

Abstract
Adolescent rats display reduced sensitivity to many dysphoria-related effects of alcohol (ethanol) including motor ataxia and sedative hypnosis, but the underlying neurobiological factors that contribute to these differences remain unknown. The cyclic adenosine monophosphate (cAMP)-dependent protein kinase A (PKA) pathway, particularly the type II regulatory subunit (RII), has been implicated in ethanol-induced molecular and behavioral responses in adults. Therefore, the current study examined cerebral cortical PKA in adolescent and adult ethanol responses. With the exception of early adolescence, PKA RIIα and RIIβ subunit levels largely did not differ from adult levels in either whole cell lysate or P2 synaptosomal expression. However, following acute ethanol exposure, PKA RIIβ P2 synaptosomal expression and activity were increased in adults, but not in adolescents. Behaviorally, intracerebroventricular administration of the PKA activator Sp-cAMP and inhibitor Rp-cAMP prior to ethanol administration increased adolescent sensitivity to the sedative-hypnotic effects of ethanol compared to controls. Sp-cAMP was ineffective in adults whereas Rp-cAMP suggestively reduced loss of righting reflex (LORR) with paralleled increases in blood ethanol concentrations. Overall, these data suggest that PKA activity modulates the sedative/hypnotic effects of ethanol and may potentially play a wider role in the differential ethanol responses observed between adolescents and adults.
Keywords: protein kinase A, cAMP, ethanol, adolescence
Introduction
Although alcohol (ethanol) is consumed worldwide, excessive use and abuse may contribute to dependence and subsequent addiction. The frequency of ethanol abuse among adolescents is highly recognized. Adolescents have heavier drinking rates than adults, and 25% of high school seniors self-report having already consumed more than five drinks in a single period in the past month (Johnston, O’Malley, Bachman, & Schulenberg, 2009; Substance Abuse and Mental Health Services Administration, 2008). Importantly, the initiation of alcohol use disorders occurs more frequently during adolescence relative to any other age group (Brown et al., 2009). In addition to increased ethanol consumption, adolescents appear to be less sensitive than adults to ethanol’s dysphoria-related effects such as sedation and intoxication, with such effects being observed across vertebrates (Behar et al., 1983; Silveri & Spear, 1998; Spear & Varlinskaya, 2010). Reduced ethanol sensitivity and increased ethanol consumption likely make adolescents more susceptible to ethanol’s neurotoxic effects at higher ethanol concentrations as well as increase the propensity for abuse and addiction later in life. In fact, research suggests that adolescent ethanol exposure may potentially “lock-in” adolescent ethanol responses into adulthood, thereby further increasing vulnerability to ethanol abuse later in life (Fleming, Acheson, Moore, Wilson, & Swartzwelder, 2012).
Despite these well-known age-related differences in ethanol responding, the underlying mechanisms are not well understood. Ethanol’s behavioral effects are mediated through the central nervous system (CNS). In fact, modulating intracellular cell-signaling pathways via G-protein coupled receptors (GPCR) and protein kinases, particularly the cAMP-dependent protein kinase (PKA), can impact a wide range of downstream targets thereby amplifying ethanol’s overall molecular effects (Diamond & Gordon, 1994). PKA is a heteromeric complex consisting of two regulatory subunits (RIα, RIIα, RIβ, RIIβ) and two catalytic subunits (Cα and Cβ) (Scott, 1991). Within each complex, the regulatory and catalytic subunits are generally homomeric (Skalhegg & Tasken, 2000). Type II regulatory subunits have received particular attention due to their expression within the CNS (Brandon, Idzerda, & McKnight, 1997). Moreover, type I regulatory subunits commonly localize to the cytoplasm whereas type II subunits are distributed near cell membranes and synapses (Coe, Dohrman, Constantinescu, Diamond, & Gordon, 1996). Such localization allows PKA to play a critical role in neurotransmission by regulating synaptic receptors as well as vesicular recruitment and release (Brandon et al., 1997; Evans & Morgan, 2003; Kuromi & Kidokoro, 2000; Lonart et al., 2003).
Multiple lines of evidence associate PKA with ethanol’s behavioral and molecular responses during adulthood. For instance, gene knockout and pharmacological inhibition of the PKA pathway reduces sensitivity to the sedative-hypnotic effects of ethanol (Lai, Kuo, & Lin, 2007; Thiele et al., 2000). Hypnotic sensitivity may be modulated by specific regions, as studies suggest the cerebral cortex is involved, as assessed by using LORR, an assay that also serves as a gauge for overt ethanol sensitivity (Blednov et al., 2011; Blednov & Harris, 2009; Franks & Lieb, 1990). In fact, studies utilizing adenylyl cyclase knockout mice suggest that cerebral cortical but not cerebellar PKA activity is involved in ethanol’s sedative-hypnotic effects (Maas et al., 2005). Furthermore, there appears to be differential effects depending on which portion of the PKA pathway is influenced, as stimulatory G-protein knockout mice display increased sensitivity to ethanol’s sedative-hypnotic effects (Wand, Levine, Zweifel, Schwindinger, & Abel, 2001). Molecularly, ethanol increases PKA P2 synaptosomal translocation, regulation, and functional responses of cortical γ-amino-butyric acidA (GABAA) receptors, as well as GABA release at the cerebellar interneuron-Purkinje cell synapse (Carlson, Kumar, Werner, Comerford, & Morrow, 2013; Kelm, Criswell, & Breese, 2008; Kumar et al., 2012). Taken together, these findings suggest that ethanol exposure is capable of activating the PKA pathway that further modulates a major neurotransmitter system implicated in ethanol action, and ultimately modulates ethanol-related behavioral responses. While both type I and type II regulatory subunits were previously thought to respond equivalently to ethanol, studies now suggest that type II subunits are preferentially involved in modulating ethanol’s molecular signaling effects (Constantinescu, Gordon, & Diamond, 2002; Constantinescu, Wu, Asher, & Diamond, 2004), and recent evidence suggests that RIIβ-subunit synaptic translocation occurs much earlier than RIIα (Kumar et al., 2012). These studies, coupled with reduced ethanol sensitivity in PKA RIIβ knockouts, suggest a more prominent role for PKA RIIβ in acute ethanol sensitivity in adults (Thiele et al., 2000).
