Francke W., Hefetz A. (2003) Ultrastructural and chemical characterization of egg surface of honey bee worker and queen-laid eggs. Chemoecology 13, 129–134 [Google Scholar]
- 27. Woyke J. (1998) Size change of Apis mellifera eggs during the incubation period. J. Apicult. Res. 37, 239–246 [Google Scholar]
- 28. Dearden P. K. (2006) Germ cell development in the honey bee (Apis mellifera); vasa and nanos expression. BMC Dev. Biol. 6, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wilson M. J., Dearden P. K. (2009) Tailless patterning functions are conserved in the honey bee even in the absence of Torso signaling. Dev. Biol. 335, 276–287 [DOI] [PubMed] [Google Scholar]
- 30. Li J., Zhang L., Feng M., Zhang Z., Pan Y. (2009) Identification of the proteome composition occurring during the course of embryonic development of bees (Apis mellifera). Insect Mol. Biol. 18, 1–9 [DOI] [PubMed] [Google Scholar]
- 31. Li J., Fang Y., Zhang L., Begna D. (2011) Honey bee (Apis mellifera ligustica) drone embryo proteomes. J. Insect Physiol. 57, 372–384 [DOI] [PubMed] [Google Scholar]
- 32. Branca R. M. M., Orre L. M., Johansson H. J., Granholm V., Huss M., Perez-Bercoff A., Forshed J., Kall L., Lehtio J. (2013) HiRIEF LC-MS enables deep proteome coverage and unbiased proteogenomics. Nat. Methods advance online publication [DOI] [PubMed] [Google Scholar]
- 33. Abraham P., Giannone R. J., Adams R. M., Kalluri U., Tuskan G. A., Hettich R. L. (2013) Putting the pieces together: high-performance LC-MS/MS provides network-, pathway-, and protein-level perspectives in Populus. Mol. Cell. Proteomics 12, 106–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang L., Fang Y., Li R., Feng M., Han B., Zhou T., Li J. (2012) Towards posttranslational modification proteome of royal jelly. J. Proteomics 75, 5327–5341 [DOI] [PubMed] [Google Scholar]
- 35. Zhang J., Xin L., Shan B., Chen W., Xie M., Yuen D., Zhang W., Zhang Z., Lajoie G. A., Ma B. (2011) PEAKS DB: De Novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell. Proteomics 11, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Conesa A., Gotz S., Garcia-Gomez J. M., Terol J., Talon M., Robles M. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 [DOI] [PubMed] [Google Scholar]
- 37. Xie C., Mao X., Huang J., Ding Y., Wu J., Dong S., Kong L., Gao G., Li C., Wei L. (2011) KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, 316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brown K. R., Jurisica I. (2007) Unequal evolutionary conservation of human protein interactions in interologous networks. Genome Biol. 8, R95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brown K. R., Jurisica I. (2005) Online predicted human interaction database. Bioinformatics 21, 2076–2082 [DOI] [PubMed] [Google Scholar]
- 40. Eisen M. B., Spellman P. T., Brown P. O., Botstein D. (1998) Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. U.S.A. 95, 14863–14868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Saldanha A. J. (2004) Java Treeview–extensible visualization of microarray data. Bioinformatics 20, 3246–3248 [DOI] [PubMed] [Google Scholar]
- 42. Yu F., Mao F., Jianke L. (2010) Royal jelly proteome comparison between A. mellifera ligustica and A. cerana cerana. J. Proteome Res. 9, 2207–2215 [DOI] [PubMed] [Google Scholar]
- 43. Stromberg S., Bjorklund M. G., Asplund C., Skollermo A., Persson A., Wester K., Kampf C., Nilsson P., Andersson A. C., Uhlen M., Kononen J., Ponten F., Asplund A. (2007) A high-throughput strategy for protein profiling in cell microarrays using automated image analysis. Proteomics 7, 2142–2150 [DOI] [PubMed] [Google Scholar]
- 44. Li J., Zhang L., Feng M., Zhang Z., Pan Y. (2009) Identification of the proteome composition occurring during the course of embryonic development of bees (Apis mellifera). Insect Mol. Biol. 18, 1–9 [DOI] [PubMed] [Google Scholar]
