

Abstract
Recently, small (<5 nm diameter) nanoparticles (NPs) have shown improved in vivo biocompatibility compared to that of larger (>10 nm) NPs. However, the fate of small NPs under physiological conditions is poorly understood and remains unexplored. Here, the long-term aggregation behavior of gold nanoparticles (AuNPs) exposed to serum proteins in a near-physiological setup is studied using continuous photon correlation spectroscopy and computer simulations. It is found that the medium, temperature, and NP concentration affect the aggregation of AuNPs, but the observed aggregates are much smaller than previously reported. Simulations show that a single layer of albumin is deposited on the NP surface, but the properties of the aggregates (size, shape, and internal structure) depend critically on the charge distribution on the proteins, which changes with the conditions of the solution. These results explain the seemingly conflicting data reported in the literature regarding the size of aggregates and the morphology of the albumin corona. The simulations suggest that controlling the concentration of NPs as well as the pH and ionic strength of the solution prior to intravenous administration may help to preserve properties of the functionalized NPs in the bloodstream.
I. Introduction
The use of nanomaterials in biomedical applications has advanced considerably in recent years. Plasmonic nanomaterials such as gold nanoparticles (AuNPs) hold great promise in medicine because of their exceptional electro-optical properties and ease of surface modification.1−3 Although AuNPs have been tested for diagnostics and therapeutics, important challenges, such as physiological fate, still need to be elucidated for routine clinical applications. A major concern is the propensity for NPs to aggregate when exposed to biological media, resulting in toxicity and inflammation.4−6 Recently, small-sized AuNPs have emerged as promising agents for imaging and clinical use7,8 because of their potential for reduced toxicity and faster clearance from the body. Little is known, however, about their behavior in the presence of biological agents under physiological conditions, and progress has been slow because of technological limitations and the complexity of the biological microenvironment. The problem is also complicated by the fact that most serum proteins, particularly albumin, are comparable in size to small NPs. A variety of experimental techniques are currently being tested to gain insight into the behavior of NPs in serum.9−14In vitro experiments have been conducted to quantify the strength of AuNP–albumin interactions, but results are sensitive to the experimental conditions, including albumin concentration, NP size, temperature, ionic strength, and acidity of the solution. Disagreements exist on whether albumin stabilizes AuNPs or instead induces/mediates aggregation: some studies suggest that albumin and AuNPs do not associate in water, whereas others suggest monolayer and even multilayer adsorption.9−11,14 To prevent aggregation and increase dispersion stability, NPs are coated with organic compounds,12,15,16 including poly(ethylene glycol) (PEG), citrate, and n-alkyl thiols of varying chain lengths, that impart biocompatibility and increase the circulation time in the bloodstream. NP size and surface chemistry also affect serum protein adsorption and may determine the biological response in vivo.17 The role of serum proteins in mediating interactions between coated NPs and in (de)stabilizing the NP aggregates is a topic of active research. Recent reports suggest, however, that the coatings may fall off once the NPs are introduced into the body, possibly as a result of steric hindrance or instability of the coating material in the biological medium.18,19 Therefore, finding the right experimental conditions under which the coating can be protected or stabilized is of great interest as well.
Experiments have commonly been conducted under nonphysiological conditions using NPs with diameters larger than ∼10 nm. The focus here is on near-physiological serum and NPs with diameters in the 2–5 nm range. Particles within this size range have been shown to facilitate body clearance and to have less toxicity than larger particles,20 although the molecular basis for decreased toxicity is not presently known. A combined experimental/simulation approach is used here to gain insight into the behavior of NPs in physiological serum, the role of albumin in NP aggregation, and the structural and hydrodynamic properties of the aggregates. Answering these questions may help to better understand the toxic response of small NPs and to pave the way toward rational design of NPs with desired properties for clinical use. In this study, continuous photon correlation spectroscopy is used, for the first time, to monitor the change in hydrodynamic size of NPs without interruption, unlike conventional end-point-based approaches. It is found that, when monitored continuously under physiological temperature and serum-like conditions, smaller NP aggregates emerge within a size range that might allow efficient body clearance. A general multiscale algorithm was developed to obtain coarse-grain models of globular proteins, which is applied here to albumin at physiological concentrations. The resulting model preserves structural features of the protein as well as the charge distribution on its surface, both found to be important for a realistic representation of the medium. The simulations provide a molecular view of the NP aggregates, their internal structures, and the role of albumin in their formation and stabilization.
II. Experimental Setup
The monodispersed bare-gold nanoparticles in water (∼5.0 × 1013 particles mL–1) were procured from Ted Pella Inc. (Redding, CA). The AuNPs were citrate-stabilized with a negative surface charge. Serum albumin was purchased from Sigma. Ultrapure water (biology grade) was purchased from K·D Medical, Inc. (Columbia, MD).
II-i. Continuous Photon Correlation Spectroscopy (CPCS)
To test the serum stability and size distribution of 5 nm AuNPs, a physiologically relevant experimental model was used. A Zetasizer Nano series, Zen3600, from Malvern with Zetasizer software 6.0 as the interface instrumentation for CPCS was used under predefined conditions to mimic an in vivo microenvironment. The instrument was set to 37 °C before the introduction of the sample. For each measurement, the AuNP dispersion was placed in a DTS0012 cell type disposable sizing cuvette. Measurement was set to 100 iterations with a 1800 s delay between measurements. The measurement angle was set to 173° backscatter, with an equilibration time of 120 s. Hydrodynamic size acquisition was set to take readings every hour. Known amounts of AuNP solution were then added to cell media containing 10% FBS, and the solution was placed in the capped cuvette holder. It was ensured that there were no air bubbles in the solution, which could otherwise interfere with the acquisition and yield inconsistent data. The advantage of this experimental setup is that once the NPs are introduced into the physiological medium they are continuously exposed to the serum proteins, thus mimicking the conditions of an in vivo environment while measurements are being performed.
