

Abstract
This work represents the first comprehensive quantitative analysis of global histone post-translational modifications (PTMs) from a virus infection, namely human cytomegalovirus (HCMV) infection. We used a nanoLC-MS/MS platform to identify and quantify the dynamic histone H3 and H4 PTMs expressed during HCMV replication in primary fibroblasts. Specifically, we examined the changes in histone PTMs over a 96 h time course to sample the immediate early (IE), early (E), and late (L) stages of viral infection. Several changes in histone H3 and H4 PTMs were observed, including a marked increase in H3K79me2 and H3K27me3K36me2, and a decrease in H4K16ac, highlighting likely epigenetic strategies of transcriptional activation and silencing during HCMV lytic infection. Heavy methyl-SILAC (hm-SILAC) was used to further confirm the histone methylation flux (especially for H3K79) during HCMV infection. We evaluated DOT1L (the H3K79 methyltransferase) mRNA levels in mock and HCMV-infected cells over a 96 h time course, and observed a significant increase in this methyltransferase as early as 24 hpi showing that viral infection up-regulates DOT1L expression, which drives H3K79me2. We then used shRNA to create a DOT1L knockdown cell population, and found that HCMV infection of the knockdown cells resulted in a 10-fold growth defect when compared with infected control cells not subjected to knockdown. This work documents multiple histone PTMs that occur in response to HCMV infection of fibroblasts, and provides a framework for evaluation of the role of epigenetic modifications in the virus-host interaction.
When viruses infect their hosts, they modulate the intracellular environment so that it is optimized to support the viral life cycle. Viruses encode factors, including proteins, noncoding RNAs, and microRNAs (miRNAs)1, which act immediately upon infection to regulate various processes including the cell cycle, host cell metabolism, nucleic acid synthesis, proliferation, and apoptosis, to name a few. In fact, many proteins and nucleic acids brought into the host cell with incoming viral particles act immediately upon viral entry to alter these cellular processes. Viruses have also engineered mechanisms to hijack host cell functions, such as nucleic acid synthesis machinery, and utilize them to their own advantage.
Human cytomegalovirus (HCMV) is a β-herpesvirus that contains a large, double-stranded DNA genome. When a human is first infected, HCMV actively replicates in many different cell types. The virus eventually spreads to CD34+ hematopoietic progenitor cells where it becomes quiescent and can remain in a latent state for the life of its host. With heightened stress or immunosuppression, however, HCMV can reactivate, reinitiating productive replication.
The DNA packaged within the capsid of a mature HCMV particle is naked, however upon infection the capsid-containing viral genome is transported through the cytoplasm to the nuclear pore, where the viral genome is released into the nucleus (1). Once in the nucleus, the viral genome becomes associated with host cell histones (2). HCMV encodes over 200 genes (3) that are transcribed in a highly coordinated cascade in productively infected cells (1). Immediate early (IE) genes are transcribed first. These genes are turned on within hours of infection, and do not require de novo protein expression. The IE proteins facilitate the transcription of early (E) genes, many of whose proteins are involved in viral DNA synthesis. Concomitant with replication of the viral genome, the late (L) genes are transcribed, and their proteins include structural components of the mature particle (1). Chromatin-modifying factors are instrumental in regulating this coordinated cascade of viral gene expression. Activating histone H3K9 and H3K14 acetylations are found at IE promoters as early as 3–6 h postinfection (hpi), prior to the acetylation at E or L promoters (4). Methylated H3K4, another activating mark, is incorporated into viral chromatin following its replication (5). Importantly, this study demonstrated selective epigenetic tagging of HCMV versus cellular chromatin. The HCMV IE1 and IE2 proteins influence chromatin modifications. IE1 interacts with the histone deactylase, HDAC3, to inhibit its activity, thereby aiding in transcriptional activation during lytic replication (6). IE2 similarly functions as a transactivator for viral genes in part through proteins that control histone function, such as the CAF1 histone chaperone complex (7) and the PCAF histone acetyltransferase (8). IE2 also functions to inhibit transcription during the late phase of infection through interaction with the histone deacetlyase, HDAC1, and the histone H3K9 methyltransferases, G9a and Suv(3–9)H1, generating repressive histone modifications (9). The major immediate-early promoter (MIEP), which controls expression of mRNAs encoding IE1 and IE2, is repressed in part through the binding of heterochromatin protein 1 (HP1) during infection of peripheral blood monocytes, a model for HCMV latency (10). Several other HCMV proteins interact with chromatin modifiers, such as the pUL29/28 interactions with the NuRD chromatin remodeling complex (11), thus underpinning the general importance of epigenetic regulation in the HCMV life cycle.
The goal of this study was to investigate the overall changes in histone post-translational modifications (PTMs) in response to HCMV infection of fibroblasts. We utilized an unbiased and comprehensive nano-liquid chromatography tandem mass spectrometry (nanoLC-MS/MS) workflow to quantitate histone H3 and H4 PTMs as well as the flux of the methylation PTMs over the course of the viral replication cycle. H3K79me2 was significantly up-regulated following infection and targeted knockdown of DOT1L, the only known methyltransferase for H3K79, decreased virus production. This study is the first quantitative analysis of global histone PTMs in response to HCMV lytic infection.
EXPERIMENTAL PROCEDURES
Cells and Viruses
Primary human embryonic lung fibroblasts (MRC5, passage 25–30) were maintained in Dulbecco's Modified Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 0.1 mm nonessential amino acids, 10 mm HEPES, 1 mm sodium pyruvate, 2 mm l-glutamine, and 100 U/ml each of penicillin and streptomycin. Cells were propagated at 37 °C in 5% CO2.
The clinical, BAC-derived HCMV isolate, TB40/E-BAC (clone #4; ref (12).), expressing the SV40-mCherry cassette (13) was utilized for HCMV infections. For MS analyses, cells were infected at a multiplicity of infection (moi) of 3.0 TCID50/cell. Herpes simplex virus-1 (HSV, strain KOS 1.1) (14) and adenovirus type 5 (Ad5) (15) were also used in this study.
DOT1L knockdown experiments employed pLKO.1puro lentiviral constructs expressing two distinct, validated shRNA sequences (Sigma, St. Louis, MO, NM-032482–180 and NM-032482–578; referred to in this study as KD1 and KD2, respectively). The pLKO.1puro Negative control shRNA (Sigma, SHC002) was used in this study (referred to herein as “scramble”). To generate lentivirus stocks, individual shuttle vectors were co-transfected into HEK 293T cells with the packaging vectors pΔ8.9 and pVSVg plasmid (16) in a 10:5:3 ratio in a total of 10 μg DNA using FuGENE6 (Roche, Indianapolis, IN) according to the manufacturer's instructions. Supernatants were collected at 24 and 48 h post-transfection, and used to transduce MRC5 cells (passage 20–24) in the presence of polybrene. At 24 h post-transduction, cells were washed of the polybrene and replenished with fresh medium. Transduced cells were then selected by the addition of puromycin.
Histone Extraction and LC-MS/MS
Histones were purified from MRC5 cells and processed for nanoLC-MS/MS analysis as previously described (17). Briefly, histones were recovered via acid extraction from nuclei and then derivatized with propionic anhydride, which appends a propionyl group to unmodified and monomethylated lysine residues. Subsequent enzymatic digestion using trypsin (1:10 weight, Promega, Madison, WI) produces Arg-C like histone peptides, and a second round of propionylation was carried out to label the N termini of the peptides.
Approximately 5 μg of histone peptides were loaded via autosampler (AS-2; Eksigent Technologies Inc., CA, USA) onto a 75 μm fused silica capillary column that was hand-packed with 130 mm of 5 μm, 200Å C18 (Michrom, Auburn, CA). Peptides were eluted over a 110 min 0.7–98% buffer B gradient (buffer A = 0.1 m acetic acid; buffer B = 95% acetonitrile in 0.1 m acetic acid) at a flow rate of ∼200 nL/min delivered by an HPLC pump (1200 series; Agilent, Santa Rosa, CA). The HPLC effluent was directly coupled to an LTQ-Orbitrap XL hybrid mass spectrometer (ThermoFisher Scientific, San Jose, CA) with a resolution of 30,000 for full MS followed by seven data-dependent and targeted MS/MS acquisitions.
