

Abstract
Peptide spectrum matching is the current gold standard for protein identification via mass-spectrometry-based proteomics. Peptide spectrum matching compares experimental mass spectra against theoretical spectra generated from a protein sequence database to perform identification, but protein sequences not present in a database cannot be identified unless their sequences are in part conserved. The alternative approach, de novo sequencing, can make it possible to infer a peptide sequence directly from a mass spectrum, but interpreting long lists of peptide sequences resulting from large-scale experiments is not trivial. With this as motivation, PepExplorer was developed to use rigorous pattern recognition to assemble a list of homologue proteins using de novo sequencing data coupled to sequence alignment to allow biological interpretation of the data. PepExplorer can read the output of various widely adopted de novo sequencing tools and converge to a list of proteins with a global false-discovery rate. To this end, it employs a radial basis function neural network that considers precursor charge states, de novo sequencing scores, peptide lengths, and alignment scores to select similar protein candidates, from a target-decoy database, usually obtained from phylogenetically related species. Alignments are performed using a modified Smith–Waterman algorithm tailored for the task at hand. We verified the effectiveness of our approach using a reference set of identifications generated by ProLuCID when searching for Pyrococcus furiosus mass spectra on the corresponding NCBI RefSeq database. We then modified the sequence database by swapping amino acids until ProLuCID was no longer capable of identifying any proteins. By searching the mass spectra using PepExplorer on the modified database, we were able to recover most of the identifications at a 1% false-discovery rate. Finally, we employed PepExplorer to disclose a comprehensive proteomic assessment of the Bothrops jararaca plasma, a known biological source of natural inhibitors of snake toxins. PepExplorer is integrated into the PatternLab for Proteomics environment, which makes available various tools for downstream data analysis, including resources for quantitative and differential proteomics.
Very often, groundbreaking discoveries with a significant impact on the biotechnological and biomedical fields have emerged from studying “non-canonical” organisms. For example, the study of Thermus aquaticus allowed us to ultimately pave the way to modern molecular biology with the characterization of that organism's thermostable DNA polymerase (1). The characterization of the green fluorescent protein in Aequoria victoria led to a revolution in cellular biology and to a Nobel Prize being awarded to Osamu Shimomura, Martin Chalfie, and Roger Tsien. In Brazil, Sergio Ferreira's work on the venom of the Brazilian poisonous snake Bothrops jararaca enabled the development of the first angiotensin-converting enzyme inhibitor drug (Captopril) for the treatment of hypertension (2).
In scenarios such as these, proteomics has the potential to allow a better understanding of the complexity of biological systems and the process of evolution than the study of the genetic code alone. It enables the characterization of molecular processes according to their protein content, facilitating new discoveries. In proteomics, the most frequently used strategy for protein identification is so-called peptide spectrum matching (PSM),1 or the comparison of experimental mass spectra obtained by fragmenting peptides in a mass spectrometer to theoretical spectra generated from a sequence database. In general, the identification process follows from the sequence whose theoretical spectrum yields the highest matching score according to some empirical or probabilistic function. Examples of search engines adopting this strategy are SEQUEST (3), X!Tandem (4), and Mascot (5).
Back in the 1990s, establishment of a cutoff score for confident identification relied mostly on user experience; for example, given a specific charge state, Washburn et al. established cross-correlation and deltaCn cutoff values for SEQUEST in order to allow the selection of a subset of confident identifications from LCQ data. This has since been termed “the Washburn criterion.” In what followed, target-decoy databases were implemented to allow for more sophisticated refinements in filtering the data (6). In 2007, Elias and Gygi published a seminal paper on the target-decoy approach to shotgun proteomics (7) that ultimately firmed this approach as a standard and motivated the development of several statistical filters capable of converging to a list of confident identifications satisfying a user-specified false-discovery rate (FDR) with significantly more sensitivity than the conservative Washburn criterion. Such statistical filters include mixtures of probabilities (8), quadratic discriminant analysis (9), semi-supervised learning with support vector machines (10), and Bayesian logic (11) using a semi-labeled decoy analysis to account for overfitting (12). With so many advances, the PSM workflow has become the gold standard, as it is very sensitive and the least error-prone method when a database is available with the corresponding proteins. The latter factor limits the application of PSM to those organisms for which accurate sequence databases have been established. If a peptide's sequence is not contained within the sequence database, it cannot be identified via the PSM method. However, efforts in developing error-tolerant PSM approaches such as implemented in Mascot have made it possible to handle minor sequence modifications constrained by a simple set of rules. Nevertheless, increasing the search space in the PSM approach leads to decreased sensitivity (13).
Even though the concept of computer-aided de novo sequencing predates that of PSM (14), advances in the quality of mass spectrometry data and the power of computer hardware have allowed it to reemerge at the heart of a highly active field. De novo sequencing is unbiased insofar as it is not constrained by a sequence database, and it is therefore complementary to PSM. However, it has remained the most error prone of the two methods (15). The challenges of de novo sequencing notwithstanding, a few recent and notable improvements in computer-aided de novo analysis are PepNovo (16), which combines graph theory with machine learning; pNovo+ (17), which is optimized for high-resolution HCD data; NovoHMM (18), relying on hidden Markov models for increased sensitivity; and PEAKS (19), which creates a spectrum graph model by performing dynamic programming on the mass values regardless of the presence of an observed fragment ion. By considering the complementarities of different fragmentation strategies (e.g. collision induced dissociation, electron transfer dissociation (20), and electron capture dissociation (21)), computational proteomics scientists have also demonstrated significant advances in de novo accuracy (22). In particular, the Bandeira group has continually pushed the limits and redefined the notion of what de novo sequencing can do by introducing the spectral networks paradigm (23–25). Briefly, this strategy can assemble mass spectra into spectral pairs by joining overlapping spectra obtained from sample aliquots digested by different enzymes. As a consequence, it reduces noise and significantly improves protein coverage. Its latest version also combines data from different fragmentation techniques.