II-ii. Transmission Electron Microscopy (TEM)
A specimen of 5 nm AuNPs for TEM imaging was obtained by depositing a 5 μL droplet from the aqueous solution onto a Quantifoil grid and leaving it to dry in air. After adsorption for 5 min, the excess solution was blotted with filter paper, washed with a few 5 μL droplets of deionized water in order to remove any dirt, and left to dry. Images were recorded in a Tecnai TF30 TEM (FEI, Hillsboro, OR) equipped with a Gatan Ultrascan 1000 CCD camera (Gatan, Pleasaton, CA). For TEM imaging of AuNPs exposed to physiological media, 5 nm AuNPs were aged in a cell serum medium solution for 24 h before preparing the sample for imaging. Sample preparation was similar as that described above for the aqueous sample.
II-iii. UV–Vis Absorption Measurement
Optical property of AuNPs dispersion was monitored using a UV–vis Spectrophotometer (Thermo Scientific, Waltham, MA). UV absorbance measurements of AuNPs in both water and serum media were carried out. Absorbance at 520 nm was used as a marker for 5 nm AuNPs.
III. Model and Simulations Setup
For statistical analysis of the system behavior, a sufficiently large number of NPs are required. Given the NP concentration, this condition determines the size of the simulation box and the level of coarsening of the proteins. An all-atom representation of the system, including water, would require at least ∼108 atoms (assuming a physiological concentration of albumin), an untreatable computational problem. The algorithm developed here reduces the size of the system to ∼104 particles.
III-i. Coarse-Grain Models of Serum Proteins
The coarsening algorithm is as follows: (i) the heavy atom i1 closest to the protein center of mass (CoM) is first identified; (ii) all of the atoms {I}1 within a distance λ from i1 are represented by a spherical particle of radius R1, mass M1, and charge Q1, centered at the CoM of the set {I}1; (iii) the heavy atom i2 ⊄ c1 = {I}1 that is closest to the protein CoM is then identified; (iv) all of the atoms {I}2 ⊄ c1 within a distance λ from i2 are represented by a spherical particle of radius R2, mass M2, and charge Q2, centered at the CoM of {I}2; (v) the process continues so that in step N the atom iN ⊄ cN = {I}1 ∪ {I}2 ∪ ... ∪ {I}N−1 closest to the protein CoM is identified; (vi) all of the atoms {I}N ⊄ cN within a distance λ from iN are then represented by a sphere of radius RN, mass MN, and charge QN, centered at the CoM of {I}N. After Nλ cycles, all of the atoms have been grouped into Nλ spheres; the parameter λ defines the level of coarsening (upper limit of the spatial resolution). The radii, masses, and charges are given, respectively, by Ri = Raa(ni/naa)1/3, Mi = ∑jmj, and Qi = ∑jqj, where i = 1, ..., Nλ and j = 1, ..., ni, and ni is the number of atoms in {I}i; where Raa ∼ 3.5 Å and naa ∼15.5 are the average radius and the number of atoms per residue of a typical protein.
The algorithm described above is general for globular proteins (or globular domains in multidomain proteins) and reversible, i.e., a coarse-grained system can be fine-grained at any stage over the course of a simulation. The method is used here to model albumin, the most abundant protein in serum. Human serum albumin is a 66.4 kDa protein with a pH-dependent conformation in solution.21 Hydrodynamic data indicate that, under physiological conditions, albumin adopts a heart-shaped conformation, as observed in the crystal.22 The coarsening process is illustrated in Figure 1. For evaluation of the charge distribution at pH 7, standard protonation states are assumed. The model preserves the overall shape of the protein and the anisotropy of the charge distribution, as both features determine the protein–protein and NP–protein interactions.
Figure 1.
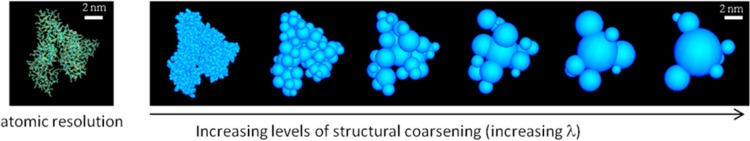
Structural coarse-graining of albumin (based on PDB: 1AO6). Reverse coarsening (fine graining) to any λ level is straightforward (used in Figures 6–8). The hydrodynamic radius RH of albumin in water has been measured at ∼3.2–3.48 nm.22,44 For the rigid model used here, n = 7 and ρi = Ri + dw, where dw is the average thickness of the hydration layer surrounding the protein. Using a single layer (dw = 2.8 Å), eq 6 yields RH = 3.33 nm. The calculated translational and rotational diffusion constants are DT ∼ 6.6 × 1011 m2 s–1 and DR ∼ 4.4 × 106 1 s–1, respectively, close to the experimental values.21 The rotational correlation times τD(1) and τD(2) (Debye’s relaxation time) of the albumin macrodipole (∼500 D21) are thus ∼33 ns and ∼0.1 μs, respectively. The model can be used to calculate dielectric and spectroscopic properties of albumin solutions, although the relaxation times indicate that long dynamics simulations would be needed to properly sample the conformational space, which justifies the choice of Monte Carlo sampling used in this study.
III-ii. Modeling of Interactions
The energy E of an aqueous solution of nanoparticles and proteins is given by
| 1 |
where the indices np and np′ refer to the NPs, P and P′ refer to the proteins, and V are the interaction energies. All of the components of the system are treated as rigid bodies, so intramolecular energy terms are omitted. The effects of water are treated implicitly. The direct interaction between AuNP cores in vacuum is typically represented23 by an attractive Hamaker potential.24 In solution, however, solvent-induced forces are much stronger, so the direct forces between NP cores can be neglected. Hydrophobic attraction plays the major role in the interactions between bare AuNPs in water. On the other hand, the interaction between coated NPs is determined mainly by the type of surfactant and the surface-coverage density, in which case electrostatics and depletion forces play a major role.5 In this study, a generic Lennard–Jones potential is used to represent the interactions between NPs, a choice justified by results from atomistic simulations.23,25−27 The mean-field effects of ions can eventually be incorporated separately, e.g., using the DLVO or related theories.28,29 Such corrections are not needed here because the ion concentration is fixed and their nonspecific effects are incorporated in the LJ parameters. Because of their small sizes, the NPs are here assumed to be spherical,30 so the NP–NP interaction energy of a monodisperse solution is given by
| 2 |
where r is the distance between the centers of np and np′, ε is the strength of the interaction, and σ is the effective NP diameter. Likewise, the interaction energy between a NP and a protein is
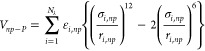 |
3 |
where ri,np is the distance between the centers of np and a sphere i of the coarse model of P. The interaction energy between two proteins is divided into electrostatic and van der Waals contributions, in the form VP–P′ = VP–P′elec + VP–P + VP′–P′elec + VP–P′. The electrostatic terms are represented by the screened Coulomb potentials implicit solvent model (SCPISM),31,32 as
| 4 |
whereas the van der Waals component is given by
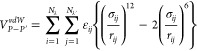 |
5 |
where Nλ and Nλ′ are the total number of spheres in P and P′, respectively, and rij are the distances between the centers of spheres i in P and j in P′. The first and second terms on the r.h.s. of eq 4 are interaction energies and self-energies, respectively, and D and R are effective screening functions and radii, both dependent on the configuration (r) of the system31,32 that capture the solvent-exclusion effects of hydration in the crowded environment.