Data Analysis
The nanoLC-MS/MS data sets were analyzed using an approach that has previously been described elsewhere (18, 19). Briefly, the optimization-based model Epiquant (http://puotl.technologypublisher.com/technology/8306) considers the MS isotopic distribution, MS/MS fragment ions and relative peptide hydrophobicity relationships to simultaneously identify and quantitate (based on integrated peak areas) all modified forms of the same histone peptide. All charge states for modified peptides were included in the analysis, and the abundance of isobaric co-eluting modified forms were deconvoluted based upon tandem MS data as previously described (18). The relative abundance of a histone PTM isoform is computed as the peak area for that isoform normalized by the sum of the peak areas for all PTM isoforms on the same histone peptide. We have previously shown this to be an accurate method for examining relative quantitative changes in PTMs between samples (19, 20), and in this study we will be investigating the fold-change of histone PTMs in HCMV-infected samples relative to mock-infected samples at different time points. Fixed modifications considered were propionylation of all unmodified and monomethylated lysine residues, and variable modifications consisted of acetylation and methylation (mono-, di-, and tri-) of lysine residues. A precursor tolerance of 10ppm was allowed with a fragment ion tolerance of 0.5Da. Missed cleavages were allowed in the search but not observed. All reported identifications were validated by manual inspection of the LC-MS/MS data (provided in supplementary Material).
Nucleic Acid and Protein Analyses
To quantify RNA levels of DOT1L, total cellular RNA was isolated using Tri-Reagent (Sigma). cDNA was generated using 1 μg of RNA with TaqMan Reverse Transcription Reagents (Roche), and equal volumes of cDNA was utilized for quantitative PCR (qPCR). The primers used to assess DOT1L are proprietary sequences from Sigma, used by the company to validate the sh-RNA constructs. Samples were analyzed in triplicate and normalized to cellular GAPDH levels, using the following primer set: FOR 5′-ACCCACTCCTCCACCTTTGAC-3′ and REV 5′-CTGTTGCTGTAGCCAAATTCGT-3′.
Quantification of extracellular viral DNA was described previously (21). The particle-to-infectious unit (IU) ratio was determined by quantification of extracellular viral DNA by qPCR, which was then divided by the infectious yield, as determined by a modified immunofluorescence assay (see below). Viral DNA was quantified using primers directed at UL123: FOR 5′-GCCTTCCCTAAGACCACCAAT-3′ and REV 5′-ATTTTCTGGGCATAAGCCATAATC-3′.
For analyses of protein expression, 2.5 × 105 cells per condition were infected and harvested at the indicated time points in the text. To assess the expression of viral proteins, cells were lysed in radioimmunoprecipitation assay (RIPA) buffer, and equal amounts of protein (assessed by Bradford assay) were used for immunoblot analyses. Primary antibodies used include anti-IE1 (clone 1B12, ref (22).), anti-pUL99 (clone 10B4–29, ref (23).), anti-UL83 (clone 8F5, ref (24).), and anti-UL82 (4F7, ref (25).), all diluted at 1:100, anti-pUL44 (Virusys, Taneytown, MD) diluted at 1:2500, and anti-tubulin (Sigma) diluted at 1:5000. For the analysis of modified histones, histones were purified as described above. Total histone protein was quantified by Bradford assay and between 1.25 μg and 10 μg were analyzed by immunoblot for total H3 (EMD Millipore, Billerica MA), diluted 1:25000 or H3K79me2 (Abcam, Cambridge, MA), diluted 1:1000. The secondary used in all cases was HRP-conjugated goat anti-mouse, diluted 1:10000 (Jackson Labs, West Grove, PA).
The detection of viral protein localization by immunofluorescence was described previously (13). In brief, 2.5 × 105 cells per condition were grown on coverslips and infected at a multiplicity of 0.5 TCID50/cell. Cells were fixed and stained at 72 hpi as described previously (13). Primary antibodies used were anti-pUL99 (clone 10B4–29, ref (23).), diluted 1:10 and anti-pUL55 (Abcam), diluted 1:100. The secondary used was Alexa Fluor 488 anti-mouse (Molecular Probes, Eugene, OR) and Hoechst (Sigma) was used to visualize the nuclei. Images were collected using a Zeiss LSM 510 confocal microscope.
Analyses of Viral Growth
Multi-step growth analyses of HCMV (moi = 0.01), HSV-1 (moi = 0.1), and adenovirus (moi = 5.0) was assessed by infecting 2.5 × 105 fibroblasts. Medium was collected at various times postinfection and used to infect fresh cultures of fibroblasts. Virus yield, expressed as infectious units/ml (IU/ml), was determined by fluorescence of a virus-specific protein, as described previously (21). Monoclonal antibodies used include HCMV anti-IE1 (clone 1B12, diluted 1:10, ref (22).), HSV-1 anti-ICP4 (26), and adenovirus anti-E2A (clone B6–8, diluted 1:75, ref (27)., a gift from Jane Flint). Nuclei were visualized with Hoechst (Sigma), and viral proteins were detected using the Alexa Fluor 488 anti-mouse secondary antibody (Molecular Probes).
RESULTS
Quantitative Mass Spectrometry Analysis of Histones H3 and H4 Modifications During the HCMV Replication Cycle
We utilized our unbiased nanoLC-MS/MS platform (Fig. 1) to identify and quantify the dynamic histone post-translational modifications (PTMs) induced during HCMV lytic infection in primary fibroblasts. Specifically, we examined the changes in histone modifications over a 96 h time course to generate dynamic snap shots corresponding to the immediate early (IE), early (E), and late (L) stages of viral infection. Infected and mock-infected control samples were harvested at 12, 24, 48, 72 and 96 h post infection (hpi). Several replicates were analyzed and quantitative analyses of more than 60 modified histone H3 (Fig. 2, supplemental Tables S1 and S2) and H4 PTM isoforms (Fig. 3, supplemental Tables S3 and S4) during the time course revealed significant up- and down-regulation of several histone PTMs. In this section, we present selected results on a peptide basis for histone PTMs that were observed to change significantly during HCMV infection relative to mock-infected samples.
Fig. 1.

Overview of the LC-MS/MS workflow for identifying and quantifying histone PTMs in HCMV-infected fibroblasts. Starting at the top left of the figure, fibroblasts are mock- or HCMV infected. After harvesting the fibroblasts, histones are extracted from chromatin fractions, chemically derivatized and digested using trypsin. The resulting histone peptides are resolved using reversed phase chromatography (nanoLC) and ionized for detection by a hybrid LTQ-Orbitrap tandem mass spectrometer. High-resolution MS1 scans provide precursor masses and intensities for each histone modification (illustrated here for the H3K9me2K14ac form), and tandem MS are collected for the MS1 peaks following data-dependent and targeted acquisition. Relative retention time, precursor mass accuracy and tandem MS spectra are used to identify each modified histone form. Extracted ion chromatograms for modified forms of the same histone peptide (e.g. the peptide containing H3K9K14 in this figure) produce chromatographic peaks, whose areas are relatively proportional to the abundance of each modified form in the sample.
Fig. 2.

Heatmap of fold changes in histone H3 PTMs over the course of the HCMV replication cycle. On the vertical axis are the different histone H3 PTMs quantified from the LC-MS data and on the horizontal axis are the various time points at which the HCMV-infected/mock-infected fibroblasts were harvested. The values correspond to log2 of the ratio of the relative PTM abundance (normalized to the sum of the abundance of all modified forms of the same histone peptide) of several infected sample replicates over the average of the mock-infected samples for each time point.
Fig. 3.

Heatmap of fold changes in histone H4 PTMs over the course of the HCMV replication cycle. On the vertical axis are the different histone H4 PTMs quantitated from the LC-MS data and on the horizontal axis are the various time points at which the HCMV-infected/mock-infected fibroblasts were harvested. The values correspond to log2 of the ratio of the relative PTM abundance (normalized to the sum of the abundance of all modified forms of the same histone peptide) of several infected sample replicates over the average of the mock-infected samples for each time point.
