

Abstract
Chloride channel gating and trafficking of the cystic fibrosis transmembrane conductance regulator (CFTR) are regulated by phosphorylation. Intrinsically disordered segments of the protein are responsible for phospho‐regulation, particularly the regulatory (R) region that is a target for several kinases and phosphatases. The R region remains disordered following phosphorylation, with different phosphorylation states sampling various conformations. Recent studies have demonstrated the crucial role that intramolecular and intermolecular interactions of the R region play in CFTR regulation. Different partners compete for the same binding segment, with the R region containing multiple overlapping binding elements. The non‐phosphorylated R region interacts with the nucleotide binding domains and inhibits channel activity by blocking heterodimerization. Phosphorylation shifts the equilibrium such that the R region is excluded from the dimer interface, facilitating gating and processing by stimulating R region interactions with other domains and proteins. The dynamic conformational sampling and transient binding of the R region to multiple partners enables complex control of CFTR channel activity and trafficking.
Keywords: binding, disordered, hub, IDP, post‐translational modification, regulation
Abbreviations
- AKAP
A kinase anchoring protein
- AMPK
AMP‐activated protein kinase
- CD
circular dichroism
- CFTR
cystic fibrosis transmembrane conductance regulator
- ER
endoplasmic reticulum
- F508del
CFTR with missing phenylalanine 508
- FTIR
fourier transform infrared spectroscopy
- MoRE
molecular recognition element
- MSD
membrane‐spanning domain
- NBD
nucleotide binding domain
- NHERF
Na+/H+ exchanger regulatory factor
- NMR
nuclear magnetic resonance
- PKA
protein kinase A
- PKC
protein kinase C
- PP2A
protein phosphatase 2A
- PP2B
protein phosphatase 2B
- PP2C
protein phosphatase 2C
- R region
regulatory region
- RACK1
receptor for activated C kinase
- RE
regulatory extension
- SAXS
small angle X‐ray scattering
- SDS‐PAGE
sodium dodecyl sulfate‐polyacrylamide gel electrophoresis
- SLC26A family
solute carrier 26A family
- WT
wild‐type
Introduction
The cystic fibrosis transmembrane conductance regulator (CFTR) is a chloride channel in the apical membrane of epithelial cells 1. Mutations in CFTR result in impaired chloride conductance across the plasma membrane and are the basis of the genetic disease cystic fibrosis 2. Almost 90% of patients contain at least one allele with a missing codon for Phe508 (F508del) located in the first nucleotide binding domain (NBD1). CFTR is regulated by phosphorylation primarily on its regulatory region (R region), which is an ~200‐residue, disordered segment of the protein 1. While CFTR is a chloride channel, it is a member of the ABC family of transporters with significant homologies in the transmembrane domains and NBDs; the R region, in contrast, is unique to CFTR. The following minireview is not intended to be a comprehensive summary of the extensive and at times contradictory R region literature. Instead, we focus on specific papers that highlight and enrich a mechanistic understanding of how phosphorylation affects regulation of CFTR by the R region.
R region structure
The N‐terminal boundary of the R region is not precisely defined, due to lack of clarity for the C‐terminal boundary of the immediately N‐terminal NBD1. Split CFTR constructs lacking the R region (1–634, 838–1480) traffic to the plasma membrane and conduct chloride, suggesting that they contain a complete functional NBD1, and thus the N‐terminal boundary of the R region is ~N635 4. This is consistent with NMR data demonstrating that the C‐terminal residues of an NBD1 construct ending at G646 comprising helix H9 (637–646) are in exchange with unfolded states and that an NBD1 construct ending at residue L636 displays stable folding 5. However, alignment of NBD sequences from homologous ABC transporters such as HisP and MalK to NBD1 suggests that the domain ends at residue S642 for an N‐terminal R region boundary of ~K643 3. The C‐terminal boundary of the R region is more defined (about D835), due to confidence in predicting the start of the immediately C‐terminal membrane‐spanning domain 2 (MSD2) 3.
The approximately 200 residues of the R region spanning these boundaries have an amino acid composition that markedly differs from the rest of CFTR, with more hydrophilic and charged residues. These features led to early recognition that the R region may be an intrinsically disordered region 6. Since then, CD spectroscopy 6, small angle X‐ray scattering (SAXS) 9 and NMR spectroscopy 10 have confirmed that the isolated R region does not have a stable three‐dimensional fold or even stably formed secondary structure. Disorder prediction software shows definitive disordered segments separated by segments that are on the discrimination limit for folded and disordered segments 9. CD data agree on a large fraction of random coil (30–95%) 6 with different amounts of α‐helical (5–46%) and β content. Different construct boundaries and experimental conditions contribute to the inherent uncertainty; constructs with earlier N‐terminal boundaries have more α‐helical content. SAXS can be used to discriminate between folded, partially folded and random coil structures, and data for the R region indicate a substantial component of random coil with some partial structure 9, incompatible with totally random coil structure 12.
While the R region is clearly disordered, it has fractional structure and clear functional roles that lead to some degree of sequence conservation among CFTR proteins of different species, from fish to human 13. Disordered protein sequences lacking a three‐dimensional fold are not under evolutionary pressure to maintain a constrained structure; consequently disordered regions like the R region are much more variable than folded protein domains such as the NBDs. Nonetheless, not only are kinase recognition sequences conserved in the R region 3, but the distances between phosphorylation sites (i.e. relative positions in the sequence) are also conserved 11. This conservation, together with the identification of a number of cystic‐fibrosis‐causing mutations within the R region (www.genet.sickkids.on.ca), highlight the importance of the R region for CFTR function and regulation, including phosphorylation and potential conserved interaction segments.
