

Abstract
This review outlines our search for the mechanism causing the early loss of islet sympathetic nerves in autoimmune diabetes. Since our previous work has documented the importance of autonomic stimulation of glucagon secretion during hypoglycaemia, the loss of these nerves may contribute to the known impairment of this specific glucagon response early in human type 1 diabetes. We therefore briefly review the contribution that autonomic activation, and sympathetic neural activation in particular, makes to the subsequent glucagon response to hypoglycaemia. We also detail evidence that animal models of autoimmune diabetes mimic both the early loss of islet sympathetic nerves and the impaired glucagon response seen in human type 1 diabetes. Using data from these animal models, we examine mechanisms by which this loss of islet nerves could occur. We provide evidence that it is not due to diabetic hyperglycaemia, but it is related to the lymphocytic infiltration of the islet. Ablating the p75 neurotrophin receptor, which is present on sympathetic axons, prevents eSIN, but, interestingly, not diabetes. Thus, we appear to have separated the immune-related loss of islet sympathetic nerves from the immune-mediated destruction of islet β-cells. Finally, we speculate on a way to restore the sympathetic innervation of the islet.
Keywords: Sympathetic, Neuropathy, P75 neurotrophin receptor, Hypoglycaemia, Type 1 diabetes, Glucagon
CLINICAL RELEVANCE: HYPOGLYCAEMIA
Marked hypoglycaemia is rare in non-diabetic subjects but increasingly common in type 1 diabetic patients who undergo intensive insulin treatment. Because hypoglycaemia is aversive, it decreases adherence to the intensive insulin therapy needed to avoid the long-term complications of this disease. In addition, marked hypoglycaemia, particularly if it is prolonged, can cause coma culminating in death. Therefore, it is important, first, to understand the mechanisms that prevent hypoglycaemia in non-diabetic individuals, and second, to identify defects in these mechanisms in diabetic subjects.
Early studies on the counterregulatory response to hypoglycaemia established the importance of glucagon in limiting both the severity and the duration of insulin-induced hypoglycaemia. The mechanisms mediating the glucagon response to hypoglycaemia have been the subject of intense study. Two factors have achieved major scientific recognition: disinhibition of the α–cell via suppression of the β-cell [1] and direct stimulation of the α–cell via activation of the autonomic nervous system [2]. Our early work has concentrated on this latter area.
AUTONOMIC STIMULATION OF GLUCAGON SECRETION: NON-DIABETIC
In 1989 [3] we first hypothesized, based on sparse literature, that the activation of the autonomic nervous system, that occurs when the brain becomes neuroglucopenic, makes a significant contribution to the subsequent glucagon response. Later studies in my laboratory, and those of Dr. Peter Havel and Dr. Bo Ahren, accumulated evidence strongly supporting this hypothesis. The first study, in dogs, showed that the surgical or pharmacological blockade of all three autonomic inputs to the islet (parasympathetic, sympathetic and adrenal medullary) impaired 75–90% of the glucagon response to marked hypoglycaemia [4]. Subsequent studies by Dr. Peter Havel in the more common laboratory animals, rats [5] and mice [6], showed similar results. Dr. Peter Havel extended this concept to primates [7] and, with Dr. Bo Ahren, to humans [8]. In 2012 [2] we summarized evidence showing that there is sequential recruitment of each arm of the autonomic nervous system as hypoglycaemia deepens. Suppression of the β-cell is maximal at the bottom of the mild hypoglycaemic range. Thus “switch-off” of the β-cell makes its major contribution to the glucagon response when hypoglycaemia is mild (70 mg/dl). In contrast, the autonomic nervous system makes its major contribution to the glucagon response when hypoglycaemia is either moderate (50 mg/dl) or marked (25 mg/dl).
Role of sympathetic nerves
Accumulated evidence suggests that islet sympathetic nerves help mediate the glucagon response to insulin-induced hypoglycaemia. For example, older studies have demonstrated that electrical activation of pancreatic sympathetic nerves potently stimulates glucagon secretion [9,10]. Later it was demonstrated that this specific neural pathway is activated during hypoglycaemia [11]. This activation is stress-specific: neither hypoxia nor hypotension activate pancreatic sympathetic nerves [11]. Further studies demonstrated that this hypoglycaemia-specific activation helps mediate the subsequent glucagon response, at least when the other autonomic inputs are ablated [12].