III-iii. Model Parametrization
Equations 2–5 contain a number of parameters that must be determined. Both ε and σ in eqs 2 and 3 depend on the size of the NP core and the physicochemical properties of the coating molecules. In a few cases, these parameters have been estimated from simulations, but these estimates are mostly in the gas phase or in nonaqueous solvents. Considering ε and σ as variables, it is possible to carry out systematic simulations of more general validity, where neither the core material nor the type of surfactant is specified. The effective size of the NPs is varied from σ = 2 to 5 nm, and the strength of the NP–NP interactions is varied from ε = 0.1 kcal mol–1 to a maximum value εmax chosen on the basis of the strength of hydrophobic attraction between bare AuNPs. The hydrophobic potential ΔG(r) depends on the change Δγ of the solvent-accessible surface area γ of the NPs, in the form ΔG = aΔγ, where a ∼ 5.5 cal mol–1 Å–1. Two NPs with diameters σ at close contact bury a surface Δγ = πσdw; thus, ΔG ∼ 2.5 kcal mol–1 (for σ = 5 nm) and ∼ 1.0 kcal mol–1 (for σ = 2 nm). The polarity/charge of the coating weakens the NP–NP interactions in pure water. In serum, however, the presence of small molecules and ions could either weaken or strengthen the interactions and even promote self-assembly into superlattices. To better capture this broader range of possibilities, the maximum strength is set at εmax ∼ 2ΔG.
The effective radius Ri in eq 4 is taken as the radius of the sphere i; the screening functions D contain Nλ parameters α, which are here assumed to be the same for all of the spheres. The dependence of α on the system configuration, charge, and temperature has been discussed.31 To estimate this parameter, a Monte Carlo (MC) simulation of an albumin binary complex is first carried out using a fully atomistic representation of the protein, and energies are calculated with the all-atom SCPISM in CHARMM.33 The initial configuration is the same as in the crystallized homodimer (1AO6). The system is heated gradually to dissociation starting from 37 °C, and the dissociation energy is estimated.31,32 Similar calculation is carried out with the coarse-grain model of the proteins, and the dissociation energy is reproduced with α = 0.195 nm–1. The parameters εi,j in eq 5 are determined by heating the coarse dimer, using a single εalb for all εi,j and σi,j = (σi + σj)/2. Assuming a vdW contribution on the order of kT (dispersion makes relatively small contributions to protein–protein interaction in water34,35), the calculations yield εalb ∼ 0.1 kcal mol–1.
Experiments have been conducted9,11 to quantify the strength of the interaction between albumin and bare AuNPs, although the main focus has been on NPs with diameters larger than ∼10 nm. Fluctuation correlation spectroscopy14 has recently been used to estimate the binding affinity between BSA and AuNPs with diameters of ∼5 nm. The dissociation constant in water at 23 °C and albumin concentration of 0.9 mM was measured at Kd ∼79 μM, which corresponds to an energy ΔG = kT ln(Kd/cØ) ∼ 5.7 kcal mol–1. Assuming a single εi,np for all of the interactions in eq 3 and setting σ = 5 nm, this dissociation energy is reproduced with εi,np = 1.9 kcal mol–1. Although the experimental conditions are different than those in physiological serum, the results provide an estimate of the expected order of magnitude of the NP–albumin interactions. These studies have also shown that increasing the NP diameter up to ∼20 nm weakens the NP–albumin interaction, although other studies suggest a reversal of this trend for particles with diameter less than ∼60 nm and yet another reversal for larger NPs. This behavior is not universal, as changing the NP coating may increase or decrease the strength of the interactions. To facilitate a systematic study in the reduced ε–σ space, these variations are represented here through a dependence of εi,np on ε using a simple mixing rule, εi,np = (εalbε)1/2.
III-iv. Simulations Setup
The system consists of a spherical container with a diameter of 0.109 μm filled with NPs at the desired molar concentration c. The effects of NP size and coating are studied by changing σ and ε. For each ε–σ–c point, two canonical MC simulations are performed at 37 °C: one in water and one in serum. The physiological concentration of albumin is ∼0.64 mM (human serum), or ∼2000 albumin molecules, corresponding to a macromolecular crowding of ∼30% in volume. The level of coarsening is such that Nλ = 7; two test simulations using Nλ = 5 and 9 showed no qualitative differences in the results. The NP concentration is varied from c = 0.1 to 2 mM, which provides adequate statistics. At the beginning of the simulations, all of the components are distributed randomly within the container, and 106 rigid-body rotations, translations, or roto-translations of one component at a time are performed to create equilibrated initial distributions. Each production run consists of an additional 106 trial moves, enough to generate stable distributions of NP aggregates, which is used here as a criterion for convergence.