Given the overt similarities between PKA modulation, particularly the RIIβ subunit, and adolescent reduced ethanol responsiveness, we hypothesize that either developmental regulation of PKA or PKA activity itself modulates adolescent ethanol-related behaviors compared to adults. We predict that either one, or both, increases across age. In the current study, we focused on the cerebral cortex as it not only modulates ethanol sedative-hypnotic responses, but also displays some of the greatest delays in neuronal maturation. Prefrontal cortical synaptic pruning extends well into mid-adolescence, and in certain studies, lasts until 30 years of age in humans (Huttenlocher & Dabholkar, 1997; Miller et al., 2012; Petanjek et al., 2011), and likely impacts adolescent ethanol action. Therefore, in the present study we investigated cerebral cortical PKA type II regulatory subunit expression and activity during adolescence as well as in response to acute ethanol exposure. The PKA pathway was also pharmacologically modified to assess its involvement in age-dependent differences in ethanol-induced loss of righting reflex.
Materials and Methods
Subjects
Behavioral and molecular experimental subjects included adolescent (P28: early adolescence, P35: middle adolescence, and P42: late adolescence) and adult (P68–P75) male Sprague-Dawley rats (discussed in Spear, 2000). All animals were housed on a 12-hour light/dark cycle (on from 7:00 AM to 7:00 PM) following standards set by the Binghamton IACUC committee. Adolescent and adult rats were raised in an in-house breeding colony. Subjects were weaned at P21 and remained housed with littermates until experimental procedures. For the kinase activity study, adult animals were received from Taconic (Hudson, NY), pair-housed and allowed 1 week for acclimation. All subjects were housed in clear Plexiglas® containers with wood shaving bedding and crinkle paper for enrichment along with ad libitum access to food and water.
Drugs and reagents
For behavioral experimentation, the PKA activator Sp-Adenosine 3′,5′-cyclic monophosphorothioate triethylammonium salt hydrate (Sp-cAMP, 50 nmol/μL) and PKA inhibitor Rp-Adenosine 3′,5′-cyclic monophosphorothioate triethylammonium salt hydrate (Rp-cAMP, 50 nmol/μL) were purchased from Sigma Aldrich (St. Louis, MO). Control animals were administered artificial cerebrospinal fluid (aCSF; 124 mM NaCl, 2.5 mM KCl, 2 mM MgSO4, 2 mM CaCl2, 26 mM NaHCO3, 1.25 mM NaH2PO4, and 10 mM glucose; pH 7.4). Ethanol (Pharmco, Brookfield, CT) was administered intraperitoneally (i.p.) as 20% ethanol in saline.
Tissue preparations
For ontogenetic cerebral cortical PKA expression analyses, non-manipulated adolescent and adult rats were rapidly decapitated at postnatal day (P) 28, 35, 42, or 75. For acute ethanol assessment, non-manipulated adolescent (P35) and adult (P75) rats were injected with a 3.5 g/kg dose of 20% ethanol or saline and rapidly decapitated at various time points post-exposure. All brains were rapidly extracted and stored at −80° C for further analysis. Brains were coronally sliced into 2-mm sections and placed in ice-cold PBS for microdissection of cortical areas. For whole cell lysate (total) analysis, samples were lysed in a homogenization buffer (1% SDS, 1 mM EDTA, and 10 mM Tris) as noted elsewhere (Grosshans, Clayton, Coultrap, & Browning, 2002). To isolate the P2 synaptosomal fractions, samples were homogenized in a 0.32 M sucrose/PBS solution. Sample vials were then centrifuged at 1,000 × G for 10 min at 4° C. The supernatant was then transferred to a separate vial and spun at 12,000 × G for 20 min at 4° C. The resulting pellet (P2) was isolated and re-suspended in PBS. Protein concentrations were determined through use of a bicinchoninic acid (BCA) protein assay.
Western blot
Samples were denatured using an SDS-glycine sample buffer and 1 M DL-dithiothreitol (DTT), heated at 95° C for 7 min and then subjected to SDS-PAGE using 8–16% Novex tris-glycine gels (Invitrogen, Carlsbad, CA). Gels were transferred to PVDF membranes and placed in 1.0% BSA blocking buffer (0.1 M Tris buffer, 1 M NaCl, 0.1% tween-20, and 1.0% BSA) overnight. Membranes were probed using primary antibodies against PKA RIIα (BD Transduction Laboratories, San Jose, CA) or PKA RIIβ (BD Transduction Laboratories) overnight followed by a goat anti-mouse horseradish peroxidase conjugated secondary antibody (Thermo Scientific, Rockford, IL). Bands were visualized using enhanced chemiluminescence (ECL GE Healthcare, Amersham, UK) under nonsaturating conditions. PKA RIIα and RIIβ antibodies have previously been shown to have specificity based on immunoblot analysis conducted with RII knockout tissue (Carlson et al., 2013; Schillace et al., 2005; Zheng et al., 2013). Blots were then exposed to a β-actin specific antibody (Millipore, Billerica, MA) for normalization. β-actin was used for normalization, as densitometric intensities did not differ across age or exposure condition. All densitometric analyses were conducted using NIH Image-J. Data were calculated as percent change versus adult (P75) controls for ontogenetic studies. Due to age-related differences in PKA expression, adolescents (P35) and adults (P75) were analyzed on separate blots and comparisons made with age-matched controls for ethanol exposure experiments. All samples were run in duplicate/triplicate to validate reproducibility between runs, with the averages being reported. Data were analyzed using a one-way ANOVA with Dunnett’s post hoc test when appropriate.
PKA Kinase activity
Adolescent (P35) and adult (P75) animal subjects were administered a 3.5 g/kg dose of 20% ethanol or equal volume saline control (i.p.) and sacrificed at 15, 30, and 60-min time points post-exposure. The brain was rapidly extracted and the cerebral cortex was carefully isolated and homogenized in lysis buffer (20 mM MOPS, 50 mM β-glycerolphosphate, 5.0 mM EGTA, 2.0 mM EDTA, 1.0 mM DTT, 1.0 mM PMSF, with 1.0% NP40, phosphatase cocktail and protease cocktail) and divided into aliquots and stored at −80° C until use. Protein concentrations were determined through use of a BCA protein assay. PKA kinase activity was measured using a PKA specific kinase activity EIA (Enzo Life Sciences, Ann Arbor, MI) and analyzing absorbance at 450 nm according to manufacturer’s instructions. Due to samples being run in duplicate as well as the number of total runs, data were calculated as percent change vs. age-related control. A separate run of only adolescent and adult vehicle controls was also done. Data were analyzed using a Student’s t test or one-way ANOVA.
Intracerebroventricular cannulation (i.c.v.)