HUMAN TYPE 1 DIABETES
Impaired glucagon response
While there are sophisticated and redundant mechanisms that usually prevent hypoglycaemia in non-diabetic individuals, hypoglycaemia is unfortunately too common in insulin-treated type 1 diabetic patients. A breakthrough in understanding why occurred in 1973 with the demonstration that the glucagon response to insulin-induced hypoglycaemia is severely impaired in type 1 diabetes [13]. Although later studies demonstrated that intensive insulin treatment causing repeated hypoglycaemia can also impair this glucagon response [14], such aggressive treatment only became common after the Diabetes Control and Complications Trial (DCCT) established in the 1990s that chronic hyperglycaemia accelerates the long-term complications of this disease [15]. Thus this early demonstration of an impaired glucagon response to insulin-induced hypoglycaemia was likely due to either the lack of disinhibition by the β-cell [16] or a previously unknown autonomic defect present in type 1 diabetes [17]. Whatever the mechanism, an impaired glucagon response to hypoglycaemia increases both its severity (depth) and duration [18].
Autonomic impairment
Although much work has been done on the contribution of β-cell loss to this glucagon impairment [16, 19], two factors led us to examine the possibility that an autonomic defect also contributes. First, in non-diabetic subjects, the β-cell is nearly turned off by the time glucose reaches mild hypoglycaemia (70 mg/dl) [20], yet most of the glucagon response occurs when glucose falls further. Thus, the large glucagon response seen as glucose reaches moderate (50 mg/dl) or marked hypoglycaemia (25 mg/dl) is due to non-β–cell mediators, likely progressive autonomic stimulation of the α–cell. Second, although available data suggest no impairment of either the adrenal medullary [21] or parasympathetic branch [21] of the autonomic nervous system early in type 1 diabetes, the integrity of the sympathetic input to the islet was unknown. Thus if an autonomic defect was present in type 1 diabetes, it was likely in the sympathetic-islet pathway. Therefore, we hypothesized [17] that a defect in this pathway contributes to the impairment of the glucagon response to insulin-induced hypoglycaemia seen early in type 1 diabetes.
Early sympathetic islet neuropathy (eSIN)
The first place we looked for such a defect was in the sympathetic innervation of the islet itself. When we examined the sympathetic nerves in pancreatic autopsy specimens from humans with prior type 1 diabetes, we found a paucity of islet sympathetic nerves [22]. To be sure that this failure to detect islet sympathetic nerves was not just due to the over-fixation common in autopsy specimens, we required positive identification of sympathetic nerves in adjacent blood vessels before accepting lack of islet sympathetic nerves as a true negative. Quantification of sympathetic nerve area within the islet revealed a 93% loss of islet sympathetic nerves in human type 1 diabetes [22]. In contrast, pancreatic autopsy specimens from non-diabetic individuals demonstrated clear sympathetic fibres, primarily in the core of the islet, as recently reported [23]. Although this study strongly suggests a major defect in an autonomic input to the islet in human type 1 diabetes, it also raises two major questions. First, is the functional impact on glucagon secretion as severe as suggested by the neuroanatomy? Second, what is the mechanism that causes this loss of islet sympathetic nerves in type 1 diabetes? Both questions were more easily addressed in animal models of autoimmune diabetes, but each animal model had its pros and cons.
ANIMAL MODELS OF AUTOIMMUNE DIABETES
eSIN
All three animal models of autoimmune diabetes that we examined [24–26] had a marked loss of islet sympathetic nerves similar to that seen in human type 1 diabetes. However, each has its advantages and disadvantages for studying either the mechanism by which these islet nerves are lost or the impact of that loss on glucagon secretion. With the acute and severe onset of diabetes in the BB rat, one can determine how early these islet sympathetic nerves are lost. One can also determine if there is progressive nerve loss with diabetes duration, as is common for other types of diabetic neuropathy [27]. Conversely, the BB diabetic rat is a poor model to determine the role of lymphocytic infiltration in this nerve loss because BB rats are lymphopenic [28]. In contrast, the NOD mouse is useful in this regard since its invasive insulitis is prominent and easily quantifiable [29]. However, the uncommon genetic background of the NOD mouse makes deleting genes in a hypothesized pathway both expensive and time consuming. A third model, the RIP-GP mouse, in whom immune-mediated diabetes is induced by viral injection [30], has a distinct advantage in that the onset of both the immune attack of the islet and diabetic hyperglycaemia can be precisely timed. In addition, it’s on a common genetic background. However, the presence of live virus precludes sophisticated studies of the impact of this nerve loss on glucagon secretion.
