

Abstract
Ventricular assist devices (VADs) are implanted in patients with end-stage heart failure to provide both short- and long-term hemodynamic support. Unfortunately, bleeding and thromboembolic complications due to the severely disturbed, dynamic flow conditions generated within these devices require complex, long-term anti-platelet and anticoagulant therapy. While several studies have examined the effectiveness of one such agent, aspirin, under flow conditions, data comparing the efficacy of in vitro and in vivo metabolized aspirin is lacking. Two sets of studies were conducted in vitro with purified human platelets circulating for 30 min in a flow loop containing the DeBakey VAD (MicroMed Cardiovascular, Houston, TX, USA): (a) 20 μM aspirin was added exogenously in vitro to platelets isolated from aspirin-free subjects, and (b) platelets were obtained from donors 2 h (n = 14) and 20 h (n = 13) after ingestion of 1,000 mg aspirin. Near real-time platelet activation state (PAS) was measured with a modified prothrombinase-based assay. Platelets exposed to aspirin in vitro and in vivo (metabolized) showed 28.2 and 25.3 % reduction in platelet activation rate, respectively, compared to untreated controls. Our results demonstrate that in vitro treatment with antiplatelet drugs such as aspirin is as effective as in vivo metabolized aspirin in testing the effect of reducing shear-induced platelet activation in the VAD. Using the PAS assay provides a practical in vitro alternative to in vivo testing of antiplatelet efficacy, as well as for testing the thrombogenic performance of devices during their research and development.
Keywords: Aspirin, Ventricular assist devices, Platelets, Thrombin
Introduction
Mechanical circulatory assist (MCS) devices such as ventricular assist devices (VADs) are often employed to augment the function of failing ventricles in patients with advanced heart failure. While VADs are effectively utilized to provide a bridge-to-transplant or as destination therapy, they unfortunately still have limitations such as mechanical failure, infection, bleeding, and thrombosis [1]. Intracorporeal continuous-flow axial rotary VADs in particular offer a compact design and simplicity in operation [2], but are characterized by pathologic flow patterns that generate elevated shear stresses and exposure times. These conditions make platelets more susceptible to shear-induced activation, resulting in enhanced coagulation and formation of thromboemboli [3]. Patients implanted with such devices are routinely prescribed antiplatelet and anticoagulant agents to limit such complications [4]. Pharmacologic antithrombotic therapy is complicated and often problematic for these patients, with the observation of concomitant MCS-associated hemorrhagic complications such as acquired von Willebrand disease (VWD), gastrointestinal and intracranial bleeding [3]. Several techniques are employed to monitor the efficacy of pharmacotherapy in reducing risk of thrombosis [5, 6], and include thromboelastography (TEG), prothrombin time (PT), activated partial thromboplastin time (aPTT), International normalized ratio (INR), aggregometry, and flow cytometry [4, 7–9], as well as point-of-care devices (platelet function analyzer [10, 11], cone-and-plate(let) analyzer [12, 13], VerifyNow [14], and PlateletMapping [14–16]). However, a standardized approach to objectively determine pharmacotherapeutic efficacy is lacking [17, 18].
Patients on continuous-flow VADs undergo pharmacotherapy that includes both anticoagulant and antiplatelet prophylaxis—the latter is our focus in the current study—to measure the hypothesized decrease in shear-induced platelet activation with the addition of antiplatelet agents in vitro. We studied the effects of aspirin [acetylsalicyclic acid (ASA)] as it is routinely prescribed to limit the effects of flow-induced platelet activation in most MCS devices and device components such as the VAD, mechanical heart valves (MHVs) and the total artificial heart (TAH) [4]. Aspirin is known to significantly reduce occurrence of adverse cardiovascular events such as stroke and myocardial infarction [19]. While aspirin is known to selectively inhibit platelet aggregation induced via the cyclooxygenase (COX-1) pathway (i.e., by agonists such as ADP, epinephrine and collagen) [20], the effect of aspirin on shear-induced platelet activation—via a different activation pathway—is poorly understood. It is speculated that aspirin inhibits thrombin generation in blood by acetylating pro-thrombin and macromolecules on the platelet membrane rather than a direct inhibition of cyclooxygenase [21]. The antithrombotic properties of aspirin post-implant of MHVs have been confirmed by a meta-analysis based on 10 separate studies wherein aspirin reduced thromboembolic events from 9 to 4 %, and a lower dose (100 mg/day) was associated with significantly reduced risk of bleeding [22].
It is known that despite the administration of aspirin, about 26 % of patients suffer from aspirin resistance and persistent platelet activation [23]. Two of the most commonly-implanted VADs in clinical trials, the recently-approved HeartMate II (Thoratec Corporation, Pleasanton, CA, USA) and the DeBakey (MicroMed Cardiovascular, Houston, TX, USA), suffer from thromboembolic incident rates of 0.9–5 and 10.7–13 %, respectively, despite prophylaxis [24–27]. While physicians have tried to optimize antiplatelet dosing [18], these efforts do not address the underlying cause of thrombotic complications, namely the flow conditions in the devices themselves. Several studies have examined the effect of aspirin administered directly [28, 29] or metabolized in vivo [30, 31] on platelet activation or aggregation under controlled in vitro fluid shear conditions. To the best of our knowledge, no in vitro examination has studied the antiplatelet effect of aspirin under the dynamic flow conditions found in VADs.