NMR is the most useful technique for providing residue‐specific information on intrinsically disordered proteins 14. Chemical shifts are sensitive to the local electrochemical environment, with α and β carbon resonances particularly responsive to secondary structure. R region secondary structure propensity estimated on the basis of chemical shifts indicates transiently populated secondary structure elements (Fig. 1) 10. The highest propensities for the α‐helix reach 30–40%, suggesting that segments 648–670, 755–778 and 801–818 sample α‐helical conformation up to 30–40% of the time. The flexible and dynamic features of the R region make crystallization of the R region impossible and no single structural representation can capture the R region conformational ensemble. However, X‐ray structures of NBD1 containing some N‐terminal residues of the R region (called the regulatory extension or RE) provide snapshots of the structural heterogeneity of this portion of the R region 15 and confirm the helical propensity of the N‐terminal segment observed by secondary structure propensity (Fig. 1). The orientation of the RE with respect to the NBD1 and the helical boundaries differs in the crystal structures (1R0X, 1XMI) (Fig. 2) while the core NBD1 structure remains nearly unchanged 15.
Figure 1.
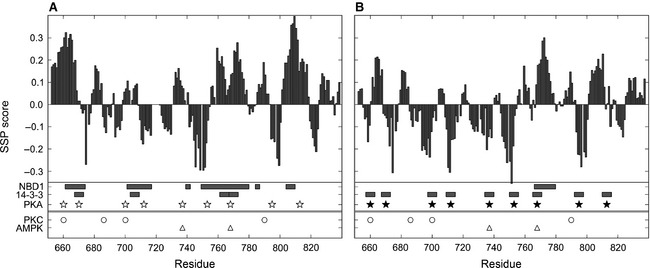
Key elements of CFTR R region in (A) the non‐phosphorylated and (B) the phosphorylated states. Bar graphs represent the secondary structure propensity (SSP). Positive values correspond to helical structure sampling, while negative values refer to extended conformations. Horizontal bars represent R region binding segments to NBD1 and 14‐3‐3 10. The lengths of 14‐3‐3 interaction segments are set to six residues to match mode I binding to 14‐3‐3. Kinase phosphorylation sites of PKA (star), PKC (circle) and AMPK (triangle) are marked. Both the secondary structure and binding experiments were carried out on non‐phosphorylated (open symbols) and PKA‐phosphorylated (solid symbols) isolated R region (654–838).
Figure 2.
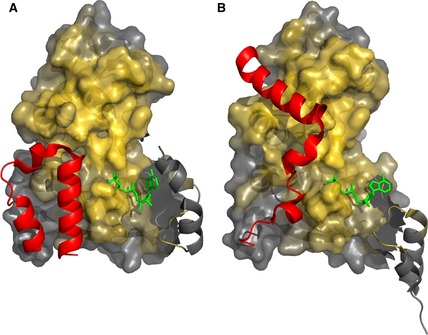
Heterogeneity of CFTR R region:NBD1 interactions. N‐terminus of the R region (red, residues 638–671) in different NBD1 crystal structures (A, 1R0X; B, 1XMI) oriented to show the NBD dimerization interface 15. The core NBD1 (surface representation) is colored as a gold gradient reflecting the proximity to atoms in the other NBD1 molecule within the NBD1:NBD1 homodimer structure (2PZE). The flexible regulatory insertion (RI) of NBD1 that is in different conformations in the two structures is shown as gray helices and is not part of the surface representation.
These studies demonstrating disorder in the isolated R region are relevant to understanding the R region within the context of full‐length CFTR. Electron microscopy of CFTR 17 has no discrete density for elements other than the MSDs and NBDs homologous to Sav1866, a bacterial ABC transporter, supporting the disordered nature of the R region. Attempts to computationally model the R region within the context of homology models of CFTR have been confounded by its disordered nature. Two published models have completely distinct conformational predictions 18 for the R region. While these structures are potential conformations that the disordered R region might sample, they are unlikely to represent a stable structure populated for a significant fraction of the time. Importantly, the sequence characteristics and functional role as a target for post‐translational modification and as a binding partner for multiple intramolecular and intermolecular targets (see below) all strongly point to the disordered nature of the R region. Regulatory regions in proteins are often disordered because they can easily adapt to different protein surfaces and are accessible to enzymes carrying out post‐translation modifications 20. Thus they can be sensitive to input from many partners and provide fine‐tuning for signaling 21. In the case of CFTR R region, phosphorylation is a dominant regulatory modification.
R region phosphorylation
Protein kinases and phosphatases play a crucial role in the regulation of CFTR primarily via controlling the level of R region phosphorylation. There is a significant body of literature on R region phosphorylation, reflecting the critical importance as well as the complexity and lack of thorough understanding of the phospho‐regulatory effects, only a small subset of which can be covered here. Several protein kinases, including protein kinase A (PKA), protein kinase C (PKC) and AMP‐activated protein kinase (AMPK), are known to phosphorylate the R region both in vitro and in vivo. Of the 19 PKA recognition sites predicted in the R region, 10 sites were confirmed in vitro (S660, S670, S686, S700, S712, S737, S753, S768, S795, S813) (Fig. 1) 1 and six of these sites (S660, S700, S712, S737, S795, S813) were found to be phosphorylated on full‐length CFTR purified from cells 2. Most of these phosphorylation sites stimulate channel activity in an additive/synergistic way; however, S737 and S768 were reported under certain conditions to be inhibitory 24. None of these sites individually seems to be essential for CFTR regulation and mutating one, two or three of them (S660, S737, S795, S813) has little affect on chloride channel activity. In contrast, one study showed that mutating these four sites abolishes most of the cAMP‐dependent response 6. Other studies showed that more sites had to be mutated to abolish the full response 25, underscoring the multi‐valency of binding interactions, either intramolecular or intermolecular, of the R region that are affected by phosphorylation.