These algorithm developments have improved de novo sequencing, shifting the bottleneck to post-sequence processing of data. This is because the output of de novo software is a long list of highly similar full and partial peptide sequence and scores. An initial attempt to overcome these limitations consisted of a tag approach that was a hybrid of de novo sequencing and database searching: short sequence tags were derived from tandem mass spectra and used to search a sequence database (26). In what followed, a modified version based on the FASTA homology search tool was proposed for homology-driven proteomics (27). This strategy was implemented as part of the CIDentify tool, whose novelty was to account, in the alignment score, for limitations of mass spectrometry sequencing such as switching between leucine and isoleucine or other combinations of amino acids having the same mass. The next steps were taken mainly by the Shevchenko group through the introduction of the MS-Blast algorithm, which relies on a different set of scores and uses the PAM30MS substitution matrix, itself tailored for mass-spectrometry-based proteomics (28, 29). For a complete review of de novo sequencing and homology searching, we suggest Ref. 30.
The current de novo post-processing paradigm presents several limitations that are similar to those of the early PSM workflow. Output files generally consist of a peptide list with corresponding scores, demanding an experienced user to assess trustworthy identifications. If the same peptide is analyzed by different mass spectrometers, different scores might be generated, which makes data comparison between different groups a challenging task. In a sense, problems are similar to those encountered when adopting the early Washburn criterion. Assembling protein information from a list of peptides is not a simple task, and usually it is not performed using state-of-the-art de novo tools. Although there are great tools for doing this at the PSM level, there is still a lack of similar tools for de novo sequencing.
To tackle the aforementioned shortcomings, and in line with our strong interest in diversity-driven proteomics (29), we present a methodology for post-processing de novo sequencing data that allows inference of protein identification through statistical mapping of de novo sequencing results to a protein sequence database. Our approach begins with the use of Gotoh's version of the Smith–Waterman algorithm, based on affine gap scoring (31) for increased scalability, to align de novo sequences against those in a target-decoy database. Then a radial basis function neural network (RBF-NN) is used to rank results according to alignment score, de novo score, precursor charge state, and peptide length. Finally, a heuristic method is used to present protein identification results in a user-friendly, interactive report. The resulting algorithm was implemented as the software PepExplorer. In essence, its goal is somewhat similar to that of post-processing tools such as DTASelect (9), Percolator (10), and SEPro (11), but with an extra layer of complexity inherent from de novo sequencing. PepExplorer can handle the output of several widely adopted de novo tools, such as PepNovo, pNovo+, and PEAKS, and accepts a generic format to enable result analysis from a broader range of tools once results are run through simple parsers. Similarly, the software accepts a series of database formats for input analysis. These features are not found in other tools. PepExplorer is freely available to the scientific community and is provided with the necessary documentation.
The effectiveness of our methodology has been verified in two distinct scenarios, the first a real but controlled experiment and the other pertaining to comprehensive profiling of the plasma components of Bothrops jararaca, a venomous viper endemic to Brazil, southern Paraguay, and northern Argentina. The first scenario's purpose was to validate the effectiveness of the tool in analyzing a published Pyrococcus furiosus dataset (11). We note that this organism is recognized by the proteomics community as well suited for benchmarking, because it allows for the rigorous testing of identification algorithms at the peptide and protein levels (32, 33). We modified the P. furiosus sequence database in such a way that no more peptides were identified via the PSM approach or another widely adopted error-tolerant search tool, Mod-A (34). We then found that we could recover protein identifications using our tool. The B. jararaca scenario has allowed us to explore uncharted territory, as this organism has an incomplete sequence database and we were therefore required to rely on those of orthologous organisms. In particular, B. jararaca plasma was chosen because it is a main research model studied at the Laboratory of Toxinology (FIOCRUZ, Brazil), and several natural inhibitors of snake toxins have already been identified/characterized from this biological matrix (35–37).
MATERIALS AND METHODS
Bothrops jararaca Plasma Sample Preparation
B. jararaca plasma was supplied to the Laboratory of Toxinology (FIOCRUZ, Brazil) during the experimental procedures described in the research project, approved by the Ethics Committee of the Butantan Institute (555/2008), of the Biomedical Science Institute of the University of São Paulo (138/2009). This project was also approved by the Brazilian Institute for Environment and Renewable Natural Resources, a Brazilian Ministry of the Environment's enforcement agency (IBAMA, License 01/2009). Protein concentration was determined via bicinchoninic acid assay (38), and 40 μg were processed to complete dryness via lyophilization. Next, 20 μl of a 0.25% (m/v) RapiGest SF (Waters) in 50 mm ammonium bicarbonate solution were added to solubilize the proteins, which were then heated for 5 min at 100 °C. Disulfide bridges were reduced with 20 mm dithiothreitol for 30 min at 60 °C and subjected to cysteine alkylation with 68 mm iodoacetamide for 15 min at room temperature in the dark. Four microliters of a 0.2-μg/μl (in 50 mm acetic acid) porcine trypsin solution (catalog number V511, Promega) were added, and incubation proceeded for 22.5 h at 37 °C followed by 45 min at 56 °C. The reaction was stopped by the addition of 2.4 μl of 5% (v/v) trifluoroacetic acid in water. For RapiGest removal, samples were incubated for 45 min at 37 °C and centrifuged at 16,000 × g for 10 min at room temperature. The supernatant was collected and desalted/concentrated with in-house-made columns packed with Poros R2 resin (Invitrogen), eluted with 60% acetonitrile in 0.1% (v/v) trifluoroacetic acid, and completely dried using a SpeedVac (Thermo Scientific) vacuum centrifuge concentrator. Samples were resuspended in 30 μl of 1% (v/v) formic acid and submitted to a 10-min ultrasonic bath cycle before analysis via nano-LC-MS/MS.