The aims of this study were twofold: (1) show that platelets directly treated with aspirin or obtained from subjects who ingested aspirin yield similar activation rates after subsequent exposure in a VAD; and (2) demonstrate that treatment with aspirin is less effective in reducing platelet activation rate (PAR) compared to device design modification. The global objective of this study is to initiate development of in vitro methodologies that compare device design changes side-by-side with traditional pharmacotherapy in order to reduce device-induced thrombogenicity and lower incidences of drug-induced complications. Our group has developed and successfully used a modified prothrombinase-based assay to measure the near real-time bulk platelet activation state (PAS) [32] of platelets in flow loops containing prosthetic heart valves [33], VADs [34], and TAH [35]. In this study, we examine the evolution of PAS of platelets during repeated passages in the DeBakey VAD after in vitro administration or in vivo metabolism of aspirin and demonstrate the similarity of the two approaches in the resulting platelet activation response. These results are compared with our recently published data from the design optimization of the DeBakey VAD to the HeartAssist 5 VAD (MicroMed Cardiovascular, Houston, TX, USA) [34] and show the distinct advantage of improving device thrombogenic performance of MCS devices by design optimization as opposed to more traditional and complex antiplatelet therapy.
Materials and methods
Study design
Two sets of studies were conducted: (a) Drug-free subjects did not take ASA prior to blood donation, with ASA added to platelets in vitro; (b) Drug-loaded subjects were administered ASA; with blood being obtained prior to ingestion of ASA and after a specified time (either 2 or 20 h). Specifically, the following three scenarios were studied: (1) in vitro ASA treatment of platelets, (2) ingestion of ASA and blood drawn at 0 h (pre-ingestion) and 2 h, and (3) ingestion of ASA and blood drawn at 0 h (pre-ingestion) and 20 h. A total of 45 healthy adult volunteers were recruited for the above three scenarios. Volunteers were screened and excluded for the following risk factors: regular aspirin or ibuprofen use or intake within 2 weeks of participation, aspirin allergy, asthma, history of gastrointestinal disorders, pregnancy, smoking, high blood pressure, high cholesterol, type I and type II diabetes, high alcoholic intake, history of myocardial infarction or stroke, and Omega-3 supplement use. Consent was obtained as per Stony Brook University IRB-approved protocol and whole blood was drawn via venipuncture into 10 % ACD-A.
In vitro drug reconstitution, in vivo antiplatelet treatment, and purified platelet preparation
For the in vitro treatment with ASA, 120 mL of whole blood was obtained, while for the in vivo ASA studies, 30 mL of whole blood was drawn from the antecubital vein prior to ASA ingestion. Participants in the in vivo treatments ingested 1,000 mg buffered aspirin (two 500 mg Ascriptin tablets, Novartis, East Hanover, NJ, USA) and were asked to return either 2 or 20 h after their initial donation for a second 30 mL donation. In addition, blood serum obtained after both donations from the volunteers who ingested ASA was analyzed for salicylate concentration.
Purified gel-filtered platelets (GFP) were prepared from whole blood as previously described and diluted to a count of 15,000 μL in HEPES-modified Tyrode’s buffer (“platelet buffer”) with 5 mM Ca2+ [32, 36]. For in vitro ASA experiments, GFP were treated with ASA dissolved in sodium bicarbonate solution (324 mg ASA, 965 mg citric acid, and 1,744 mg sodium hydrogen carbonate in 50 mL double-distilled H2O) 10 min prior to experiments. Control platelet mixtures were prepared with the addition of the solvent vehicle and without ASA 10 min prior to exposure in the VAD.
Exposure in VAD and measurement of PAS
GFP mixtures were exposed for 30 min at 37 °C in a flow loop containing a DeBakey VAD (MicroMed Cardiovascular Inc., Houston, TX, USA), with the inlet and outlets connected via short 0.5″ I.D. Tygon R3603 loop tubing and 0.25″ I.D. Tygon R3603 flow resistor tubing of 47″ length (Fig. 1). VAD pump conditions were set at 4 L/min cardiac output and 9,500 rpm, corresponding to average physiological and clinical operating ranges, and controlled using the MicroMed Clinical Data Acquisition System (CDAS). These settings correspond to a pressure rise of ~70–80 mmHg across the pump [34].
Fig. 1.
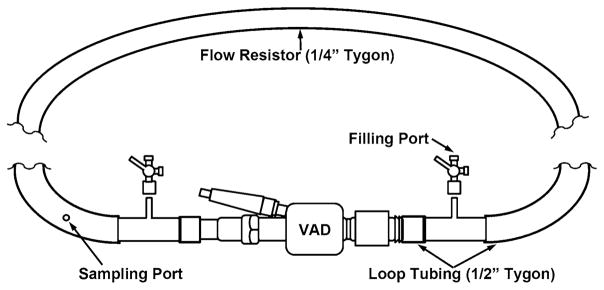
Flow loop with MicroMed DeBakey VAD. Platelets were recirculated through a VAD operating at 9,500 rpm and a cardiac output of 4 L/min for 30 min. Platelet samples were withdrawn every 10 min through a silicone port upstream of the VAD. (Adapted with permission from Girdhar et al. [34])
Platelet samples were taken at t = 0, 10, 20, and 30 min for the chemically modified prothrombinase PAS assay [32, 37] through a silicone sampling port upstream of the VAD. Briefly, the PAS assay uses acetylated prothrombin to measure the rate of thrombin generation. The use of acetylated prothrombin blocks feedback action of generated thrombin on the platelets, and ensures linear kinetics during the assay and quantitative measurement of PAS. The results of this assay correlate well with P-selectin [33] and Annexin V [37] expression, as quantified with flow cytometry. PAS values were normalized against the activity of fully activated platelets, obtained by sonication (10 W for 10 s, Branson Sonifier 150 with microprobe, Branson, MO, USA). PAS values are therefore expressed as a fraction of the maximum thrombin-generating capacity, with a maximum of 1.0. This assay allows for a 1:1 correlation between the applied shear stress and thrombin generation and activation changes as low as 0.1 % can be detected [32]. The PAR for each experiment was obtained from the slopes of best-fit lines fit to normalized PAS values.