IV. Results and Discussion
IV-i. Experimental Results
A survey of the literature indicates that physicochemical characterizations for toxicological interpretation are typically performed under nonphysiological conditions, most commonly at room temperature, and at the beginning and end of the exposure times.9,36 Because the behavior of NP varies with temperature and protein crowding, NP–protein interactions must be studied as close to physiological conditions as possible. Once NPs are introduced into the body, they are in continuous contact with the proteins. Therefore, to probe the real-time dynamics of the NP–protein system, data should be collected continuously. CPCS is used here to probe the hydrodynamic behavior of 5 nm diameter, citrate-reduced and capped AuNPs at 37 °C in a near-physiological fluid (serum proteins, pH 7.4). Measurements are performed continuously once the AuNPs are introduced into serum. In plain water at 37 °C, the NPs form clusters with an average hydrodynamic radius RH of ∼10 nm (Figure 3A), but TEM data show that the size of the NP core is not affected (Figure 3B). This contrasts with the situation in serum, where a 2-fold increase in the average size of the aggregates is observed, along with a significant shift in the size distribution (Figure 3C,D). Data collected at 24 °C confirm the sensitivity of hydrodynamic measurements to the temperature of the solution (Figure 2). The size distribution at lower temperatures is generally shifted toward larger values of RH and displays multiple peaks at long exposure times. This is consistent with stronger effective NP–NP and/or NP–protein interactions and reduced NP mobility. A dependence of aggregate size on the AuNP concentration in serum is also apparent: concentrated solutions tend to broaden and shift the distribution toward larger RH values (Figure 4). However, the optical property is not affected by the medium (Figure 5). These data show the extent to which measured properties of AuNPs in biological fluids are sensitive to temperature and NP concentration. Contrary to published studies, only minimal variations were observed in the values of RH for the small particles considered in this study.
Figure 3.

Hydrodynamic size and aggregation behavior of 5 nm AuNPs in water and serum media. (A) Continuous photon correlation spectroscopy histogram of 5 nm AuNPs in water at 37 °C; the inset shows the AuNP solution being measured. (B) TEM image shows individually dispersed AuNPs with a core size of 5 nm. (C) Hydrodynamic size of 5 nm AuNPs increases when exposed to serum, as is evident from the histogram; the inset shows the AuNP solution being measured. (D) TEM image shows an increased core size of AuNPs, and the data corroborates with the hydrodynamic data. These data show that both the hydrodynamic and core size of the AuNPs are dependent of the media to which they are exposed.
Figure 2.

Histograms showing hydrodynamic size distribution of 5 nm AuNPs at 4, 12, and 24 h time points from top to bottom. Size measurements of AuNP solutions were carried out in the presence of serum proteins at RT and 37 °C and were continuously measured for 24 h. Data shows absolutely no change in hydrodynamic size when measured in serum at RT (4 h, 10 nm; 12 h, 10 nm; 24 h, 10 nm), whereas there was an observable change in the AuNP size when measured in serum (4 h, 19 nm; 12 h, 18 nm; 24 h, 18 nm) as well as a broad size distribution.
Figure 4.
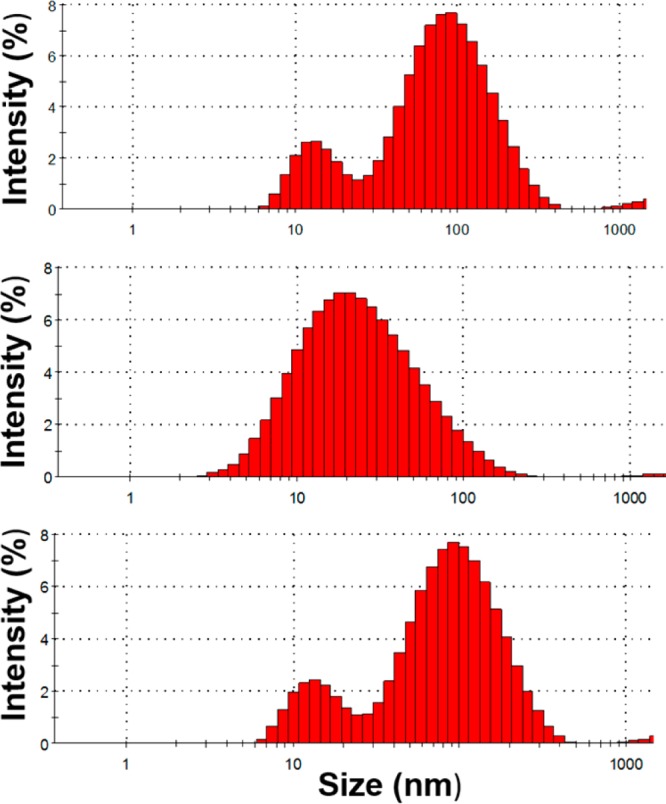
Histogram showing hydrodynamic size distribution of 5 nm AuNPs at 4, 12, and 24 h time points from top to bottom. A highly concentrated AuNP solution was exposed to serum proteins at 37 °C and continuously measured for 24 h. Data shows an uneven, inconsistent (4 h, 54 nm; 12 h, 18 nm; 24 h, 57 nm), and broad size distribution.
Figure 5.

UV–vis absorption spectroscopy of 5 nm AuNPs. (A) UV spectra of 5 nm AuNPs in water showing a peak at 520 nm. (B) UV spectra of 5 nm AuNPs exposed to serum media showing the characteristic peak at 520 nm. Media did not influence the optical property of the AuNPs.
IV-ii. Computational Results
Current instrumentation has a number of limitations37 that prevent obtaining a detailed molecular picture of the aggregation mechanism and the role of serum proteins in the formation/stabilization of aggregates. Consequently, it is difficult to predict the behavior of NPs with different sizes, surfactants, and concentrations. The computer simulations carried out here are designed to fill this experimental gap and provide molecular insight into the aggregation process. The model used, although a simplification of complex physiological serum, allows a realistic representation of the medium with the desired level of structural detail, including the anisotropy of the charge distribution on the albumin proteins.
The simulations are carried out in the σ–ε–c space, where neither the NP material nor the nature of the surfactant is specified a priori. Thus, specific behaviors can be identified in different regions of the σ–ε–c space, which can then be used for reverse-engineering NPs with desired aggregation properties.28,38 Molecular features inferred from in vitro experiments, such as the number of proteins bound to the NPs and the modes of association, depend on the properties of the NPs and the conditions of the solution.9−12,14,39 To identify the origin of this sensitivity, simulations are first carried out in a diluted solution of bare AuNPs of 5 nm in diameter under conditions resembling physiological serum. Albumin contains many acidic and basic amino acids on its surface and can easily bind inorganic ions and fatty acids that change its overall charge. Two charge distributions are considered here: one corresponding to physiological (blood) pH 7 (total charge −15 e), and one corresponding to the isoelectric pH 4.7 (IEP). Most experimental conditions fall between these limits.