Mass Spectrometry Analysis
The sample was analyzed in technical triplicate via LC-MS/MS. Tryptic digests were separated via reversed-phase capillary liquid chromatography coupled to nano-electrospray high-resolution mass spectrometry for identification. For each sample, 2 μl of desalted tryptic peptide digest were initially applied to a 2-cm-long (100-μm internal diameter) trap column packed with 5-μm, 200 Å Magic C18 AQ matrix (Michrom Bioresources) and then separated on a 30-cm-long (75-μm internal diameter) column that was packed with the same matrix, directly on a self-pack 15-μm PicoFrit empty column (New Objective). Chromatography was carried out on an EASY-nLC II instrument (Thermo Scientific). Samples were loaded onto the trap column at 2000 nL/min while chromatographic separation occurred at 200 nL/min. Mobile phase A consisted of 0.1% (v/v) formic acid in water, and mobile phase B consisted of 0.1% (v/v) formic acid in acetonitrile. Gradient conditions were as follows: 2% to 40% B during 162 min; up to 80% B in 4 min; and maintenance at this concentration for 2 min. Eluted peptides were directly introduced to the LTQ XL/Orbitrap mass spectrometer (Thermo, San Jose, CA) for analysis. The source voltage was set at 1.9 kV, the capillary temperature at 200 °C, and the tube lens voltage at 100 V. Fourier transform MS full and multi-stage MS automatic gain control target values were set at 500,000 and 200,000, respectively. MS1 spectra were acquired on the Orbitrap analyzer (300 to 1700 m/z) at a 60,000 resolution (for m/z 445.1200). We acquired tandem mass spectra from the 10 most intense ions by means of HCD fragmentation (minimum signal of 10,000 required; isolation width of 2.0; normalized collision energy of 45.0; and activation time of 30 s) followed by MS2 acquisition on the Orbitrap analyzer at 15,000 resolution. The dynamic exclusion option was enabled and set with the following values for each parameter: repeat count = 1; repeat duration = 30 s; exclusion list size = 500; exclusion duration = 45 s; and exclusion mass width = 10 ppm. Charge state rejection was enabled for unassigned charges and for those equal to 1.
Preparation of Sequence Databases Used for Similarity-driven Identification and PSM
Reference sequences for P. furiosus were obtained from UniProt, and those for Reptilia together with Amphibia are from the NCBI RefSeq; all were downloaded in June 2013. The sequences obtained from the Reptilia and Amphibia databases were merged into a single structure and then joined by 127 sequences of common mass spectrometry contaminants, as well as, for each database entry, a reversed version of the corresponding sequence (a decoy sequence). The final P. furiosus and Reptilia plus Amphibia databases had 4347 and 302,287 sequences, respectively.
Three P. furiosus proof-of-concept databases were generated by repeatedly adding “mutations” and insertions to the sequence database. These databases are referenced as PFU_Gap25_Substitution15, PFU_Gap20_Substitution10, and PFU_Gap15_Substitution8. In the PFU_Gap25_Substitution15 database, for example, an amino acid was inserted at every 25th position, and every 15th amino acid was replaced by some other, randomly chosen amino acid. These databases provide increasing distance from the original database and thereby presented the algorithms with different levels of difficulty. As no new proteins were added to obtain any of them, each of these databases has the same number of sequences as the initial P. furiosus database. Our goal has been to modify the native sequences from an organism's database to simulate the appearance of different, but phylogenetically close, organisms that would render PSM useless.
Peptide Spectrum Matches and Quality Assessment
The mass spectra were exported to the MS2 format (39) from the RAW files using PatternLab's RawReader module. The ProLuCID (40) search engine was used to compare experimentally obtained spectra against theoretical spectra generated from a sequence database and select the most similar. Briefly, the search was limited to fully tryptic peptide candidates, as we imposed only carbamidomethylation as a fixed modification. The search engine accepted peptide candidates within a 50-ppm tolerance from the measured precursor m/z and 550 ppm for the MS2, and we used XCorr and ZScore as the primary and secondary search engine scores, respectively.
The validity of the peptide spectrum matches was assessed using the Search Engine Processor (SEPro) (11). Identifications were grouped by charge state (≤+2 and >+2), resulting in two distinct subgroups. For each result, the ProLuCID XCorr, DeltaCN, and ZScore values were used to generate a Bayesian discriminator. The identifications were sorted in nondecreasing order according to the discriminator score. A cutoff score was established to accept an FDR of 1% based on the number of labeled decoys. This procedure was independently performed on each data subset, resulting in a false-positive rate that was independent of charge state. Additionally, an amino acid sequence at least six residues long was required. Results were post-processed to only accept matches with less than 5 ppm and proteins supported by at least two spectral counts. This last filter led to a 0% FDR in the search results.
De Novo Sequencing
De novo sequencing was performed using PEAKS Studio 6.0 (Bioinformatics Solutions Inc., ON, Canada). The parent mass error tolerance was 7 ppm, and the fragment mass error tolerance was 0.05 Da. Carbamidomethylation of cysteine was considered as a fixed modification. Acceptable results required an average local confidence score of at least 70% and a total local confidence score of at least 5 and were exported to a CSV file using the export option built into the software.
Blind Post-translational Modification Search with Mod-A
Mod-A was used to search the original and modified versions of the PFU dataset using its automatic precursor mass detection mode and allowing for arbitrary modifications in the peptides. The parameter files used by Mod-A are included in the supplemental material.
PepExplorer Algorithm
The PepExplorer algorithm was coded in C# 4.5. It has a graphical user interface but can also be executed from the command prompt, which enables it to work in cluster environments. The algorithm's workflow can be summarized in four steps: de novo result parsing, sequence alignment, result filtering, and result presentation (Fig. 1). Below we detail each of these steps. All parameters can be adjusted using the graphical user interface (Fig. 2).
Fig. 1.

A de novo tool is used to generate candidate sequences from mass spectra. The output from the de novo tool, together with a target-decoy database, serves as input to PepExplorer. PepExplorer uses the Smith–Waterman algorithm to align the de novo sequences against the target-decoy database (1). An RBF neural network is employed to rank the de novo alignments according to a confidence score that takes into account the de novo sequencing score, the alignment score, and the number of amino acids contained in the peptide (2). Finally, a dynamic report is generated (3).
Fig. 2.

The PepExplorer graphical user interface.