Altered Global Levels of Histone H3 PTMs
H3K9K14
For this peptide, we considered combinations of methylations (mono-, di- or tri-) or acetylations on K9 and K14. As presented in Fig. 4A, there is a uniform increase in H3K9me2K14ac and H3K9me3K14ac (K14ac containing forms are highlighted in red), whereas H3K9me2K14un and H3K9me3K14un (K14un containing forms are highlighted in blue), are generally unchanged during the time course. This suggests substrate specificity for H3K9me2/me3 methylation during infection. Specifically, H3K9me2/me3 increases only in combination with H3K14ac.
Fig. 4.
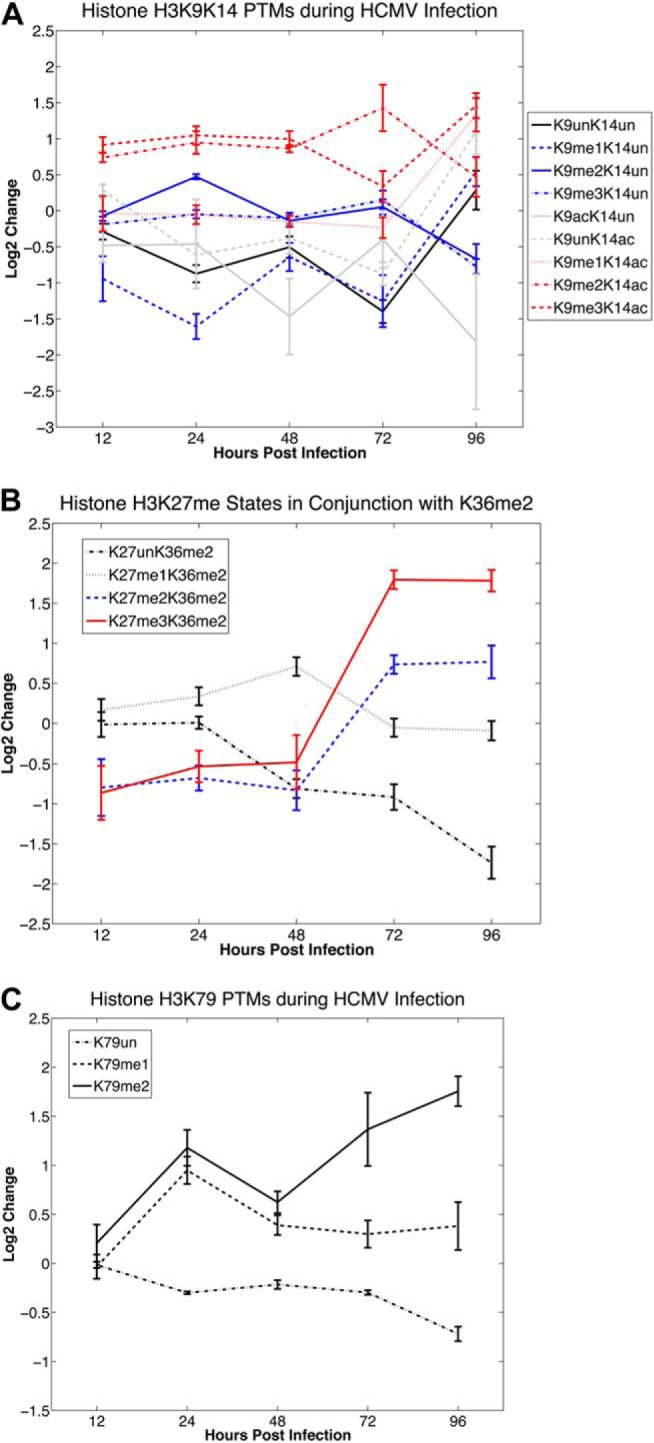
Time course of fold changes in histone H3 PTMs following HCMV infection. The values correspond to log2 of the average ratio of the relative PTM abundance (normalized to the sum of the abundance of all modified forms of the same histone peptide) for infected samples over the mock-infected samples for each time point. Error bars corresponding to ±2 standard deviations around the average values are included for each time point. A, Temporal changes in PTMs on the H3K9K14 peptide during HCMV infection relative to mock infection. B, Temporal changes in methylation on H3K27 in conjunction with H3K36me2 on the same peptide during HCMV infection relative to mock infection. C, Temporal changes in H3K79 methylation during HCMV infection relative to mock infection.
H3K27K36
Our analyses of this peptide included methylations (mono-, di- or tri-) or acetylations on K27 and methylations (mono- or di-) on K36 (Figs. 2 and 4B). There was substantial down-regulation of H3K27ac throughout the time course of HMCV infection (Fig. 2). However, the most striking changes we observed in the H3K27K36 peptides were for those containing a dimethylation mark on H3K36. H3K27unK36me2 significantly decreases during the infection time course, almost monotonically (Fig. 4B). In contrast, H3K27me3K36me2 and H3K27me2K36me2 are significantly down-regulated in the early stages (12 and 24 hpi) of infection, but are then significantly up-regulated during the late stage of the infection (72 and 96 hpi). These data suggest that substrates containing H3K27unK36me2 become progressively methylated on H3K27 as a function of time.
To further examine the increase in H3K27 methylation in conjunction with H3K36me2, we utilized heavy methyl-SILAC (hm-SILAC) (28, 29) to track the incorporation of new methylation PTMs on the H3K27K36 peptide. In this approach, 13CD3-methionine is introduced into methionine-depleted cell culture media and is metabolically converted into S-adenosyl methionine (SAM) with a heavy methyl donor group, which is distinguished from existing methylation marks based on the incurred mass shift (see example in Fig. 5A). Thus, we can track the real time incorporation of new methylation PTMs using mass spectrometry to calculate global methyltransferase activity for each histone substrate. In Fig. 5B, we present the results of hm-SILAC labeling for H3K27me3K36me2, where the different labeled states corresponding to this form are summarized by the number of heavy methyl groups incorporated (i.e. H3K27me3K36me2:#, where # corresponds to the number of heavy methyl groups on this modified form). The original H3K27me3K36me2 (H3K27me3K36me2:0) is substantially less in infected samples. The most significant incorporations of the heavy (or new) methylations are observed for H3K27me3K36me2:4, H3K27me3K36me2:3 and H3K27me3K36me2:2 in that order (over four-, 2.3-, and two-fold, respectively). Interestingly, the H3K27me3K36me2 forms containing 4 heavy methyl marks (H3K27me3K36me2:4 in Fig. 5B) is over four-fold higher in infected samples compared with mock. Upon examination of the tandem MS for H3K27me3K36me2:3, it is observed that this state primarily corresponds to 1 heavy methyl on K27 and 2 heavy methyls on K36. Similarly, inspection of the tandem MS for H3K27me3K36me2:4 reveals this state primarily corresponds to two heavy methyls on K27 and 2 heavy methyls on K36, and a less abundant state consisting of three heavy methyls on K27 and 1 heavy methyl on K36 (annotated tandem mass spectra provided in supplementary Material).
Fig. 5.
