

Abstract
TASK-1 is a two-pore domain potassium channel that is important to modulating cell excitability, most notably in the context of neuronal pathways. In order to leverage TASK-1 for therapeutic benefit, its physiological role needs better characterization; however, designing selective inhibitors that avoid the closely related TASK-3 channel has been challenging. In this study, a series of bis-amide derived compounds were found to demonstrate improved TASK-1 selectivity over TASK-3 compared to reported inhibitors. Optimization of a marginally selective hit led to analog 35 which displays a TASK-1 IC50 = 16 nM with 62-fold selectivity over TASK-3 in an orthogonal electrophysiology assay.
Keywords: TASK1, KCNK3, selective potassium channel inhibitor, bis-amide
Maintenance of the negative resting membrane potential of excitable cells depends significantly on the widely distributed family of two-pore domain potassium channels (K2P). At the basal membrane potential, these channels remain constitutively open, thus enabling potassium ion passage and generating a background conductance that regulates cell excitability.1–6 The family shows modulatory susceptibility to various stimuli such as pH, temperature, neurotransmitters, and pharmaceuticals, but appears relatively time-independent and nonresponsive to changes in voltage.7,8 These factors, coupled with variable expression levels, impart cells with the ability to finely tune ionic gradients involved with cellular response. The contributions of voltage-gated (Kv) and inwardly rectifying (Kir) potassium channels in the establishment and maintenance of resting membrane potential cannot be ignored; however, the distinctive biophysical properties of the K2P channels along with their modulation by physiochemical stimuli favor them in this role.4
Since their identification nearly twenty years ago,1–3 the K2P subfamily of TASK (TWIK-related-acid-sensitive-K+) channels has garnered much attention, in part, due to their high density in tissues impacted by disease and the hypothesis that selective manipulation of these channels may provide unique opportunities for therapeutic intervention.9 Efforts to elucidate specific roles for a particular TASK channel and its relationship to abberant cellular behavior have been complicated by the lack of selective chemical probes; however, insight has been achieved with gene knockout mouse models. For example, genetic deletion of murine TASK-1 (K2P3.1, KCNK3) channels has been shown to result in the development of severe hyperaldosteronism.10–12 In separate studies, the block or knockout of TASK-1 was associated with increased cell death following stroke-related ischemia, suggesting that these channels provided a neuroprotective effect.9,13–15 These results were complimented by studies in which TASK-1 null mice suffered less neurodegeneration in a multiple sclerosis inflammation/autoimmune model.16,17 The intermediacy of TASK-1 channels has also been demonstrated in pulmonary arterial hypertension by means of inhibition by endothelin-1.18–20 Collectively, these outcomes have illuminated and driven efforts to precisely decipher the contributions of TASK-1 channels to various pharmacological conundrums. However, the realization of selective chemical modulators is another approach that could aid in deconvoluting redundant and complex cellular circuitry related to TASK conductance.
Selectivity is one of the main obstacles to identifying compounds that are useful in the physiological examination of TASK channels.21 For instance, though several TASK family members are only distantly related,22–25 TASK-1 shares > 50% amino acid sequence identity with TASK-3 (K2P9.1, KCNK9),26,27 and has demonstrated similar co-expression patterns,28,29 sometimes even resulting in heterodimerization.7,30 Nonetheless, promising inhibitory TASK-1 compounds31–34 have emerged, including the recent reports of biaryl derivative 1 (A-1899)35 and Merck’s aminopyrimidines, represented by compound 236 (Fig. 1). The Decher laboratory described A-1899 (1) with a TASK-1IC50 = 7 nM (CHO cells) and 10-fold selectivity over TASK-3. Compound 2 showed reversed, 10-fold selectivity towards TASK-3 with a disclosed TASK-1IC50 = 300 nM. Importantly, these moderately selective TASK-1 benchmarks have been embraced as useful pharmacological tools37–39 though the search continues for high affinity, highly discriminating TASK-1 inhibitors to aid in the unambiguous interrogation of TASK-1 related mechanisms. With this in mind, our team initiated a project aimed at identifying potent and selective TASK-1 inhibitors.
Figure 1.
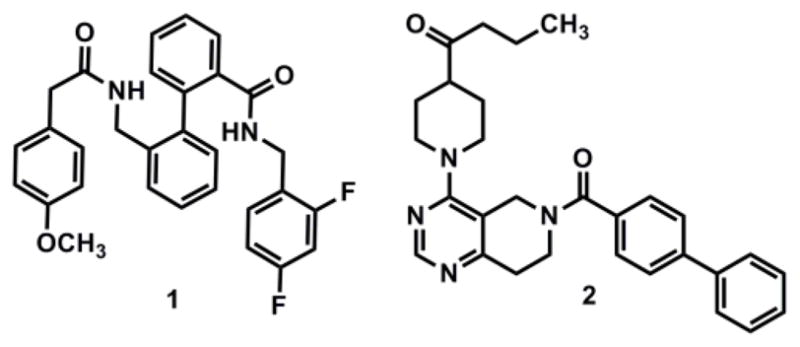
Reference TASK-1 and TASK-3 inhibitors
As part of the NIH Molecular Libraries Probe Production Centers Network (MLPCN), a high throughput screen of the NIH Molecular Libraries Small Molecule Repository (MLSMR) was performed with 339,662 compounds by the Johns Hopkins Ion Channel Center (JHICC). This campaign, executed to discover selective TASK-3 inhibitors, included a counterscreen for TASK-1 selectivity.40 TASK activity was assessed using CHO cells expressing either TASK-1 or TASK-3 in a fluorescence-based, thallium flux (TF) assay.41,42 For the most promising hit compounds, activity for each channel was confirmed in separate QPatch automated electrophysiology assays directly measuring whole cell voltage in the same cell line.43 Confirmed hits were then counterscreened against potassium voltage-gated channels KCNQ2 (Kv7.2), and KCNH2 (hERG), along with the Kir2.1 channel, resulting in a few validated chemotypes as potential candidates for further structure-activity relationship (SAR) optimization. Bis-amide 3 (Fig. 2) was representative of one TASK-1 selective hit scaffold obtained from this endeavor, exhibiting TF assay IC50 values of 0.027 μM and 0.50 μM for TASK-1 and TASK-3, respectively (19-fold selectivity), and QPatch assay IC50 values of 0.075 μM and 0.69 μM for TASK-1 and TASK-3, respectively (9-fold selectivity). No inhibition was observed for the other channels surveyed (IC50 values > 30 μM for KCNQ2, hERG, and Kir2.1 channels), and the hit rate against other PubChem assays was less than 0.5%. Given its attractive TASK-1 activity profile and synthetic feasibility, analogs were made with the shaded regions of optimization in mind (Fig. 2).