Ventricular administration of compounds has been shown to quickly penetrate multiple brain regions, including cortical areas (Gozzi et al., 2005; Proescholdt, Hutto, Brady, & Herkenham, 2000). Adolescent (P28) and adult (P68) subjects were anesthetized with 2.5–3.0% isoflurane and placed in a stereotaxic device. A 12.5-mm long steel cannula (Plastics One) was targeted to a lateral ventricle using the following coordinates from bregma: AP, −0.5 mm; ML, ±1.2 mm; DV, 2.0 mm for adolescent rats, and AP, −0.8 mm; ML, ±1.5 mm; DV, 2.5 mm for adult rats, and fitted with a guide cap. Subjects were administered Buprenex immediately post-surgery as well as 24 h later. Following surgery, all subjects were single-housed with added crinkle paper as enrichment for 1 week prior to testing. All animals were handled daily which included checking weights and cannula patency. Cannula placement was assessed 24 h following completion of behavioral testing by infusing India ink into the guide cannula following euthanasia and examining coronal slices immediately following extraction for ink dispersion. Animals with improper placement were removed from subsequent analysis. Our success rate for accurate cannula placement was ~97%.
Loss of righting reflex
The hypnotic effects of ethanol were assessed using the LORR paradigm as described elsewhere (Werner et al., 2006). Briefly, all animals were cannulated using the procedures outlined above. After 1 week (P35 and P75), subjects were infused with 100 nmol of Sp-cAMP, Rp-cAMP, or aCSF control at a rate of 1 μL/minute (2 μL total), with the injector needle left in for an additional minute to ensure that solutions did not backflow into the cannula. After 15 min, subjects were administered a hypnotic dose of ethanol (4.0 g/kg, i.p.). A slightly higher hypnotic dose was used for LORR than molecular analyses as data collected in this lab suggest that adolescent subjects experiencing surgery are persistently less sensitive to the 3.5 g/kg dose used for immunoblot analyses. Subjects were placed into plastic sleep troughs until able to right themselves. An animal was considered to have regained its righting reflex if capable of righting itself three consecutive times within 60 sec. The latency to lose and regain the righting reflex was recorded. Tail blood samples were taken immediately following recovery of righting reflex and stored at −80° C until further blood ethanol concentration (BEC) analyses. Adolescent and adult loss of righting duration were analyzed separately using a one-way ANOVA with Dunnett’s post hoc test when appropriate due to baseline differences in ethanol responding as has been done elsewhere (Varlinskaya & Spear, 2012). BECs were analyzed with a priori comparisons based on LORR latencies.
Results
PKA RII subunit expression varies during early adolescence
To assess ontogenetic PKA RII isoform expression, we compared early-, mid- and late-adolescents to adults (P28, P35, P42, and P75, respectively) in cortical whole cell lysates. Analysis of PKA RIIα expression yielded an effect of age (F(3,56) = 4.90, p < 0.001). Further analysis revealed ~30% less RIIα in early adolescents than in adults (p < 0.0001, Fig. 1A). For PKA RIIβ, an effect of age was also observed (F(3,57) = 5.56, p < 0.01), with PKA RIIβ subunit expression slightly elevated during mid-adolescence compared to adults (~14%, p < 0.01, Fig. 1B). No observable differences were noted during late adolescence for either subunit. Due to changes in total expression during adolescence, basal expression of type II regulatory subunits were further examined in P2 synaptosomal preparations. Analysis of PKA RIIα expression in cortical P2 synaptosomes revealed an effect of age (F(3,28) = 12.23, p < 0.001), with early- and mid-adolescent rats having ~35% and ~27% less PKA RIIα compared to adults, respectively (both p < 0.0001, Fig. 1C). Analysis of PKA RIIβ did not reveal any effect of age (Fig. 1D). Taken together, these results suggest that mainly PKA RIIα subunit expression is regulated during adolescence.
Figure 1.
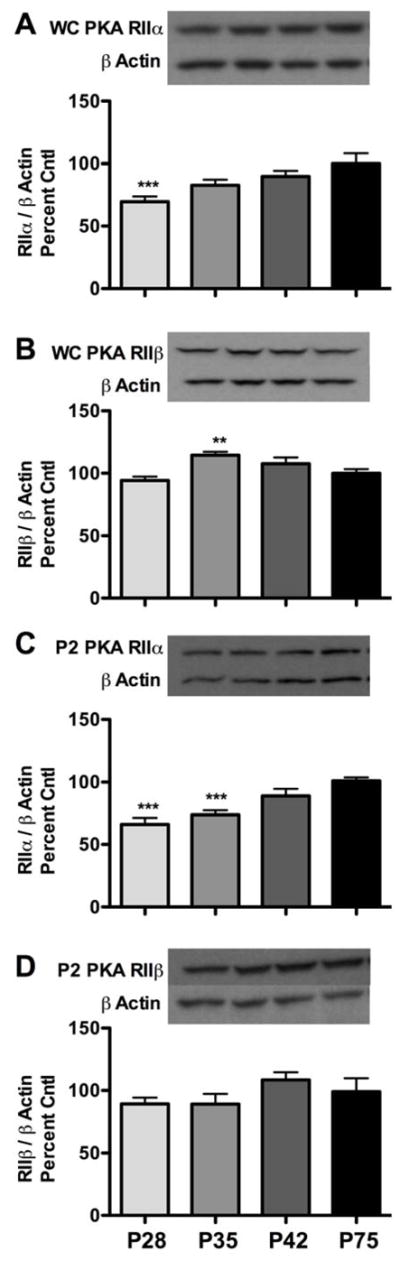
Ontogenetic expression of PKA RII subunits in total and cortical P2 synaptosomal fractions. (A and C) PKA RIIα subunit levels in early- (P28), mid- (P35), and late-adolescents (P42) compared to adult (P75) rats in both total and P2 synaptosomal fractions, respectively. (B and D) PKA RIIβ subunit levels in early-, mid-, and late-adolescents compared to adult (P75) rats in both total and P2 synaptosomal fractions, respectively. Representative blots are shown for each. Data are presented as mean ± SEM. WC = whole cell lysate. **p < 0.01, ***p < 0.0001 compared to adult controls; n = 14–16 per group for A and B and n = 7–8 per group for C and D.
Adult, but not mid-adolescent PKA RIIβ expression is increased in cortical P2 synaptosomes following ethanol exposure
We next wanted to determine if PKA type II regulatory subunits translocate to the P2 synaptosomal fraction following acute ethanol exposure in adolescents as previously reported in adults (Kumar et al., 2012). Given that various stages of adolescence differ in ethanol sensitivity compared to adults (Silveri & Spear, 1998), all subsequent ethanol studies utilized mid-adolescents (P35) and adults (P75). No differences were detected for PKA RIIα (Fig. 2A and B). However, for PKA RIIβ subunit expression, analysis of adults revealed an effect of ethanol exposure (F(2,33) = 5.14, p < 0.05). Further analysis indicated that ethanol increased P2 synaptosomal PKA RIIβ subunit expression in adults by 32.4 ± 13.0 and 37.0 ± 7.1% at 30 and 60 min, respectively, compared to controls (p < 0.05 for both, Fig. 2D). No effect of ethanol exposure was found for adolescents (Fig. 2C). Taken together, these data suggest that only adult PKA RIIβ P2 synaptosomal expression is increased following ethanol exposure.