Impaired glucagon response
It is important to note that both the BB diabetic rat [31] and the diabetic NOD mouse [25] have a marked impairment of their glucagon response to hypoglycaemia. Since the BB diabetic rat has a normal glucagon response to epinephrine [24], it models the selective glucagon secretory defect seen in human type 1 diabetes [32]. Further, all three animal models demonstrate an impaired glucagon response to activation of sympathetic nerves. For example, in BB diabetic rats, electrical stimulation of the sympathetic trunk projecting to the pancreas elicits a markedly impaired glucagon response compared to non-diabetic BB rats [24]. In NOD mice, the glucagon response to tyramine, a drug which releases norepinephrine from sympathetic nerves, was impaired in diabetic animals compared to non-diabetic controls [25]. Finally, in RIP-GP mice, in which the nerve loss seen after viral injection was reproduced by a sympathetic neurotoxin, the glucagon response to tyramine was also impaired [26]. These impaired glucagon responses were not due either to β–cell loss or diabetic hyperglycaemia, since chemically-induced diabetes, in the absence of islet nerve loss, produced no impairment of the glucagon response to tyramine in the NOR mouse, a close relative of the NOD mouse [25] or the glucagon response in Wistar rats, the background strain of the BB rat (unpublished observations). These impaired glucagon responses were due solely to loss of islet sympathetic nerves, since they were mimicked by chemically induced nerve loss, in the absence of diabetes [25,26,33].
POTENTIAL MECHANISMS FOR eSIN
Chronic hyperglycaemia
The Diabetes Control and Complications Trial (DCCT) has linked chronic hyperglycaemia to diabetic neuropathy by demonstrating a slower progression in patients who had better glucose control [15] due to intensive insulin therapy. Indeed, even in animal models of diabetes it takes many months of poorly controlled hyperglycaemia to produce experimental neuropathy. For example, in the BB diabetic rat, diabetic autonomic neuropathy begins to develop only after 6 months of diabetes [34]. In contrast, the early sympathetic islet neuropathy seen in BB diabetic rats occurs as early as 5 days after diabetes [35] and does not progress after 1–3 months of diabetes [35]. Further, our close examination of islets from non-diabetic NOD mice revealed that those few islets that were heavily infiltrated had nerve loss before the advent of diabetes [25]. Finally, in the RIP-GP mouse, the islet had lost 60% of their islet sympathetic nerves two days before the onset of hyperglycaemia [26]. Thus, although chronic diabetic hyperglycaemia is a major contributor to other types of diabetic neuropathy, it is not a contributor to the loss of islet sympathetic nerves that occurs in autoimmune diabetes.
Invasive Insulitis
Several lines of evidence have linked the loss of islet sympathetic nerves to the lymphocytic infiltration of the islet. First, in human type 1 diabetes [22] and autoimmune models thereof [26,35] the loss of islet sympathetic nerves is coupled with retention of sympathetic nerves in the surrounding exocrine pancreas. Second, islet sympathetic nerves are retained in human type 2 diabetes and in animal models of chemically, rather than immune, induced diabetes [25,35]. Third, in RIP-GP mice the onset of infiltration coincides with the onset of nerve loss [26]. Fourth, in non-diabetic NOD mice the minority of islets that are heavily infiltrated have fewer sympathetic nerves than the majority that are lightly infiltrated [25]. Fifth, blocking lymphocytic infiltration of the NOD islet prevents the loss of their sympathetic nerves [25].
After the development of NOD diabetes, nerve loss appears to progress with the duration of disease, but so does the degree of infiltration [25]. More importantly there is a very strong correlation between the amount of islet infiltration and the amount of nerve loss [25] (see Fig. 1). However, the molecular mechanism by which the invading lymphocytes cause loss of islet sympathetic nerves is unclear.
Figure 1. Invasive insulitis is associated with loss of islet sympathetic nerves.
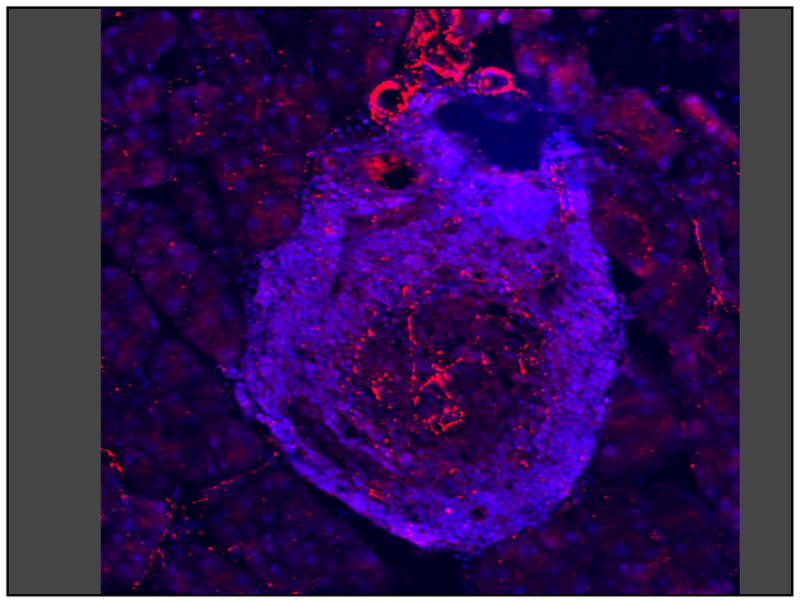
A single pancreatic islet from a diabetic NOD mouse showing invasive insulitis (small blue cells) and sympathetic axons with varicosities (red lines and dots). Sympathetic neural density is decreased only in the infiltrated part of the islet.