There are six PKC consensus sequences located in the R region (T682, S686, S707, S790, T791, S809), of which four have been phosphorylated in the isolated R region in vitro (S660, S686, S700, S790) (Fig. 1) and one (S686) was verified on the full‐length protein as well 23. PKC phosphorylation on the R region does not stimulate the channel activity but instead enhances the effect of PKA phosphorylation. Interestingly, high concentrations of free calcium (0.5 mm) inhibit PKC phosphorylation of the isolated R region 23, an effect that could be on the enzymatic activity or possibly, indirectly, on the accessibility or structure of the R region itself.
AMPK, the cellular energy level activated kinase, phosphorylates the R region on two sites (S737, S768) (Fig. 1) that were previously described as inhibitory sites 24, of which S768 has much greater impact than S737 28. Phosphorylation of these residues, which may fix CFTR in a state that is insensitive to PKA or PKC phosphorylation 29, decreases the open probability of the channel by altering the channel opening but not the closing, resulting in longer inter‐burst intervals but a similar length of each opening 30. In contrast, another study found that PKA phosphorylation could overcome the AMPK inhibitory effect 28.
No single phosphatase has been demonstrated to be both necessary and sufficient to completely downregulate CFTR channel activity, suggesting that CFTR can be dephosphorylated by multiple phosphatases, including the protein phosphatase 2 (PP2) isoforms PP2A, PP2B and PP2C and alkaline phosphatase 31. Exposing CFTR to PP2A, PP2C and alkaline phosphatase reduces CFTR activity more than 90% 32; however, PP2C deactivates the most rapidly. The various rates suggest that different phosphatases may act on functionally distinct PKA sites 33. PP2A and PP2C both appear to be biologically relevant phosphatases. PP2C can dephosphorylate CFTR in vitro 34, can bind directly to the R region and to full‐length CFTR and can deactivate CFTR chloride channel 33. PP2A directly associates with CFTR segment 1451–1476 31, it can dephosphorylate the R region in vitro and a direct interaction of its regulator subunit (RP65) with the R region segment 672–855 has been demonstrated 36.
CFTR, the three kinases PKA, PKC and AMPK and at least two phosphatases, PP2A and PP2C, are part of a large macromolecular complex anchored to the cytoskeleton at the apical membrane. The catalytic subunit of AMPK tightly interacts with another disordered CFTR segment, residues 1420–1457, through a region of the kinase that normally binds the two AMPK regulator subunits 32. Deleting this CFTR C‐terminal segment has not been found to affect the biosynthesis of the channel, despite the fact that it contains two important endocytosis motifs 38 required for the interaction with AMPK 32. The B regulatory subunit of PP2A interacts with CFTR residues 1451–1476 31, which contain a highly conserved acidic cluster 38. The extreme C‐terminal PDZ motif of CFTR (1477–1480) anchors it within the NHERF (Na+/H+ exchanger regulatory factor)–ezrin–actin cytoskeleton complex 39 via PDZ‐domain‐containing NHERFs. A fraction of cellular PKA is co‐localized with CFTR via ezrin, an A kinase anchoring protein (AKAP) 39. Disruption of PKA–AKAP interactions dramatically reduced the ability of wild‐type (WT) CFTR to respond to cAMP modulation. The RACK1 (receptor for activated C kinase) scaffold protein co‐localizes with CFTR, interacting with both the PDZ1 domain of NHERF1 and the activated PKCε on two distinct sites 40. Moreover, RACK1 is part of the PKCε‐RACK1‐NHERF1 dimer–tubulin complex directly connected to the tubulin cytoskeleton. Although cell‐specific CFTR targeting of PP2B has not been shown, the localization and expression of AKAP79, a scaffold protein that binds PP2B, PKA and PKC, support the importance of PP2B in CFTR regulation 41. In addition to ensuring that PKA and PKC are in close proximity to CFTR, both the NHERF1–ezrin–actin and the RACK1‐NHERF1 dimer–tubulin complexes may function to stabilize CFTR surface expression 42.
R region structural changes upon phosphorylation
Investigations by SDS‐PAGE, fluorescence, CD, FTIR and NMR spectroscopy provide evidence for the R region becoming generally more flexible and less structured upon phosphorylation, although conflicting results have been published. Data on the hydrodynamic properties include decreased R region mobility on SDS‐PAGE following PKA phosphorylation 23 interpreted as an apparent molecular weight that is greater than that expected from the addition of the extra phosphates and pointing to a more random and expanded structure. The R region (654–838) contains almost identical numbers of positively (28 K/R/H) and negatively (29 D/E) charged residues, but phosphorylation increases the number of negative charges and results in charge repulsion by the increased amount of negative charge. The change in the charge balance alone, however, does not explain the absence of mobility shift upon PKC phosphorylation 23 or S768 phosphorylation or the major shift in response to S737 phosphorylation 44. Whilst computational studies 11 and the previously mentioned SDS‐PAGE data 23 suggest an increase in R region size following phosphorylation, SAXS data 9 point to a decrease in the radius of gyration.