Figure 2.
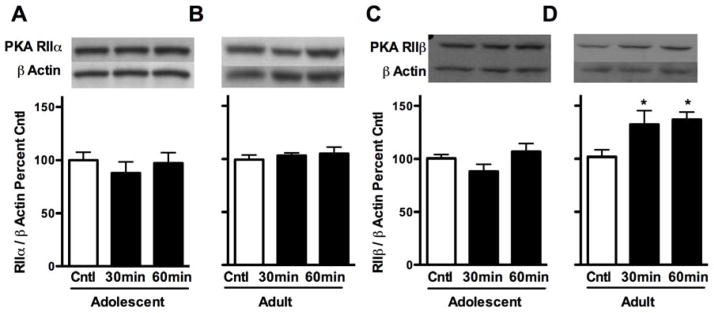
Effects of 3.5 g/kg ethanol exposure on PKA RII subunit expression in cerebral cortical P2 synaptosomal fractions in adolescent (P35) and adult rats (P75). (A and B) PKA RIIα subunit expression 30 and 60 minutes following ethanol exposure in adolescents and adults, respectively. (C and D) PKA RIIβ subunit expression 30 and 60 minutes following ethanol exposure in adolescents and adults, respectively. Representative blots are shown for each. Data are presented as mean ± SEM. *p < 0.05 compared to age-matched controls; n = 8 per group for adolescents and n = 10–13 for adults.
Adult, but not mid-adolescent PKA activity is increased following acute high-dose ethanol exposure
As PKA RII subunit translocation is an indirect measure of PKA activity, we directly assessed whether increased adult PKA kinase activity paralleled our P2 synaptosomal analysis. Initial analysis of PKA activity in mid-adolescents and adults revealed no basal differences (Fig. 3A). No effect of time post-ethanol exposure was found for adolescents (Fig. 3B). For adults, however, we found a strong trend toward an effect of time post-ethanol exposure (F(3,20) = 2.76, p = 0.06) (Fig. 3C). This potential finding was further analyzed by collapsing data from the three time points post-ethanol as a means to more broadly assess the impact of ethanol with time as a factor per se. The analysis revealed that ethanol increased PKA activity by 49.3% irrespective of time (p < 0.01) in adults, but not adolescents. Overall, these data indicate that ethanol increases PKA activity in adults whereas adolescent PKA activity remains unchanged.
Figure 3.
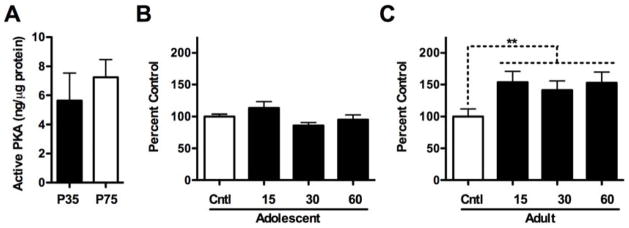
Effects of 3.5 g/kg ethanol exposure on PKA kinase activity following ethanol exposure. (A) Analysis of basal PKA activity in adolescents (P35) and adults (P75). (B and C) Effects of ethanol on PKA activity in adolescents and adults, respectively. **p < 0.01 compared to age-matched controls; n = 6 per group.
Age-dependent effects of PKA activity on ethanol-induced LORR in mid-adolescents and adults
Pharmacological modulation of PKA has been shown to alter ethanol-induced sleep times in adults (Kumar et al., 2012; Lai et al., 2007). Compared to adults, however, mid-adolescents are less sensitive to the sedative-hypnotic effects of high-dose ethanol exposure (Silveri & Spear, 1998). Due to the observed age-dependent increases in kinase activity following acute ethanol exposure, we next assessed whether pharmacologically modulating PKA activity prior to ethanol would alter adolescent LORR sensitivity. Analysis of adolescent LORR duration revealed an effect of treatment (F(2,30) = 4.94, p < 0.05). Further analysis revealed that either increasing or decreasing PKA activity increased LORR duration in adolescents by ~46% and 58%, respectively, compared to controls (p < 0.05, Fig. 4A). The effects of central Sp-cAMP are likely independent of ethanol clearance as a priori comparisons suggested a trend for lower BECs in Sp-cAMP-treated subjects compared to vehicle controls (169.5 ± 7.3 and 183.7 ± 6.7, respectively, p = 0.08) after regaining righting reflex. Interestingly, LORR differed between vehicle and Rp-cAMP-treated adolescent rats, whereas BECs did not (190.1 ± 13.3; p > 0.30). For adults, LORR did not differ following administration of Sp- or Rp-cAMP; however, a suggestive decrease was noted for Rp-cAMP-treated adults (Fig. 4B). In agreement, Rp-cAMP-treated adults had higher BECs compared to vehicle controls (p < 0.05), upon regaining their righting reflex (Veh, 182.9 ± 3.3; Rp-cAMP, 212.9 ± 13.0). Sp-cAMP-treated adults did not differ from vehicle controls (Sp-cAMP, 185.6 ± 12.7; p > 0.80). Sp- and Rp-cAMP did not cause hypnotic effects alone at either age (not shown). Overall, these data suggest that elevating PKA activity increases adolescent ethanol sensitivity, whereas reducing PKA activity decreased adult sensitivity.
Figure 4.
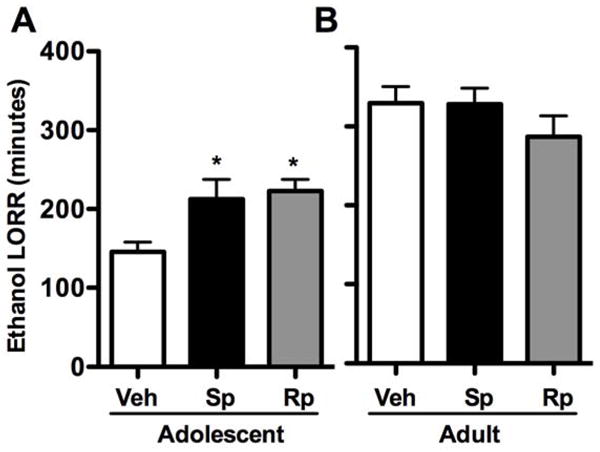
Effects of PKA activity on ethanol-induced loss of righting reflex. aCSF (Veh), PKA activator Sp-cAMP (Sp), or PKA inhibitor Rp-cAMP (Rp) was administered intracerebroventricularly 15 minutes prior to a 4.0 g/kg ethanol dose in (A) adolescents (P35) and (B) adults (P75). Data are presented as mean ± SEM. *p < 0.05 compared to age-matched vehicle controls; n = 8–12 per group.