- 45. Dearden P. K., Wilson M. J., Sablan L., Osborne P. W., Havler M., McNaughton E., Kimura K., Milshina N. V., Hasselmann M., Gempe T., Schioett M., Brown S. J., Elsik C. G., Holland P. W., Kadowaki T., Beye M. (2006) Patterns of conservation and change in honey bee developmental genes. Genome Res. 16, 1376–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Miura E., Kato Y., Matsushima R., Albrecht V., Laalami S., Sakamoto W. (2007) The balance between protein synthesis and degradation in chloroplasts determines leaf variegation in Arabidopsis yellow variegated mutants. Plant Cell 19, 1313–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hinault M. P., Ben-Zvi A., Goloubinoff P. (2006) Chaperones and proteases: cellular fold-controlling factors of proteins in neurodegenerative diseases and aging. J. Mol. Neurosci.: MN 30, 249–265 [DOI] [PubMed] [Google Scholar]
- 48. Eden E., Geva-Zatorsky N., Issaeva I., Cohen A., Dekel E., Danon T., Cohen L., Mayo A., Alon U. (2011) Proteome half-life dynamics in living human cells. Science 331, 764–768 [DOI] [PubMed] [Google Scholar]
- 49. Rothman S. (2010) How is the balance between protein synthesis and degradation achieved? Theor. Biol. Med. Model. 7, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mitchell P., Moyle J. (1967) Chemiosmotic hypothesis of oxidative phosphorylation. Nature 213, 137–139 [DOI] [PubMed] [Google Scholar]
- 51. Schultz B. E., Chan S. I. (2001) Structures and proton-pumping strategies of mitochondrial respiratory enzymes. Annu. Rev. Bioph. Biom. 30, 23–65 [DOI] [PubMed] [Google Scholar]
- 52. Curtiss J., Heilig J. S. (1995) Establishment of Drosophila imaginal precursor cells is controlled by the Arrowhead gene. Development 121, 3819–3828 [DOI] [PubMed] [Google Scholar]
- 53. Gala A., Fang Y., Woltedji D., Zhang L., Han B., Feng M., Li J. (2013) Changes of proteome and phosphoproteome trigger embryo-larva transition of honey bee worker (Apis mellifera ligustica). J. Proteomics 78, 428–446 [DOI] [PubMed] [Google Scholar]
- 54. De La Luz Sierra M., Yang F., Narazaki M., Salvucci O., Davis D., Yarchoan R., Zhang H. H., Fales H., Tosato G. (2004) Differential processing of stromal-derived factor-1alpha and stromal-derived factor-1beta explains functional diversity. Blood 103, 2452–2459 [DOI] [PubMed] [Google Scholar]
- 55. Sisson J. C., Field C., Ventura R., Royou A., Sullivan W. (2000) Lava lamp, a novel peripheral golgi protein, is required for Drosophila melanogaster cellularization. J. Cell Biol. 151, 905–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kurzik-Dumke U., Gundacker D., Renthrop M., Gateff E. (1995) Tumor suppression in Drosophila is causally related to the function of the lethal(2) tumorous imaginal discs gene, a dnaJ homolog. Dev. Gen. 16, 64–76 [DOI] [PubMed] [Google Scholar]
- 57. Keller R. (2006) Mechanisms of elongation in embryogenesis. Development 133, 2291–2302 [DOI] [PubMed] [Google Scholar]
- 58. van Vliet C., Thomas E. C., Merino-Trigo A., Teasdale R. D., Gleeson P. A. (2003) Intracellular sorting and transport of proteins. Prog. Biophys. Mol. Bio. 83, 1–45 [DOI] [PubMed] [Google Scholar]
- 59. Sun B., Wang W., Salvaterra P. M. (1998) Functional analysis and tissue-specific expression of Drosophila Na+,K+-ATPase subunits. J. Neurochem. 71, 142–151 [DOI] [PubMed] [Google Scholar]
- 60. Zheng A., Li J., Begna D., Fang Y., Feng M., Song F. (2011) Proteomic analysis of honey bee (Apis mellifera L.) pupae head development. PLoS One 6, e20428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hernandez L. G., Lu B., da Cruz G. C., Calabria L. K., Martins N. F., Togawa R., Espindola F. S., Yates J. R., Cunha R. B., de Sousa M. V. (2012) Worker honey bee brain proteome. J. Proteome Res. 11, 1485–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Woltedji D., Fang Y., Han B., Feng M., Li R., Lu X., Li J. (2013) Proteome analysis of hemolymph changes during the larval to pupal development stages of honey bee workers (Apis mellifera ligustica). J. Proteome Res. 12, 5189–5198 [DOI] [PubMed] [Google Scholar]