De Novo Result Parsing
PepExplorer currently contains parsers for three widely adopted de novo sequencing algorithms: PepNovo, pNovo+, and Peaks. PepExplorer treats the de novo algorithm with an abstraction layer that allows for the inclusion of new parsers upon request. The software also allows one to analyze a list of peptides by copying and pasting them in the corresponding text box found in the de novo output box (Fig. 2). However, in this scenario its neural network runs in a simplified mode and does not consider precursor charge states, scan numbers, or de novo score quality.
Sequence Alignment
PepExplorer relies on Gotoh's version of the Smith–Waterman algorithm (31), built into its core for aligning peptide sequences against a target-decoy sequence database specified by the user. The user can specify several alignment parameters, such as the open gap and extend gap penalties, the number of de novo sequence results to be considered per spectrum, and a substitution matrix of choice. For this study these values were 13, 5, 1, and the PAM30MS matrix, respectively.
These default open gap and extend gap parameters resulted from a grid search also made available in PepExplorer through the “Advanced Analysis” menu. This enables the algorithm to explore the landscape of combinations of these two parameters within user-predefined bounds and report the combination yielding the greatest number of alignments under a user-defined FDR. For this work, we performed the grid search in the PFU dataset allowing both the open gap and the extend gap penalties to vary from 2 to 30. The grid search results are available as part of the online supplementary files in the software's website.
Result Filtering with the RBF-NN
Each obtained sequence alignment is internally treated by PepExplorer as an alignment object containing the following properties: peptide length, de novo score, precursor charge, number of gaps, identifier, similarity, and alignment scores. All these parameters are used for result quality assessment, together with complementary information relevant to report assembly, such as scan number, raw file name, and details on the sequence alignment.
As a first step, these alignment objects are separated into two lists: those originating from peptide ions with charge state less than or equal to 2, and those from peptide ions with charge states greater than 2. Each of these lists is handled by a different RBF-NN. This enables convergence to a list of alignment objects satisfying an FDR that is independent of precursor charge state.
Given a list of alignment objects, the RBF-NN is based on defining six clusters; to this end, PepExplorer relies on the k-means++ algorithm (41) applied to the normalized values (i.e. between 0 and 1) of the alignment score, the de novo score, and the peptide length of each alignment object. We note that k-means++ employs a “careful seeding” to address the NP-hard problem of minimizing the average squared distance between points in the same cluster. The “careful seeding” is performed by choosing the first cluster center randomly from among the data points to be clustered. Subsequent cluster centers are chosen from locations coinciding with the remaining data points with probability proportional to each point's squared distance to the closest existing center. The algorithm then continues with the established k-means optimization procedure. The initial “careful seeding” is justified by the resultant faster convergence and better solutions. PepExplorer runs k-means++ 50 times in search of the best solution.
The RBF-NN is then trained to capture the nonlinear relationship between target and decoy alignment objects. The network we used was a single-hidden-layer feed-forward neural network whose three input nodes forwarded the input signal to the hidden nodes directly, with no weights. The kernel transfer function used in the jth hidden node was
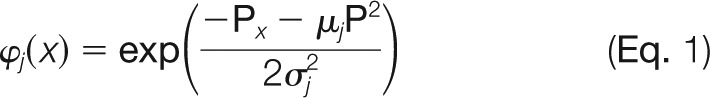 |
where μj is the jth cluster center determined by k-means++ and σj is a width parameter given by the smallest Euclidean distance between any two cluster centers. The latter is used to better capture the localness and thus the smoothness and continuity of the fitted function. The connections from the six hidden nodes to the single output node are weighted, and the value of the output node is given by
 |
where wj is the connection weight between the jth hidden node and the output node. During training, y(x) is either +1 or −1, depending respectively on whether the alignment object in question corresponds to a target sequence or a decoy sequence (alignment objects mapping to sections of sequences found in both target and decoy sequences are not considered). The weights of the RBF-NN equations are determined by means of linear regression using a least-squares objective function. All identifications are sorted in a nondecreasing order according to the classification function. Finally, a cutoff score can be established to achieve an FDR based on the decoy identifications.
Result Presentation
Results are presented in the form of a dynamic, interactive report that allows the user to sort them according to a criterion of choice and interact with the report by setting parameters of interest. The report can quickly adjust to a user-specified FDR or provide a list of maximum-parsimony alignments, as all alignments are stored to enable the algorithm to quickly converge to various settings. Among the threshold parameters we highlight the global FDR, the minimum alignment count (the closest to spectral count), the maximum alignment parsimony, the use of distinct RBF-NN for precursors of different charge states, and the minimum identifier. The report is provided as two interactive panels, the upper one being related to protein information and the lower to identification data. The upper panel provides information such as protein identifier, protein length, coverage percentage, sequence count, alignment count, and description. When a protein is selected, detailed information is made available in the lower panel of all alignments that mapped to it such as the scan number, file name, de novo score, precursor charge state, identifier, similarity, number of gaps, alignment score, sequence found in the database, and sequence provided by the de novo sequencing tool (Fig. 3). When a row of interest is selected in this lower panel, a new window displaying the sequence alignment is made available. In this window, when a row is selected in the upper panel with the protein information, a graphical coverage report is displayed (Fig. 4). This report is integrated with the cloud service of PatternLab for Proteomics (42), enabling the use of the Infer Domains function to instantly access predicted on-demand protein domains inferred with HMMER3 over Pfam-A (43).
Fig. 3.

Graphical user interface of the results browser. The results browser is composed of two panels. The upper panel displays information related to protein identification. When the user clicks on a protein of interest, further details on the peptides and their corresponding alignments are displayed in the lower panel.
Fig. 4.

Example of report provided by PepExplorer for each identified protein. A, the graphical report of the protein sequence coverage shows the extension of the area covered by predicted peptides. B, the result from the domain inferred by the cloud service running HMMER3 over Pfam-A on the fly is shown.
RESULTS
PFU Proof of Concept
A Venn diagram showing the overlap of the protein identifications from ProLuCID/SEPro, Mod-A, and PepExplorer on the unmodified PFU database is found in Fig. 5. We recall that only proteins having two or more spectral counts were considered.
Fig. 5.