Discussion
In the present study, we observed decreased cortical PKA RIIα during early- and mid-adolescence, but not RIIβ; however, differences were not detected during late adolescence when altered ethanol behavioral responses are still reported (Silveri & Spear, 1998). Analysis of cortical PKA translocation to the synapse following a high-dose ethanol exposure revealed that PKA RIIβ was increased in adults, but not in adolescents. Direct assessment of cortical PKA kinase activity also revealed that adult, but not adolescent, PKA activity was increased following high-dose ethanol exposure. Taken together, the expression and activity data suggest that adolescent cortical PKA activity is not increased by acute high-dose ethanol exposure. Further analysis indicated that increasing central PKA activity increased ethanol-induced LORR sensitivity for adolescents, whereas reducing PKA activity reduced sensitivity in adult animals.
Characterization of cortical PKA regulatory subunits RIIα and RIIβ during early-, mid-, and late-adolescent periods in both cortical whole cell lysates and P2 synaptosomal fractions revealed some developmental differences. PKA RIIα regulatory subunit expression was noted to be lower in both preparations during early adolescent periods. Although it is possible that reductions in total PKA RIIα subunits could contribute to decreased ethanol sensitivity during early adolescence, such a possibility is mitigated because total PKA RIIα levels are similarly reduced during mid-adolescence. Furthermore, neither PKA RIIα nor RIIβ expression during late adolescence differed in expression compared to adults when age-related differences in ethanol-related behaviors are still prominent (Silveri & Spear, 1998). In addition, although total RIIβ expression was slightly elevated during mid-adolescence compared to adults, this minor increase in magnitude likely has minimal biological significance as neither basal PKA activity nor RIIβ P2 synaptosomal expression differed between adolescents and adults. In fact, RIIβ P2 synaptosomal expression did not differ across adolescence relative to adults, further suggesting that basal PKA activity related to RIIβ during adolescence may be similar to adults. Taken together, this does not agree with our initial hypothesis regarding differences in basal PKA expression accounting for ontogenetic differences.
Given the overall absence of differences in baseline PKA expression throughout adolescence, we next investigated the second part of our hypothesis by determining whether ethanol-induced regulation of P2 synaptosomal PKA RII levels and PKA activity differed between adolescents and adults. Similar to previous in vivo and in vitro work (Carlson et al., 2013; Kumar et al., 2012), we found increases in P2 synaptosomal PKA RIIβ following ethanol exposure in adults, but no changes in PKA RIIα compared to controls. Conversely, neither PKA RIIα nor RIIβ subunit expression differed in adolescents following ethanol exposure, suggesting that adolescent PKA translocation is not induced by high-dose ethanol exposure. Such changes in adult RIIβ but not RIIα 60 min after ethanol exposure replicates previously published data (Kumar et al., 2012). Given that our prior work indicates that increases in RIIα are delayed after the time points examined in the current study, it is possible that adolescents may exhibit the same effect, but further studies would have to be conducted to determine whether such increases also occur in adolescents. However, earlier increases in PKA RIIβ than in RIIα indirectly suggests that RIIβ has a greater role in initial ethanol sensitivity. This is further supported by knockouts being less sensitive (Thiele et al., 2000), which reduces the rationale to assess RIIα at later time points. In parallel, catalytic subunit levels could also be assessed as they are directly involved in PKA activity; however, such levels would likely mirror RIIβ expression as previous work has shown that catalytic subunits act in a transient, proximal fashion to regulatory subunits and anchoring proteins that also serve to protect them from proteolytic degradation (reviewed in Brandon et al., 1997; Mailliard & Diamond, 2004; Thiele et al., 2000; Veglia & Cembran, 2013).
PKA P2 synaptosomal expression is an indirect representation of PKA activity; therefore, we directly measured PKA kinase activity. The current study indicated no differences in baseline PKA activity between adolescent and adult rats. This result was expected as no differences in baseline PKA RIIβ P2 synaptosomal expression were observed during mid-adolescence and adulthood. Following ethanol exposure, PKA activity was increased in adults. This parallels our P2 synaptosomal results and is in agreement with previous observations (Coe et al., 1996). Interestingly, adolescent PKA was not increased by acute ethanol exposure. To our knowledge, these results are the first to suggest ontogenetic differences in ethanol-induced activation of the PKA pathway. Because of the observed increase in adult P2 synaptosomal PKA RIIβ translocation following ethanol exposure in the current study and in prior studies, it is possible that the PKA RIIβ subunit is a major contributor to ethanol-induced alterations in PKA activity (Carlson et al., 2013; Kumar et al., 2012). Furthermore, it is unlikely that this increased kinase activity is driven by type I regulatory subunits due to their cytoplasmic localization (Constantinescu et al., 2004). Overall, our present PKA P2 synaptosomal expression and activity results support our hypothesis that ethanol-induced PKA activity differs across age, but more work is needed to further test this hypothesis. It is also possible that these effects will be age- and region-specific as Kelm et al. (2008) demonstrated that PKA plays a role in ethanol-induced increases in spontaneous GABA release in pre-adolescent rat cerebellar recordings. Moreover, our PKA activity results may be dependent on subcortical regions such as the prefrontal cortex, as recent work has suggested that mid-adolescent PKA activity is reduced compared to post-pubertal animals (Heng, Markham, Hu, & Tseng, 2011). However, it is likely that age-related differences in PKA responding are independent of a systemic stress response. Although intraperitoneal administration, as well as higher doses of ethanol may increase stress regulatory systems (e.g., Boyd, Kumar, O’Buckley, Porcu, & Morrow, 2010), we have recently reported that ethanol increases P2 synaptosomal PKA in primary cultured cortical neurons (Carlson et al., 2013). Also, recent work demonstrates that adolescents and adults exhibit similar increases in corticosterone in response to ethanol, albeit at lower doses than used in the current study (Willey, Anderson, Morales, Ramirez, & Spear, 2012). Taken together, ethanol’s influence on PKA is likely due to upstream signaling events.
While PKA activation involves cAMP production following stimulatory G-protein coupled receptor activation of adenylyl cyclases, it remains unclear whether a specific adenylyl cyclase or differential enhancement of stimulatory GPCRs such as adenosine A2 and dopamine D1 receptors may be contributing to adolescent and adult ethanol-related PKA activity (Diamond & Gordon, 1994, 1997; Nagy, Diamond, Casso, Franklin, & Gordon, 1990). Differences in PKA activity may also be associated with age-related differences in phosphodiesterases. Alternatively, age-related differences in PKA inhibitor (PKI-α) may also be a factor and should be explored, particularly as PKI-α has been suggested to play a role in PKA-mediated gene regulation following chronic intermittent ethanol exposure (Repunte-Canonigo, Lutjens, van der Stap, & Sanna, 2007). Therefore, it is critical that future studies examine these possibilities in relation to age-dependent differences in PKA activity following ethanol exposure.