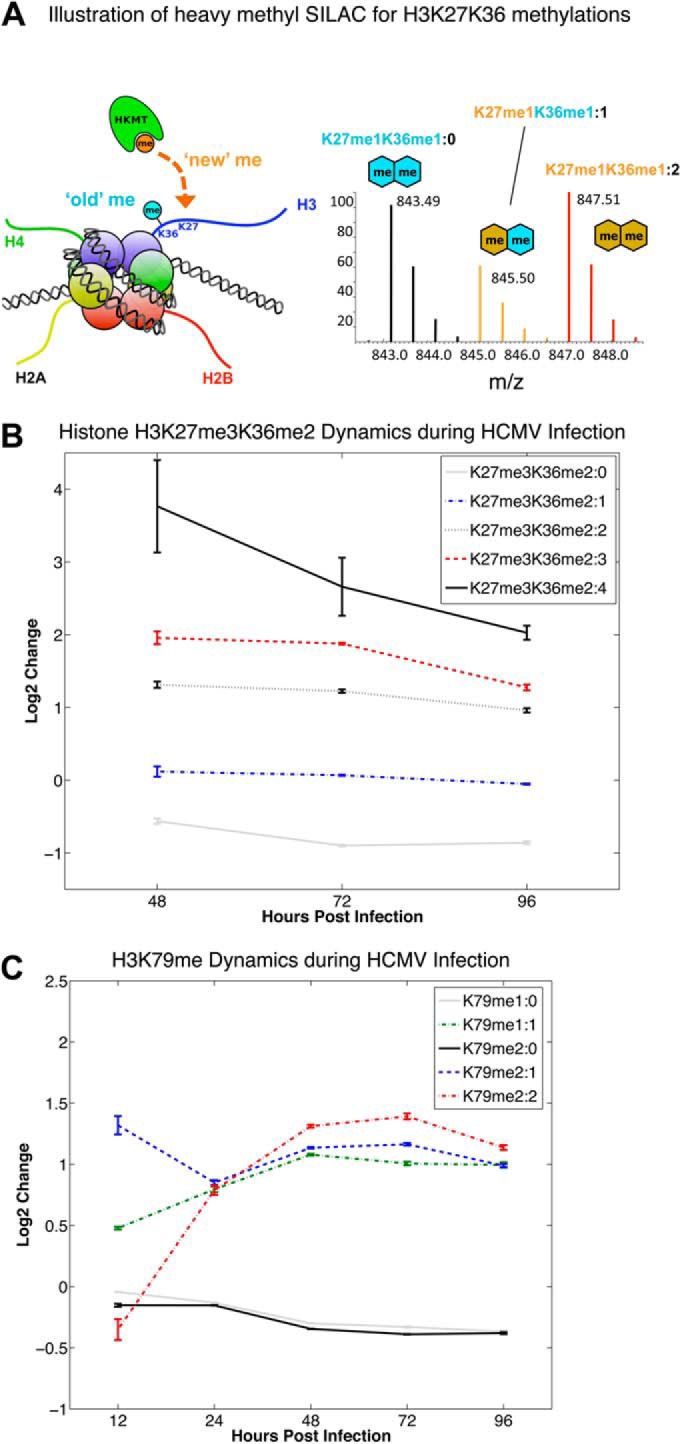
Time course of fold changes in the histone methylation flux for select histone H3 modified forms following HCMV infection. A, Illustration of the heavy methyl-SILAC (hm-SILAC) approach, where heavy methyl groups (13CD3) are dynamically transferred by HKMTs onto histones in cells and result in a mass shift of +4 Da from existing populations of the same modified histone form in the mass spectrum. H3K27me1K36me1 is presented as an example, where H3K27me1K36me1:0 denotes 0 heavy methylation PTMs (i.e. both methyl groups are ‘old’), H3K27me1K36me1:1 denotes 1 heavy methylation PTM, and H3K27me1K36me1:2 denotes both methylation PTMs are newly incorporated heavy methyl groups. In the case of 1 heavy methylation PTM, the tandem MS ions must be examined to confirm if it is on H3K27 or H3K36. The corresponding mass shifts are illustrated in a mass spectrum for the z = 2+ precursor ions. In B and C, the values correspond to log2 of the average ratio of the relative PTM abundance (normalized to the sum of the abundance of all isotopically labeled states of the same modified form) of infected samples over the mock-infected samples for each time point. Error bars corresponding to ± 2 standard deviations around the average values are included for each time point. B, Temporal changes in the incorporation of ‘new’ methylation PTMs on the H3K27me3K36me2 modified form during HCMV infection relative to mock infection. C, Temporal changes in the incorporation of “new” methylation PTMs on the H3K79me1 and H3K79me2 modified forms during HCMV infection relative to mock infection.
H3K79
We analyzed the H3K79 peptide, accounting for mono-, di- or tri-methylation of K79 and found that the methylation status of this lysine increased during HCMV infection (Fig. 4C). In particular, H3K79me2 increased significantly throughout the infection time course, reaching almost a fourfold increase at 96 hpi. We also observed an almost two-fold increase in H3K79me1 by 24 hpi, although this methylation state returned to nominal values for the remainder of the time course.
To evaluate whether the increase we observed in H3K79me2 (Fig. 4C) resulted from the addition of new methylation PTMs, we utilized the hm-SILAC technique (Fig. 5A) to distinguish existing versus new methyl groups as HCMV infection progresses. As shown in Fig. 5C, existing “old” H3K79me1/me2 (i.e. H3K79me1:0 and H3K79me2:0) do not increase over the course of infection. In contrast, newly added methylations, or fully labeled H3K79me1:1 and H3K79me2:2 exhibit a twofold increase in infected samples. Taken together, these data suggest that the increase in H3K79me during HCMV infection in fibroblasts is because of the addition of new methyl groups to this peptide.
Altered Global Levels of Histone H4 PTMs
H4K5K8K12K16
Our evaluation of H4K5K8K12K16 included combinatorial acetylations on K5, K8, K12 and K16. The temporal changes of these marks for infected over mock cells during the course of infection are presented in supplemental Fig. S1. We generally observed nominal values for H4 monoacetylation, with the notable exception of H4K16ac, which we found significantly decreased in infected samples across the time course we evaluated. Interestingly, we observed a significant difference between those peptides containing K16ac versus K16un (supplemental Fig. S1A). Consistent with our findings presented in supplemental Fig. S1A, we observed that all H4 diacetylated species containing H4K16ac generally decreased during infection (highlighted in black, supplemental Fig. S1B). In contrast, those H4 diacetyl forms not acetylated on H4K16 were generally more abundant, though we did not observe a significant up-regulation during infection. However, K5acK12ac significantly increased in the late stages of infection (72 and 96 hpi). We did not detect any significant changes in the H4–3ac (supplemental Fig. S1C) species during the course of infection, perhaps because of their low abundance and difficulty in quantitating.
H4K20
We examined the H4K20 peptide by considering mono-, di- and tri-methylation on K20. In contrast to many of the H3 modified forms, the H4K20 modified forms exhibit sharp transitions during the infection time course (Fig. 3 and supplemental Fig. S1D). In the early stages of infection (reaching an apex at 24 hpi), there is a general increase in H4K20me2 and H4K20me3, and these two modified states subsequently decrease to nominal values by later stages of infection (72 and 96 hpi). Conversely, the unmodified version of H4K20 (i.e.: H4K20un) is significantly down-regulated at 12 and 24 hpi in infected samples but then sharply increases to over two-fold at 48 hpi. By the end of the time course (i.e. 96 hpi) all H4K20 modified forms return to their approximate nominal values.
HCMV Infection Results in the Up-regulation of H3K79 and its Methyltransferase, DOT1L
To begin to elucidate the biological consequences of the virus-induced modifications, we focused on H3K79, as there is only one known methyltransferase for modifications at this site. H3K79me2 significantly increased following infection (Figs. 4C and 6A). Although we observed an increase in H3K79me1 at 24 hpi, this PTM returned to near-baseline values for the remainder of the time course (Fig. 4C). The increase of H3K79me2 was confirmed by purifying histones from mock or HCMV-infected fibroblasts and assessing the levels of this PTM by immunoblot analysis (Fig. 6B).
Fig. 6.