Statistical analysis
Differences in PAR were obtained by subtracting the PAR calculated for ASA-treated platelet experiments from PAR obtained for paired control experiments conducted on the same day in a similar manner with untreated platelets. Percentage change in the PAR was calculated by dividing this difference by the control PAR and multiplying by 100. Paired samples Student’s t-tests were used to compare the ASA-treated and control PAR values for each set of experiments, while differences between the PAR for control and ASA-treated experiment were compared to a value of 0, which represents the condition where PAR for ASA-treated and untreated platelets is identical. Significance was achieved for p < 0.05. Results are presented as the mean ± standard error of the mean (SEM), unless otherwise stated.
Results
Platelet activation was measured using the modified pro-thrombinase method for platelets flowing in the MicroMed DeBakey VAD operating at a cardiac output of 4 L/min and 9,500 rpm for 30 min. Three sets of experiments were conducted: (1) platelets treated in vitro with 20 μM ASA (n = 15), (2) platelets treated in vivo with 1,000 mg ASA and tested 2 h after ingestion (n = 14), and (3) platelets treated in vivo with 1,000 mg ASA and tested 20 h after ingestion (n = 13). One volunteer from the 20 h in vivo ASA treatment experiments did not complete the study, while one volunteer each from the two in vivo ASA treatment studies yielded PARs more than two standard deviations from mean values, and data from their participation is not considered in the subsequent analysis. For each ASA-treated platelet experiment, a paired control experiment with untreated platelets and the solvent vehicle control was performed on the same day with identical VAD operating conditions.
Significant reduction in PAR was observed for platelets treated in vitro with 20 μM ASA (Fig. 2). Mean PAR decreased 0.94 ± 0.42 (×10−4) min−1, or 28.2 %, after in vitro ASA treatment compared to paired untreated controls (p < 0.05, Table 1). In vivo ASA-treated platelets showed a 0.45 ± 0.15 (×10−4) min−1, or 25.3 %, reduction in PAR 2 h after treatment when compared with paired control experiments (p < 0.01, Fig. 3; Table 1). This contrasts with a 0.01 ± 0.35 (×10−4) min−1, or 0.6 %, increase in PAR 20 h after treatment when compared with paired control experiments (p > 0.5, Fig. 4; Table 1). Salicylate concentration increased to 6.73 ± 0.56 mg/dL 2 h after ASA ingestion (p < 0.001, Fig. 5a), but returned to control baseline levels 20 h post-ingestion (0.13 ± 0.07 mg/dL, p > 0.5, Fig. 5b; Table 1).
Fig. 2.
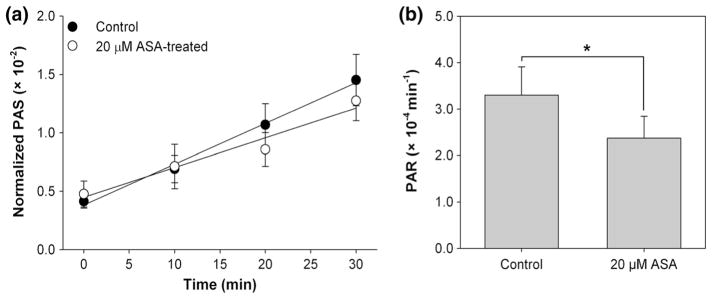
Platelet activation post-in vitro ASA treatment. a Evolution of PAS for 20 μM ASA-treated platelets and untreated platelets recirculated for 30 min through the VAD showed a b 28.2 % decrease in the PAR after ASA treatment, determined from the slope of lines fit to PAS values (n = 15, p < 0.05). Error bars represent the SEM of PAS or PAR, respectively
Table 1.
PARs and salicylate concentrations for ASA-treated platelets in the DeBakey VAD
| Experiment | PARa (×10−4 min−1) | ΔPARa (×10−4 min−1) | p vs. control | [SAL]b (mg/dL) | p vs. control |
|---|---|---|---|---|---|
| In vitro ASA treatment (n= 15) | |||||
| Control | 3.30 ± 0.61 | – | – | ||
| 20 μM ASA | 2.37 ± 0.47 | 0.94 ± 0.42 | 0.04 | ||
| In vivo 2 h ASA treatment (n = 14) | |||||
| Control | 1.78 ± 0.20 | – | – | 0.03 ± 0.02 | – |
| 2 h post-treatment | 1.33 ± 0.17 | 0.45 ± 0.15 | < 0.01 | 6.73 ± 0.56 | < 0.001 |
| In vivo 20 h ASA treatment (n = 13) | |||||
| Control | 1.62 ± 0.32 | – | – | 0.12 ± 0.06 | – |
| 20 h post-treatment | 1.63 ± 0.33 | −0.01 ± 0.35 | > 0.5 | 0.13 ± 0.07 | > 0.5 |
PAR, ΔPAR, and (SAL) values are presented as mean ± SEM
Mean PAR values are obtained from the slope of lines fit to PAS values from the 30 min platelet exposure in the VAD. The difference in PAR, ΔPAR, between the aspirin-treated platelets and untreated platelets are reported as the means of ΔPAR for each experimental pair
Salicylate concentrations (SAL), were obtained during each blood donation and prior to experiments
Fig. 3.