Due to the converging evidence indicating that ethanol exposure is not sufficient to alter cortical PKA expression or activity in adolescents, we pharmacologically modulated PKA activity in the brain. In agreement with previously published works (Silveri & Spear, 1998), ethanol’s hypnotic effects were substantially reduced in adolescents compared to adults. However, increasing central PKA activity increased ethanol’s hypnotic effects in adolescents, but not in adults. The lack of effect in adults suggests that the dose of ethanol used here (4.0 g/kg) may have resulted in a ceiling effect in PKA activity as our previous results indicate that PKA activators increased LORR duration at a lower dose (Kumar et al., 2012). Moreover, our results are also in agreement with other reports investigating ethanol sensitivity following PKA modulation (Lai et al., 2007). Interestingly, increasing PKA activity did not fully abrogate differences in ethanol responses between adolescents and adults. More than likely, variations in other molecular pathways, such as other protein kinases, may contribute to the remaining gap in ethanol age-dependent hypnotic sensitivity. In support of this, our recent work indicating that cortical PKC activity also contributes to adolescent hypnotic responses further implicates the kinome in developmental ethanol-related behavior (Santerre, Gigante, Landin, & Werner, 2013).
Intriguingly, central inhibition of PKA also increased LORR duration in adolescents, whereas a suggestive decrease in LORR coupled with significant increases in BECs was observed in adults. Such effects potentially suggest that the relation of PKA activity is not linear, but rather is a U-shaped response curve as reviewed elsewhere (see Howe, 2011). Given that basal PKA activity is similar between adolescents and adults, it is likely that some PKA activity is necessary for homeostatic functioning, and inhibiting this activity increases sensitivity to ethanol exposure. Conversely, dissimilar effects were likely seen in adults as PKA inhibition partially reversed ethanol-induced increases in PKA activity. These marginal reductions in adult sedative-hypnotic sensitivity are likely related to the dose of Rp-cAMP employed in the current study compared to effective doses used elsewhere (Chu, Ito, Li, & Morozov, 2012; ffrench-Mullen, Danks, & Spence, 1994; Li et al., 2013) and that central inhibition of PKA dose-dependently reduced ethanol-induced LORR elsewhere (Lai et al., 2007).
Interestingly, adolescent and adult BECs at righting reflex recovery were comparable, suggesting pharmacokinetic differences between ages. Although similar waking BECs between adolescents and adults have been noted (Silveri & Spear, 2002), other studies have reported age-related differences in BECs at waking (Broadwater, Varlinskaya, & Spear, 2011; Santerre et al., 2013; Silveri & Spear, 1998). The reasons behind this discrepancy are not clear, but work assessing ontogenetic ethanol metabolic rates in detail indicate that non-metabolic factors likely play a prominent role (Silveri & Spear, 2000). Furthermore, it is unclear why central administration of a PKA inhibitor would potentially reduce ethanol metabolism in adolescents, as BEC’s for Rp-cAMP-treated subjects were similar to controls. Presumably related to PKA’s U-shaped activity-response curve, reductions in basal CNS PKA activity in specific regions, such as in the hypothalamus, may play a role.
Although the behavioral results correlate with age-related differences in cortical PKA expression and activity, a main limitation of this study is that we cannot exclude the possibility that other regions exposed to the PKA activator also contribute to ethanol’s sedative-hypnotic effects. In addition, given that a slightly higher dose was needed for the behavioral analysis, due to preliminary studies showing differences in adolescent ethanol sensitivity following surgery, we cannot preclude that such procedures may play a role in behavioral responding. However, ethanol-induced PKA RIIβ regulation and PKA activity experiments were conducted in naïve animals, which lessens the impact of surgical procedures on PKA activity. Nonetheless, this phenomenon is currently being investigated.
It is worth noting that the overall physiological impact of PKA activation and its potential involvement in promoting ethanol intoxication and sedation remains unclear. The ability to sense adverse levels of intoxication may serve as a protective mechanism to prevent the deleterious effects associated with high levels of ethanol exposure. The current results support the possibility that PKA hyperactivity may serve as a molecular intoxication sensor that is repressed during adolescence; again, further work is needed to test this possibility. In summary, this study further confirms ethanol-induced increases in PKA activity in adults, and suggests that the inability to increase PKA activity may have a role in ethanol-related behavioral effects during adolescence.
Acknowledgments
This work was supported by National Institute of Health grants AA019367 (Linda P. Spear – faculty recruitment of David F. Werner) and AA017823 (DFW) and the Developmental Exposure Alcohol Research Center. We would also like to thank Dr. A. Leslie Morrow, Linda Spear, and Terrence Deak for thoughtful discussions during the drafting of this manuscript and Eric Truxell for editing.