Figure 2.
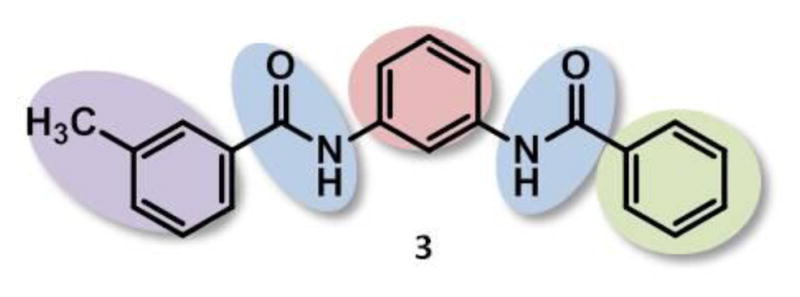
Hit structure 3 with SAR focus highlighted.
The University of Kansas Specialized Chemistry Center (KU SCC) prepared 117 compounds in the SAR campaign using standard amide coupling chemistry (Scheme 1). Treatment of 1,3-nitroanilines with substituted benzoyl chlorides 4 afforded nitro amides which were subsequently reduced with Raney nickel and sodium borohydride to generate anilines 5. Aniline intermediates were then coupled with differentially substituted aryl or alkyl chlorides to generate the desired analogs 6. Reverse amides were also synthesized for the 1,3-orientation about the central phenyl ring. Analogous chemistry was utilized except for the exchange of amino benzoic esters in place of a nitroaniline in the first step, followed by saponification and implementation of routine amino acid coupling conditions.
Scheme 1.
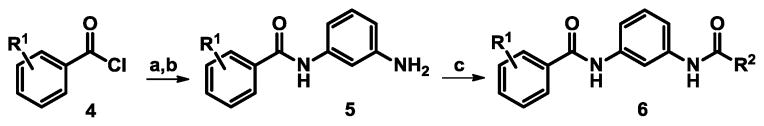
General synthesis for the 1,3-bis-amide scaffold
Reagents: (a) 1,3-nitroaniline, triethylamine, acetonitrile, 70 °C, 3 h, 52 – 92%. (b) Raney nickel, NaBH4, methanol, 0 °C, 2.5 h, 85 – 91%. (c) aryl or alkyl chloride, triethylamine, acetonitrile, 70 °C, 3 h, 15 – 67%.
Optimization was primarily driven by the cellular TASK-1 and TASK-3 TF assays, and all compounds were profiled at 30 μM against the parental TASK-1 cells, as well as KCNQ2, hERG, and Kir2.1 channels. Selected analogs were subsequently evaluated in TASK-1 and TASK-3 confirmatory electrophysiology assays. For the first set of analogs, the parent scaffold’s unsubstituted phenyl group remained fixed while changes to the 3-methyl phenyl ring were explored (Table 1).
Table 1.