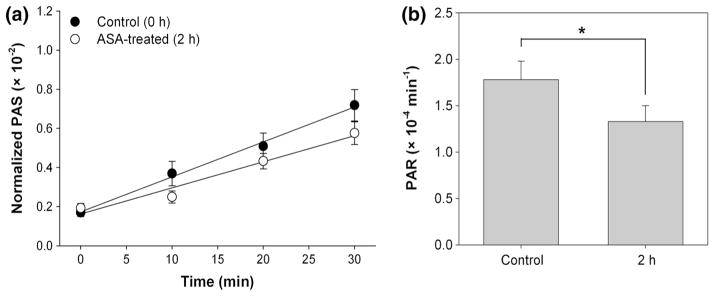
Platelet activation 2 h post-in vivo ASA treatment. a Evolution of PAS for ASA-treated platelets and untreated platelets recirculated for 30 min through the VAD showed a b 25.3 % decrease in the PAR after ASA treatment, determined from the slope of lines fit to PAS values (n = 14, p < 0.01). Error bars represent the SEM of PAS or PAR, respectively
Fig. 4.
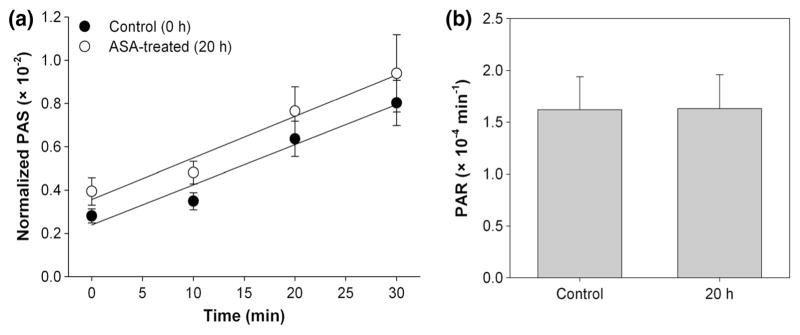
Platelet activation 20 h post-in vivo ASA treatment. a Evolution of PAS for ASA-treated platelets and untreated platelets recirculated for 30 min through the VAD showed a b 0.6 % increase in the PAR after ASA treatment, determined from the slope of lines fit to PAS values (n = 13, p > 0. 5). Error bars represent the SEM of PAS or PAR, respectively
Fig. 5.
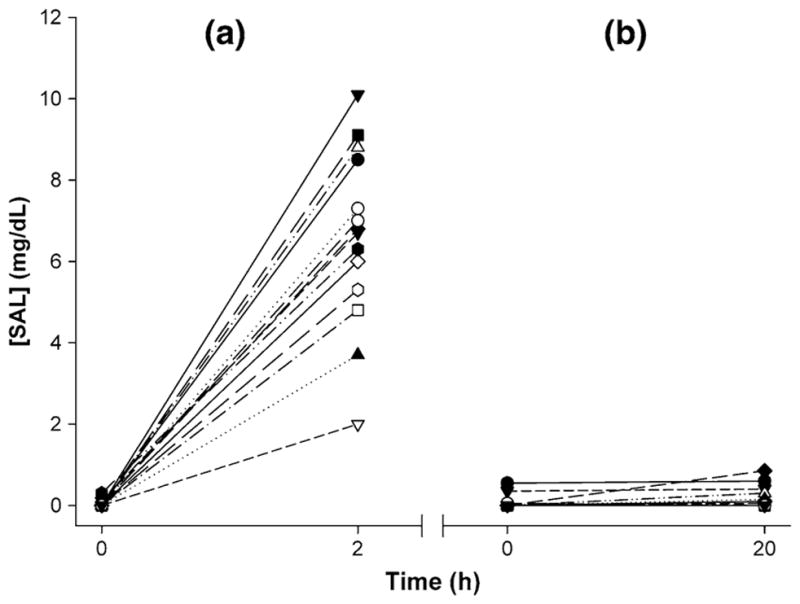
Salicylate concentration (SAL) a 2 h and b 20 h post-in vivo ASA treatment. Blood samples showed a mean increase of 6.70 mg/dL SAL 2 h post-ingestion (n = 14, p < 0.001) and 0.01 mg/dL 20 h after ingestion (n = 13, p > 0.5)
However, the reduction in platelet activation achieved with ASA is threefold less than that achieved by design optimization of the original DeBakey VAD to the Heart-Assist 5 VAD [34].
Discussion
We investigated whether aspirin added to platelets in vitro (pro-drug form) or in vivo (metabolized form) prior to the in vitro recirculation experiments in the DeBakey VAD decrease shear-induced platelet activation due to repeated passages through the VAD. We demonstrate for the first time that direct treatment with aspirin is as effective as in vivo metabolized aspirin in reducing platelet activation due to the dynamic flow conditions in the DeBakey VAD. We have shown that a maximum over-the-counter dosage of 1,000 mg aspirin yields a 25.3 % drop in PAR 2 h after ingestion when exposed in a flow loop containing the DeBakey VAD, but this activation rate returns to baseline levels 20 h after ingestion (Table 1), emphasizing the need for daily aspirin administration. Direct treatment with 20 μM aspirin yields a 28.2 % drop in PAR under similar experimental conditions (Table 1).