Advanced algorithms exist to calculate hydrodynamic parameters of bodies of arbitrary shapes.40 Hydrodynamic radii (RH) are estimated here from a generalization of the Kirkwood equation for a cluster of n spherical units, as41
| 6 |
where ρi is the hydrodynamic radius of unit i, rij is the distance between i and j, and ⟨ ⟩ is an ensemble average over all the conformations of the cluster.42 Figure 6 shows the hydrodynamic radii of AuNP/albumin aggregates obtained from the simulations. The remarkable differences in behavior between both charge distributions are qualitative as well as quantitative and are attributed only to the electrostatic interactions between the proteins. At pH 7 (Figure 6A), the aggregates contain a small number of proteins bound to a central particle (typically two or three and never more than five; cf. histogram in the inset of Figure 6B). Because of the electrostatic repulsion between the proteins, they tend to accommodate in specific geometric patterns that minimize the system’s free energy by maximizing the average protein–protein distances. This conformational requirements yield characteristic peaks in the RH distributions, which are expected to broaden or coalesce in a polydisperse solution. In the conventional representation of albumin as an equilateral triangular prism, the base of the prism is typically in contact with the NP surface, whereas the tip tends to point away from it; this distribution is similar to that proposed for polymer-coated FePt and CdSe/ZnS nanoparticles.39 Therefore, for a given number of bound proteins, this arrangement maximizes RH. At the IEP, the number of albumins in the aggregates increases significantly (Figure 6B), with six or seven proteins most frequently bound to the central particle, although up to 13 can be accommodated. In the absence of electrostatic repulsion between the proteins, their spatial distributions are now more compact, which tends to decrease RH for a given number of bound proteins. No preferential binding modes can be identified in this case, because the weak protein–protein interactions perturb any arrangement that would otherwise be selected. The variability of binding modes yields a single, broad peak in the RH distribution centered at ∼6.5 nm. These results show how the properties of the albumin layer (and, presumably, of other protein corona) depend on the pH and the ionic strength of the solution, as both determine the charge distribution on the proteins.
Figure 6.
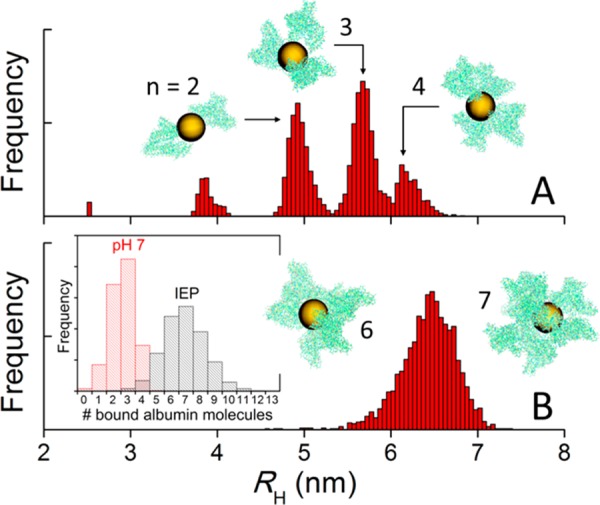
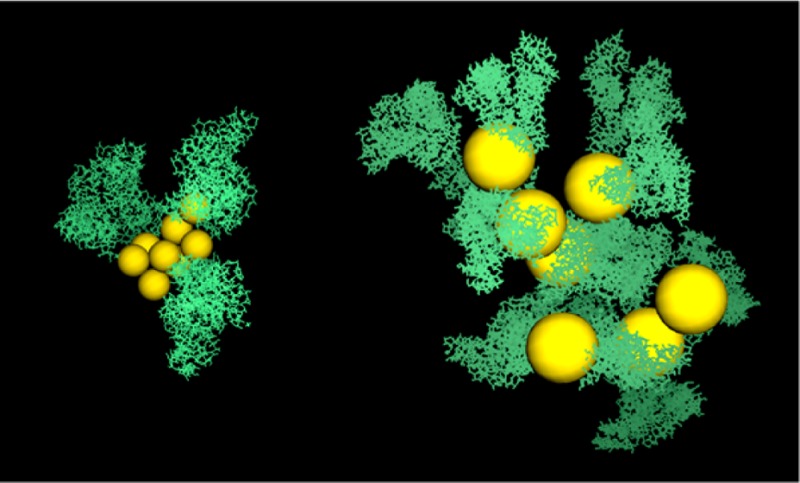
Hydrodynamic radii (RH) and morphology of AuNP/albumin complexes at physiological pH (A) and at the isoelectric point (IEP) (B) in a diluted nanoparticle solution at 37 °C and physiological albumin concentration, obtained from canonical Monte Carlo simulations. The nanoparticles have a diameter of 5 nm. The geometric arrangement of proteins and the number (n) of proteins bound to the nanoparticle (histograms in inset of panel B) depend on the charge distribution on the protein, which is controlled by the pH and the ionic strength of the solution. The aggregates shown in the insets are snapshots taken from the equilibrated simulations and are drawn to scale (yellow, AuNPs; green, atomic representation of albumin obtained after fine graining).
The qualitative features of the single-AuNP/albumin aggregates hold for NPs of different sizes and surfactants. Increasing the NP concentration, however, leads to the formation of stable multi-NP/albumin aggregates. The structural features of such aggregates, the conditions under which they emerge, and the role of albumin in their formation and stabilization are studied next. Figure 7 shows the general effects of albumin on a concentrated solution of NPs at physiological concentration and temperature. Two characteristic NP sizes are shown, with diameters of 2 and 5 nm, at a concentration of 2 × 1017 particles mL–1. The particles are coated with a surfactant that yields an effective NP–NP attraction of 2.5 kcal mol–1. In pure water at 37 °C, large clusters containing up to ∼40 (for 2 nm) and ∼60 (for 5 nm) particles are formed at equilibrium (Figures 7A and 8A). The peak centered at ∼10 nm in the RH distribution of the 5 nm NPs (Figure 8D) is consistent with the CPCS data (Figure 3A). When the NPs are diluted in serum, however, only small NP clusters are observed (Figures 7B and 8B), mainly dimers, although trimers and tetramers can be detected in trace amounts, as evidenced by the peaks in the size distributions (Figures 7E and 8E). This dissolution mechanism of NP clusters in serum was observed in all of the simulations (Figure 9) and is likely to be a general feature of NPs with sizes comparable to that of albumin. It seems that both excluded-volume effects of the crowded medium and electrostatic repulsion between the crowding agents play a role. This is supported by two observations: a small fraction of multimers remains in the case of 2 nm NPs (Figure 7E), but not in the solution of 5 nm NPs, as the smaller particles can more easily accommodate in the interstitial space between the proteins. In addition, the particles tend to form slightly larger clusters at the IEP (Figure 9), as uncharged proteins can be more easily displaced to create transient cavities to form larger clusters; at pH 7, this would be energetically unfavorable because the displaced proteins would need to move closer to other charged proteins.