- 63. Feng M., Fang Y., Li J. (2009) Proteomic analysis of honey bee worker (Apis mellifera) hypopharyngeal gland development. BMC Genomics 10, 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jianke L., Mao F., Begna D., Yu F., Aijuan Z. (2010) Proteome comparison of hypopharyngeal gland development between Italian and royal jelly producing worker honey bees (Apis mellifera L.). J. Proteome Res. 9, 6578–6594 [DOI] [PubMed] [Google Scholar]
- 65. Feng M., Fang Y., Han B., Zhang L., Lu X., Li J. (2013) Novel aspects of understanding molecular working mechanisms of salivary glands of worker honey bees (Apis mellifera) investigated by proteomics and phosphoproteomics. J, Proteomics 87, 1–15 [DOI] [PubMed] [Google Scholar]
- 66. Fleig R., Sander K. (1986) Embryogenesis of the honey bee Apis mellifera L. (Hymenoptera: Apidea): a SEM study. Int. J. Insect Morphol. 15, 449–462 [Google Scholar]
- 67. Xinpei Y., Boxiong Z., Mengkui X., Guohua Y., Qilong C., Xiaofen T. (2005) Composition and changes of yolk proteins from silkworm Bombyx mori during embryonic development stages. Chin. J. Agri. Biotechnol. 2, 99–106 [Google Scholar]
- 68. Yamahama Y., Seno K., Hariyama T. (2008) Changes in lipid droplet localization during embryogenesis of the silkworm, Bombyx mori. Zoolog. Sci. 25, 580–586 [DOI] [PubMed] [Google Scholar]
- 69. Tram U., Riggs B., Sullivan W. (2001) Cleavage and Gastrulation in Drosophila Embryos. eLS, John Wiley & Sons, Ltd [Google Scholar]
- 70. Schmitzova J., Klaudiny J., Albert S., Schroder W., Schreckengost W., Hanes J., Judova J., Simuth J. (1998) A family of major royal jelly proteins of the honey bee Apis mellifera L. Cell Mol. Life Sci. 54, 1020–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Moritz R. F. A., Southwick E. E. (1992) Bees a superorganisms. An evolutionary reality, 1 Ed., Springer-Verlag, Berlin [Google Scholar]
- 72. Garcia L., Saraiva Garcia C. H., Calabria L. K., Costa Nunes da Cruz G., Sanchez Puentes A., Bao S. N., Fontes W., Ricart C. A., Salmen Espindola F., Valle de Sousa M. (2009) Proteomic analysis of honey bee brain upon ontogenetic and behavioral development. J. Proteome Res. 8, 1464–1473 [DOI] [PubMed] [Google Scholar]
- 73. Amdam G., Fennern E., Havukainen H. (2012) Vitellogenin in Honey Bee Behavior and Lifespan. In: Galizia C. G., Eisenhardt D., Giurfa M., eds. Honeybee Neurobiology and Behavior, pp. 17–29, Springer Netherlands [Google Scholar]
- 74. Bottke W., Burschyk M., Volmer J. (1988) On the origin of the yolk protein ferritin in snails. Roux's Arch Dev. Biol. 197, 377–382 [DOI] [PubMed] [Google Scholar]
- 75. Bhat K. M., Farkas G., Karch F., Gyurkovics H., Gausz J., Schedl P. (1996) The GAGA factor is required in the early Drosophila embryo not only for transcriptional regulation but also for nuclear division. Development 122, 1113–1124 [DOI] [PubMed] [Google Scholar]
- 76. Krupenko N. I., Dubard M. E., Strickland K. C., Moxley K. M., Oleinik N. V., Krupenko S. A. (2010) ALDH1L2 is the mitochondrial homolog of 10-formyltetrahydrofolate dehydrogenase. J. Biol. Chem. 285, 23056–23063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Scheffzek K., Kliche W., Wiesmuller L., Reinstein J. (1996) Crystal structure of the complex of UMP/CMP kinase from Dictyostelium discoideum and the bisubstrate inhibitor P1-(5′-adenosyl) P5-(5′-uridyl) pentaphosphate (UP5A) and Mg2+ at 2.2 A: implications for water-mediated specificity. Biochemistry 35, 9716–9727 [DOI] [PubMed] [Google Scholar]