Standard antiplatelet protocols for individuals implanted with VADs involve commencing administration of 81–325 mg ASA daily 2–5 days postoperatively [7], with maximum dosage of 650 mg/day [4]. However, doses as low as 40 mg are sufficient to cause significant prolongation of bleeding time [38]. Despite daily aspirin therapy, up to 40 % of VAD patients encounter aspirin resistance, and increase in dosage reduces platelet aggregation in most patients [23, 39]. Investigators have attributed this to high platelet turnover after surgery [40], particularly among the high fraction of young platelets unresponsive to aspirin [41], elevated thromboxane production [42], and uninhibited thrombin generation [43], among other potential etiologies. For this reason, some VAD patients concomitantly take dipyridamole, as it has the effect of reducing platelet aggregation by inhibition of adenosine uptake and inhibition of phosphodiesterase pathways more than aspirin alone [4, 44] and does appear to significantly increase the risk of bleeding [45]. We observed that platelets treated directly with 20 μM dipyridamole showed 31.2 % decrease in activation rate [46], showing reduction similar to that of aspirin-treated platelets, and it may be inferred that concurrent treatment is likely to reduce this activation rate further. This will be investigated in our future studies.
We used isolated platelets in order to examine the direct effect of aspirin and fluid shear stress on thrombin generation without the subsequent feedback, platelet aggregation, and clot formation. These platelets were diluted to a concentration of 15,000 μL due to the large volume required for the flow loop and limited blood volume obtained from volunteers. These limitations may not directly extrapolate to the response of physiological whole blood consisting of red blood cells, white blood cells, platelets at tenfold higher concentration, and plasma proteins, which have the ability to amplify the activation response, and lead to aggregation and adhesion downstream of the VAD. Flow cytometry provides detailed information about expression and amount of platelet membrane glycoproteins, as well as the extent of individual platelet activation, and is able to rapidly process large amounts of individual platelets in small volumes [47]. In addition, flow cytometry has been utilized to measure platelet activation after VAD implantation [9, 23, 48]. However, the PAS assay was employed in our study as it provides near-real time information on bulk platelet thrombin generation and shows the direct effect of fluid shear stress. This assay has also been successfully used in measuring the PAS of platelet-red blood cell mixtures flowing through the DeBakey and HeartAsssist 5 VADs, without and with treatment with dipyridamole [34, 46]. In addition, treating platelets directly with 20 μM solubilized aspirin was slightly more effective than 2 h in vivo metabolism of 1,000 mg buffered aspirin (Table 1). This may allow a quicker and more efficient in vitro approach of studying the shear-induced activation of aspirin-treated platelets in blood recirculating devices.
The passage time of platelets in the healthy human or in patients implanted with axial flow VADs operating at 4 L/min is ~60 s, whereas the passage time in the flow loop utilized in this study is ~1.8 s, and the platelets are thus exposed to the equivalent of 16.7 h of exposure during the experiment. While the PARs observed during the experimental exposure are low, extrapolating their activity over the 5–7 days average lifetime of a platelet in combination with their irreversible damage history and subsequent shear-induced sensitization [49] yields platelets that are quite prothrombotic, even after activation rate reduction observed in our experiments post-treatment with high-dose aspirin.
The development of safe and effective VAD therapy is a pressing issue necessitated by the emergence of complications such thrombosis and bleeding, due to pathological device-induced flow conditions and side effects of pharmacotherapy [4, 17]. Resolution of these complications is paramount for patients implanted with such devices for destination therapy, particularly when viable heart transplants are not possible or available [50, 51]. Traditionally, VAD patients are placed on concurrent antiplatelet and anticoagulant therapy, but this approach carries an inherent risk of bleeding [4]. While several studies have proposed optimal anticoagulation and antiplatelet protocols for axial flow VAD patients, with warfarin (INR of 2.5) and 100 mg/day aspirin resulting in the lowest bleeding-thrombosis rates [18], such protocols do not reduce these incidences to truly acceptable levels, nor do they mitigate device-induced complications, such as acquired VWD [52]. Design optimization techniques, such as the device thrombogenicity emulation (DTE) methodology developed by our group [53], offer an alternative approach to reduce device-induced risk of thrombosis and bleeding, and in turn lower the need for complex antiplatelet therapy. Redesigning the DeBakey VAD (i.e. to the HeartAssist 5), where several geometric modifications were made using this approach, yielded 88.3–91.9 % reduction in shear-induced platelet activation without the addition of anti-platelet agents [34]. Our results indicate that in vitro treatment with antiplatelet agents coupled with device design optimization [34] may reduce PARs even further. This suggests that using a thorough design optimization technique, with computational fluid dynamics simulations and in vitro validation of design changes, may result in VADs with less disturbed hemodynamics. This in turn leads to lower thrombogencity and risk of acquired VWD development, potentially reducing dependence on complex antiplatelet pharmacotherapy.
Acknowledgments
The authors thank Dr. Sheela George for her assistance with experiments and the Stony Brook University Medical Center Chemistry Lab for salicylate measurements. This work was supported by the National Institute of Biomedical Imaging and Bio-engineering Quantum Grant (Award No. 5U01EB012487-00, Dr. Bluestein).
Footnotes
Disclosures Drs. Bluestein and Slepian were members of the Scientific Advisory Board of MicroMed Cardiovascular, Inc at the time of conduct of this investigation. The other authors report no conflicts of interest.
Contributor Information
Jawaad Sheriff, Department of Biomedical Engineering, Stony Brook University, T15-090 Health Sciences Center, Stony Brook, NY 11794-8151, USA.
Gaurav Girdhar, Department of Biomedical Engineering, Stony Brook University, T15-090 Health Sciences Center, Stony Brook, NY 11794-8151, USA.
Wei-Che Chiu, Department of Biomedical Engineering, Stony Brook University, T15-090 Health Sciences Center, Stony Brook, NY 11794-8151, USA.
Jolyon Jesty, Department of Biomedical Engineering, Stony Brook University, T15-090 Health Sciences Center, Stony Brook, NY 11794-8151, USA.