TASK thallium flux assay data for 1,3-bis-amides bearing 3-methylphenyl replacements
| entry | analog | structure | thallium flux (TF) assay dataa | selectivity | ||
|---|---|---|---|---|---|---|
|
| ||||||
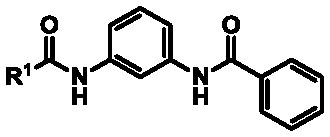
|
TASK-1 channel | TASK-3 channel | TASK3 IC50/TASK1 IC50 | |||
|
| ||||||
| R1 | %b | IC50 (μM) | IC50 (μM) | |||
| 1 | 3 | 3-CH3-phenyl | 97.4 | 0.027 | 0.50 | 19 |
| 2 | 7 | 2-CH3-phenyl | 91.9 | 0.10 | 6.5 | 65 |
| 3 | 8 | 4-CH3-phenyl | 56.2 | > 3 | > 30 | NA |
| 4 | 9 | phenylc | 100 | 0.021 | 23 | > 1100 |
| 5 | 10 | 2-OCH3-phenyl | 94.9 | 0.22 | 1.5 | 6.6 |
| 6 | 11 | 3-OCH3-phenyl | 96.7 | 0.33 | 4.3 | 13 |
| 7 | 12 | 4-OCH3-phenyl | 97.9 | > 3 | 2.1 | 0.47 |
| 8 | 13 | 3-F-phenyl | 78.8 | 0.045 | > 30 | > 670 |
| 9 | 14 | 4-F-phenyl | 77.2 | 0.31 | 20 | 64 |
| 10 | 15 | 2-Cl-phenyl | 97.3 | 0.50 | 7.1 | 14 |
| 11 | 16 | 3-Cl-phenyl | 99.8 | 0.052 | 2.4 | 46 |
| 12 | 17 | 4-Cl-phenyl | 26.3 | > 3 | > 30 | NA |
| 13 | 18 | 2-CF3-phenyl | 94.7 | 0.13 | 2.5 | 19 |
| 14 | 19 | 3-CF3-phenyl | 94.6 | 0.16 | >30 | > 190 |
| 15 | 20 | 4-CF3-phenyl | 33.7 | > 3 | > 30 | NA |
| 16 | 21 | 4-t-butyl-phenyl | 22.0 | > 3 | 3.2 | NA |
| 17 | 22 | 4-N(CH3)2-phenyl | 87.3 | 0.26 | > 30 | > 110 |
| 18 | 23 | 2-furyl | 93.2 | 0.037 | 5.9 | 160 |
| 19 | 24 | 3-furyl | 72.9 | 0.80 | 25 | 32 |
| 20 | 25 | 2-thiophene | 95.0 | 0.029 | 0.97 | 33 |
| 21 | 26 | 3-thiophene | 95.6 | 0.16 | 5.6 | 35 |
| 22 | 27 | 2-pyridyl | 94.3 | 0.086 | 2.5 | 29 |
| 23 | 28 | 3-pyridyl | 65.6 | 1.29 | >30 | >23 |
| 24 | 29 | 4-pyridyl | 91.3 | 0.25 | >30 | >120 |
| 25 | 30 | cyclohexyl | 94.8 | 0.079 | 5.48 | 69.4 |
Data was averaged from n ≥ 2 experiments.
Percent inhibition of TASK-1 at 3μM.
TASK1 parental cell inhibition was observed (84% at 3μM, IC50 = 0.24 μM).
NA = not applicable.
Overall, retention of TASK-1 potency pivoted on the steric requirement of the substituted phenyl ring rather than the substituent’s electronic nature. For instance, 4-substituted phenyl analogs showed a > 10-fold loss of TASK activity compared to the 3-methylphenyl containing parent 3, regardless of the para substituent’s electron density (compounds 33, 37, 39, 41 and 44–45). In most cases, 3-phenyl substitution was favored over other patterns in terms of the greatest gains in combined potency and TASK-1 selectivity (compounds 18, 21 and 24). The symmetrical phenyl analog 9 looked promising with comparable potency to the hit and little TASK-3 liability; however, it also inhibited non-induced parental cells with an IC50 value = 0.24 μM. Heterocyclic replacements were also examined. The 2-furyl and 2-thiophene derivatives were more potent than their respective 3-aryl counterparts (23–26). Pyridine variants (27–29), while less potent, afforded two of three analogs with respectable potency that were devoid of TASK-3 liability. Several compounds were identified with notably improved TASK-1 selectivity compared to hit 3, and no significant inhibition of KCNQ2, hERG, or Kir2.1 channels was observed (> 30 μM).
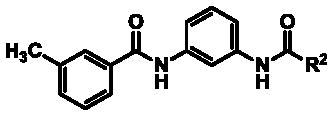
Preliminary SAR analysis revealed that, despite the near symmetrical nature of the scaffold, the dissimilar terminal aryl moieties dramatically influenced potency and selectivity. As a result, analogs were generated in which the 3-methylphenyl group of hit 3 was held constant while replacements for the unsubstituted phenyl ring were surveyed (Table 2).
Table 2.
TASK thallium flux assay data for 1,3-bis-amides bearing phenyl group replacements
| entry | analog | structure | thallium flux (TF) assay dataa,b | selectivity | ||
|---|---|---|---|---|---|---|
|
| ||||||
|
TASK-1 channel | TASK-3 channel | TASK-3 IC50/TASK-1 IC50 | |||
|
| ||||||
| R2 | %c | IC50 (μM) | IC50 (μM) | |||
| 1 | 31 | 2-CH3-phenyl | 95.5 | 0.091 | 0.96 | 11 |
| 2 | 32 | 3-CH3-phenyl c | 95.6 | 0.030 | 0.15 | 5.0 |
| 3 | 33 | 2-i-propyl-phenyl | 94.5 | 0.33 | 1.7 | 5.2 |
| 4 | 34 | 4-t-butyl-phenyl | 96.6 | 0.19 | >30 | >160 |
| 5 | 35 | 2-OCH3-phenyl | 96.6 | 0.004 | 0.39 | 100 |
| 6 | 36 | 3-OCH3-phenyl | 92.0 | 0.031 | 2.2 | 69 |
| 7 | 37 | 4-OCH3-phenyl | 69.3 | 1.3 | >30 | >23 |
| 8 | 38 | 2,5-di-OCH3-phenyl | 93.2 | 0.15 | 2.3 | 15 |
| 9 | 39 | 2-OCH2CH3-phenyld | 100 | 0.009 | 0.41 | 46 |
| 10 | 40 | 2-OCF3-phenyl | 93.4 | 0.029 | 0.75 | 26 |
| 11 | 41 | 2-isopropoxyphenyl | 100 | 0.21 | 2.7 | 13 |
| 12 | 42 | 2-Cl-phenyl | 92.0 | 0.023 | 1.34 | 58 |
| 13 | 43 | 3-Cl-phenyl | 92.2 | 0.003 | 0.48 | 160 |
| 14 | 44 | 4-Cl-phenyl | 77.9 | 0.15 | >30 | >200 |
| 15 | 45 | 3-F-phenyl | 93.7 | 0.025 | 0.60 | 24 |
| 16 | 46 | 2-CF3-phenyl | 91.9 | 0.014 | 1.4 | 100 |
| 17 | 47 | 3-CF3-phenyl | 79.3 | 0.010 | 5.5 | 550 |
| 18 | 48 | 4-CF3-phenyl | 20.2 | >3 | >30 | NA |
| 19 | 49 | 4-N(CH3)2-phenyl | 100 | 0.15 | >30 | >200 |
| 20 | 50 | 2-furyl | 92.8 | 0.14 | 2.7 | 19 |
| 21 | 51 | 2-thiophene | 94.1 | 0.005 | 0.33 | 66 |
| 22 | 52 | 4-pyridyl | 96.0 | 0.095 | > 30 | > 310 |
Data was averaged from n ≥ 2 experiments.