Data on changes in secondary structure include CD experiments demonstrating an increased random coil character for the isolated R region upon PKA phosphorylation 45 and FTIR measurements indicating no secondary structure change in the context of full‐length CFTR 46. In contrast, Marasini and coworkers showed a reduction in random coil character and highly increased α‐helical content in isolation using CD 7. Drawing conclusions from the data is complicated by the different kinases and targeted phosphorylation sites on the R region. PKC phosphorylation has been reported to have no measurable effect on secondary structure 45. The finding that S737 phosphorylation has a very large effect while S768 has no impact on the secondary structure propensity of nearby segments explains the different consequences of phosphorylation of the two sites for SDS‐PAGE mobility. Likewise, that PKC phosphorylation of S686 and S790 has little effect on the amount of secondary structure is consistent with the comparable CD signals of the non‐phosphorylated and PKC‐phosphorylated R region 45. Residue‐specific and phosphorylation‐site‐specific information from NMR on the secondary structure of the highly PKA‐phosphorylated isolated R region indicates an overall reduction in helical structure with an increase in β content, potentially due to the repulsion between the phosphate and the partially negatively charged C‐termini of helical elements (Fig. 1) 10. Clarification of the consequences on structural propensities and hydrodynamic behavior may rely on future site‐specific structural data from NMR on samples phosphorylated by different kinases at defined sites.
R region interactions
Intramolecular interactions
The R region is much longer than required to bridge the distance between NBD1 and MSD2 and the absence of a compact fold enables the R region to reach any part of CFTR and into the space around the channel to contact other proteins (Fig. 3). This allows CFTR to have a large interactome, with the R region functioning as a protein interaction hub. The R region has been reported to interact with all intracellular portions of CFTR including the N‐terminus 47, intracellular domains 48, NBD1 10, NBD2 and the C‐terminus 49 in a phosphorylation‐dependent manner. The different phosphorylation states of the R region have distinct functions during channel gating and trafficking. The non‐phosphorylated R region generally acts to inhibit chloride channel activity, probably by sterically blocking NBD heterodimerization 10. NMR binding experiments showed that helical segments of the non‐phosphorylated R region interact with NBD1 and that phosphorylation drastically reduces the affinity of these interactions 10. One site of R region interaction with NBD1, based on crystal structures of NBD1 containing the RE 15, is across the heterodimer interface (Figs 1 and 2), pointing to phosphorylation being required for unhindered NBD heterodimerization and channel gating. Constructs that lack the R region segment 760–783, which samples helical conformations and contains the S768 phosphorylation site, were constitutively active (i.e. PKA independent) 50. However, the open probability and the chloride current of split channels lacking R region segments 760–783 or 708–835 are only 30–40% of the WT channel 50, suggesting additional regulatory mechanisms.
Figure 3.
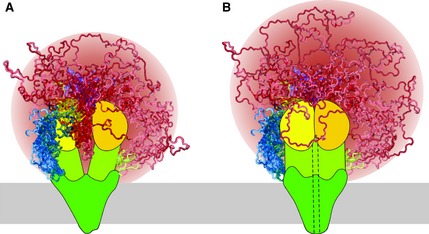
Schematic models of (A) non‐phosphorylated/closed channel and (B) PKA‐phosphorylated/open channel states of CFTR. Shown are the N‐terminal segment (blue), membrane‐spanning domains (dark green), the intracellular domains (yellow‐green), NBD1 (yellow), NBD2 (gold), R region (red) and the C‐terminal segment (purple). Disordered elements (N‐terminus, R region and C‐terminus) are shown as a superposition of multiple possible conformations. Helix 9 of NBD1 (residues 635–643, yellow) and the C‐terminal elbow helix (residues 842–855, green) are also not in a fixed position to demonstrate that they can sample multiple conformations and may be considered part of the R region. The dashed line (in B) schematically illustrates the open channel pore, the membrane is represented as a gray bar and the R region sampled space is colored as a gradient (red of various intensities). PKA phosphorylation sites are not marked. R region sampling in the two phosphorylation states is similar, although the phosphorylated R region samples space farther from the core of CFTR as it is excluded from the NBD interface.
NMR studies of NBD1 with the RE demonstrated that interactions between the NBD1 core and the regulatory elements, the regulatory insertion (RI) and RE (N‐terminal portion of the R region) are disrupted upon phosphorylation 51. The release of these elements upon phosphorylation for F508del NBD1 with the RE, however, is significantly smaller than for WT NBD1, suggesting aberrant R region interactions in the mutant CFTR that are ‘stickier’ and continue to block heterodimerization 51. Recent NMR studies of NBD1 demonstrate that the mutant H620Q and CFFT‐001, a dual corrector‐potentiator for CFTR 5, both enhance release of helices H8 and H9 from the NBD1 core. H8 is immediately N‐terminal to and H9 is arguably the beginning of the R region and both are coupled to R region segments that interact with the heterodimer interface. Release of H8 and H9 by H620Q or CFFT‐001 is thus expected to potentiate CFTR activity, at least in part, by reducing NBD1:R region interactions to promote NBD dimerization 52.
In addition to reducing the inhibitory effect of the non‐phosphorylated R region on channel gating, phosphorylation of the R region appears to stimulate chloride conductance. The location of the R region within the protein is not critical, as moving it to the C‐terminus gives the same effect 50. Phosphorylation strongly increases the interaction of the R region with other parts of CFTR, particularly within the portion of the protein C‐terminal to the R region 52. These findings suggest that the mechanism behind the phosphorylated R region stimulatory effect probably involves association of the phosphorylated R region with other parts of CFTR, possibly the N‐ or C‐terminus of CFTR, the intracellular domains or all of these regions.