Marvin J. Slepian, Department of Biomedical Engineering, Stony Brook University, T15-090 Health Sciences Center, Stony Brook, NY 11794-8151, USA. Departments of Medicine and Biomedical Engineering, Sarver Heart Center, University of Arizona, Tucson, AZ, USA
Danny Bluestein, Email: danny.bluestein@stonybrook.edu, Department of Biomedical Engineering, Stony Brook University, T15-090 Health Sciences Center, Stony Brook, NY 11794-8151, USA.
References
- 1.Christiansen S, Klocke A, Autschbach R. Past, present, and future of long-term mechanical cardiac support in adults. J Card Surg. 2008;23(6):664–676. doi: 10.1111/j.1540-8191.2008.00696.x. [DOI] [PubMed] [Google Scholar]
- 2.Bellumkonda L, Bonde P. Ventricular assist device therapy for heart failure–past, present, and future. Int Anesthesiol Clin. 2012;50(3):123–145. doi: 10.1097/AIA.0b013e31826233a9. [DOI] [PubMed] [Google Scholar]
- 3.Eckman PM, John R. Bleeding and thrombosis in patients with continuous-flow ventricular assist devices. Circulation. 2012;125(24):3038–3047. doi: 10.1161/CIRCULATIONAHA.111.040246. [DOI] [PubMed] [Google Scholar]
- 4.Ensor CR, Paciullo CA, Cahoon WD, Jr, Nolan PE., Jr Pharmacotherapy for mechanical circulatory support: a comprehensive review. Ann Pharmacother. 2011;45(1):60–77. doi: 10.1345/aph.1P459. [DOI] [PubMed] [Google Scholar]
- 5.Harrison P, Frelinger AL, 3rd, Furman MI, Michelson AD. Measuring antiplatelet drug effects in the laboratory. Thromb Res. 2007;120(3):323–336. doi: 10.1016/j.thromres.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 6.Braunwald E, Angiolillo D, Bates E, Berger PB, Bhatt D, Cannon CP, Furman MI, Gurbel P, Michelson AD, Peterson E, Wiviott S. Assessing the current role of platelet function testing. Clin Cardiol. 2008;31(3 Suppl 1):I10–I16. doi: 10.1002/clc.20361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wever-Pinzon O, Stehlik J, Kfoury AG, Terrovitis JV, Diakos NA, Charitos C, Li DY, Drakos SG. Ventricular assist devices: pharmacological aspects of a mechanical therapy. Pharmacol Ther. 2012;134(2):189–199. doi: 10.1016/j.pharmthera.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Majeed F, Kop WJ, Poston RS, Kallam S, Mehra MR. Prospective, observational study of antiplatelet and coagulation biomarkers as predictors of thromboembolic events after implantation of ventricular assist devices. Nat Clin Pract Cardiovasc Med. 2009;6(2):147–157. doi: 10.1038/ncpcardio1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slaughter MS, Sobieski MA, 2nd, Graham JD, Pappas PS, Tatooles AJ, Koenig SC. Platelet activation in heart failure patients supported by the HeartMate II ventricular assist device. Int J Artif Organs. 2011;34(6):461–468. doi: 10.5301/IJAO.2011.8459. [DOI] [PubMed] [Google Scholar]
- 10.Kundu SK, Heilmann EJ, Sio R, Garcia C, Davidson RM, Ostgaard RA. Description of an in vitro platelet function analyzer–PFA-100. Semin Thromb Hemost. 1995;21(Suppl 2):106–112. doi: 10.1055/s-0032-1313612. [DOI] [PubMed] [Google Scholar]
- 11.Mammen EF, Comp PC, Gosselin R, Greenberg C, Hoots WK, Kessler CM, Larkin EC, Liles D, Nugent DJ. PFA-100 system: a new method for assessment of platelet dysfunction. Semin Thromb Hemost. 1998;24(2):195–202. doi: 10.1055/s-2007-995840. [DOI] [PubMed] [Google Scholar]
- 12.Varon D, Dardik R, Shenkman B, Kotev-Emeth S, Farzame N, Tamarin I, Savion N. A new method for quantitative analysis of whole blood platelet interaction with extracellular matrix under flow conditions. Thromb Res. 1997;85(4):283–294. doi: 10.1016/s0049-3848(97)00014-5. [DOI] [PubMed] [Google Scholar]
- 13.Varon D, Lashevski I, Brenner B, Beyar R, Lanir N, Tamarin I, Savion N. Cone and plate(let) analyzer: monitoring glycoprotein IIb/IIIa antagonists and von Willebrand disease replacement therapy by testing platelet deposition under flow conditions. Am Heart J. 1998;135(5 Pt 2 Su):S187–S193. doi: 10.1016/s0002-8703(98)70248-0. [DOI] [PubMed] [Google Scholar]
- 14.Alstrom U, Granath F, Oldgren J, Stahle E, Tyden H, Siegbahn A. Platelet inhibition assessed with VerifyNow, flow cytometry and PlateletMapping in patients undergoing heart surgery. Thromb Res. 2009;124(5):572–577. doi: 10.1016/j.thromres.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 15.Michelson AD, Frelinger AL, 3rd, Furman MI. Current options in platelet function testing. Am J Cardiol. 2006;98(10A):4N–10N. doi: 10.1016/j.amjcard.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 16.Michelson AD. Methods for the measurement of platelet function. Am J Cardiol. 2009;103(3 Suppl):20A–26A. doi: 10.1016/j.amjcard.2008.11.019. [DOI] [PubMed] [Google Scholar]
- 17.Von Ruden SA, Murray MA, Grice JL, Proebstle AK, Kopacek KJ. The pharmacotherapy implications of ventricular assist device in the patient with end-stage heart failure. J Pharm Pract. 2012;25(2):232–249. doi: 10.1177/0897190011431635. [DOI] [PubMed] [Google Scholar]