One possibility is that a subset of T lymphocytes are directed against an antigen present on sympathetic nerves, analogous to the prevalent view that a subset of T lymphocytes are directed against an antigen contained in islet β–cells resulting in their destruction and subsequent diabetes. The primary argument against sympathetic-specific T lymphocytes is indirect: sympathetic nerves are lost only in the islet, not in the surrounding exocrine pancreas. This islet-selective nerve loss has been demonstrated in human type 1 diabetes [22] and in the two animal models in which it has been examined: the BB diabetic rat [35] and the RIP-GP mouse [26]. Since ectopic β–cells, transplanted to the kidney capsule, are quickly destroyed by circulating T lymphocytes, one would predict a similar destruction of exocrine sympathetic nerves if sympathetic-specific T lymphocytes were the cause of the loss of sympathetic nerves within the islet. Since many sympathetic axons found in the exocrine pancreas also extend into the islet, it is difficult to imagine that the same axon can express a different antigen and therefore escape T-lymphocyte-mediated destruction when it happens to be outside of the islet.
Neurotrophins
Nerve Growth Factor (NGF)
Since the traditional diabetes-related mechanisms of nerve or tissue damage were unlikely to cause the loss of islet sympathetic nerves, we began to focus on the sympathetic nerves themselves searching for non-traditional mechanisms that could cause rapid and local nerve loss. Since sympathetic nerves are known to express TrkA, the receptor for NGF, and are known to be dependent upon NGF, not only for their development but also for their maintenance, we hypothesized that a loss of NGF might cause eSIN. Interestingly, in vitro evidence strongly suggests that islet β–cells are a major source of islet NGF [36]. Thus it appeared possible that immune-mediated destruction of islet β–cells, by itself, might cause eSIN by severely depleting the islet of this important sympathetic neurotrophin. However, if this hypothesis were correct, then any type of severe β–cell destruction should also cause eSIN. But the severe β–cell loss caused by streptozotocin in rats [35] or alloxan in mice [25] caused no loss of islet sympathetic nerves whatsoever. Additionally, although the amount of β–cell loss in human type 2 diabetes is less severe than that in type 1 diabetes, there was no loss of islet sympathetic nerves associated with the β–cell loss of type 2 diabetes [22]. Thus we conclude that loss of β–cell-derived NGF and thereby insufficient stimulation of TrkA is not, by itself, a cause of eSIN.
p75 neurotrophin receptor (NTR)
Sympathetic nerves, however, have another type of neurotrophin receptor, the p75NTR, whose activation has been associated with localized and rapid loss of sympathetic axons [37]. It should be noted that this loss of sympathetic axons is not a result of apoptosis of the neuronal cell body but rather a localized pruning of an axonal segment which leaves both the parent axon and neuronal cell body intact. This axonal pruning is prominent during development when sympathetic nerves are growing towards their targets. Axons that reach their targets first are thought to secrete an agonist, which activates the p75NTR on those redundant sympathetic axons that have failed to reach their target [37]. Such localized pruning of sympathetic axons also occurs in adults: there is a cyclic denervation of the uterus that is coordinated with oestrus [38]. An agonist for the p75NTR has been implicated in this denervation [39]. Finally, a similar process occurs in pathology. There is an acute loss of sympathetic axons in the heart adjacent to the infarct zone, which appears to be mediated by activation of the p75NTR on their sympathetic axons [40]. These studies raise the possibility that activation of the p75NTR may cause the acute loss of islet sympathetic nerves seen when the islet is under immune attack.
We therefore knocked out the p75NTR in the RIP-GP mice by crossbreeding and induced an immune attack of the islet by injection of the lymphocytic choriomeningitis virus. Deletion of the p75NTR gene had little effect on baseline sympathetic innervation of the islet but prevented the marked loss of islet sympathetic nerves that occurs at the onset of infiltration in RIP-GP mice [26]. Although the knockout was not restricted to sympathetic nerves, it did not affect the amount of T-lymphocyte infiltration, the onset of diabetic hyperglycaemia or the magnitude of diabetic hyperglycaemia [26]. Thus, we appear to have separated the immune-related loss of islet sympathetic nerves from the immune-mediated loss of islet β–cells. Further, the loss of islet sympathetic nerves was not simply delayed since after 3 weeks of diabetes, islet sympathetic nerves were still retained in p75NTR knockout mice [26].