Intermolecular interactions
Ko and coworkers reported reciprocal regulation between the solute carrier 26A (SLC26A) family (A3, A6) and CFTR 54. SLC26A3, a chloride–bicarbonate exchanger, co‐localizes with CFTR at the apical membrane, markedly increasing CFTR channel open probability, in a manner associated with direct interaction between the R region and the STAS domain of the SLC26A3. Binding of the STAS and R region is enhanced by PKA‐mediated phosphorylation of the R domain 54, pointing to another stimulatory effect of R region phosphorylation. One possible explanation for the SLC26A3‐mediated increase in CFTR activity may be that the presence of the STAS domain shifts the R region binding equilibria towards association with STAS and away from interactions with CFTR NBDs that block productive ATP‐dependent gating.
Since CFTR folding and processing is inefficient, even most WT channels do not reach the plasma membrane. F508del exacerbates the folding problem, so that most F508del CFTR is retained in the endoplasmic reticulum (ER) and only a small portion of the translated protein reaches the plasma membrane 55. Although the PKA‐phosphorylated F508del CFTR has been reported in one study to have the same open probability, its activation is at least seven times slower and its half‐life at the plasma membrane is much shorter than that of WT 52. Notably, pharmacologically ER‐trapped WT CFTR does not display the same degradation pattern as F508del 57 suggesting different intramolecular and intermolecular interactions, affecting both gating and processing.
The R region interacts with other proteins during transit of the channel from the ER to the plasma membrane and the post‐translational maturation of CFTR is enhanced by phosphorylation 58. CFTR plasma membrane levels increase upon PKA stimulation and can be blocked by the PKA inhibitor H‐89. These effects are associated with PKA‐mediated phosphorylation of the R region and its interactions with 14‐3‐3 proteins 58. 14‐3‐3 is a protein‐interacting adaptor molecule that regulates ER exit in a phosphorylation‐sensitive manner. Although 14‐3‐3 prefers to bind to short phosphorylated linear motifs, it can also recognize non‐phosphorylated substrates. All R region PKA phosphorylation motifs seem to be involved in regulation by 14‐3‐3 (Fig. 1) 58. The interaction between 14‐3‐3 and the phosphorylated R region might also increase heterodimerization of the NBDs through competition with the R region binding equilibria, thereby increasing the folding stability of CFTR and enhancing processing.
Most disordered proteins with a significant role in regulation contain molecular recognition elements (MoREs), short segments that are important for recognition of binding partners 59. MoREs are enriched in hydrophobic residues (W, L, F, Y) as well as charged residues, especially arginine and aspartate 59. The R region contains many predicted MoREs with different length distributions, with many related to the phosphorylation sites (almost all PKA sites contain leucines or phenylalanines and arginines). Experimental results show that the R region has three long and three short interaction segments for NBD1 and nine short for 14‐3‐3 (Fig. 1), reasonably consistent with the MoRE predictions. The observation that there are a larger number of R region binding partners than interaction segments suggests interaction hot spots with non‐exclusive binding preferences. Different proteins probably compete for the same segments with the phosphorylation status of the R region and the available partners determining the actual interactions.
R region in action
An emerging picture of the R region is as a disordered hub, sampling multiple conformations rapidly and forming transient interactions with different parts of CFTR and with other partners. By doing this, it gathers, transduces and distributes information from various sources. Phosphorylation events shift the conformational ensemble equilibrium. There is likely to be overlap in the conformational space that can be sampled by non‐, partially or highly phosphorylated R region. Figure 3 demonstrates the conformational heterogeneity of the R region in a model of non‐phosphorylated CFTR, representing a closed channel state. In this state the NBDs are probably separated; therefore the N‐terminus of the R region easily moves between the NBDs and samples similar conformations to those found in the NBD1 X‐ray structures (Fig. 2). This segment can also interact with other parts of the NBDs or other partners farther from the dimerization interface. The rest of the R region also samples a significant volume of space with the ability to make contact points with the NBDs. Figure 3 illustrates a model of phosphorylated CFTR, representing an open channel state, with the NBDs tightly associated. In a highly PKA‐phosphorylated state, the R region is expected to sample a similar volume of space as in non‐phosphorylated CFTR, although it is probably excluded from the NBD dimerization interface and therefore can extend farther away from the core of CFTR (Fig. 3). While this does not agree with hydrodynamic data from SAXS for the isolated R region, the increased β character observed by NMR, the reduction of its interactions with the NBDs and increased binding for the SLC26A3/6 STAS domains and 14‐3‐3 suggest that the phosphorylated R region may sample more extended conformations within the context of full‐length CFTR.
That the R region can sample similar space in either phosphorylation state and that in vivo CFTR has some degree of basal phosphorylation suggest that most of the binding partners interact with a variety of phosphorylation states of the R region, but with modulated affinities. One example is the interaction with 14‐3‐3, which interacts more weakly with the non‐phosphorylated state and more strongly following phosphorylation 58. Thus, R region phosphorylation is not a sharp switch but rather functions to precisely tune competing interactions.