Footnotes
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Behar D, Berg CJ, Rapoport JL, Nelson W, Linnoila M, Cohen M, et al. Behavioral and physiological effects of ethanol in high-risk and control children: a pilot study. Alcoholism: Clinical and Experimental Research. 1983;7:404–410. doi: 10.1111/j.1530-0277.1983.tb05495.x. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Borghese CM, McCracken ML, Benavidez JM, Geil CR, Osterndorff-Kahanek E, et al. Loss of ethanol conditioned taste aversion and motor stimulation in knockin mice with ethanol-insensitive α2-containing GABA(A) receptors. The Journal of Pharmacology and Experimental Therapeutics. 2011;336:145–154. doi: 10.1124/jpet.110.171645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Harris RA. Deletion of vanilloid receptor (TRPV1) in mice alters behavioral effects of ethanol. Neuropharmacology. 2009;56:814–820. doi: 10.1016/j.neuropharm.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd KN, Kumar S, O’Buckley TK, Porcu P, Morrow AL. Ethanol induction of steroidogenesis in rat adrenal and brain is dependent upon pituitary ACTH release and de novo adrenal StAR synthesis. Journal of Neurochemistry. 2010;112:784–796. doi: 10.1111/j.1471-4159.2009.06509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon EP, Idzerda RL, McKnight GS. PKA isoforms, neural pathways, and behaviour: making the connection. Current Opinion in Neurobiology. 1997;7:397–403. doi: 10.1016/s0959-4388(97)80069-4. [DOI] [PubMed] [Google Scholar]
- Broadwater M, Varlinskaya EI, Spear LP. Chronic intermittent ethanol exposure in early adolescent and adult male rats: effects on tolerance, social behavior, and ethanol intake. Alcoholism: Clinical and Experimental Research. 2011;35:1392–1403. doi: 10.1111/j.1530-0277.2011.01474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown S, McGue M, Maggs J, Schulenberg J, Hingson R, Swartzwelder S, et al. Underage alcohol use: summary of developmental processes and mechanisms: ages 16–20. Alcohol Research & Health. 2009;32:41–52. [PMC free article] [PubMed] [Google Scholar]
- Carlson SL, Kumar S, Werner DF, Comerford CE, Morrow AL. Ethanol activation of protein kinase A regulates GABAA α1 receptor function and trafficking in cultured cerebral cortical neurons. The Journal of Pharmacology and Experimental Therapeutics. 2013;345:317–325. doi: 10.1124/jpet.112.201954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu HY, Ito W, Li J, Morozov A. Target-specific suppression of GABA release from parvalbumin interneurons in the basolateral amygdala by dopamine. The Journal of Neuroscience. 2012;32:14815–14820. doi: 10.1523/JNEUROSCI.2997-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe IR, Dohrman DP, Constantinescu A, Diamond I, Gordon AS. Activation of cyclic AMP-dependent protein kinase reverses tolerance of a nucleoside transporter to ethanol. The Journal of Pharmacology and Experimental Therapeutics. 1996;276:365–369. [PubMed] [Google Scholar]
- Constantinescu A, Gordon AS, Diamond I. cAMP-dependent protein kinase types I and II differentially regulate cAMP response element-mediated gene expression: implications for neuronal responses to ethanol. The Journal of Biological Chemistry. 2002;277:18810–18816. doi: 10.1074/jbc.M112107200. [DOI] [PubMed] [Google Scholar]
- Constantinescu A, Wu M, Asher O, Diamond I. cAMP-dependent protein kinase type I regulates ethanol-induced cAMP response element-mediated gene expression via activation of CREB-binding protein and inhibition of MAPK. The Journal of Biological Chemistry. 2004;279:43321–43329. doi: 10.1074/jbc.M406994200. [DOI] [PubMed] [Google Scholar]
- Diamond I, Gordon AS. The role of adenosine in mediating cellular and molecular responses to ethanol. EXS. 1994;71:175–183. doi: 10.1007/978-3-0348-7330-7_18. [DOI] [PubMed] [Google Scholar]
- Diamond I, Gordon AS. Cellular and molecular neuroscience of alcoholism. Physiological Reviews. 1997;77:1–20. doi: 10.1152/physrev.1997.77.1.1. [DOI] [PubMed] [Google Scholar]
- Evans GJ, Morgan A. Regulation of the exocytotic machinery by cAMP-dependent protein kinase: implications for presynaptic plasticity. Biochemical Society Transactions. 2003;31:824–827. doi: 10.1042/bst0310824. [DOI] [PubMed] [Google Scholar]
- ffrench-Mullen JM, Danks P, Spence KT. Neurosteroids modulate calcium currents in hippocampal CA1 neurons via a pertussis toxin-sensitive G-protein-coupled mechanism. The Journal of Neuroscience. 1994;14:1963–1977. doi: 10.1523/JNEUROSCI.14-04-01963.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming RL, Acheson SK, Moore SD, Wilson WA, Swartzwelder HS. In the rat, chronic intermittent ethanol exposure during adolescence alters the ethanol sensitivity of tonic inhibition in adulthood. Alcoholism: Clinical and Experimental Research. 2012;36:279–285. doi: 10.1111/j.1530-0277.2011.01615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks NP, Lieb WR. Mechanisms of general anesthesia. Environmental Health Perspectives. 1990;87:199–205. doi: 10.1289/ehp.9087199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozzi A, Schwarz AJ, Reese T, Crestan V, Bertani S, Turrini G, et al. Functional magnetic resonance mapping of intracerebroventricular infusion of a neuroactive peptide in the anaesthetised rat. Journal of Neuroscience Methods. 2005;142:115–124. doi: 10.1016/j.jneumeth.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Grosshans DR, Clayton DA, Coultrap SJ, Browning MD. Analysis of glutamate receptor surface expression in acute hippocampal slices. Science’s STKE, 2002. 2002:l8. doi: 10.1126/stke.2002.137.pl8. [DOI] [PubMed] [Google Scholar]
- Heng LJ, Markham JA, Hu XT, Tseng KY. Concurrent upregulation of postsynaptic L-type Ca(2+) channel function and protein kinase A signaling is required for the periadolescent facilitation of Ca(2+) plateau potentials and dopamine D1 receptor modulation in the prefrontal cortex. Neuropharmacology. 2011;60:953–962. doi: 10.1016/j.neuropharm.2011.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe AK. Cross-talk between calcium and protein kinase A in the regulation of cell migration. Current Opinion in Cell Biology. 2011;23:554–561. doi: 10.1016/j.ceb.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher PR, Dabholkar AS. Regional differences in synaptogenesis in human cerebral cortex. The Journal of Comparative Neurology. 1997;387:167–178. doi: 10.1002/(sici)1096-9861(19971020)387:2<167::aid-cne1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Johnston LD, O’Malley PM, Bachman JG, Schulenberg JE. Overview of Key Findings, 2008. Bethesda, MD: National Institute on Drug Abuse; 2009. Monitoring the Future: National Results on Adolescent Drug Use. (NIH Publication No. 09-7401) [Google Scholar]
- Kelm MK, Criswell HE, Breese GR. The role of protein kinase A in the ethanol-induced increase in spontaneous GABA release onto cerebellar Purkinje neurons. Journal of Neurophysiology. 2008;100:3417–3428. doi: 10.1152/jn.90970.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Ren Q, Beckley JH, O’Buckley TK, Gigante ED, Santerre JL, et al. Ethanol activation of protein kinase A regulates GABA(A) receptor subunit expression in the cerebral cortex and contributes to ethanol-induced hypnosis. Frontiers in Neuroscience. 2012;6:44. doi: 10.3389/fnins.2012.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuromi H, Kidokoro Y. Tetanic stimulation recruits vesicles from reserve pool via a cAMP-mediated process in Drosophila synapses. Neuron. 2000;27:133–143. doi: 10.1016/s0896-6273(00)00015-5. [DOI] [PubMed] [Google Scholar]