- 18.Rossi M, Serraino GF, Jiritano F, Renzulli A. What is the optimal anticoagulation in patients with a left ventricular assist device? Interact Cardiovasc Thorac Surg. 2012 doi: 10.1093/icvts/ivs297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324(7329):71–86. doi: 10.1136/bmj.324.7329.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Armstrong PC, Truss NJ, Ali FY, Dhanji AA, Vojnovic I, Zain ZN, Bishop-Bailey D, Paul-Clark MJ, Tucker AT, Mitchell JA, Warner TD. Aspirin and the in vitro linear relationship between thromboxane A2-mediated platelet aggregation and platelet production of thromboxane A2. J Thromb Haemost. 2008;6(11):1933–1943. doi: 10.1111/j.1538-7836.2008.03133.x. [DOI] [PubMed] [Google Scholar]
- 21.Szczeklik A, Krzanowski M, Gora P, Radwan J. Anti-platelet drugs and generation of thrombin in clotting blood. Blood. 1992;80(8):2006–2011. [PubMed] [Google Scholar]
- 22.Massel D, Little SH. Risks and benefits of adding anti-platelet therapy to warfarin among patients with prosthetic heart valves: a meta-analysis. J Am Coll Cardiol. 2001;37(2):569–578. doi: 10.1016/s0735-1097(00)01135-9. [DOI] [PubMed] [Google Scholar]
- 23.Houel R, Mazoyer E, Boval B, Kirsch M, Vermes E, Drouet L, Loisance DY. Platelet activation and aggregation profile in prolonged external ventricular support. J Thorac Cardiovasc Surg. 2004;128(2):197–202. doi: 10.1016/j.jtcvs.2003.11.059. [DOI] [PubMed] [Google Scholar]
- 24.Boyle AJ, Russell SD, Teuteberg JJ, Slaughter MS, Moazami N, Pagani FD, Frazier OH, Heatley G, Farrar DJ, John R. Low thromboembolism and pump thrombosis with the HeartMate II left ventricular assist device: analysis of outpatient anti-coagulation. J Heart Lung Transplant. 2009;28(9):881–887. doi: 10.1016/j.healun.2009.05.018. [DOI] [PubMed] [Google Scholar]
- 25.Miller LW, Pagani FD, Russell SD, John R, Boyle AJ, Aaronson KD, Conte JV, Naka Y, Mancini D, Delgado RM, MacGillivray TE, Farrar DJ, Frazier OH. Use of a continuous-flow device in patients awaiting heart transplantation. N Engl J Med. 2007;357(9):885–896. doi: 10.1056/NEJMoa067758. [DOI] [PubMed] [Google Scholar]
- 26.Goldstein DJ. Worldwide experience with the MicroMed DeBakey Ventricular Assist Device as a bridge to transplantation. Circulation. 2003;108(Suppl 1):II272–II277. doi: 10.1161/01.cir.0000087387.02218.7e. [DOI] [PubMed] [Google Scholar]
- 27.Goldstein DJ, Zucker M, Arroyo L, Baran D, McCarthy PM, Loebe M, Noon GP. Safety and feasibility trial of the MicroMed DeBakey ventricular assist device as a bridge to transplantation. J Am Coll Cardiol. 2005;45(6):962–963. doi: 10.1016/j.jacc.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 28.Nakamura T, Uchiyama S, Yamazaki M, Iwata M. Effects of dipyridamole and aspirin on shear-induced platelet aggregation in whole blood and platelet-rich plasma. Cerebrovasc Dis. 2002;14(3–4):234–238. doi: 10.1159/000065669. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto J, Taka T, Nakajima S, Ueda M, Sugimoto E, Sasaki Y, Muraki T, Seki J, Watanabe S. A shear-induced in vitro platelet function test can assess clinically relevant anti-thrombotic effects. Platelets. 1999;10(2–3):178–184. doi: 10.1080/09537109976266. [DOI] [PubMed] [Google Scholar]
- 30.Tschopp M, Reinhart WH. Platelet aggregation under high shear conditions during and after a 28-day administration of 100 mg acetylsalicylic acid in healthy volunteers. Clin Hemorheol Microcirc. 2008;38(1):45–50. [PubMed] [Google Scholar]
- 31.Turner NA, Moake JL, Kamat SG, Schafer AI, Kleiman NS, Jordan R, McIntire LV. Comparative real-time effects on platelet adhesion and aggregation under flowing conditions of in vivo aspirin, heparin, and monoclonal antibody fragment against glycoprotein IIb-IIIa. Circulation. 1995;91(5):1354–1362. doi: 10.1161/01.cir.91.5.1354. [DOI] [PubMed] [Google Scholar]
- 32.Jesty J, Bluestein D. Acetylated prothrombin as a substrate in the measurement of the procoagulant activity of platelets: elimination of the feedback activation of platelets by thrombin. Anal Biochem. 1999;272(1):64–70. doi: 10.1006/abio.1999.4148. [DOI] [PubMed] [Google Scholar]
- 33.Claiborne TE, Girdhar G, Gallocher-Lowe S, Sheriff J, Kato YP, Pinchuk L, Schoephoerster RT, Jesty J, Bluestein D. Thrombogenic potential of Innovia polymer valves versus Carpentier-Edwards Perimount Magna aortic bioprosthetic valves. ASAIO J. 2011;57(1):26–31. doi: 10.1097/MAT.0b013e3181fcbd86. [DOI] [PubMed] [Google Scholar]