Many CFTR interacting proteins at the plasma membrane are located in the same subcellular compartment as CFTR. Moreover, most of these are components of the same macromolecular complex anchored to the cytoskeleton. Activators, inhibitors, modifiers and other members of the complex enable CFTR to be readily responsive to different environmental conditions. The proposed view of the R region as a dynamic hub suggests that it plays a dominant role, scanning the surrounding protein surfaces and making transient interactions with affinities and lifetimes dependent on its phosphorylation state. The plasticity of this disordered region of CFTR is crucial for the complex regulatory control of CFTR channel activity and its other biological functions in maintaining ion balance at the cell membrane.
Acknowledgements
This work was supported by grants to J.D.F.‐K. from Cystic Fibrosis Canada, the Cystic Fibrosis Foundation and the Natural Sciences and Engineering Research Council of Canada.
References
- 1.Gadsby DC & Nairn AC (1999) Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol Rev 79, S77–S107 [DOI] [PubMed] [Google Scholar]
- 2.Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR & Smith AE (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63, 827–834 [DOI] [PubMed] [Google Scholar]
- 3.Ostedgaard LS, Baldursson O & Welsh MJ (2001) Regulation of the cystic fibrosis transmembrane conductance regulator Cl‐channel by its R domain. J Biol Chem 276, 7689–7692 [DOI] [PubMed] [Google Scholar]
- 4.Csanády L, Chan KW, Seto‐Young D, Kopsco DC, Nairn AC & Gadsby DC (2000) Severed channels probe regulation of gating of cystic fibrosis transmembrane conductance regulator by its cytoplasmic domains. J Gen Physiol 116, 477–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hudson RP, Chong PA, Protasevich II, Vernon R, Noy E, Bihler H, An JL, Kalid O, Sela‐Culang I, Mense Met al (2012) Conformational changes relevant to channel activity and folding within the first nucleotide binding domain of the cystic fibrosis transmembrane conductance regulator. J Biol Chem 287, 28480–28494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ostedgaard LS, Baldursson O, Vermeer DW, Welsh MJ & Robertson AD (2000) A functional R domain from cystic fibrosis transmembrane conductance regulator is predominantly unstructured in solution. Proc Natl Acad Sci USA 97, 5657–5662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dulhanty AM & Riordan JR (1994) A two‐domain model for the R domain of the cystic fibrosis transmembrane conductance regulator based on sequence similarities. FEBS Lett 343, 109–114 [DOI] [PubMed] [Google Scholar]
- 8.Marasini C, Galeno L & Moran O (2012) Thermodynamic study of the native and phosphorylated regulatory domain of the CFTR. Biochem Biophys Res Commun 423, 549–552 [DOI] [PubMed] [Google Scholar]
- 9.Marasini C, Galeno L & Moran O (2013) A SAXS‐based ensemble model of the native and phosphorylated regulatory domain of the CFTR. Cell Mol Life Sci 70, 923–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker JMR, Hudson RP, Kanelis V, Choy W‐Y, Thibodeau PH, Thomas PJ & Forman‐Kay JD (2007) CFTR regulatory region interacts with NBD1 predominantly via multiple transient helices. Nat Struct Mol Biol 14, 738–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hegedus T, Serohijos AWR, Dokholyan NV, He L & Riordan JR (2008) Computational studies reveal phosphorylation‐dependent changes in the unstructured R domain of CFTR. J Mol Biol 378, 1052–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mertens HDT & Svergun DI (2010) Structural characterization of proteins and complexes using small‐angle X‐ray solution scattering. J Struct Biol 172, 128–141 [DOI] [PubMed] [Google Scholar]
- 13.Chen JM, Scotet V & Ferec C (2000) Definition of a ‘functional R domain’ of the cystic fibrosis transmembrane conductance regulator. Mol Genet Metab 71, 245–249 [DOI] [PubMed] [Google Scholar]
- 14.Felli IC & Pierattelli R (2012) Recent progress in NMR spectroscopy: toward the study of intrinsically disordered proteins of increasing size and complexity. IUBMB Life 64, 473–481 [DOI] [PubMed] [Google Scholar]
- 15.Lewis HA, Buchanan SG, Burley SK, Conners K, Dickey M, Dorwart M, Fowler R, Gao X, Guggino WB, Hendrickson WAet al (2004) Structure of nucleotide‐binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J 23, 282–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewis HA, Zhao X, Wang C, Sauder JM, Rooney I, Noland BW, Lorimer D, Kearins MC, Conners K, Condon Bet al (2005) Impact of the deltaF508 mutation in first nucleotide‐binding domain of human cystic fibrosis transmembrane conductance regulator on domain folding and structure. J Biol Chem 280, 1346–1353 [DOI] [PubMed] [Google Scholar]
- 17.Zhang L, Aleksandrov LA, Riordan JR & Ford RC (2011) Domain location within the cystic fibrosis transmembrane conductance regulator protein investigated by electron microscopy and gold labelling. Biochim Biophys Acta 1808, 399–404 [DOI] [PubMed] [Google Scholar]