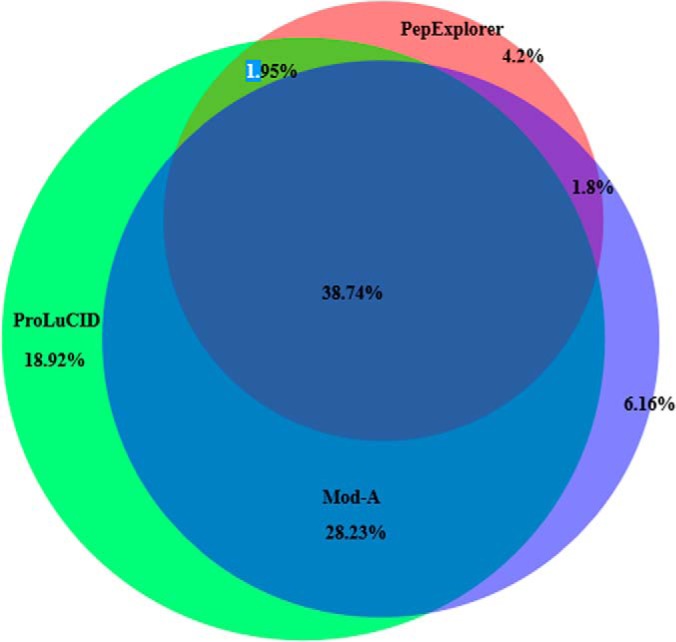
A Venn diagram comparing the protein identification overlap of ProLuCID, Mod-A, and PepExplorer in the PFU dataset using the unmodified database.
We further manually examined the non-decoy proteins uniquely identified by PepExplorer; because we individually analyzed each case, based on spectral quality, alignment scores, and coverage, we feel comfortable in considering them as correctly identified, even though they were not found in our gold-standard search, ProLuCID.
The results of the application of these tools to the modified versions of the PFU dataset are discriminated in Table I.
Table I. Performance of ProLuCID, Mod-A, and PepExplorer on the four PFU datasets. The first number represents the number of proteins having at least two spectral counts identified under a 1% FDR. The number in parentheses is the average sequence coverage.
| Number of proteins (Average Coverage) |
||||
|---|---|---|---|---|
| PFU | PFU_Gap_25_Substitution_15 | PFU_Gap_20_Substitution_10 | PFU_Gap_15_Substitution_8 | |
| ProLuCID | 585 (0.16) | 45 (0.04) | 7 (0.04) | 0 |
| Mod-A | 499 (0.16) | 63 (0.06) | 45 (0.05) | 0 |
| PepExplorer | 311 (0.17) | 190 (0.17) | 143 (0.17) | 102 (0.17) |
All results are made available as part of the supplemental material or at the PepExplorer website.
Bothrops jararaca Plasma Proteomic Assessment
PepExplorer generated 3862 alignments (1% FDR), corresponding to 1333 peptides mapping to 199 proteins arranged into 86 protein groups. The ProLuCID/SEPro pipeline identified 349 spectra corresponding to 83 peptides mapping to 17 proteins arranged into 12 protein groups (0% FDR). All protein groups identified by ProLuCID were present in the PepExplorer results. Moreover, all but five proteins identified by ProLuCID had their identifiers contained in the PepExplorer results. These five remaining identifications shared peptides or had at least 80% identity with one protein provided by PepExplorer. The detailed lists of identifications, SEPro files, and PepExplorer files are provided in the supplemental material.
A 100% overlap between our similarity-driven approach and a PSM approach might not occur because of the convergence strategy adopted by PepExplorer, as it will opt for proteins having greater numbers of alignment mappings to converge to a maximum-parsimony list. When we compared the average sequence coverages obtained for the same proteins identified by the PepExplorer and PSM approaches, we found an approximately 64% increase with the former approach (supplemental Table S1).
Recently, De Morais-Zani and co-workers (44) analyzed the plasma composition of juvenile and adult B. jararaca snakes seeking ontogenetic variability. They used an experimental strategy consisting of two-dimensional electrophoresis separation followed by mass spectrometry analysis and protein identification by PSM, using MASCOT as the search engine. The authors were able to report eight plasma protein groups, with one of them possibly due to sample contamination during collection (β-actin). With the exception of transferrin, all plasma proteins reported in that study were also detected in our PSM approach (ProLuCID/SEPro); furthermore, we were also able to identify other proteins such as fibronectin 1, α-2-macroglobulin, apolipoprotein B100, fibrinogen β chain, and small serum protein (supplemental Table S1). One possible explanation for our extended list of PSM identifications might be our experimental approach (shotgun proteomics) as opposed to theirs (two-dimensional electrophoresis).
Finally, when we compared the PepExplorer results (for proteins displaying a sequence count greater than two) we were able to identify all the plasma protein families mentioned above and additional ones, namely, γ phospholipase inhibitor type IV, plasminogen, ceruplasmin, IgG Fc-binding protein-like, complement C4-B-like, inter-α-trypsin inhibitor heavy chains H4- and H3-like, Ig λ light chain variable region, calnexin, multiple EGF-like domains protein 6, collagen α-1(XXIV) chain, kininogen-1, anionic trypsin-like, fibrinogen α chain-like, Ig γ-1 chain C region, and heparin cofactor 2 (supplemental Table S1). Supplemental Fig. S1 exemplifies peptide identifications provided by PepExplorer that were missed in the PSM approach.
DISCUSSION
Error-tolerant, similarity-driven tools have as an ultimate goal the listing of “true” identifications. However, defining what it means for an identification to be true is far from trivial, so in general one seeks to define trueness based on how the sequences in question differ according to some measure relating to the evolutionary distance between them. Among the first successful attempts at quantifying an evolutionary distance, we highlight the point accepted mutation (PAM) divergence, defined for two given sequences as the average number of accepted point mutations per 100 amino acids required in order to convert one sequence into the other without any insertions or deletions (45). The PAM matrices are substitution matrices that summarize an expected evolutionary change at the amino-acid level through log-odds substitution ratios. Theoretically, this approach is designed to compare sequences that are within a known evolutionary PAM divergence in evolution. Conversely, it is common experience that PAM matrices are, in general, very effective in finding “true alignments” that reflect biological phenomena even though PAM divergences do not always correspond to true evolutionary distances. In the experiment at hand, we chose one of the so-called low-order PAM matrices (e.g. PAM30MS), which theoretically should favor “closer” sequences and therefore such true alignments. Future versions and tools should incorporate strategies for automatically selecting substitution matrices tailored for the problem at hand. This could ultimately help in determining a subset of sequences for maximizing the sensitivity of the algorithm. We argue that the current version of PepExplorer helps by showing which peptides (and ultimately proteins) can be taken into consideration confidently enough. However, selecting an adequate substitution matrix remains an issue for the user's careful consideration.