Figure 7.

Effect of albumin on a concentrated monodisperse solution of nanoparticles with a diameter of 2 nm at 37 °C and pH 7. (A) NPs form compact clusters in pure water. (B) Clusters dissolve in the presence of albumin at physiological concentrations. (C) Disperse clusters are actually part of large, morphologically irregular cluster/albumin aggregates stabilized by the proteins that bind to the outer layer of the compact clusters. (D) Atomistic representation of a representative aggregate in the 2 nm NPs serum solution (snapshot from the simulation). Hydrodynamic radii of the NP clusters and aggregates are displayed in panels D–F (n and N are the number of albumin molecules and the number of NPs in the aggregates, respectively). At the IEP, the clusters are slightly larger and the aggregates are more compact, as expected from reduced electrostatic repulsion between proteins (cf. Figure 9).
Figure 8.

Same as in Figure 7 except that the 5 nm NP solution is evaluated. In this case, the larger aggregates are porous, highly irregular structures containing both NP monomers and clusters (multimers) stabilized by a network of proteins (G and H, snapshots from the simulations; both aggregates shown contain the same number of NPs as that in Figure 7D but a very different number of proteins). Monomers are omitted in panel B but are shown in panel C as part of the aggregates.
Figure 9.
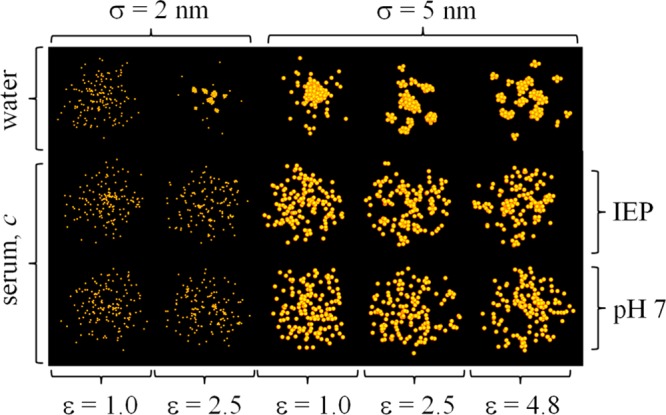
Snapshot of the equilibrated simulations for the ε–σ–c points discussed in the text for a NP concentration c ∼ 2 × 1017 particles mL–1. Specific behaviors can be identified in different regions of the ε–σ–c space, which can be used to guide the design of NPs (size and coating material) for given solution conditions.
A closer inspection shows, however, that the seemingly dispersed NP clusters in serum are actually part of extended cluster/albumin aggregates of highly irregular morphologies (Figures 7C and 8C) that are stabilized by the proteins. The presence of such aggregates tends to shift the hydrodynamic size distribution toward values larger than those in pure water (Figures 7F and 8F), a result also consistent with CPCS data (Figure 3). A detailed structural analysis reveals the presence of two kinds of aggregates that are most critically dependent on core size and coating material: (1) aggregates formed by small NP clusters that bind albumin directly to their outer surfaces, thereby creating a protein corona that further stabilizes the compact core of the cluster (Figure 8G) (this kind of aggregate is more common for the smaller NPs and/or lower concentrations), and (2) aggregates formed by unclustered NPs (monomers) and small clusters that are brought together and stabilized by albumin, which is now an integral part of the aggregates (Figure 8G,H); these aggregates are common for larger NPs and/or higher NP concentrations. These mixed aggregates are likely the colloids observed in the CPCS and TEM experiments (Figure 3), as they could survive long enough for detection and contain a sufficient amount of AuNP for optical contrast. The porosity of these aggregates results from the extended network of charged proteins in their interior that mediate and stabilize the cluster–cluster interactions. These aggregates are expected to be more labile than aggregates that contain compact clusters, which tend to have higher cohesive energy. These observations may have clinical significance: rational design of porous, labile aggregates may resolve two obstacles for successful therapeutic applications, namely, toxicity (even the largest aggregates contain a rather small number of NPs) and degradation of the bioactive coating (albumin, which acts as a glue to maintain the integrity of the aggregates, prevents the medium from accessing the buried NP surfaces, at least until the aggregates dissolve). Similar results were observed in simulations at the IEP, although, in this case, the aggregates are less porous and tend to have smaller hydrodynamic radii. This is a direct consequence of the reduced electrostatic repulsion between proteins and is consistent with the results obtained at high dilution (Figure 6). It is finally noted that over the course of the simulations large aggregates that develop in close proximity tend to merge and break down at equilibrium. These are telltale signs of speciation events that may occur during the system dynamics (not studied here), which could also be controlled through a judicious choice of the system parameters.