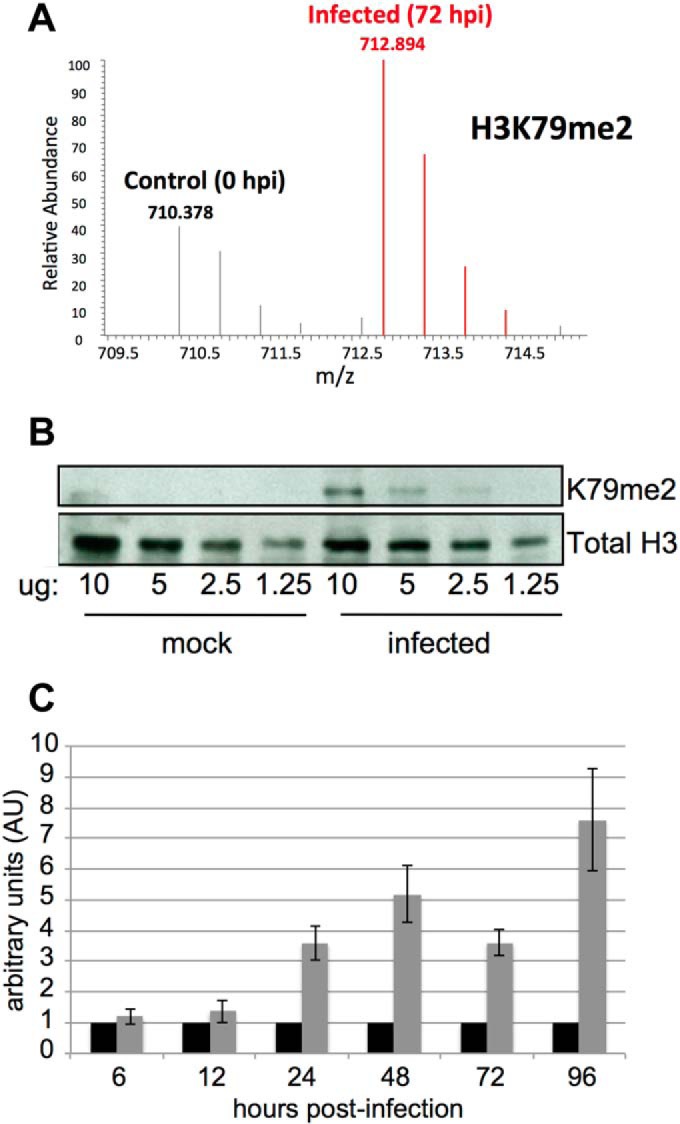
H3K79me2 and its methyltransferase DOT1L are up-regulated in response to HCMV infection. A, H3K79me2 is up-regulated by 72 hpi at a multiplicity of 3.0 TCID50/cell. Data is presented as fold change of infected fibroblasts as compared with mock-infected cells. B, Fibroblasts were mock or HCMV infected as in (A) and harvested at 72 hpi. Protein from purified histones were loaded at the indicated concentrations and probed with an antibody specific for H3K79me2. As a loading control, the samples were immunoblotted with an antibody directed at total H3 protein. C, The methyltransferase, DOT1L, specific for H3K79me2 methylation, is up-regulated in response to infection. Fibroblasts were infected as in (A) and total RNA was isolated at the indicated time points. DOT1L levels were measured by RT-qPCR. Each sample was analyzed in triplicate and in each case, normalized to cellular GAPDH.
Currently, DOT1L is the only methyltransferase known to modify H3K79, and thus we asked if DOT1L RNA levels increased during HCMV infection. By comparing DOT1L RNA levels in mock and HCMV-infected cells over a 96 h time course, we observed a significant increase in this methyltransferase as early as 24 hpi (Fig. 6C), concurrent with the spike in H3K79me2 PTM (Fig. 4C). Although we have not directly measured DOT1L protein, these findings are consistent with the interpretation that viral infection up-regulates DOT1L activity, which drives the H3K79me2 modification.
DOT1L shRNA-mediated Knockdown Reduces Herpesvirus Yield
To probe the necessity of increased DOT1L and H3K79me2, we generated MRC5 cells expressing DOT1L-specific shRNAs. We generated two independent populations, as well as a negative control population transduced with a lentiviral construct expressing a scrambled or nontargeting shRNA sequence. We observed an ∼70% reduction in DOT1L RNA levels in each cell population, MRC5 knockdown 1 (KD1) and MRC5 knockdown 2 (KD2), compared with the scrambled and nontargeting knockdown (Fig. 7A). HCMV infection of the DOT1L knockdown cells resulted in a nearly 10-fold growth defect starting at 8–12 dpi when compared with infected negative control cells (Fig. 7B; 8 dpi p value < 0.05, 12 dpi p value < 0.01 for each of the infected knockdown cells relative to the control). Importantly, this defect is not because of the production of defective viral particles in the knockdown cells, as the particle-to-IU ratio was similar for virus produced in the DOT1L shRNA knockdown as compared with control cells (supplemental Fig. S2A). To determine if the effect of DOT1L knockdown was specific to HCMV, we infected these cells with the α-herpesvirus, herpes simplex virus-1 (HSV-1). The production of HSV-1 in KD1 and KD2 cells was reduced at 24 hpi and more significantly at 48 hpi when compared with the infected negative control cells (Fig. 7C), although to a lesser extent than observed with HCMV. DOT1L knockdown did not affect the production of adenovirus progeny (Fig. 7D), demonstrating that its activity is not universally required for optimal replication of DNA viruses.
Fig. 7.

HCMV and HSV-1, but not adenovirus, generate less infectious extracellular progeny in DOT1L knockdown cells. A, DOT1L expression and H3k79me2 levels are decreased in response to shRNA knockdown in fibroblasts. Two independently generated DOT1L cell populations were generated by transducing lentiviruses expressing two separate shRNA sequences targeting DOT1L (KD1 and KD2). As a control, a separate cell population was generated using a lentivirus containing a scrambled sequence (scramble). RNA from the three cell populations was collected and analyzed for DOT1L expression by RT-qPCR. Each sample was analyzed in triplicate and in each case, normalized to cellular GAPDH. Each of the knockdown cell populations as well as the scramble knockdown population were infected with either (B) HCMV (multiplicity of 0.01 PFU/ml) (C) HSV-1 (multiplicity of 0.1 PFU/ml), and (D) adenovirus (multiplicity of 5.0 PFU/ml) and harvested at the indicated time points.
We next asked at what point during the viral life cycle DOT1L activity is required. We assessed representative viral proteins corresponding to each of the three gene subclasses in DOT1L knockdown and negative control infected cells (IE: IE1; E: pUL44; L: pUL99, pUL82 and pUL83) and found no difference in viral protein levels between the DOT1L knockdown and control cells (supplemental Fig. S2B and S2C). Immunofluorescent analysis confirmed that two late viral proteins, pUL99 and gB, localized normally to the perinuclear viral assembly compartment (30) (supplemental Fig. S2D). Although it is conceivable that analysis of a larger set of viral proteins could reveal an abnormality, our study of representative proteins, including “leaky late” (pUL82 and pUL83) and “true late” (pUL99) proteins, failed to detect a change in protein level and confirmed their normal localization to a virus-induced structure that is formed late in the replication cycle. Assuming all viral protein levels are normal, our data is consistent with the interpretation that the defect is very late in the infectious cycle, perhaps during assembly of the virus particle.
DISCUSSION
This is the first study to comprehensively analyze the global histone modification changes in response to HCMV infection. We observed significant changes in a variety of PTMs on both H3 and H4 histones. Additionally, we found the dynamics of certain combinatorial modifications, such as H3K27me3K36me2 (Fig. 4B), were altered by HCMV infection. This approach highlights the importance of considering combinations of histone modifications, as antibody-based methods would not capture the substrate specific responses observed here. Although the methods used here do not distinguish PTMs on viral versus cellular DNA, it is nevertheless clear that HCMV infection results in a profound change in the global modification pattern on H3 and H4 histones.