The results provided herein can be used to compare three paradigms for performing spectral identification: PSM, an error-tolerant/blind post-translational modification approach, and a similarity-driven approach. The strategies are shown to be complementary, each having advantages and disadvantages. For example, the PSM approach was found to be the most sensitive one on the PFU dataset. This happened because we were dealing with a model organism, and thus fully (and tightly) relying on the restrictions provided in the sequence database would yield the best sensitivity. However, its performance rapidly degraded as more distractions and modifications were inserted into the database. Although Mod-A did not outperform PSM on the original PFU database, it was able to retain significantly more identifications as more distractions were inserted in the database. Mod-A most likely did not outperform the PSM approach on the original PFU dataset because the latter takes into account many more possibilities, resulting in a larger search space and sacrificing sensitivity (13). However, it would not be surprising if Mod-A outperformed PSM with higher organisms, as it will tolerate amino acid substitutions and unanticipated post-translational modifications. Indeed, taking into account multiple post-translational modifications can also quickly degrade the performance of de novo tools, and for this reason Mod-A will always provide results that are complementary to those of PepExplorer. Finally, PepExplorer presented the least sensitive results on the original PFU dataset, as de novo approaches are know to be error prone. However, the alignment paradigm is able to effectively retain the results as distractions are included in the database.
Finally, we would like to point out some potential applications of PepExplorer. Our algorithm is used to pinpoint a subset of de novo results that are similar to the database at hand. Yet there can be several de novo results, having a very high de novo sequencing score, that are not included in the PepExplorer output. These results should be given special attention: what PepExplorer discards could actually turn out to be truly novel molecules, given the high confidence of the de novo results.
CONCLUDNG REMARKS
PepExplorer is recommended for large-scale shotgun proteomic experiments, that is, those in which a considerable number of spectra are generated, as in the datasets presented. Its use is not recommended for analyzing small collections of spectra such as those obtained when analyzing a two-dimensional gel spot. In such cases MS-BLAST (28) should be used instead.
A key realization brought about by modern biotechnology has been that underneath the myriad unknown organisms lies great potential (46). Current strategies inspired by this realization include exploring extreme biomes for so-called extremophiles, a peculiar class of organisms that are generally responsible for the biosynthesis of molecular components useful for pharmaceutical or industrial applications. Perhaps one of the best examples has been the discovery of Thermus aquaticus and its heat-resistant polymerases, elected by Science in 1989 as the “molecules of the year” (47) and which have since aided in the development of biotechnology tools and ultimately facilitated the engineering of more effective drugs. The molecular characterization of venoms has also resulted in the engineering of new drugs (48). In conclusion, the literature is full of examples demonstrating the vast richness of biomolecular components and drug candidates that are naturally produced by different organisms already existing in our fauna and flora.
Recent advances in proteomic technologies are significantly impacting similarity-driven proteomics and, consequently, the exploration of novel organisms. Recently, Coon and coworkers benchmarked a new hybrid mass spectrometer, the Orbitrap Fusion (Thermo). The authors mention events in which they identified up to 19 sequences within less than a second, enabling them to achieve 90% coverage of the yeast proteome in one hour (49). Through this, the authors have raised the bar, in terms of the number of proteins identified per minute, to 70. High scanning rates coupled with ever-increasing resolving power are ingredients to boost the performance of de novo sequencing algorithms. As the general quality of predicted peptides is increasing, we foresee de novo sequencing playing a key role in the efficient handling of data from organisms with no available genomic information.
The field of genomics is also constantly going through significant advances. For example, next-generation sequencers are enabling the single-cell transcriptome (50) and personal genomics (51). Indeed, the coupling of “omics” sciences such as proteomics and metabolomics with next-generation sequencers will pave the way to true systems biology approaches, as these strategies are complementary to each other. The ever-growing amount of data on sequenced organisms, powered by next-generation sequencers, adds to similarity-driven approaches, as even more organisms will have their genomic information available. However, instrument time, expertise in data analysis, and financial resources are current bottlenecks for many groups.
Here we described a new methodology for dealing with de novo sequencing approaches, taking into account rigorous statistical criteria. We clearly demonstrated its efficiency in a controlled but real experiment with the PFU modified database and then presented the most comprehensive proteomic profile of B. jararaca plasma. Efforts such as the present work are necessary, as they expand the possibilities of what can be achieved in proteomics and in the study of organism biology. In the near future we plan to automate the integration of data between different strategies like PSM and de novo, aiming at a wider perspective for mass-spectral analyses.
Availability of PepExplorer, the Raw Data, and Results
PepExplorer and supplementary files, including the B. jararaca raw data and all the results described in this work, are made freely available for academic purposes at our website. In order to view the full PSM results, installation of SEPro is required. PepExplorer is required for viewing results.
Supplementary Material
Footnotes
Author contributions: F.V.L., R.H.V., M.J., and P.C.C. designed research; F.V.L., R.H.V., D.L.B., J.P., R.M., J.R.Y., V.C.B., M.J., and P.C.C. performed research; F.V.L., R.H.V., J.P., J.R.Y., and P.C.C. contributed new reagents or analytic tools; F.V.L., R.H.V., D.L.B., J.P., R.M., V.C.B., M.J., and P.C.C. analyzed data; F.V.L., R.H.V., V.C.B., and P.C.C. wrote the paper; R.M. extensively tested the software; M.J. provided key insights making this research possible.