V. Conclusions
Recently, small AuNPs (∼2–5 nm) have shown extraordinary potential for biomedical applications as inert imaging agents with better body clearance. For their successful clinical translation, the physiological fate of NPs needs to be characterized. Given the size of the NPs, it is a technical challenge to predict the nature of their behavior under physiological conditions. Using continuous photon correlation spectroscopy, it was shown here that AuNP aggregates develop under near-physiological conditions, but they are small and may thus show improved clearance from the body. A multiscaling approach was used to model albumin at physiological concentrations (∼30% in volume), which allowed a systematic computational study and collection of statistically meaningful data. The algorithm is general and can be equally applied to other serum proteins, such as histone, fibrinogen, and globulins, to simulate increasingly realistic serum environments. The limitation is imposed by the computer resources, which determines the level of coarsening, and by the availability of experimental data on NP–protein interactions for use in the model parametrization.19,43 It was found that, unlike aggregates formed in water, which tend to be rather compact and stable, albumin is an integral component of the aggregates, which are structurally porous. The stability of the aggregates is determined by the NP–albumin interactions, which can be controlled by a judicious choice of the coating molecules and density. It was found that the distribution of albumin in the aggregates depends critically on the charge distribution on the proteins, which can be controlled through the pH and the ionic strength of the solution. Experimental data, such as the zeta potential and DLS spectra, are typically interpreted on the basis of simple models of the colloids (e.g., unstructured spherical particles with smooth surfaces). Attention to the irregular morphology and the internal structure of the aggregates found in this study may help to develop improved models for data interpretation. The structural features of the aggregates may lead to reduced toxicity and prevent or delay coating degradation by protecting the NP surfaces buried in the aggregate interior. The simulations suggest that adjusting the concentration of small NPs and the conditions of the albumin solution prior to intravenous administration may help to preserve the properties of the functionalized NPs in the bloodstream and thus to rationally design clinically useful nanomaterials.
Acknowledgments
This study utilized the high-performance computer capabilities of the Biowulf PC/Linux cluster at the NIH. This work was supported, in part, by the Intramural Research Program (IRP) of the National Institute of Biomedical Imaging and Bioengineering (NIBIB), and Center for Information Technology at the National Institutes of Health (NIH). A.B. was supported by a postdoctoral fellowship from NIH-NIBIB/NIST NRC. The authors thank Peter Steinbach for reading the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Dreaden E. C.; Alkilany A. M.; Huang X.; Murphy C. J.; El-Sayed M. A. The Golden Age: Gold Nanoparticles for Biomedicine. Chem. Soc. Rev. 2012, 41, 2740–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykman L.; Khlebstov N. Gold Nanoparticles in Biomedical Applications: Recent Advances and Perspectives. Chem. Soc. Rev. 2012, 41, 2256–2582. [DOI] [PubMed] [Google Scholar]
- Shang L.; Nienhaus G. U. Gold Nanoclusters as Novel Optical Probes for in Vitro and in Vivo Fluorescence Imaging. Biophys. Rev. 2012, 4, 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monopoli M. P.; Aberg C.; Salvati A.; Dawson K. A. Biomolecular Coronas Provide the Biological Identity of Nanosized Materials. Nat. Nanotechnol. 2012, 7, 779–786. [DOI] [PubMed] [Google Scholar]
- Nel A. E.; Madler L.; Velegol D.; Xia T.; Hoek E. M. V.; Somasundaran P.; Klaessig F.; Castranova V.; Thompson M. Understanding Biophysicochemical Interactions at the Nano–Bio Interface. Nat. Mater. 2009, 8, 543–557. [DOI] [PubMed] [Google Scholar]
- Tenzer S. Rapid Formation of Plasma Protein Corona Critically Affects Nanoparticle Pathophysiology. Nat. Nanotechnol. 2013, 8, 772–781. [DOI] [PubMed] [Google Scholar]
- Zhou C.; Long M.; Qin Y.; Sun X.; Zheng J. Luminescent Gold Nanoparticles with Efficient Renal Clearance. Angew. Chem, Int. Ed. 2011, 50, 3168–3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C.; Hao G.; Thomas P. G.; Liu J.; Yu M. H.; Sun S.; Orhan K.; Sun X.; Zheng J. Near-Infrared Emitting Radioactive Gold Nanoparticles with Molecular Pharmacokinetics. Angew. Chem, Int. Ed. 2012, 51, 10118–10122. [DOI] [PubMed] [Google Scholar]
- Lacerda S. H.; Park J. J.; Meuse C.; Pristinski D.; Becker M. L.; Karim A.; Douglas J. F. Interaction of Gold Nanoparticles with Common Human Blood Proteins. ACS Nano 2010, 4, 365–379. [DOI] [PubMed] [Google Scholar]
- Gobizova V. V.; Sergeeva I. A.; Petrova G. P.; Priezzhev A. V.; Khlebtsov N. G. Interaction of Albumin and G-Globulin Molecules with Gold Nanoparticles in Water Solutions. Moscow Univ. Phys. Bull. 2011, 66, 449–452. [Google Scholar]
- Tsai D. H.; DelRio F. W.; Keene A. M.; Tyner K. M.; MacCuspie R. I.; Cho T. J.; Zachariah M. R.; Hackley V. A. Adsorption and Conformation of Serum Albumin Protein on Gold Nanoparticles Investigated Using Dimensional Measurements and in Situ Spectroscopic Methods. Langmuir 2011, 27, 2464–2477. [DOI] [PubMed] [Google Scholar]
- Dominguez-Medina S.; McDonough S.; Swanglap P.; Landes C. F.; Link S. In Situ Measurement of Bovine Serum Albumin Interaction with Gold Nanospheres. Langmuir 2012, 28, 9131–9139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedervall T.; Lynch I.; Lindman S.; Berggard T.; Thulin E.; Nilsson H.; Dawson K. A. Understanding the Nanoparticle–Protein Corona Using Methods To Quantify Exchange Rates and Affinities of Proteins for Nanoparticles. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 2050–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli I.; Alam S.; Patel B.; Mukhopadhyay A. Interaction and Diffusion of Gold Nanoparticles in Bovine Serum Albumin Solutions. Appl. Phys. Lett. 2013, 102, 2037051. [Google Scholar]