Percent inhibition of TASK-1 at 3μM.
hERG IC50 = 12.4 μM.
NA = not applicable.
Interestingly, 4-phenyl substituted analogs in this series showed some erosion of TASK-1 potency compared to hit 3; however, TASK-3 inhibition was abolished in each case (> 30 μM, Table 2, compounds 34, 37, 44, 48–49 and 52). While bulkier 2-substituted phenyl groups were not well tolerated (compounds 33 and 41), smaller and more polar 2- and 3-substituted derivatives produced a robust boost in TASK-1 potency without a commensurate response in TASK-3 inhibition, thus generating analogs with low nanomolar TASK-1 potency and > 100-fold selectivity (Table 2, compounds 35, 43, and 46–47, and 52). As with previous analogs, no significant inhibition of KCNQ2, hERG, or Kir2.1 channels was observed with any of these compounds (> 30 μM) unless otherwise noted.
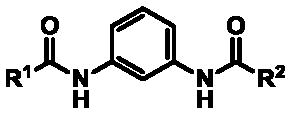
Given the improvements observed with 2-alkoxyphenyl analogs 35 and 39, additional compounds featuring this element were explored (Table 3). Many of these potently inhibited TASK1 (IC50 ≤ 11 nM) with better selectivity over TASK-3 than what was observed for the hit compound 3. While inhibition of TASK-3 was not completely decoupled from enhancement of TASK-1 potency, in most cases, the margin between the two was expanded. One of the highlights of this group included the symmetrical 2-methoxyphenyl bis-amide 53 whose profile benefitted from a modest improvement in TASK-1 and a more substantial loss of TASK-3 activity, resulting in selectivity > 190-fold. Attempts were made to capitalize on the finding that incorporation of a pyridine appeared to abolish TASK-3 liability (see compounds 28–29, Table 1, and 52 in Table 2); however, the limitations in TASK-1 potency could not be overcome by the installation of a 2-methoxy substituent (compound 61, Table 3).
Table 3.
TASK thallium flux assay and selectivity data for 1,3-bis-amides bearing dual terminal phenyl group replacements
| entry | analog | structure | thallium flux (TF) assay dataa,b | selectivity | |||
|---|---|---|---|---|---|---|---|
|
| |||||||
|
TASK-1 channel | TASK-3 channel | TASK-3 IC50/TASK-1 IC50 | ||||
|
| |||||||
| R1 | R2 | %c | IC50 (μM) | IC50 (μM) | |||
| 1 | 53 | 2-OCH3- phenyl | 2-OCH3- phenyl | 96.6 | 0.010 | 1.88 | 190 |
| 2 | 54 | 2-OCH3- phenyl | 2-OCF3- phenyl | 92.1 | 0.004 | 0.41 | 100 |
| 3 | 55 | 2-OCH3- phenyl | 2-OEt- phenyl | 92.2 | 0.002 | 0.066 | 33 |
| 4 | 56 | 2-OCH3- phenyl | 2-CH3- phenyl | 100 | 0.073 | 2.2 | 31 |
| 5 | 57 | 2-OCH3- phenyl | 2-i-propyl phenyl | 100 | 0.007 | 1.0 | 140 |
| 7 | 58 | 2-OCF3- phenyl | 2-i-propyl phenyl | 100 | 0.011 | 0.73 | 66 |
| 8 | 59 | 2-OCF3- phenyl | 2-isopropoxy phenyl | 93.4 | 0.001 | 0.11 | 110 |
| 9 | 60 | 2-OEt- phenyl | 2-CF3- phenyl | 92.8 | 0.002 | 0.30 | 150 |
| 10 | 61 | 2-OCH3- 4-pyridyl | 3-CH3- phenyl | 90.0 | 0.18 | 2.43 | 13.5 |
Data was averaged from n ≥ 2 experiments.
Percent inhibition of TASK-1 at 3μM.
Attention was shifted to the connectivity of the amide groups to the central phenyl ring, though none were found to be superior to those augmentations already discussed. The corresponding 1,2-bis-amide featuring the same substituted phenyl moieties as hit 3 showed a 10-fold loss in TASK-1 activity (IC50 = 0.26 μM; TASK-3 IC50 = 17.8 μM). Reversing the amide on either side of the parent structure 3 led to a > 40-fold loss in TASK-1 activity (data not shown). While replacing the 3-tolyl amide of 3 with 3-tolylsulfonamide eliminated TASK-3 activity (IC50 > 30 μM), TASK-1 potency was significantly diminished (IC50 = 1.5 μM).