Discovering Correlative Changes in H3 and H4 PTMs
We applied clustering analysis to the data in Figs. 2 and 3 to identify any underlying correlative behavior in the fold changes of histone PTMs during the HCMV infection time course. The clustering of H3 PTM isoforms is presented in supplemental Fig. S3 and resulted in a broad classification of isoforms as either increasing or decreasing (left and right dendrograms in supplemental Fig. S3, respectively). Within this group of increasing histone H3 PTM isoforms, H3K79me1 and H3K79me2 (both substrates for DOT1L) are clustered together, as are hypermethylated H3K27K36 isoforms (namely, H3K27me3K36me(1/2) and H3K27me2K36me2). These correlative changes are further discussed below. Within the group of decreasing histone H3 PTM isoforms (right dendrogram in supplemental Fig. S3), we observe a very strong clustering of unmodified histone H3 substrates (i.e. H3K4un, K3K18unK23un, H3K27unK36un, and H3K79un), which is most likely because of the shutting down of histone synthesis upon cell cycle arrest during HCMV infection. Another highly correlated cluster in this group of decreasing H3 PTMs includes all H3K36 unmodified substrates; namely, H3K27me(1/2/3)K36un and H3K27acK36un. These changes, combined with the aforementioned observed increase in hypermethylated H3K27K36 isoforms, suggest aberrant methyltransferase activity on these histone H3 substrates. An intriguing trend observed from this analysis is the frequent clustering of isoforms differing by a single methyl mark. In particular, we observed clustering of H3K9me(2/3)K14ac, H3K79me(1/2), H3K27me(1/2/3)K36un, H3K9me(2/3)K14un, H3K27me(2/3)K36me2, and H3K27me3K36(me1/2). These correlative changes might point to sites of progressive methylation or demethylation as the result of HCMV infection.
The clustering of H4 PTM isoforms is presented in supplemental Fig. S4 and resulted in a broad classification of substrates based on the absence or presence of H4K16ac (left and right dendrograms in supplemental Fig. S4, respectively). Within the group of substrates lacking H4K16ac, H4K12ac was found to cluster with marks H4K20me2 and H4K20me3. Interestingly, within the group of histone H4 PTM substrates containing K16ac (right dendrogram in supplemental Fig. S4), these isoforms were further classified into highly correlated clusters with or without H4K5ac (i.e. H4K5ac and H4K5un, respectively). In general, this analysis reveals that all H4K16ac-containing isoforms are decreasing during the HCMV infection time course.
Epigenetic Regulation of HCMV Infection via DOT1L
To begin to dissect the implications of the HCMV-induced PTMs, we focused on H3K79me, as methylation on this substrate was significantly higher in HCMV-infected cells (Fig. 4C, supplemental Fig. S3). Unlike other histone H3 methylation sites (i.e. H3K4, H3K9, H3K27, H3K36), the H3K79 site has only one known methyltransferase, DOT1L, which can catalyze mono-, di- and trimethylation (31). Thus, manipulation of DOT1L presents a more lucrative target to investigate because of its lack of competitive and compensative methylation from other histone lysine methyltransferases (HKMTs). It is interesting to note from a drugability perspective that DOT1L is the only HKMT without a SET domain and bears more of a structural resemblance to protein arginine methyltransferases (PRMT) (32), although no arginine substrates for DOT1L have been reported.
DOT1L possess a variety of functions in mammalian cells, including telomeric silencing, transcriptional elongation and DNA repair (reviewed in (33)). The role of DOT1L in transcription has been verified by several ChIP-seq studies, which demonstrated that H3K79 methylation is consistently found within the bodies of transcribed genes in yeast, mouse, fly and human genomes. Furthermore, several DOT1L-containing multimeric protein complexes contain P-TEFb (34), which phosphorylates Ser2 on the CTD tail of RNA Pol II to facilitate transcription elongation (35). As H3K79 methylation levels do not change upon DNA damage (36), it is tempting to speculate that increased methylation at this site during HCMV infection reflects a role for DOT1L-mediated H3K79 methylation in the replication of the viral genome. Recent findings demonstrated the association of H3K79me2 with sites of DNA replication, and found that this PTM functions to regulate the timing of DNA replication at replication origins (37). Furthermore, H3K79me2 markedly increased after 24 hpi (Fig. 4C), coincident with the onset of viral DNA replication. Therefore inhibition of H3K79me2 by blocking DOT1L could function to alter the accurate timing and regulation of viral DNA synthesis.
Epigenetic Signatures Associated with Gene Silencing
We also observed several changes in histone PTMs that are associated with various mechanisms of transcriptional repression. For instance, we detected a substantial decrease in H4K16ac (supplemental Fig. S1 and S3), which is required for the release of RNA Pol II from promoter-proximal pausing and the transition from initiation to elongation (38). hMOF (KAT8) is to date the only known histone acetyltransferase specifically for this site (39, 40), and is part of both the nonspecific lethal (NSL) and male specific lethal (MSL) protein complexes (39, 41, 42). Interestingly, H4K20me3 blocks hMOF-dependent H4K16ac (38), and because we consistently observe a sharp increase in H4K20me3, it is attractive to hypothesize that this mechanism functions in our system as well (see supplemental Figs. S1A and S1D). Alternatively, an up-regulation in SIRT2, the only known specific histone deactylase (HDAC) for H4K16ac (43), could result in the global decrease in H4K16ac, but the effect of HCMV infection on SIRT2 activity is currently unknown. Interestingly, an earlier MS study of histone H4 in human cancer models observed an early global loss of H4K16ac and H4K20me3 (44), also implying that the marks have potentially important epigenetic roles in the host response to disease or infection.
We also observed a substantial increase in H3K27 methylation on substrates containing H3K36me2 during the late stages of viral infection (Figs. 4B and 5B). H3K36me2 is a mark commonly found throughout the body of actively transcribed genes (45). In contrast, H3K27me3 is a mark associated with silenced genes and is maintained by the Polycomb repressive complexes (the mark is “written” by Ezh2 of the PRC2 complex (46–48) and is “read” by the chromobox proteins, Cbx, of the PRC1 complex, which maintains silencing (49, 50)). From this data, one could hypothesize an infection-dependent Polycomb repressive mechanism for silencing actively transcribed genes (marked by H3K36me2) through the deposition of H3K27me3. However, the hmSILAC experiments revealed H3K27me3K36me2:3 (i.e. states containing 3 new methyl marks) and H3K27me3K36me2:4 (i.e. states containing 4 new methyl marks) both contain two new methylations on K36. This implies that the initial substrates for H3K27me3K36me2 are unmodified on K36 (i.e. H3K27me1K36un or H3K27me2K36un, respectively). Therefore, there appears a degree of substrate specificity and cross-talk between the methyltransferases and demethylases for K27 and K36. Lastly, we found that H3K27ac, a histone PTM shown to mark the enhancer elements of active genes (51), dramatically decreased throughout the entire time course (Fig. 2). Indeed, from the clustering analysis (supplemental Fig. S3) it was observed that H3K27me(1/2)K36un and H3K27acK36un are closely correlated within the group of decreasing H3 PTM isoforms. These repressive epigenetic marks could be indicative of the silencing of the host transcriptional program in response to the cell cycle arrest in G1/S that is induced by the virus.
Linking Transcriptomic Data to Histone PTMs
To compare our readouts for the changes in histone PTMs across the HCMV life cycle with the corresponding expression levels of chromatin-modifying enzymes, we analyzed existing microarray data from a study of HCMV-infected fibroblasts (52). In this study, the mRNA levels for over 12,600 genes were measured and normalized by taking the ratio of infected relative to mock for several time points up to 48 hpi. The difference call for each gene was evaluated at every time point, and transcripts determined not to significantly change during HCMV infection were removed from further analysis. We then focused our attention to those genes involved in chromatin modifying or remodeling processes, which resulted in a list of 76 relevant genes. The microarray data for these genes were clustered (53) to elucidate similar responses to viral infection, and the results are presented in supplemental Fig. S5.
Interestingly, of the subset of genes that are substantially up-regulated between 12 and 48 hpi in supplemental Fig. S5, two are Polycomb group (PcG) proteins: (1) Ezh2, the methyltransferase for H3K27, was up-regulated fivefold; and (2) hPc2 (Chromobox homolog 4, Cbx4), which contains an N-terminal chromodomain that preferentially binds H3K27me3 to maintain epigenetic silencing (54), was up-regulated 10-fold (this increase in Cbx4 expression was notably reported in the original microarray study). The increase in Ezh2 expression correlates with our proteomic observation that H3K27me3 also increases during infection (Fig. 4B). The concurrent increase in Cbx4 expression levels supports our hypothesis that a Polycomb repression mechanism is indeed activate during HCMV infection.