* We acknowledge CNPq, FAPERJ, CAPES (Grant No. 063/2010 - Edital Toxinologia), and PDTIS for financial support and use of core facilities. J.R.Y. acknowledges support from NIH Grant Nos. P41 GM103533 and R01 MH067880.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- PSM
- peptide spectrum matching
- FDR
- false-discovery rate
- RBF-NN
- radial basis function neural network
- SEPro
- Search Engine Processor
- PAM
- point accepted mutation.
REFERENCES
- 1. Saiki R. K., Gelfand D. H., Stoffel S., Scharf S. J., Higuchi R., Horn G. T., Mullis K. B., Erlich H. A. (1988) Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239, 487–491 [DOI] [PubMed] [Google Scholar]
- 2. Opie L. H., Kowolik H. (1995) The discovery of captopril: from large animals to small molecules. Cardiovasc. Res. 30, 18–25 [PubMed] [Google Scholar]
- 3. Eng J. K., McCormack A. L., Yates J. R. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 4. Craig R., Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformat. 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 5. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 6. Moore R. E., Young M. K., Lee T. D. (2002) Qscore: an algorithm for evaluating SEQUEST database search results. J. Am. Soc. Mass Spectrom. 13, 378–386 [DOI] [PubMed] [Google Scholar]
- 7. Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 8. Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 9. Cociorva D., Tabb D., Yates J. R. (2007) Validation of tandem mass spectrometry database search results using DTASelect. Curr. Protoc. Bioinformat. Chapter 13, Unit 13.4 [DOI] [PubMed] [Google Scholar]
- 10. Käll L., Canterbury J. D., Weston J., Noble W. S., MacCoss M. J. (2007) Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 [DOI] [PubMed] [Google Scholar]
- 11. Carvalho P. C., Fischer J. S. G., Xu T., Cociorva D., Balbuena T. S., Valente R. H., Perales J., Yates J. R., 3rd, Barbosa V. C. (2012) Search engine processor: filtering and organizing peptide spectrum matches. Proteomics 12, 944–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barboza R., Cociorva D., Xu T., Barbosa V. C., Perales J., Valente R. H., França F. M. G., Yates J. R., 3rd, Carvalho P. C. (2011) Can the false-discovery rate be misleading? Proteomics 11, 4105–4108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Borges D., Perez-Riverol Y., Nogueira F. C. S., Domont G. B., Noda J., da Veiga Leprevost F., Besada V., França F. M. G., Barbosa V. C., Sánchez A., Carvalho P. C. (2013) Effectively addressing complex proteomic search spaces with peptide spectrum matching. Bioinformat. 29, 1343–1344 [DOI] [PubMed] [Google Scholar]
- 14. Biemann K., Cone C., Webster B. R., Arsenault G. P. (1966) Determination of the amino acid sequence in oligopeptides by computer interpretation of their high-resolution mass spectra. J. Am. Chem. Soc. 88, 5598–5606 [DOI] [PubMed] [Google Scholar]
- 15. Pevtsov S., Fedulova I., Mirzaei H., Buck C., Zhang X. (2006) Performance evaluation of existing de novo sequencing algorithms. J. Proteome Res. 5, 3018–3028 [DOI] [PubMed] [Google Scholar]
- 16. Frank A., Pevzner P. (2005) PepNovo: de novo peptide sequencing via probabilistic network modeling. Anal. Chem. 77, 964–973 [DOI] [PubMed] [Google Scholar]
- 17. Chi H., Sun R.-X., Yang B., Song C.-Q., Wang L.-H., Liu C., Fu Y., Yuan Z.-F., Wang H.-P., He S.-M., Dong M.-Q. (2010) pNovo: de novo peptide sequencing and identification using HCD spectra. J. Proteome Res. 9, 2713–2724 [DOI] [PubMed] [Google Scholar]
- 18. Fischer B., Roth V., Roos F., Grossmann J., Baginsky S., Widmayer P., Gruissem W., Buhmann J. M. (2005) NovoHMM: a hidden Markov model for de novo peptide sequencing. Anal. Chem. 77, 7265–7273 [DOI] [PubMed] [Google Scholar]
- 19. Ma B., Zhang K., Hendrie C., Liang C., Li M., Doherty-Kirby A., Lajoie G. (2003) PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 17, 2337–2342 [DOI] [PubMed] [Google Scholar]
- 20. Coon J. J., Ueberheide B., Syka J. E. P., Dryhurst D. D., Ausio J., Shabanowitz J., Hunt D. F. (2005) Protein identification using sequential ion/ion reactions and tandem mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 102, 9463–9468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zubarev R. A., Horn D. M., Fridriksson E. K., Kelleher N. L., Kruger N. A., Lewis M. A., Carpenter B. K., McLafferty F. W. (2000) Electron capture dissociation for structural characterization of multiply charged protein cations. Anal. Chem. 72, 563–573 [DOI] [PubMed] [Google Scholar]
- 22. Zubarev R. A., Zubarev A. R., Savitski M. M. (2008) Electron capture/transfer versus collisionally activated/induced dissociations: solo or duet? J. Am. Soc. Mass Spectrom. 19, 753–761 [DOI] [PubMed] [Google Scholar]
- 23. Bandeira N. (2007) Spectral networks: a new approach to de novo discovery of protein sequences and posttranslational modifications. Biotechniques 42, 687, 689,, 691 passim [DOI] [PubMed] [Google Scholar]
- 24. Guthals A., Bandeira N. (2012) Peptide identification by tandem mass spectrometry with alternate fragmentation modes. Mol. Cell. Proteomics 11, 550–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guthals A., Clauser K. R., Bandeira N. (2012) Shotgun protein sequencing with meta-contig assembly. Mol. Cell. Proteomics 11, 1084–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mann M., Wilm M. (1994) Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal. Chem. 66, 4390–4399 [DOI] [PubMed] [Google Scholar]
- 27. Taylor J. A., Johnson R. S. (1997) Sequence database searches via de novo peptide sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 11, 1067–1075 [DOI] [PubMed] [Google Scholar]