- 34.Girdhar G, Xenos M, Alemu Y, Chiu WC, Lynch BE, Jesty J, Einav S, Slepian MJ, Bluestein D. Device thrombogenicity emulation: a novel method for optimizing mechanical circulatory support device thromboresistance. PLoS One. 2012;7(3):e32463. doi: 10.1371/journal.pone.0032463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slepian MJ, Alemu Y, Soares JS, Smith RG, Einav S, Bluestein D. The Syncardia(™) total artificial heart: in vivo, in vitro, and computational modeling studies. J Biomech. 2013;46(2):266–275. doi: 10.1016/j.jbiomech.2012.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schulz-Heik K, Ramachandran J, Bluestein D, Jesty J. The extent of platelet activation under shear depends on platelet count: differential expression of anionic phospholipid and factor Va. Pathophysiol Haemost Thromb. 2005;34(6):255–262. doi: 10.1159/000093104. [DOI] [PubMed] [Google Scholar]
- 37.Jesty J, Yin W, Perrotta P, Bluestein D. Platelet activation in a circulating flow loop: combined effects of shear stress and exposure time. Platelets. 2003;14(3):143–149. doi: 10.1080/0953710031000092839. [DOI] [PubMed] [Google Scholar]
- 38.Buerke M, Pittroff W, Meyer J, Darius H. Aspirin therapy: optimized platelet inhibition with different loading and maintenance doses. Am Heart J. 1995;130(3 Pt 1):465–472. doi: 10.1016/0002-8703(95)90353-4. [DOI] [PubMed] [Google Scholar]
- 39.Houel R, Mazoyer E, Kirsch M, Boval B, Drouet L, Loisance DY. Resistance to aspirin after external ventricular assist device implantation. J Thorac Cardiovasc Surg. 2003;126(5):1636–1637. doi: 10.1016/S0022. [DOI] [PubMed] [Google Scholar]
- 40.Zimmermann N, Kienzle P, Weber AA, Winter J, Gams E, Schror K, Hohlfeld T. Aspirin resistance after coronary artery bypass grafting. J Thorac Cardiovasc Surg. 2001;121(5):982–984. doi: 10.1067/mtc.2001.111416. [DOI] [PubMed] [Google Scholar]
- 41.Guthikonda S, Lev EI, Patel R, DeLao T, Bergeron AL, Dong JF, Kleiman NS. Reticulated platelets and uninhibited COX-1 and COX-2 decrease the antiplatelet effects of aspirin. J Thromb Haemost. 2007;5(3):490–496. doi: 10.1111/j.1538-7836.2007.02387.x. [DOI] [PubMed] [Google Scholar]
- 42.Eikelboom JW, Hirsh J, Weitz JI, Johnston M, Yi Q, Yusuf S. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation. 2002;105(14):1650–1655. doi: 10.1161/01.cir.0000013777.21160.07. [DOI] [PubMed] [Google Scholar]
- 43.Boeer K, Reinhofer M, Losche W. Validation of a procedure to assess ASA-response in patients with decreased, initial TRAP induced aggregation. Platelets. 2010;21(5):314–319. doi: 10.3109/09537101003763442. [DOI] [PubMed] [Google Scholar]
- 44.Rajah SM, Penny AF, Crow MJ, Pepper MD, Watson DA. The interaction of varying doses of dipyridamole and acetyl salicylic acid on the inhibition of platelet functions and their effect on bleeding time. Br J Clin Pharmacol. 1979;8(5):483–489. doi: 10.1111/j.1365-2125.1979.tb01031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eisert WG. Dipyridamole in antithrombotic treatment. Adv Cardiol. 2012;47:78–86. doi: 10.1159/000338053. [DOI] [PubMed] [Google Scholar]
- 46.Bluestein D, Einav S, Slepian MJ. Device thrombogenicity emulation: a novel methodology for optimizing the thromboresistance of cardiovascular devices. J Biomech. 2013;46(2):338–344. doi: 10.1016/j.jbiomech.2012.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michelson AD. Flow cytometry: a clinical test of platelet function. Blood. 1996;87(12):4925–4936. [PubMed] [Google Scholar]
- 48.Loffler C, Straub A, Bassler N, Pernice K, Beyersdorf F, Bode C, Siegenthaler MP, Peter K. Evaluation of platelet activation in patients supported by the Jarvik 2000* high-rotational speed impeller ventricular assist device. J Thorac Cardiovasc Surg. 2009;137(3):736–741. doi: 10.1016/j.jtcvs.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 49.Sheriff J, Bluestein D, Girdhar G, Jesty J. High-shear stress sensitizes platelets to subsequent low-shear conditions. Ann Biomed Eng. 2010;38(4):1442–1450. doi: 10.1007/s10439-010-9936-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lietz K. Destination therapy: patient selection and current outcomes. J Card Surg. 2010;25(4):462–471. doi: 10.1111/j.1540-8191.2010.01050.x. [DOI] [PubMed] [Google Scholar]
- 51.Lietz K, Miller LW. Destination therapy: current results and future promise. Semin Thorac Cardiovasc Surg. 2008;20(3):225–233. doi: 10.1053/j.semtcvs.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 52.Kurien S, Hughes KA. Anticoagulation and bleeding in patients with ventricular assist devices: walking the tightrope. AACN Adv Crit Care. 2012;23(1):91–98. doi: 10.1097/NCI.0b013e31824124d0. [DOI] [PubMed] [Google Scholar]
- 53.Xenos M, Girdhar G, Alemu Y, Jesty J, Slepian M, Einav S, Bluestein D. Device Thrombogenicity Emulator (DTE)—design optimization methodology for cardiovascular devices: a study in two bileaflet MHV designs. J Biomech. 2010;43(12):2400–2409. doi: 10.1016/j.jbiomech.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]