- 18.Serohijos AWR, Hegedus T, Aleksandrov AA, He L, Cui L, Dokholyan NV & Riordan JR (2008) Phenylalanine‐508 mediates a cytoplasmic–membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci USA 105, 3256–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mornon J‐P, Lehn P & Callebaut I (2009) Molecular models of the open and closed states of the whole human CFTR protein. Cell Mol Life Sci 66, 3469–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM & Obradović Z (2002) Intrinsic disorder and protein function. Biochemistry 41, 6573–6582 [DOI] [PubMed] [Google Scholar]
- 21.Iakoucheva LM, Brown CJ, Lawson JD, Obradović Z & Dunker AK (2002) Intrinsic disorder in cell‐signaling and cancer‐associated proteins. J Mol Biol 323, 573–584 [DOI] [PubMed] [Google Scholar]
- 22.Ma J, Zhao J, Drumm ML, Xie J & Davis PB (1997) Function of the R domain in the cystic fibrosis transmembrane conductance regulator chloride channel. J Biol Chem 272, 28133–28141 [DOI] [PubMed] [Google Scholar]
- 23.Picciotto MR, Cohn JA, Bertuzzi G, Greengard P & Nairn AC (1992) Phosphorylation of the cystic fibrosis transmembrane conductance regulator. J Biol Chem 267, 12742–12752 [PubMed] [Google Scholar]
- 24.Wilkinson DJ, Strong TV, Mansoura MK, Wood DL, Smith SS, Collins FS & Dawson DC (1997) CFTR activation: additive effects of stimulatory and inhibitory phosphorylation sites in the R domain. Am J Physiol 273, L127–L133 [DOI] [PubMed] [Google Scholar]
- 25.Chang XB, Tabcharani JA, Hou YX, Jensen TJ, Kartner N, Alon N, Hanrahan JW & Riordan JR (1993) Protein kinase A (PKA) still activates CFTR chloride channel after mutagenesis of all 10 PKA consensus phosphorylation sites. J Biol Chem 268, 11304–11311 [PubMed] [Google Scholar]
- 26.Chappe V, Hinkson DA, Howell LD, Evagelidis A, Liao J, Chang X‐B, Riordan JR & Hanrahan JW (2004) Stimulatory and inhibitory protein kinase C consensus sequences regulate the cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci USA 101, 390–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jia Y, Mathews CJ & Hanrahan JW (1997) Phosphorylation by protein kinase C is required for acute activation of cystic fibrosis transmembrane conductance regulator by protein kinase A. J Biol Chem 272, 4978–4984 [DOI] [PubMed] [Google Scholar]
- 28.Kongsuphol P, Cassidy D, Hieke B, Treharne KJ, Schreiber R, Mehta A & Kunzelmann K (2009) Mechanistic insight into control of CFTR by AMPK. J Biol Chem 284, 5645–5653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.King JD, Fitch AC, Lee JK, McCane JE, Mak D‐OD, Foskett JK & Hallows KR (2009) AMP‐activated protein kinase phosphorylation of the R domain inhibits PKA stimulation of CFTR. Am J Physiol 297, C94–C101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vais H, Zhang R & Reenstra WW (2004) Dibasic phosphorylation sites in the R domain of CFTR have stimulatory and inhibitory effects on channel activation. Am J Physiol 287, C737–C745 [DOI] [PubMed] [Google Scholar]
- 31.Thelin WR, Kesimer M, Tarran R, Kreda SM, Grubb BR, Sheehan JK, Stutts MJ & Milgram SL (2005) The cystic fibrosis transmembrane conductance regulator is regulated by a direct interaction with the protein phosphatase 2A. J Biol Chem 280, 41512–41520 [DOI] [PubMed] [Google Scholar]
- 32.Hallows KR, Raghuram V, Kemp BE, Witters LA & Foskett JK (2000) Inhibition of cystic fibrosis transmembrane conductance regulator by novel interaction with the metabolic sensor AMP‐activated protein kinase. J Clin Invest 105, 1711–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dahan D, Evagelidis A, Hanrahan JW, Hinkson DA, Jia Y, Luo J & Zhu T (2001) Regulation of the CFTR channel by phosphorylation. Eur J Physiol 443, S92–S96 [DOI] [PubMed] [Google Scholar]
- 34.Luo J, Pato MD, Riordan JR, Hanrahan JW, Physiol AJ & Circ H (1998) Differential regulation of single CFTR channels by PP2C, PP2A, and other phosphatases. Am J Physiol 274, 1397–1410 [DOI] [PubMed] [Google Scholar]
- 35.Zhu T, Dahan D, Evagelidis A, Zheng S, Luo J & Hanrahan JW (1999) Association of cystic fibrosis transmembrane conductance regulator and protein phosphatase 2C. J Biol Chem 274, 29102–29107 [DOI] [PubMed] [Google Scholar]
- 36.Vastiau A, Cao L, Jaspers M, Owsianik G, Janssens V, Cuppens H, Goris J, Nilius B & Cassiman J‐J (2005) Interaction of the protein phosphatase 2A with the regulatory domain of the cystic fibrosis transmembrane conductance regulator channel. FEBS Lett 579, 3392–3396 [DOI] [PubMed] [Google Scholar]
- 37.King JD, Lee J, Riemen CE, Neumann D, Xiong S, Foskett JK, Mehta A, Muimo R & Hallows KR (2012) Role of binding and nucleoside diphosphate kinase A in the regulation of the cystic fibrosis transmembrane conductance regulator by AMP‐activated protein kinase. J Biol Chem 287, 33389–33400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ostedgaard LS, Randak C, Rokhlina T, Karp P, Vermeer D, Ashbourne Excoffon KJ & Welsh MJ (2003) Effects of C‐terminal deletions on cystic fibrosis transmembrane conductance regulator function in cystic fibrosis airway epithelia. Proc Natl Acad Sci USA 100, 1937–1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monterisi S, Favia M, Guerra L, Cardone RA, Marzulli D, Reshkin SJ, Casavola V & Zaccolo M (2012) CFTR regulation in human airway epithelial cells requires integrity of the actin cytoskeleton and compartmentalized cAMP and PKA activity. J Cell Sci 125, 1106–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liedtke CM, Raghuram V, Yun CC & Wang X (2004) Role of a PDZ1 domain of NHERF1 in the binding of airway epithelial RACK1 to NHERF1. Am J Physiol 286, C1037–C1044 [DOI] [PubMed] [Google Scholar]