- 28. Shevchenko A., Sunyaev S., Loboda A., Shevchenko A., Bork P., Ens W., Standing K. G. (2001) Charting the proteomes of organisms with unsequenced genomes by MALDI-quadrupole time-of-flight mass spectrometry and BLAST homology searching. Anal. Chem. 73, 1917–1926 [DOI] [PubMed] [Google Scholar]
- 29. Junqueira M., Carvalho P. C. (2012) Tools and challenges for diversity-driven proteomics in Brazil. Proteomics 12, 2601–2606 [DOI] [PubMed] [Google Scholar]
- 30. Ma B., Johnson R. (2012) De novo sequencing and homology searching. Mol. Cell. Proteomics 11, O111.014902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gotoh O. (1982) An improved algorithm for matching biological sequences. J. Mol. Biol. 162, 705–708 [DOI] [PubMed] [Google Scholar]
- 32. Vaudel M., Burkhart J. M., Breiter D., Zahedi R. P., Sickmann A., Martens L. (2012) A complex standard for protein identification, designed by evolution. J. Proteome Res. 11, 5065–5071 [DOI] [PubMed] [Google Scholar]
- 33. Yates J. R., 3rd, Park S. K. R., Delahunty C. M., Xu T., Savas J. N., Cociorva D., Carvalho P. C. (2012) Toward objective evaluation of proteomic algorithms. Nat. Methods 9, 455–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Na S., Bandeira N., Paek E. (2012) Fast multi-blind modification search through tandem mass spectrometry. Mol. Cell. Proteomics 11, M111.010199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Estevão-Costa M. I., Rocha B. C., de Alvarenga Mudado M., Redondo R., Franco G. R., Fortes-Dias C. L. (2008) Prospection, structural analysis and phylogenetic relationships of endogenous gamma-phospholipase A(2) inhibitors in Brazilian Bothrops snakes (Viperidae, Crotalinae). Toxicon 52, 122–129 [DOI] [PubMed] [Google Scholar]
- 36. Tanaka-Azevedo A. M., Tanaka A. S., Sano-Martins I. S. (2003) A new blood coagulation inhibitor from the snake Bothrops jararaca plasma: isolation and characterization. Biochem. Biophys. Res. Commun. 308, 706–712 [DOI] [PubMed] [Google Scholar]
- 37. Valente R. H., Dragulev B., Perales J., Fox J. W., Domont G. B. (2001) BJ46a, a snake venom metalloproteinase inhibitor. Isolation, characterization, cloning and insights into its mechanism of action. Eur. J. Biochem. FEBS 268, 3042–3052 [DOI] [PubMed] [Google Scholar]
- 38. Smith P. K., Krohn R. I., Hermanson G. T., Mallia A. K., Gartner F. H., Provenzano M. D., Fujimoto E. K., Goeke N. M., Olson B. J., Klenk D. C. (1985) Measurement of protein using bicinchoninic acid. Anal. Biochem. 150, 76–85 [DOI] [PubMed] [Google Scholar]
- 39. McDonald W. H., Tabb D. L., Sadygov R. G., MacCoss M. J., Venable J., Graumann J., Johnson J. R., Cociorva D., Yates J. R., 3rd (2004) MS1, MS2, and SQT-three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identifications. Rapid Commun. Mass Spectrom. 18, 2162–2168 [DOI] [PubMed] [Google Scholar]
- 40. Xu T., Venable J. D., Park S., Cociorva D., Lu B., Liao L., Wohlschlegel J., Hewel J., Yates J. R. (2006) ProLuCID, a fast and sensitive tandem mass spectra-based protein identification program. Mol. Cell. Proteomics 5, S174 [Google Scholar]
- 41. Arthur D., Vassilvitskii S. (2007) in Proceedings of the Eighteenth Annual ACM-SIAM Symposium on Discrete Algorithms, SODA ‘07, pp. 1027–1035, Society for Industrial and Applied Mathematics, Philadelphia, PA [Google Scholar]
- 42. Leprevost F. V., Lima D. B., Crestani J., Perez-Riverol Y., Zanchin N., Barbosa V. C., Carvalho P. C. (2013) Pinpointing differentially expressed domains in complex protein mixtures with the cloud service of PatternLab for Proteomics. J. Proteomics 89, 179–182 [DOI] [PubMed] [Google Scholar]
- 43. Eddy S. R. (2009) A new generation of homology search tools based on probabilistic inference. Genome Inform. Int. Conf. Genome Inform. 23, 205–211 [PubMed] [Google Scholar]
- 44. De Morais-Zani K., Grego K. F., Tanaka A. S., Tanaka-Azevedo A. M. (2013) Proteomic analysis of the ontogenetic variability in plasma composition of juvenile and adult Bothrops jararaca snakes. Int. J. Proteomics 2013, 135709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dayhoff M. O. (1979) Atlas of Protein Sequence and Structure, National Biomedical Research Foundation, Washington, D.C [Google Scholar]
- 46. Brock T. D. (1997) The value of basic research: discovery of Thermus aquaticus and other extreme thermophiles. Genetics 146, 1207–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guyer R. L., Koshland D. E., Jr. (1989) The Molecule of the Year. Science 246, 1543–1546 [DOI] [PubMed] [Google Scholar]
- 48. Fox J. W., Serrano S. M. T. (2007) Approaching the golden age of natural product pharmaceuticals from venom libraries: an overview of toxins and toxin-derivatives currently involved in therapeutic or diagnostic applications. Curr. Pharm. Des. 13, 2927–2934 [DOI] [PubMed] [Google Scholar]
- 49. Hebert A. S., Richards A. L., Bailey D. J., Ulbrich A., Coughlin E. E., Westphall M. S., Coon J. J. (2014) The one hour yeast proteome. Mol. Cell. Proteomics 13, 339–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tang F., Barbacioru C., Wang Y., Nordman E., Lee C., Xu N., Wang X., Bodeau J., Tuch B. B., Siddiqui A., Lao K., Surani M. A. (2009) mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 [DOI] [PubMed] [Google Scholar]
- 51. Vernot B., Stergachis A. B., Maurano M. T., Vierstra J., Neph S., Thurman R. E., Stamatoyannopoulos J. A., Akey J. M. (2012) Personal and population genomics of human regulatory variation. Genome Res. 22, 1689–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}