- Lai CC, Kuo TI, Lin HH. The role of protein kinase A in acute ethanol-induced neurobehavioral actions in rats. Anesthesia and Analgesia. 2007;105:89–96. doi: 10.1213/01.ane.0000263030.13249.36. [DOI] [PubMed] [Google Scholar]
- Li P, Sun HJ, Han Y, Wang JJ, Zhang F, Tang CS, et al. Intermedin enhances sympathetic outflow via receptor-mediated cAMP/PKA signaling pathway in nucleus tractus solitarii of rats. Peptides. 2013;47:1–6. doi: 10.1016/j.peptides.2013.05.002. [DOI] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Südhof TC, Linden DJ. Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell. 2003;115:49–60. doi: 10.1016/s0092-8674(03)00727-x. [DOI] [PubMed] [Google Scholar]
- Maas JW, Jr, Vogt SK, Chan GC, Pineda VV, Storm DR, Muglia LJ. Calcium-stimulated adenylyl cyclases are critical modulators of neuronal ethanol sensitivity. The Journal of Neuroscience. 2005;25:4118–4126. doi: 10.1523/JNEUROSCI.4273-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailliard WS, Diamond I. Recent advances in the neurobiology of alcoholism: the role of adenosine. Pharmacology & Therapeutics. 2004;101:39–46. doi: 10.1016/j.pharmthera.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Miller DJ, Duka T, Stimpson CD, Schapiro SJ, Baze WB, McArthur MJ, et al. Prolonged myelination in human neocortical evolution. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:16480–16485. doi: 10.1073/pnas.1117943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy LE, Diamond I, Casso DJ, Franklin C, Gordon AS. Ethanol increases extracellular adenosine by inhibiting adenosine uptake via the nucleoside transporter. The Journal of Biological Chemistry. 1990;265:1946–1951. [PubMed] [Google Scholar]
- Petanjek Z, Judaš M, Šimic G, Rasin MR, Uylings HB, Rakic P, et al. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:13281–13286. doi: 10.1073/pnas.1105108108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proescholdt MG, Hutto B, Brady LS, Herkenham M. Studies of cerebrospinal fluid flow and penetration into brain following lateral ventricle and cisterna magna injections of the tracer [14C]inulin in rat. Neuroscience. 2000;95:577–592. doi: 10.1016/s0306-4522(99)00417-0. [DOI] [PubMed] [Google Scholar]
- Repunte-Canonigo V, Lutjens R, van der Stap LD, Sanna PP. Increased expression of protein kinase A inhibitor alpha (PKI-alpha) and decreased PKA-regulated genes in chronic intermittent alcohol exposure. Brain Research. 2007;1138:48–56. doi: 10.1016/j.brainres.2006.09.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santerre JL, Gigante ED, Landin JD, Werner DF. Molecular and behavioral characterization of adolescent protein kinase C following high dose ethanol exposure. Psychopharmacology. 2013;231:1809–1820. doi: 10.1007/s00213-013-3267-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schillace RV, Andrews SF, Galligan SG, Burton KA, Starks HJ, Bouwer HG, et al. The role of protein kinase A anchoring via the RII alpha regulatory subunit in the murine immune system. Journal of Immunology. 2005;174:6847–6853. doi: 10.4049/jimmunol.174.11.6847. [DOI] [PubMed] [Google Scholar]
- Scott JD. Cyclic nucleotide-dependent protein kinases. Pharmacology & Therapeutics. 1991;50:123–145. doi: 10.1016/0163-7258(91)90075-w. [DOI] [PubMed] [Google Scholar]
- Silveri MM, Spear LP. Decreased sensitivity to the hypnotic effects of ethanol early in ontogeny. Alcoholism: Clinical and Experimental Research. 1998;22:670–676. doi: 10.1111/j.1530-0277.1998.tb04310.x. [DOI] [PubMed] [Google Scholar]
- Silveri MM, Spear LP. Ontogeny of ethanol elimination and ethanol-induced hypothermia. Alcohol. 2000;20:45–53. doi: 10.1016/s0741-8329(99)00055-5. [DOI] [PubMed] [Google Scholar]
- Silveri MM, Spear LP. The effects of NMDA and GABAA pharmacological manipulations on ethanol sensitivity in immature and mature animals. Alcoholism: Clinical and Experimental Research. 2002;26:449–456. [PubMed] [Google Scholar]
- Skalhegg BS, Tasken K. Specificity in the cAMP/PKA signaling pathway. Differential expression, regulation, and subcellular localization of subunits of PKA. Frontiers in Bioscience. 2000;5:D678–D693. doi: 10.2741/skalhegg. [DOI] [PubMed] [Google Scholar]
- Spear LP. The adolescent brain and age-related behavioral manifestations. Neuroscience and Biobehavioral Reviews. 2000;24:417–463. doi: 10.1016/s0149-7634(00)00014-2. [DOI] [PubMed] [Google Scholar]
- Spear LP, Varlinskaya EI. Sensitivity to ethanol and other hedonic stimuli in an animal model of adolescence: implications for prevention science? Developmental Psychobiology. 2010;52:236–243. doi: 10.1002/dev.20457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Substance Abuse and Mental Health Services Administration. Results from the 2007 National Survey on Drug Use and Health: National Findings. Rockville, MD: Office of Applied Studies; 2008. [Google Scholar]
- Thiele TE, Willis B, Stadler J, Reynolds JG, Bernstein IL, McKnight GS. High ethanol consumption and low sensitivity to ethanol-induced sedation in protein kinase A-mutant mice. The Journal of Neuroscience. 2000;20:RC75. doi: 10.1523/JNEUROSCI.20-10-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varlinskaya EI, Spear LP. Increases in anxiety-like behavior induced by acute stress are reversed by ethanol in adolescent but not adult rats. Pharmacology, Biochemistry, and Behavior. 2012;100:440–450. doi: 10.1016/j.pbb.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veglia G, Cembran A. Role of conformational entropy in the activity and regulation of the catalytic subunit of protein kinase A. The FEBS Journal. 2013;280:5608–5615. doi: 10.1111/febs.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wand G, Levine M, Zweifel L, Schwindinger W, Abel T. The cAMP-protein kinase A signal transduction pathway modulates ethanol consumption and sedative effects of ethanol. The Journal of Neuroscience. 2001;21:5297–5303. doi: 10.1523/JNEUROSCI.21-14-05297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner DF, Blednov YA, Ariwodola OJ, Silberman Y, Logan E, Berry RB, et al. Knockin mice with ethanol-insensitive alpha1-containing gamma-aminobutyric acid type A receptors display selective alterations in behavioral responses to ethanol. The Journal of Pharmacology and Experimental Therapeutics. 2006;319:219–227. doi: 10.1124/jpet.106.106161. [DOI] [PubMed] [Google Scholar]
- Willey AR, Anderson RI, Morales M, Ramirez RL, Spear LP. Effects of ethanol administration on corticosterone levels in adolescent and adult rats. Alcohol. 2012;46:29–36. doi: 10.1016/j.alcohol.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng R, Yang L, Sikorski MA, Enns LC, Czyzyk TA, Ladiges WC, et al. Deficiency of the RIIβ subunit of PKA affects locomotor activity and energy homeostasis in distinct neuronal populations. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E1631–E1640. doi: 10.1073/pnas.1219542110. [DOI] [PMC free article] [PubMed] [Google Scholar]