- 41.Fischer H, Illek B & Machen TE (1998) Regulation of CFTR by protein phosphatase 2B and protein kinase C. Eur J Physiol 436, 175–181 [DOI] [PubMed] [Google Scholar]
- 42.Li J, Dai Z, Jana D, Callaway DJE & Bu Z (2005) Ezrin controls the macromolecular complexes formed between an adapter protein Na+/H+ exchanger regulatory factor and the cystic fibrosis transmembrane conductance regulator. J Biol Chem 280, 37634–37643 [DOI] [PubMed] [Google Scholar]
- 43.Auerbach M & Liedtke CM (2007) Role of the scaffold protein RACK1 in apical expression of CFTR. Am J Physiol 293, C294–C304 [DOI] [PubMed] [Google Scholar]
- 44.Csanády L, Seto‐Young D, Chan KW, Cenciarelli C, Angel BB, Qin J, McLachlin DT, Krutchinsky AN, Chait BT, Nairn ACet al (2005) Preferential phosphorylation of R‐domain serine 768 dampens activation of CFTR channels by PKA. J Gen Physiol 125, 171–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dulhanty AM & Riordan JR (1994) Phosphorylation by cAMP‐dependent protein kinase causes a conformational change in the R domain of the cystic fibrosis transmembrane conductance regulator. Biochemistry 33, 4072–4079 [DOI] [PubMed] [Google Scholar]
- 46.Grimard V, Li C, Ramjeesingh M, Bear CE, Goormaghtigh E & Ruysschaert J‐M (2004) Phosphorylation‐induced conformational changes of cystic fibrosis transmembrane conductance regulator monitored by attenuated total reflection‐Fourier transform IR spectroscopy and fluorescence spectroscopy. J Biol Chem 279, 5528–5536 [DOI] [PubMed] [Google Scholar]
- 47.Naren AP, Cormet‐Boyaka E, Fu J, Villain M, Blalock JE, Quick MW & Kirk KL (1999) CFTR chloride channel regulation by an interdomain interaction. Science 286, 544–548 [DOI] [PubMed] [Google Scholar]
- 48.Bozoky Z, Krzeminski M, Baker JM, Muhandiram R, Birtley J, Al‐Zahrani A, Thomas PJ, Frizzell R, Ford RC & Forman‐Kay JD (2012) Phospho‐dependent interactions of the regulatory R region of CFTR and structural models of dynamic R region interactions within full‐length CFTR and with 14‐3‐3. Pediatr Pulmonol 35 (Suppl), 226 [Google Scholar]
- 49.Wang G & Duan DD (2012) Regulation of activation and processing of the cystic fibrosis transmembrane conductance regulator (CFTR) by a complex electrostatic interaction between the regulatory domain and cytoplasmic loop 3. J Biol Chem 287, 40484–40492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baldursson O, Ostedgaard LS, Rokhlina T, Cotten JF & Welsh MJ (2001) Cystic fibrosis transmembrane conductance regulator Cl‐channels with R domain deletions and translocations show phosphorylation‐dependent and ‐independent activity. J Biol Chem 276, 1904–1910 [DOI] [PubMed] [Google Scholar]
- 51.Kanelis V, Hudson RP, Thibodeau PH, Thomas PJ & Forman‐Kay JD (2010) NMR evidence for differential phosphorylation‐dependent interactions in WT and DeltaF508 CFTR. EMBO J 29, 263–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chappe V, Irvine T, Liao J, Evagelidis A & Hanrahan JW (2005) Phosphorylation of CFTR by PKA promotes binding of the regulatory domain. EMBO J 24, 2730–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seavilleklein G, Amer N, Evagelidis A, Chappe F, Irvine T, Hanrahan JW & Chappe V (2008) PKC phosphorylation modulates PKA‐dependent binding of the R domain to other domains of CFTR. Am J Physiol 295, C1366–C1375 [DOI] [PubMed] [Google Scholar]
- 54.Ko SBH, Zeng W, Dorwart MR, Luo X, Kim KH, Millen L, Goto H, Naruse S, Soyombo A, Thomas PJet al (2004) Gating of CFTR by the STAS domain of SLC26 transporters. Nat Cell Biol 6, 343–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lukacs GL, Mohamed A, Kartner N, Chang XB, Riordan JR & Grinstein S (1994) Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J 13, 6076–6086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang F, Zeltwanger S, Hu S & Hwang TC (2000) Deletion of phenylalanine 508 causes attenuated phosphorylation‐dependent activation of CFTR chloride channels. J Physiol 524, 637–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tosoni K, Stobbart M, Cassidy DM, Venerando A, Pagano MA, Luz S, Amaral MD, Kunzelmann K, Pinna LA, Farinha CMet al (2013) CFTR mutations altering CFTR fragmentation. Biochem J 449, 295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liang X, Da Paula AC, Bozóky Z, Zhang H, Bertrand CA, Peters KW, Forman‐Kay JD & Frizzell RA (2012) Phosphorylation‐dependent 14‐3‐3 protein interactions regulate CFTR biogenesis. Mol Biol Cell 23, 996–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fuxreiter M, Tompa P & Simon I (2007) Local structural disorder imparts plasticity on linear motifs. Bioinformatics 23, 950–956 [DOI] [PubMed] [Google Scholar]