Brain derived neurotrophic factor (BDNF)
We next sought evidence that BDNF was the agonist activating this p75NTR during autoimmune attack of the islet. The knockout strategy we applied to the p75NTR could not be applied to BDNF since deletion of the BDNF gene is neonatally lethal [41]. However, others have shown that deletion of just one BDNF allele not only allows survival but also impairs the ability of tissues to increase their BDNF production upon stimulation [39]. We therefore knocked out one BDNF allele in RIP-GP mice by crossbreeding. Again, the loss of one BDNF allele did not affect baseline sympathetic innervation of the islet. However, it totally prevented the loss of islet sympathetic nerves when the islet was under immune attack (unpublished observations). Therefore, an increase of BDNF within the infiltrated islet likely activates the p75NTR causing the loss of islet sympathetic nerves.
REMAINING QUESTIONS
While this study both implicates BDNF as the activating agonist of the p75NTR and provides a novel mechanism for the loss of islet sympathetic nerves in autoimmune diabetes, it leaves three major questions unanswered. First, what cell types within the islet secrete the BDNF that causes the nerve loss? The possibilities include the endocrine or vascular cells of the islet, the islet sympathetic nerve themselves or the lymphocytes which invade the islet. There is precedent for each. Stimulated production of BDNF by a target tissue has been clearly demonstrated: oestrogen stimulates the uterus to make BDNF [39], which in turn initiates its cyclic sympathetic denervation [38]. Sympathetic nerves, stimulated by NGF, can also secrete BDNF as demonstrated by their pruning of adjacent sympathetic nerves that are NGF deficient [37]. Finally, there are scattered reports that lymphocytes can also secrete BDNF [42–44]. The methods needed to determine which cell type in the islet makes BDNF include immunohistochemistry for hemagglutinin-tagged BDNF [45], Western blots [45], ELISAs [39] and, finally, the genetic deletion of one BDNF allele from each one of these tissues in turn.
Second, what is the direct stimulus increasing islet BDNF during the immune attack? Answering the first question would simplify the search for the stimulus, but even without such information, the stimulation is likely related to the infiltration of the islet. Since cytokines are produced when the islet is infiltrated, they are attractive candidates for the stimulator that increases islet BDNF and causes nerve loss. Unfortunately, the number of candidate cytokines is large. It is therefore likely that after the cellular source of islet BDNF is identified, in vitro experiments exposing that cell type to a variety of cytokines will either provide a definitive answer or markedly narrow the field.
Third, what is the actual contribution of this islet nerve loss to the impaired glucagon response to hypoglycemia seen in autoimmune diabetes? The ability to selectively prevent the loss of islet sympathetic nerves during the development of diabetes, by deleting either the p75NTR or one allele of BDNF, provides a unique opportunity to answer this important scientific question. However, as mentioned above, the presence of live BSL2-class virus in RIP-GP mice that were made diabetic by LCMV injection, makes it unsafe to perform the sophisticated in vivo studies of glucagon secretion needed to answer this question in this model. However, there is a potential solution. The vonHerrath group has discovered how to stimulate a strong immune attack of the islet, without using live virus [46]. We anticipate that this non-viral, immune-mediated attack of the islet will also cause eSIN and that knocking out the p75NTR will also prevent eSIN but not the subsequent diabetes. If so, one could determine in this non-viral model the degree of impairment of the glucagon response to hypoglycemia in diabetic mice who have either lost or retained their islet sympathetic nerves. The expected additional impairment in diabetic mice with eSIN should then be attributable solely to the loss of their islet sympathetic nerves.
THERAPEUTIC INTERVENTION
While these studies of the mechanism of islet nerve loss are inherently scientifically interesting, they may have little clinical utility for preventing eSIN since patients diagnosed with type 1 diabetes mellitus likely have already lost their islet sympathetic nerves. What may be of clinical benefit is a strategy to restore the sympathetic innervation of the islet. Because their parent axons in the exocrine pancreas appear intact [35], it may be possible to regrow islet sympathetic nerves. Such an approach is supported by studies showing that sympathetic nerves eventually reinnervate transplanted islets [47]. However, it might also be necessary to both reverse diabetic hyperglycaemia and provide a source of islet NGF. For example, diabetic hyperglycaemia seems to retard nerve growth [48] and NGF is a known stimulus for the regrowth of islet sympathetic nerves [49]. Further, it is likely that islet NGF is depleted by the autoimmune destruction of β–cells since they are a major source of islet NGF [36]. A sympathetically re-innervated islet should have an improved glucagon response to insulin-induced hypoglycaemia. Therefore, individuals so treated should be less susceptible to severe and prolonged iatrogenic hypoglycaemia. Given the aversive nature of hypoglycaemia, such an intervention should increase adherence to the intensive insulin therapy shown to prevent the debilitating long-term complications of this disease [15].