- 78. Zhai B., Villen J., Beausoleil S. A., Mintseris J., Gygi S. P. (2008) Phosphoproteome analysis of Drosophila melanogaster embryos. J. Proteome Res. 7, 1675–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fotedar R., Diederich L., Fotedar A. (1996) Apoptosis and the cell cycle. Prog. Cell Cycle Res. 2, 147–163 [DOI] [PubMed] [Google Scholar]
- 80. Nakatsu F., Ohno H. (2003) Adaptor protein complexes as the key regulators of protein sorting in the post-Golgi network. Cell Struct. Funct. 28, 419–429 [DOI] [PubMed] [Google Scholar]
- 81. Alberts B., Johnson A., Lewis J., Raff M., Walter K. R. P. (2002) Molecular Biology of the Cell, Garland Science, New York [Google Scholar]
- 82. Stein D., Cho Y. S., Stevens L. M. (2013) Localized serine protease activity and the establishment of Drosophila embryonic dorsoventral polarity. Fly 7, 161–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Vertuani S., Angusti A., Manfredini S. (2004) The antioxidants and pro-antioxidants network: an overview. Curr. Pharm. Des. 10, 1677–1694 [DOI] [PubMed] [Google Scholar]
- 84. Seehuus S. C., Norberg K., Gimsa U., Krekling T., Amdam G. V. (2006) Reproductive protein protects functionally sterile honey bee workers from oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 103, 962–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wulfkuhle J. D., Petersen N. S., Otto J. J. (1998) Changes in the F-actin cytoskeleton during neurosensory bristle development in Drosophila: the role of singed and forked proteins. Cell Motil. Cytoskel. 40, 119–132 [DOI] [PubMed] [Google Scholar]
- 86. Llimargas M., Strigini M., Katidou M., Karagogeos D., Casanova J. (2004) Lachesin is a component of a septate junction-based mechanism that controls tube size and epithelial integrity in the Drosophila tracheal system. Development 131, 181–190 [DOI] [PubMed] [Google Scholar]
- 87. Yi P., Johnson A. N., Han Z., Wu J., Olson E. N. (2008) Heterotrimeric G proteins regulate a noncanonical function of septate junction proteins to maintain cardiac integrity in Drosophila. Dev. Cell 15, 704–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ahmad S. M., Tansey T. R., Busser B. W., Nolte M. T., Jeffries N., Gisselbrecht S. S., Rusan N. M., Michelson A. M. (2012) Two forkhead transcription factors regulate the division of cardiac progenitor cells by a Polo-dependent pathway. Dev. Cell 23, 97–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gao F. B., Bogert B. A. (2003) Genetic control of dendritic morphogenesis in Drosophila. Trends Neurosci. 26, 262–268 [DOI] [PubMed] [Google Scholar]
- 90. Kuhnlein R. P. (2012) Thematic review series: Lipid droplet synthesis and metabolism: from yeast to man. Lipid droplet-based storage fat metabolism in Drosophila. J. Lipid Res. 53, 1430–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Nelson J. (1915) The embryology of the honey bee, Kessinger Publishing, Whitefish, Montana [Google Scholar]
- 92. Herz J., Bock H. H. (2002) Lipoprotein receptors in the nervous system. Annu. Rev. Biochem. 71, 405–434 [DOI] [PubMed] [Google Scholar]
- 93. Lovegrove B., Simoes S., Rivas M. L., Sotillos S., Johnson K., Knust E., Jacinto A., Hombria J. C. (2006) Coordinated control of cell adhesion, polarity, and cytoskeleton underlies Hox-induced organogenesis in Drosophila. Curr. Biol. 16, 2206–2216 [DOI] [PubMed] [Google Scholar]
- 94. Enslee E. C., Riddiford L. M. (1981) Blastokinesis in embryos of the bug, Pyrrhocoris apterus. A light and electron microscopic study 1. Normal blastokinesis. J. Embryol. Exp. Morphol. 61, 35–49 [PubMed] [Google Scholar]
- 95. Heisenberg C. P. (2009) Dorsal closure in Drosophila: cells cannot get out of the tight spot. Bioessays 31, 1284–1287 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.