The histone acetyltransferase (HAT), p300, was also significantly up-regulated (in two independent transcripts) between 12 and 48 hpi (supplemental Fig. S5). This HAT is implicated in acetylating many protein (both histone and nonhistone) substrates, and it acetylates histones in positions H3K14 and H3K23 (55). This observation also correlates with our findings, as in Fig. 4A we observed H3K14ac, in conjunction with H3K9me2/me3, to generally increase during infection. These findings are consistent with an earlier ChIP-based study detailing an increase in H3K14ac on viral promoters and nonpromoter regions (4).
Transcripts that were significantly down-regulated in the microarray during HCMV infection notably include those in the SWI/SNF complex, such as BAF57 and hSNF2a (the ATP-dependent chromatin remodeling subunit), as well as histone variants H4 and H2A. The decrease in histone variant expression is not surprising given that the virus causes the host cells to arrest in G1/S, and the expression of these histone variants are replication-dependent. Unfortunately, microarray entries for SIRT2 (HDAC for H4K16ac) and Suv420H2 (HKMT for H4K20) were not available for comparison with our proteomic data.
Supplementary Material
Footnotes
Author contributions: C.O., P.A.D., T.S., and B.A.G. designed research; C.O. and P.A.D. performed research; C.O., P.A.D., T.S., and B.A.G. analyzed data; C.O., P.A.D., T.S., and B.A.G. wrote the paper.
* PAD would like to acknowledge funding from an NIH F32 NRSA Fellowship (F32GM093490) and Imperial College London. BAG acknowledges funding from an NIH Innovator grant (DP2OD007447) from the Office of the Director. CMO was supported by fellowships from the New Jersey Commission on Cancer Research (09-1960-CCR-EO) and the American Cancer Society (119028-PF-10−164-01-MPC). TS received funds from an NIH grant (AI87672).
 This article contains supplemental Figs. S1 to S5 and Tables S1 to S4.
This article contains supplemental Figs. S1 to S5 and Tables S1 to S4.
1 The abbreviations used are:
- miRNA
- microRNA
- CAF1
- chromatin assembly factor 1
- Cbx
- chromobox protein
- ChIP
- chromatin immunoprecipitation
- crs
- cis repression sequence
- DOT1L
- DOT1 (disruptor of telomeric silencing)-like histone H3 methyltransferase (S. cerevisiae) E – early
- EZH2
- enhancer of zeste homolog 2 (Drosophila)
- HAT
- histone acetyltransferase
- HDAC
- histone deacetylase
- HCMV
- human cytomegalovirus
- HKMTs
- histone lysine methyltransferases
- hMOF
- MOZ, YBF2/SAS3, SAS2 and TIP60 protein 1, KAT8
- hpi
- hours post infection
- HSV-1
- herpes simplex virus-1
- IE
- immediate early
- KAT
- K(lysine) acetyltransferase L – late
- LC-MS/MS
- liquid chromatography tandem mass spectrometry
- MIEP
- major immediate early promoter
- NuRD
- nucleosome remodeling and deacetylase complex, p300 - E1A-associated protein p300; KAT3B
- PCAF
- p300- and CBP-associated factor; KAT2B
- PTM
- post-translational modification
- PRC1/2
- polycomb repressor complex ½
- SET domain
- Su(var), Enhancer of zeste and Trithorax domain.
REFERENCES
- 1. Mocarski ESaC C.T. (2001) Cytomegaloviruses and their replication. In Fields Virology. Knipe DM, Howley P.M., Griffin D.E., Lamb R.A., Martin M.A., Roizman B., Straus S.E. (Eds.), vol. 2, 4 ed Philadelphia: Lippincott-Raven; pp. 2629–2673 [Google Scholar]
- 2. Nitzsche A., Paulus C., Nevels M. (2008) Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J. Virol. 82, 11167–11180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Murphy E., Yu D., Grimwood J., Schmutz J., Dickson M., Jarvis M. A., Hahn G., Nelson J. A., Myers R. M., Shenk T. E. (2003) Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. U.S.A. 100, 14976–14981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cuevas-Bennett C., Shenk T. (2008) Dynamic histone H3 acetylation and methylation at human cytomegalovirus promoters during replication in fibroblasts. J. Virol. 82, 9525–9536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nitzsche A., Steinhausser C., Mucke K., Paulus C., Nevels M. (2012) Histone H3 lysine 4 methylation marks postreplicative human cytomegalovirus chromatin. J. Virol. 86, 9817–9827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nevels M., Paulus C., Shenk T. (2004) Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc. Natl. Acad. Sci. U.S.A. 101, 17234–17239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee S. B., Lee C. F., Ou D. S., Dulal K., Chang L. H., Ma C. H., Huang C. F., Zhu H., Lin Y. S., Juan L. J. (2011) Host-viral effects of chromatin assembly factor 1 interaction with HCMV IE2. Cell Res. 21, 1230–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bryant L. A., Mixon P., Davidson M., Bannister A. J., Kouzarides T., Sinclair J. H. (2000) The human cytomegalovirus 86-kilodalton major immediate-early protein interacts physically and functionally with histone acetyltransferase P/CAF. J. Virol. 74, 7230–7237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reeves M., Murphy J., Greaves R., Fairley J., Brehm A., Sinclair J. (2006) Autorepression of the human cytomegalovirus major immediate-early promoter/enhancer at late times of infection is mediated by the recruitment of chromatin remodeling enzymes by IE86. J. Virol. 80, 9998–10009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Murphy J. C., Fischle W., Verdin E., Sinclair J. H. (2002) Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J. 21, 1112–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Terhune S. S., Moorman N. J., Cristea I. M., Savaryn J. P., Cuevas-Bennett C., Rout M. P., Chait B. T., Shenk T. (2010) Human cytomegalovirus UL29/28 protein interacts with components of the NuRD complex which promote accumulation of immediate-early RNA. PLoS Pathog. 6, e1000965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sinzger C., Hahn G., Digel M., Katona R., Sampaio K. L., Messerle M., Hengel H., Koszinowski U., Brune W., Adler B. (2008): Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. The J. Gen. Virol. 89, 359–368 [DOI] [PubMed] [Google Scholar]
- 13. O'Connor C. M., Shenk T. (2011) Human cytomegalovirus pUS27 G protein-coupled receptor homologue is required for efficient spread by the extracellular route but not for direct cell-to-cell spread. J. Virol. 85, 3700–3707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Macdonald S. J., Mostafa H. H., Morrison L. A., Davido D. J. (2012) Genome sequence of herpes simplex virus 1 strain KOS. J. Virol. 86, 6371–6372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jones N., Shenk T. (1978) Isolation of deletion and substitution mutants of adenovirus type 5. Cell 13, 181–188 [DOI] [PubMed] [Google Scholar]
- 16. Zufferey R., Nagy D., Mandel R. J., Naldini L., Trono D. (1997) Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol 15, 871–875 [DOI] [PubMed] [Google Scholar]
- 17. Leroy G., Chepelev I., Dimaggio P. A., Blanco M. A., Zee B. M., Zhao K., Garcia B. A. (2012) Proteogenomic characterization and mapping of nucleosomes decoded by Brd and HP1 proteins. Genome Biol. 2012, 13, R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. DiMaggio P. A., Jr., Young N. L., Baliban R. C., Garcia B. A., Floudas C. A. (2009) A mixed integer linear optimization framework for the identification and quantification of targeted post-translational modifications of highly modified proteins using multiplexed electron transfer dissociation tandem mass spectrometry. Mol. Cell. Proteomics 8, 2527–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu Y., DiMaggio P. A., Jr., Perlman D. H., Zakian V. A., Garcia B. A. (2013) Novel phosphorylation sites in the S. cerevisiae Cdc13 protein reveal new targets for telomere length regulation. J. Proteome Res. 12, 316–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leroy G., Dimaggio P. A., Chan E. Y., Zee B. M., Blanco M. A., Bryant B., Flaniken I. Z., Liu S., Kang Y., Trojer P., et al. (2013) A quantitative atlas of histone modification signatures from human cancer cells. Epigenetics Chromatin 6, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Terhune S, Torigoi E, Moorman N, Silva M, Qian Z, Shenk T, Yu D: (2007) Human cytomegalovirus UL38 protein blocks apoptosis. J. Virol. 