Acknowledgments
This work was supported by the Medical Research Service of the Department of Veterans Affairs and National Institutes of Health grants R01-DK-50154 and P30-DK-017047. The authors wish to thank Trish Banik for technical assistance and Pam Henderson for administrative assistance (both of the VA Puget Sound Health Care System).
Footnotes
Conflict of Interest
The authors do not declare any conflict of interest relevant to this manuscript.
References
- 1.Cryer PE. Glucagon in the pathogenesis of hypoglycemia and hyperglycemia in diabetes. Endocrinology. 2012;153:1039–1048. doi: 10.1210/en.2011-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taborsky GJ, Jr, Mundinger TO. The role of the autonomic nervous system in mediating the glucagon response to hypoglycemia. Endocrinology. 2012;153:1055–1062. doi: 10.1210/en.2011-2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Havel PJ, Taborsky GJ., Jr The contribution of the autonomic nervous system to changes of glucagon and insulin secretion during hypoglycemic stress. Endocrine Rev. 1989;10:332–350. doi: 10.1210/edrv-10-3-332. [DOI] [PubMed] [Google Scholar]
- 4.Havel PJ, Veith RC, Dunning BE, Taborsky GJ., Jr Role for autonomic nervous system to increase pancreatic glucagon secretion during marked insulin-induced hypoglycemia in dogs. Diabetes. 1991;40:1107–1114. doi: 10.2337/diab.40.9.1107. [DOI] [PubMed] [Google Scholar]
- 5.Havel PJ, Parry SJ, Stern JS, et al. Redundant parasympathetic and sympathoadrenal mediation of increased glucagon secretion during insulin-induced hypoglycemia in conscious rats. Metabolism. 1994;43:860–866. doi: 10.1016/0026-0495(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 6.Havel PJ, Akpan JO, Curry DL, Stern JS, Gingerich RL, Ahren B. Autonomic control of pancreatic polypeptide and glucagon secretion during neuroglucopenia and hypoglycemia in mice. Am J Physiol. 1993;265:246–254. doi: 10.1152/ajpregu.1993.265.1.R246. [DOI] [PubMed] [Google Scholar]
- 7.Havel PJ, Valverde C. Autonomic mediation of glucagon secretion during insulin-induced hypoglycemia in rhesus monkeys. Diabetes. 1996;45:960–966. doi: 10.2337/diab.45.7.960. [DOI] [PubMed] [Google Scholar]
- 8.Havel PJ, Ahren B. Activation of autonomic nerves and the adrenal medulla contributes to increased glucagon secretion during moderate insulin-induced hypoglycemia in women. Diabetes. 1997;46:801–807. doi: 10.2337/diab.46.5.801. [DOI] [PubMed] [Google Scholar]
- 9.Marliss EB, Girardier L, Seydoux J, et al. Glucagon release induced by pancreatic nerve stimulation in the dog. J Clin Invest. 1973;52:1246–1259. doi: 10.1172/JCI107292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahren B, Veith RC, Taborsky GJ., Jr Sympathetic nerve stimulation versus pancreatic norepinephrine infusion in the dog: 1) Effects on basal release of insulin and glucagon. Endocrinology. 1987;121:323–331. doi: 10.1210/endo-121-1-323. [DOI] [PubMed] [Google Scholar]
- 11.Havel PJ, Veith RC, Dunning BE, Taborsky GJ., Jr Pancreatic noradrenergic nerves are activated by neuroglucopenia but not hypotension or hypoxia in the dog: Evidence for stress-specific and regionally-selective activation of the sympathetic nervous system. J Clin Invest. 1988;82:1538–1545. doi: 10.1172/JCI113763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Havel PJ, Mundinger TO, Taborsky GJ., Jr Pancreatic sympathetic nerves contribute to increased glucagon secretion during severe hypoglycemia in dogs. Am J Physiol. 1996;270:20–26. doi: 10.1152/ajpendo.1996.270.1.E20. [DOI] [PubMed] [Google Scholar]
- 13.Gerich J, Langlois M, Noacco C, Karam J, Forsham P. Lack of glucagon response to hypoglycemia in diabetes: evidence for an intrinsic pancreatic alpha cell defect. Science. 1973;182:171–173. doi: 10.1126/science.182.4108.171. [DOI] [PubMed] [Google Scholar]
- 14.Heller SR, Cryer PE. Reduced neuroendocrine and symptomatic responses to subsequent hypoglycemia after 1 episode of hypoglycemia in nondiabetic humans. Diabetes. 1991;40:223–226. doi: 10.2337/diab.40.2.223. [DOI] [PubMed] [Google Scholar]