With a number of analogs demonstrating promising TASK-1 selectivity and potency in the TF assay, data confirmation was sought in the electrophysiology assay for a select subset of compounds (Table 4). With the exception of compounds 47 and 52, TASK-1 data between the TF and QPatch assays was reasonably congruent (IC50 ≤ 5-fold difference). A stronger correlation between the two assays was observed for TASK-3, differing by ≤ 2.6-fold except for compound 39. Though the QPatch analysis revealed reduced TASK-1 selectivity compared to the TF assay, several bis-amides were found to have improved selectivity compared to reported compound A-1899 (1). Compound 35 was noteworthy in that it retained low nanomolar activity (IC50 = 16 nM) in the TASK-1 QPatch assay with 62-fold selectivity over TASK-3. Considering the potency and selectivity profiles in both assays, compound 35 was chosen as a flagship probe (ML365) which was worthy of more elaborate characterization.
Table 4.
Correlation of TASK TF and QPatch assay data
| entry | analog | thallium flux (TF) assay dataa | TF assay selectivity | QPatch assay dataa | QPatch selectivity | ||
|---|---|---|---|---|---|---|---|
|
| |||||||
| TASK-1 channel | TASK-3 channel | TASK-3 IC50/TASK-1 IC50 | TASK-1 channel | TASK-3 channel | TASK-3 IC50/TASK-1 IC50 | ||
|
|
|
||||||
| IC50 (μM) | IC50 (μM) | IC50 (μM) | IC50 (μM) | ||||
| 1 | 3 | 0.027 | 0.50 | 19 | 0.075 | 0.69 | 9 |
| 2 | 35 | 0.004 | 0.39 | 100 | 0.016 | 0.99 | 62 |
| 3 | 39 | 0.009 | 0.41 | 46 | 0.011 | 0.038 | 34 |
| 4 | 40 | 0.029 | 0.75 | 26 | 0.060 | 0.99 | 16 |
| 5 | 47 | 0.010 | 5.5 | 550 | 0.37 | 3.94 | 11 |
| 6 | 52 | 0.095 | > 30 | > 310 | 2.14 | 12.5 | 6 |
| 7 | 53 | 0.010 | 1.88 | 190 | 0.030 | 0.71 | 24 |
| 8 | 60 | 0.002 | 0.30 | 150 | 0.010 | 0.25 | 25 |
Data was averaged from n ≥ 2 experiments.
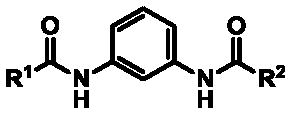
The bis-amide series was constructed to refine the overall activity profile; however, some architecture was assembled with physiochemical properties in mind. The initial hit compound (3) was poorly soluble in aqueous phosphate buffer saline (PBS) and at all three pH levels of pION buffer.44,45 Incorporation of alkyl ethers and pyridine derivatives improved aqueous solubility compared to the hit compound 3 (Table 5). Solubility of compound 35 (probe ML365) was also assessed in assay media. For the TF assay medium46 compound 35 was determined to have a kinetic solubility reading of 0.47 μg/mL, or 1.30 μM. Solubility of 35 in QPatch assay medium47 was found to be 0.17 μg/mL, or 0.47 μM. Though these are modest values, the solubility for 35 was 325- and 29-fold over the compound’s IC50 values for the TF and QPatch assays, respectively.
Table 5.
Solubility data for select bis-amide TASK-1 inhibitors
| entry | analog |
|
pION buffer pH 0/6.2/7.4 | PBS buffer pH 7.4 | |
|---|---|---|---|---|---|
|
| |||||
| R1 | R2 | μg/mL (μM) | μg/mL (μM) | ||
| 1 | 3 | phenyl | 3-CH3- phenyl | <0.01 for all (0.030 for all) | 0.27 (0.82) |
| 2 | 35 | 2-OCH3- phenyl | 3-CH3- phenyl | <0.01/0.06/0.07 (0.028/0.17/0.19) | 0.010 (0.028) |
| 3 | 52 | 4-pyridyl | 3-CH3- phenyl | 30.9/31.4/31.4 (93.2/94.8/94.8) | 8.2 (24.7) |
| 4 | 60 | 2-OEt- phenyl | 2-CF3- phenyl | 0.07/0.05/0 (0.16/0.12/0) | 0.38 (0.89) |
| 5 | 61 | 2-OCH3- 4-pyridyl | 3-CH3- phenyl | 43.4/46.7/44.3 ((120.1/129.2/122.6) | 0.36 (1.0) |
Probe ML365 (35) was also evaluated for aqueous stability in PBS, for susceptibility to nucleophilic addition and formation of conjugates when treated with thiol-bearing dithiothreitol (DTT).48 For the aqueous stability experiment, 100% of ML365 remained after 48 hours. When exposed to a 5-fold concentration of DTT for 8 hours, 100% of ML365 (35) was found without any detectable adduct formation or dimerization.49
In summary, a TASK-3 directed screening campaign revealed a promising TASK-1-selective bis-amide scaffold as part of a counterscreening effort. Guided by two differentiated cellular assays, optimization led to several potent TASK-1 inhibitors with the benefit of substantially improved selectivity over the related TASK-3 channel and no liability on KCNQ2, hERG, and Kir2 channels. Compared to reported inhibitors which have maximally shown 10-fold TASK-1 selectivity, compound 35 (ML365) demonstrated > 60-fold selectivity for TASK-1 over the related TASK-3 channel when assessed by an orthogonal electrophysiological assay. Ultimately, the discovery of 35 and its analogs represents an important milestone in the development of useful agents with sufficient specificity to better define the pharmacological role of the TASK-1 channel. The preliminary SAR supports that future endeavors focused on additional bis-amide derivatives featuring key functionality will likely lead to compounds with improved solubility and superior TASK-1 selectivity.