81, 3109–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhu H, Shen Y, Shenk T: (1995) Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J. Virol. 69, 7960–7970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Silva M. C., Yu Q. C., Enquist L., Shenk T. (2003) Human cytomegalovirus UL99-encoded pp28 is required for the cytoplasmic envelopment of tegument-associated capsids. J. Virol. 77, 10594–10605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nowak B., Gmeiner A., Sarnow P., Levine A. J., Fleckenstein B. (1984) Physical mapping of human cytomegalovirus genes: identification of DNA sequences coding for a virion phosphoprotein of 71 kDa and a viral 65-kDa polypeptide. Virology 134, 91–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kalejta R. F., Bechtel J. T., Shenk T. (2003) Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol. Cell. Biol. 23, 1885–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Showalter S. D., Zweig M., Hampar B. (1981) Monoclonal antibodies to herpes simplex virus type 1 proteins, including the immediate-early protein ICP 4. Infect. Immun. 34, 684–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reich N. C., Sarnow P., Duprey E., Levine A. J. (1983) Monoclonal antibodies which recognize native and denatured forms of the adenovirus DNA-binding protein. Virology 128, 480–484 [DOI] [PubMed] [Google Scholar]
- 28. Ong S. E., Mittler G., Mann M. (2004) Identifying and quantifying in vivo methylation sites by heavy methyl SILAC. Nat. Methods 1, 119–126 [DOI] [PubMed] [Google Scholar]
- 29. Zee B. M., Levin R. S., Xu B., LeRoy G., Wingreen N. S., Garcia B. A. (2010) In vivo residue-specific histone methylation dynamics. J. Biol. Chem. 285, 3341–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sanchez V., Greis K. D., Sztul E., Britt W. J. (2000) Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J. Virol. 74, 975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frederiks F., Tzouros M., Oudgenoeg G., van Welsem T., Fornerod M., Krijgsveld J., van Leeuwen F. (2008) Nonprocessive methylation by Dot1 leads to functional redundancy of histone H3K79 methylation states. Nat. Struct. Mol. Biol. 15, 550–557 [DOI] [PubMed] [Google Scholar]
- 32. Campagna-Slater V., Mok M. W., Nguyen K. T., Feher M., Najmanovich R., Schapira M. (2011) Structural chemistry of the histone methyltransferases cofactor binding site. J. Chem. Inf. Model 51, 612–623 [DOI] [PubMed] [Google Scholar]
- 33. Nguyen A. T., Zhang Y. (2011) The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 25, 1345–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mueller D., Bach C., Zeisig D., Garcia-Cuellar M. P., Monroe S., Sreekumar A., Zhou R., Nesvizhskii A., Chinnaiyan A., Hess J. L., Slany R. K. (2007) A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 110, 4445–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marshall N. F., Price D. H. (1995) Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J. Biol. Chem. 270, 12335–12338 [DOI] [PubMed] [Google Scholar]
- 36. Huyen Y., Zgheib O., Ditullio R. A., Jr., Gorgoulis V. G., Zacharatos P., Petty T. J., Sheston E. A., Mellert H. S., Stavridi E. S., Halazonetis T. D. (2004) Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 432, 406–411 [DOI] [PubMed] [Google Scholar]
- 37. Fu H., Maunakea A. K., Martin M. M., Huang L., Zhang Y., Ryan M., Kim R., Lin C. M., Zhao K., Aladje M. I. (2013) Methylation of histone H3 on lysine 79 associates with a group of replication origins and helps limit DNA replication once per cell cycle. PLoS Genet. 9, e1003542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kapoor-Vazirani P., Kagey J. D., Vertino P. M. (2011) SUV420H2-mediated H4K20 trimethylation enforces RNA polymerase II promoter-proximal pausing by blocking hMOF-dependent H4K16 acetylation. Mol. Cell. Biol. 31, 1594–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Smith E. R., Cayrou C., Huang R., Lane W. S., Cote J., Lucchesi J. C. (2005) A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol. Cell. Biol. 25, 9175–9188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Taipale M., Rea S., Richter K., Vilar A., Lichter P., Imhof A., Akhtar A. (2005) hMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Mol. Cell. Biol. 25, 6798–6810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Horikoshi N., Kumar P., Sharma G. G., Chen M., Hunt C. R., Westover K., Chowdhury S., Pandita T. K. (2013) Genome-wide distribution of histone H4 Lysine 16 acetylation sites and their relationship to gene expression. Genome Integr. 4, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shogren-Knaak M., Ishii H., Sun J. M., Pazin M. J., Davie J. R., Peterson C. L. (2006) Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847 [DOI] [PubMed] [Google Scholar]
- 43. Vaquero A., Scher M. B., Lee D. H., Sutton A., Cheng H. L., Alt F. W., Serrano L., Sternglanz R., Reinberg D. (2006) SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 20, 1256–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fraga M. F., Ballestar E., Villar-Garea A., Boix-Chornet M., Espada J., Schotta G., Bonaldi T., Haydon C., Ropero S., Petrie K., Iyer N. G., Perez-Rosado A., Calvo E., Lopez J. A., Cano A., Calasanz M. J., Colomer D., Piris M. A., Ahn N., Imhof A., Caldas C., Jenuwein R., Esteller M. (2005) Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 37, 391–400 [DOI] [PubMed] [Google Scholar]
- 45. Wagner E. J., Carpenter P. B. (2012) Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 13, 115–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R. S., Zhang Y. (2002) Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298, 1039–1043 [DOI] [PubMed] [Google Scholar]
- 47. Czermin B., Melfi R., McCabe D., Seitz V., Imhof A., Pirrotta V. (2002) Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111, 185–196 [DOI] [PubMed] [Google Scholar]
- 48. Muller J., Hart C. M., Francis N. J., Vargas M. L., Sengupta A., Wild B., Miller E. L., O'Connor M. B., Kingston R. E., Simon J. A. (2002) Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111, 197–208 [DOI] [PubMed] [Google Scholar]
- 49. Levine S. S., Weiss A., Erdjument-Bromage H., Shao Z., Tempst P., Kingston R. E. (2002) The core of the polycomb repressive complex is compositionally and functionally conserved in flies and humans. Mol. Cell. Biol. 22, 6070–6078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Simon J. A., Kingston R. E. (2009) Mechanisms of polycomb gene silencing: knowns and unknowns. Nat. Rev. Mol. Cell Biol. 10, 697–708 [DOI] [PubMed] [Google Scholar]
- 51. Creyghton M. P., Cheng A. W., Welstead G. G., Kooistra T., Carey B. W., Steine E. J., Hanna J., Lodato M. A., Frampton G. M., Sharp P. A., Boyer L. A., Young R. A., Jaenisch R. (2010) Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 107, 21931–21936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Browne E. P., Wing B., Coleman D., Shenk T. (2001) Altered cellular mRNA levels in human cytomegalovirus-infected fibroblasts: viral block to the accumulation of antiviral mRNAs. J. Virol. 75, 12319–12330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. DiMaggio P. A., Jr., McAllister S. R., Floudas C. A., Feng X. J., Rabinowitz J. D., Rabitz H. A. (2008) Biclustering via optimal re-ordering of data matrices in systems biology: rigorous methods and comparative studies. BMC Bioinformatics 9, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ismail I. H., Gagne J. P., Caron M. C., McDonald D., Xu Z., Masson J. Y., Poirier G. G., Hendzel M. J. (2012) CBX4-mediated SUMO modification regulates BMI1 recruitment at sites of DNA damage. Nucleic Acids Res. 40, 5497–5510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Szerlong H. J., Prenni J. E., Nyborg J. K., Hansen J. C. (2010) Activator-dependent p300 acetylation of chromatin in vitro: enhancement of transcription by disruption of repressive nucleosome-nucleosome interactions. J. Biol. Chem. 285, 31954–31964 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.