- Thanh N. T. K.; Green L. A. W. Functionalisation of Nanoparticles for Biomedical Applications. Nano Today 2010, 5, 213–230. [Google Scholar]
- Manson J.; Kumar D.; Meenan B. J.; Dixon D. Polyethylene Glycol Functionalized Gold Nanoparticles: The Influence of Capping Density on Stability in Various Media. Gold Bull. 2011, 44, 99–105. [Google Scholar]
- Walkey C. D.; Olsen J. B.; Guo H.; Emili A.; Chan W. C. Nanoparticle Size and Surface Chemistry Determine Serum Protein Adsorption and Macrophage Uptake. J. Am. Chem. Soc. 2012, 134, 2139–47. [DOI] [PubMed] [Google Scholar]
- Xie J.; Lee S.; Chen X. Nanoparticle-Based Theranostic Agents. Adv. Drug Delivery Rev. 2010, 62, 1064–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J. J. The Interaction of Proteins with Solid Surfaces. Curr. Opin. Struct. Biol. 2004, 14, 110–115. [DOI] [PubMed] [Google Scholar]
- Chen Y. S.; Hung Y. C.; Liau I.; Huang G. S. Assesment of the in Vivo Toxicity of Gold Nanoparticles. Nanoscale Res. Lett. 2009, 4, 858–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters T., Jr.All About Albumin: Biochemistry, Genetics, and Medical Applications; Academic Press: San Diego, 1996. [Google Scholar]
- Ferrer M. L.; Duchowicz R.; Carrasco B.; Garcia de la Torre J.; Acuna A. U. The Conformation of Serum Albumin in Solution: A Combined Phosphorescence Depolarization-Hydrodynamic Modeling Study. Biophys. J. 2001, 80, 2422–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapotschnikow P.; Pool R.; Vlugt T. J. H. Molecular Simulations of Interacting Nanocrystals. Nano Lett. 2008, 8, 2930–2934. [DOI] [PubMed] [Google Scholar]
- Hamaker H. C. The London–Van Der Waals Attraction between Spherical Particles. Physica 1937, 10, 1058–1072. [Google Scholar]
- Patel N.; Egorov S. A. Interactions between Sterically Stabilized Nanoparticles in Supercritical Fluids: A Simulation Study. J. Chem. Phys. 2007, 126, 054706. [DOI] [PubMed] [Google Scholar]
- Cui M.; Yang X. Molecular Simulation of Gold Nanoparticle Dispersion and Aggregation in Supercritical CO2. J. Mater. Sci. 2012, 48, 891–899. [Google Scholar]
- Jimenez A.; Sarsa A.; Blazquez M.; Pineda T. A Moleular Dynamics Study of the Surfactant Surface Density of Alkanethiol Self-Assembled Monolayer on Gold Nanoparticles as a Function of the Radius. J. Phys. Chem. C 2010, 114, 21309–21314. [Google Scholar]
- Kim T.; Lee K.; Gong M. S.; Joo S. W. Control of Gold Nanoparticle Aggregation by Manipulation of Interparticle Interactions. Langmuir 2005, 21, 9524–9528. [DOI] [PubMed] [Google Scholar]
- Hassan S. A.; Mehler E. L.. In Silico Approaches to Structure and Function of Cell Components and Their Assemblies: Molecular Electrostatics and Solvent Effects. In Comprehensive Biophysics; Egelman E., Ed.; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Petkov V.; Peng Y.; Williams G. J.; Huang B.; Tomalia D.; Ren Y. Structure of Gold Nanoparticles Suspended in Water Studied by X-ray Diffraction and Computer Simulations. Phys. Rev. B 2005, 72, 1954402. [Google Scholar]
- Hassan S. A.; Steinbach P. J. Water-Exclusion and Liquid-Structure Forces in Implicit Solvation. J. Phys. Chem. B 2011, 115, 14668–146682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone A.; Pant H.; Hassan S. A. Specific and Non-Specific Protein Association in Solution: Computation of Solvent Effects and Prediction of First-Encounter Modes for Efficient Configurational Bias Monte Carlo Simulations. J. Phys. Chem. B 2013, 117, 12360–12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks C. L. 3rd; Mackerell A. D. Jr.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; et al. Charmm: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paliwal A.; Asthagiri D.; Abras D.; Lenhoff A. M.; Paulaitis M. E. Light-Scattering Studies of Protein Solutions: Role of Hydration in Weak Protein–Protein Interactions. Biophys. J. 2005, 89, 1564–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan S. A.Implicit Treatment of Solvent Dispersion Forces in Protein Simulations J. Comput. Chem. [Online early access]. DOI: 10.1002/jcc.23655. Published Online: June 12, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimbone M.; Calcagno L.; Messina G.; Paeri P.; Compagnini G. Dynamic Light Scattering and UV–Vis Spectroscopy of Gold Nanoparticles Solution. Mater. Lett. 2011, 65, 2906–2909. [Google Scholar]
- Tomaszewska E.; Soliwoda K.; Kadziola K.; Tkacz-Szczesna B.; Celishowski G.; Cichomski M.; Szmaja W.; Grobelny J. Detection Limits of DLS and UV–Vis Spectroscopy in Characterization of Polydisperse Nanoparticles Colloids. J. Nanomater. 2013, 2013, 313081. [Google Scholar]
- Treuel L.; Brandholt S.; Maffre P.; Wiegele S.; Shang L.; Nienhaus G. U. Impact of Protein Modification on the Protein Corona on Nanoparticles and Nanoparticle–Cell Interactions. ACS Nano 2014, 8, 503–513. [DOI] [PubMed] [Google Scholar]
- Rocker C.; Potzl M.; Zhang F.; Parak W. J.; Nienhaus G. U. A Quantitative Fluorescence Study of Protein Monolayer Formation on Colloidal Nanoparticles. Nat. Nanotechnol. 2009, 4, 577–580. [DOI] [PubMed] [Google Scholar]
- Garcia de la Torre J.; Navarro S.; Lopez-Martinez C.; Diaz F. G.; Lopez-Cascales J. J. Hydro: A Computer Program for the Prediction of Hydrodynamic Properties of Macromolecules. Biophys. J. 1994, 67, 530–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan S. A. Morphology of Ion Clusters in Aqueous Electrolytes. Phys. Rev. E 2008, 77, 031501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan S. A. Computer Simulation of Ion Cluster Speciation in Concentrated Aqueous Solutions at Ambient Conditions. J. Phys. Chem. B 2008, 112, 10573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainrub A.; Pettitt B. M. Accurate Prediction of Binding Thermodynamics for DNA on Surfaces. J. Phys. Chem. B 2011, 115, 13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda S.; Nishinari K. Intermolecular Forces in Bovine Serum Albumin Solutions Exhibiting Solidlike Mechanical Behaviors. Biomacromolecules 2000, 1, 757–763. [DOI] [PubMed] [Google Scholar]