Supplementary Material
The synthesis50 of compound 35 is generally representative of most analogs discussed in this manuscript; however, supplementary material associated with this article, including general synthetic methods and biological assay protocols that are not available in cited references, can be found in the online version, at: X
Supplementary Material
Acknowledgments
The authors gratefully acknowledge funding from the National Institutes of Health, grant numbers R03 MH090849-01, R03 MH090837-01 and R03 DA031670-01 to M. Wu and partial funding in support of chemistry performed at the University of Kansas Specialized Chemistry Center (NIH U54HG005031) and screening performed at the Johns Hopkins Ion Channel Center (NIH U54 MH084691). The authors also express their sincere appreciation to Dr. Douglas A. Bayliss, University of Virginia Health System, for the donation of stably KCNK3-expressing CHO cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Goldstein SAN, Bockenhauer D, O’Kelly I, Zilberberg N. Nat Rev Neurosci. 2001;2:175. doi: 10.1038/35058574. [DOI] [PubMed] [Google Scholar]
- 2.Talley EM, Sirois JE, Lei Q, Bayliss DA. The Neuroscientist. 2003;9:46. doi: 10.1177/1073858402239590. [DOI] [PubMed] [Google Scholar]
- 3.Lesage F, Lazdunski M. Am J Physiol Renal Physiol. 2000;279:F793. doi: 10.1152/ajprenal.2000.279.5.F793. [DOI] [PubMed] [Google Scholar]
- 4.Bayliss DA, Barrett PQ. Trends Pharmacol Sci. 2008;29:566. doi: 10.1016/j.tips.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bayliss DA, Talley EM, Sirois JE, Lei Q. Respiration Physiol. 2001;129:159. doi: 10.1016/s0034-5687(01)00288-2. [DOI] [PubMed] [Google Scholar]
- 6.Kim D. Curr Pharm Des. 2005;11:2717. doi: 10.2174/1381612054546824. [DOI] [PubMed] [Google Scholar]
- 7.Talley EM, Bayliss DA. J Biol Chem. 2002;277:17733. doi: 10.1074/jbc.M200502200. [DOI] [PubMed] [Google Scholar]
- 8.Patel AJ, Honore E. Trends Neurosci. 2001;24:339. doi: 10.1016/s0166-2236(00)01810-5. [DOI] [PubMed] [Google Scholar]
- 9.Es-Salah-Lamoureux Z, Steele DF, Fedida D. Trends Pharmacol Sci. 2010;31:587. doi: 10.1016/j.tips.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Heitzmann D, Derand R, Jungbauer S, Bandulik S, Sterner C, Schweda F, El Wakil A, Lalli E, Guy N, Mengual R, Reichold M, Tegtmeier I, Bendahhou S, Gomez-Sanchez CE, Aller MI, Wisden W, Weber A, Lesage F, Warth R, Barhanin J. EMBO J. 2008;27:179. doi: 10.1038/sj.emboj.7601934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies LA, Hu C, Guagliardo NA, Sen N, Chen X, Talley EM, Carey RM, Bayliss DA, Barrett PQ. Proc Natl Acad Sci. 2008;105:2203. doi: 10.1073/pnas.0712000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guagliardo NA, Yao J, Bayliss DA, Barrett PQ. Mol and Cell Endocrinol. 2011;336:47. doi: 10.1016/j.mce.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muhammad S, Aller MI, Maser-Gluth C, Schwaninger M, Wisden W. Neurosci. 2010;167:758. doi: 10.1016/j.neuroscience.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 14.Bittner S, Budde T, Wiendl H, Meuth SG. Brain Pathol (Zurich, Switzerland) 2010;20:999. doi: 10.1111/j.1750-3639.2010.00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meuth SG, Kleinschnitz C, Broicher T, Austinat M, Braeuninger S, Bittner S, Fischer S, Bayliss DA, Budde T, Stoll G, Wiendl H. Neurobiol Dis. 2009;33:1. doi: 10.1016/j.nbd.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meuth SG, Bittner S, Meuth P, Simon OJ, Budde T, Wiendl H. J Biol Chem. 2008;283:14559. doi: 10.1074/jbc.M800637200. [DOI] [PubMed] [Google Scholar]
- 17.Bittner S, Meuth SG, Gobel K, Melzer N, Herrmann AM, Simon OJ, Weishaupt A, Budde T, Bayliss DA, Bendszus M, Wiendl H. Brain. 2009;132:2501. doi: 10.1093/brain/awp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olschewski A, Li Y, Tang B, Hanze J, Eul B, Bohle RM, Wilhelm J, Morty RE, Brau ME, Weir EK, Kwapiszewska G, Klepetko W, Seeger W, Olschewski H. Circ Res. 2006;98:1072. doi: 10.1161/01.RES.0000219677.12988.e9. [DOI] [PubMed] [Google Scholar]
- 19.Tang B, Li Y, Nagaraj C, Morty RE, Gabor S, Stacher E, Voswinckel R, Weissmann N, Leithner K, Olschewski H, Olschewski A. J Resp Cell Mol Biol. 2009;41:476. doi: 10.1165/rcmb.2008-0412OC. [DOI] [PubMed] [Google Scholar]