- 15.DCCT; The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 16.Zhou H, Zhang T, Oseid E, Harmon J, Tonooka N, Robertson RP. Reversal of defective glucagon responses to hypoglycemia in insulin-dependent autoimmune diabetic BB rats. Endocrinology. 2007;148:2863–2869. doi: 10.1210/en.2006-1375. [DOI] [PubMed] [Google Scholar]
- 17.Taborsky GJ, Jr, Ahren B, Havel PJ. Autonomic mediation of glucagon secretion during hypoglycemia: implications for impaired alpha-cell responses in type 1 diabetes. Diabetes. 1998;47:995–1005. doi: 10.2337/diabetes.47.7.995. [DOI] [PubMed] [Google Scholar]
- 18.Gerich J, Davis J, Lorenzi M, et al. Hormonal mechanisms of recovery from insulin-induced hypoglycemia in man. Am J Physiol. 1979;236:380–385. doi: 10.1152/ajpendo.1979.236.4.E380. [DOI] [PubMed] [Google Scholar]
- 19.Raju B, Cryer PE. Loss of the decrement in intraislet insulin plausibly explains loss of the glucagon response to hypoglycemia in insulin-deficient diabetes: documentation of the intraislet insulin hypothesis in humans. Diabetes. 2005;54:757–764. doi: 10.2337/diabetes.54.3.757. [DOI] [PubMed] [Google Scholar]
- 20.Banarer S, McGregor VP, Cryer PE. Intraislet hyperinsulinemia prevents the glucagon response to hypoglycemia despite an intact autonomic response. Diabetes. 2002;51:958–965. doi: 10.2337/diabetes.51.4.958. [DOI] [PubMed] [Google Scholar]
- 21.White NH, Gingerich RL, Levandoski LA, Cryer PE, Santiago JV. Plasma pancreatic polypeptide response to insulin-induced hypoglycemia as a marker for defective glucose counterregulation in insulin-dependent diabetes mellitus. Diabetes. 1985;34:870–875. doi: 10.2337/diab.34.9.870. [DOI] [PubMed] [Google Scholar]
- 22.Mei Q, Foulis AK, Fligner C, et al. Selective Loss of sympathetic nerves from the islet in human type 1 diabetes: a potential mechanism for impaired glucagon responses to hypoglycemia. Diabetes. 2006;55 (Suppl 1):A15. [Google Scholar]
- 23.Rodriguez-Diaz R, Abdulred MH, Formoso A, et al. Autonomic axons in the human endocrine pancreas show unique innervation patterns. Cell Metabol. 2011;14:45–54. doi: 10.1016/j.cmet.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mundinger TO, Mei Q, Figlewicz DP, Lernmark A, Taborsky GJ., Jr Impaired glucagon response to sympathetic nerve stimulation in the BB diabetic rat: effect of early sympathetic islet neuropathy. Am J Physiol Endocrinol Metab. 2003;285:E1047–E1054. doi: 10.1152/ajpendo.00136.2003. [DOI] [PubMed] [Google Scholar]
- 25.Taborsky GJ, Jr, Mei Q, Hackney DJ, Figlewicz DP, LeBoeuf R, Mundinger TO. Loss of islet sympathetic nerves and impairment of glucagon secretion: relationship to invasive insulitis. Diabetologia. 2009;52:2602–2611. doi: 10.1007/s00125-009-1494-5. [DOI] [PubMed] [Google Scholar]
- 26.Taborsky GJ, Jr, Mei Q, Bornfeldt KE, Hackney DJ, Mundinger TO. The p75 neurotrophin receptor is required for the major loss of sympathetic nerves from islets under autoimmune attack. Diabetes. 2014;63:2369–2379. doi: 10.2337/db13-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DCCT. Factors in development of diabetic neuropathy. Baseline analysis of neuropathy in feasibility phase of Diabetes Control and Complications Trial (DCCT). The DCCT Research Group. Diabetes. 1988;37:476–481. [PubMed] [Google Scholar]
- 28.Marliss EB, Nakhooda AF, Poussier P, Sima AAF. The diabetic syndrome of the BB Wistar rat: possible relevance to type I (insulin-dependent) diabetes in man. Diabetologia. 1982;22:225–232. doi: 10.1007/BF00281296. [DOI] [PubMed] [Google Scholar]
- 29.Signore A, Pozzilli P, Gale EA, Andreani D, Beverley PC. The natural history of lymphocyte subsets infiltrating the pancreas of NOD mice. Diabetologia. 1989;32:282–289. doi: 10.1007/BF00265543. [DOI] [PubMed] [Google Scholar]
- 30.Oldstone MB, Nerenberg M, Southern P, Price J, Lewicki H. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: role of anti-self (virus) immune response. Cell. 1991;65:319–331. doi: 10.1016/0092-8674(91)90165-u. [DOI] [PubMed] [Google Scholar]