- 20.Seyler C, Duthil-Straub E, Zitron E, Gierten J, Scholz EP, Fink RHA, Karle CA, Becker R, Katus HA, Thomas D. British J Pharmacol. 2012;165:1467. doi: 10.1111/j.1476-5381.2011.01626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beltran LR, Dawid C, Beltran M, Gisselmann G, Degenhardt K, Mathie K, Hofmann T, Hatt H. Front Pharmacol. 2013;4:141. doi: 10.3389/fphar.2013.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reyes R, Duprat F, Lesage F, Fink M, Salinas M, Farman N, Lazdunski M. J Biol Chem. 1998;273:30863. doi: 10.1074/jbc.273.47.30863. [DOI] [PubMed] [Google Scholar]
- 23.Decher N, Maier M, Dittrich W, Gassenhuber J, Bruggemann A, Busch AE, Steinmeyer K. FEBS Lett. 2001;492:84. doi: 10.1016/s0014-5793(01)02222-0. [DOI] [PubMed] [Google Scholar]
- 24.Girard C, Duprat F, Terrenoire C, Tinel N, Fosset M, Romey G, Lazdunski M, Lesage F. Biochem Biophys Res Comm. 2001;282:249. doi: 10.1006/bbrc.2001.4562. [DOI] [PubMed] [Google Scholar]
- 25.Kim D, Gnatenco C. Biophys Res Comm. 2001;284:923. doi: 10.1006/bbrc.2001.5064. [DOI] [PubMed] [Google Scholar]
- 26.Kim Y, Bang H, Kim D. J Biol Chem. 2000;275:9340. doi: 10.1074/jbc.275.13.9340. [DOI] [PubMed] [Google Scholar]
- 27.Meadows HJ, Randall AD. Neuropharmacol. 2001;40:551. doi: 10.1016/s0028-3908(00)00189-1. [DOI] [PubMed] [Google Scholar]
- 28.Karschin C, Wischmeyer E, Preisig-Muller R, Rajan S, Derst C, Grzeschik KH, Daut J, Karschin A. Mol Cell Neurosci. 2001;18:632. doi: 10.1006/mcne.2001.1045. [DOI] [PubMed] [Google Scholar]
- 29.Talley EM, Solórzano G, Lei Q, Kim D, Bayliss DA. J Neurosci. 2001;21:7491. doi: 10.1523/JNEUROSCI.21-19-07491.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Czirják G, Enyedi P. J Biol Chem. 2002;277:5426. doi: 10.1074/jbc.M107138200. [DOI] [PubMed] [Google Scholar]
- 31.Mathie A, Veale EL. Curr Opin Invest Drugs. 2007;8:555. [PubMed] [Google Scholar]
- 32.Maingret F, Patel AJ, Lazdunski M, Honore E. EMBO J. 2001;20:47. doi: 10.1093/emboj/20.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brendel J, Goegelein H, Wirth K, Kamm W, Gmbh S-AD. WO 2007124849 Ed Germany. 2007
- 34.Brendel J, Goegelein H, Wirth K, Kamm W [DE] S-ADGF, editor. United States: 2009149496. US. 2009
- 35.Streit AK, Netter MF, Kempf F, Walecki M, Rinné S, Bollepalli MK, Preisig-Müller R, Renigunta V, Daut J, Baukrowitz T, Sansom MSP, Stansfeld PJ, Decher N. J Biol Chem. 2011;286:13977. doi: 10.1074/jbc.M111.227884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coburn CA, Luo Y, Cui M, Wang J, Soll R, Dong J, Hu B, Lyon MA, Santarelli VP, Kraus RL, Gregan Y, Wang Y, Fox SV, Binns J, Doran SM, Reiss DR, Tannenbaum PL, Gotter AL, Meinke PT, Renger JJ. ChemMedChem. 2012;7:123. doi: 10.1002/cmdc.201100351. [DOI] [PubMed] [Google Scholar]
- 37.Schiekel J, Lindner M, Hetzel A, Wemhoner K, Renigunta V, Schlichthorl G, Decher N, Oliver D, Daut J. Cardiovas Res. 2013;97:97. doi: 10.1093/cvr/cvs285. [DOI] [PubMed] [Google Scholar]
- 38.Cotten JF. Anesth Analg. 2013;116:810. doi: 10.1213/ANE.0b013e318284469d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bittner S, Bauer MA, Ehling P, Bobak N, Breuer J, Herrmann AM, Golfels M, Wiendl H, Budde T, Meuth SG. Exp Neurol. 2012;238:149. doi: 10.1016/j.expneurol.2012.08.021. [DOI] [PubMed] [Google Scholar]
- 40.A manuscript describing the screening and development efforts leading to a TASK-3 selective probe (ML308) is in preparation.
- 41.Weaver CD, Harden D, Dworetzky SI, Robertson B, Knox RJ. J Biomol Screen. 2004;9:671. doi: 10.1177/1087057104268749. [DOI] [PubMed] [Google Scholar]
- 42.Center JHIC. PubChem. Vol. 212. National Center for Biotechnology Information; Mar 28, 2013. p. 2013.p. AID 652212. https://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=652. [Google Scholar]
- 43.Kutchinsky J, Friis S, Asmild M, Taboryski R, Pedersen S, Vestergaard RK, Jacobsen RB, Krzywkowski K, Schroder RL, Ljungstrom T, Helix N, Sorensen CB, Bech M, Willumsen NJ. Assay Drug Dev Technol. 2003;1:685. doi: 10.1089/154065803770381048. [DOI] [PubMed] [Google Scholar]
- 44.Solubility data was obtained from the Sanford Burnham Medical Institute Exploratory Pharmacology Core under the direction of Dr. Layton Smith. Assays were performed by A. Mangravita-Novo.