- 31.Jacob RJ, Dziura J, Morgen JP, Shulman GI, Sherwin RS. Time course of the defective alpha-cell response to hypoglycemia in diabetic BB rats. Metabolism. 1996;45:1422–1426. doi: 10.1016/s0026-0495(96)90125-0. [DOI] [PubMed] [Google Scholar]
- 32.Benson JW, Jr, Johnson DG, Palmer JP, Werner PL, Ensinck JW. Glucagon and catecholamine secretion during hypoglycemia in normal and diabetic man. J Clin Endocrinol Metab. 1977;44:459–464. doi: 10.1210/jcem-44-3-459. [DOI] [PubMed] [Google Scholar]
- 33.Mundinger TO, Mei Q, Lernmark A, Taborsky GJ., Jr The degree of islet neuropathy seen in BB diabetes is sufficient to impair the glucagon response to sympathetic nerve stimulation. Diabetes. 2002;51 (Suppl 2):A148. [Google Scholar]
- 34.Yagihashi S, Sima AA. Diabetic autonomic neuropathy. The distribution of structural changes in sympathetic nerves of the BB rat. Am J Pathol. 1985;121:138–147. [PMC free article] [PubMed] [Google Scholar]
- 35.Mei Q, Mundinger TO, Lernmark A, Taborsky GJ., Jr Early, selective, and marked loss of sympathetic nerves from the islets of biobreeder diabetic rats. Diabetes. 2002;51:2997–3002. doi: 10.2337/diabetes.51.10.2997. [DOI] [PubMed] [Google Scholar]
- 36.Rosenbaum T, Vidaltamayo R, Sanchez-Soto MC, Zentella A, Hiriart M. Pancreatic B cells synthesize and secrete nerve growth factor. Proc Natl Acad Sci U S A. 1998;95:7784–7788. doi: 10.1073/pnas.95.13.7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh KK, Park KJ, Hong EJ, et al. Developmental axon pruning mediated by BDNF-p75NTR-dependent axon degeneration. Nat Neurosci. 2008;11:649–658. doi: 10.1038/nn.2114. [DOI] [PubMed] [Google Scholar]
- 38.Zoubina E, Smith PG. Axonal degeneration and regeneration in rat uterus during the estrous cycle. Autonom Neurosci. 2000;84:176–185. doi: 10.1016/S1566-0702(00)00209-5. [DOI] [PubMed] [Google Scholar]
- 39.Krizsan-Agbas D, Pedchenko T, Hasan W, Smith PG. Oestrogen regulates sympathetic neurite outgrowth by modulating brain derived neurotrophic factor synthesis and release by the rodent uterus. Eur J Neurosci. 2003;18:2760–2768. doi: 10.1111/j.1460-9568.2003.03029.x. [DOI] [PubMed] [Google Scholar]
- 40.Lorentz CU, Parrish DC, Alston EN, et al. Sympathetic denervation of peri-infarct myocardium requires the p75 neurotrophin receptor. Exp Neurol. 2013;249:111–119. doi: 10.1016/j.expneurol.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- 42.Kerschensteiner M, Gallmeier E, Behrens L, et al. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J Exp Med. 1999;189:865–870. doi: 10.1084/jem.189.5.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kruse N, Cetin S, Chan A, Gold R, Luhder F. Differential expression of BDNF mRNA splice variants in mouse brain and immune cells. J Neuroimmunol. 2007;182:13–21. doi: 10.1016/j.jneuroim.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 44.Edling AE, Nanavati T, Johnson JM, Tuohy VK. Human and murine lymphocyte neurotrophin expression is confined to B cells. J Neurosci Res. 2004;77:709–717. doi: 10.1002/jnr.20176. [DOI] [PubMed] [Google Scholar]
- 45.Yang J, Siao CJ, Nagappan G, et al. Neuronal release of proBDNF. Nat Neurosci. 2009;12:113–115. doi: 10.1038/nn.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinic MM, von Herrath MG. Real-time imaging of the pancreas during development of diabetes. Immunol Rev. 2008;221:200–213. doi: 10.1111/j.1600-065X.2008.00581.x. [DOI] [PubMed] [Google Scholar]
- 47.Korsgren O, Jansson L, Andersson A, Sundler F. Reinnervation of transplanted pancreatic islets. Transplantation. 1993;56:138–143. [PubMed] [Google Scholar]
- 48.Mehra S, Tavakoli M, Kallinikos PA, et al. Corneal confocal microscopy detects early nerve regeneration after pancreas transplantation in patients with type 1 diabetes. Diabetes Care. 2007;30:2608–2612. doi: 10.2337/dc07-0870. [DOI] [PubMed] [Google Scholar]
- 49.Raivich G, Kreutzberg GW. Nerve growth factor and regeneration of peripheral nervous system. Clin Neurol Neurosurg. 1993;95:S84–S88. doi: 10.1016/0303-8467(93)90041-e. [DOI] [PubMed] [Google Scholar]