- 45.Kinetic solubility assessment was performed in Pion Prisma HT Universal buffer (pION) and in phosphate buffer saline (PBS) at room temperature (23 °C). PBS is 137 mM NaCl, 2.7 mM KCl, 10 mM sodium phosphate dibasic, and 2 mM potassium phosphate monobasic.
- 46.TF assay medium conditions: 1x HBSS, pH 7.4
- 47.QPatch assay medium conditions:14 mM NaCl, 140 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, and 10 mM HEPES, pH 7.4
- 48.Compound 35 was dissolved at 10 micromolar in 1:1 PBS/acetonitrile at pH 7.4 (1% DMSO) and incubated at room temperature with no nucleophile present or 50 micromolar dithiotreitol.
- 49.Assays were performed by Patrick Porubsky at the University of Kansas Specialized Chemistry Center. Experimental details can be found in the supplemental material.
- 50.Synthesis of compound 35: Step 1, preparation of 3-methyl-N-(3-nitrophenyl)benzamide: To a vial containing acetonitrile (10 mL) was added 3-nitroaniline (0.50 g, 3.62 mmol), NEt3 (0.56 mL, 3.98 mmol), and 3-methylbenzoyl chloride (0.50 mL, 3.80 mmol). The vial was capped, and the resulting solution was heated at 75 °C for 3 h. After cooling to room temperature, the solution was concentrated, the residue was dissolved in CH2Cl2 (10 mL) (2 and then it was washed with saturated aqueous NaHCO3 x 5 mL). The separated organic extract was dried (MgSO4), filtered and adsorbed onto silica. Purification by MPLC (0 – 30% EtOAc:hexanes) afforded 3-methyl-N-(3-nitrophenyl)benzamide (0.86 g, 3.36 mmol, 93% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.67 (s, 1H), 8.81 (t, J = 2.2 Hz, 1H), 8.21 (ddd, J = 8.2, 2.2, 1.0 Hz, 1H), 7.97 (ddd, J = 8.2, 2.3, 1.0 Hz, 1H), 7.86 – 7.75 (m, 2H), 7.67 (t, J = 8.2 Hz, 1H), 7.52 – 7.41 (m, 2H), 2.42 (d, J = 0.8 Hz, 3H). Step 2, preparation of N-(3-aminophenyl)-3-methylbenzamide: To a 500 mL round-bottom flask was added 3-methyl-N-(3-nitrophenyl)benzamide (0.86 g, 3.36 mmol) in MeOH and CH2Cl2 (1.5:1, 16.5 mL total). The reaction was cooled to 0 °C and Raney Nickel (0.020 g, 0.34 mmol) was added to the flask. After portionwise addition of sodium borohydride (0.32 g, 8.39 mmol), the reaction mixture was allowed to warm to room temperature and then stirred for 3 h. The solution was filtered through Celite to remove any residual Raney Nickel, and the collected filtrate was diluted with CH2Cl2 (15 mL). After washing with water (2x 15 mL), the CH2Cl2 layer separated, dried (MgSO4), filtered and adsorbed onto silica. Purification by MPLC (0–10% MeOH:CH2Cl2) afforded N-(3-aminophenyl)-3- methylbenzamide (0.69 g, 3.03 mmol, 90% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.89 (s, 1H), 7.77 – 7.65 (m, 2H), 7.44 – 7.30 (m, 2H), 7.11 (t, J = 2.1 Hz, 1H), 6.96 (t, J = 7.9 Hz, 1H), 6.85 (ddd, J = 8.0, 2.0, 1.0 Hz, 1H), 6.31 (ddd, J = 7.9, 2.2, 1.1 Hz, 1H), 2.40 (s, 3H). Step 3, preparation of 2-methoxy-N-(3-(3-methylbenzamido)phenyl)benzamide (35). To a vial containing acetonitrile (10 mL) was added N-(3-aminophenyl)-3-methylbenzamide (0.30 g, 1.32 mmol), NEt3 (0.28 mL, 1.98 mmol) and 2-methoxybenzoyl chloride (0.22 mL, 1.45 mmol), and the resulting solution was capped and heated at 80 °C for 5 h. After cooling to room temperature, the solution was concentrated, and the residue was dissolved in CH2Cl2 (10 mL). The organic (2x 8 extract was washed with saturated aqueous NaHCO3 mL), separated, and dried (MgSO4). Filtration and concentration generated a residue that was purified by reverse-phase MPLC (10 – 100% acetonitrile:water) to afford 2-methoxy-N-(3-(3-methylbenzamido)phenyl)benzamide (0.16 g, 0.43 mmol, 33 % yield). 1H NMR (400 MHz, CDCl3) δ 9.86 (s, 1H), 8.25 (dd, J1 = 7.8 Hz, J2 = 1.9 Hz, 1H), 8.14 (br. s, 1H), 8.12 (br. t, J = 2.0 Hz, 1H), 7.67 (br. s, 1H), 7.66 – 7.59 (m, 2H), 7.51 – 7.46 (m, 1H), 7.39 – 7.30 (m, 4H), 7.14 – 7.09 (m, 1H), 7.03 (br. d, J = 8.4 Hz, 1H), 4.05 (s, 3H), 2.59 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 166.00, 163.37, 157.19, 138.89, 138.74, 138.56, 134.86, 133.31, 132.50, 132.42, 129.50, 128.56, 127.70, 124.06, 121.59, 121.56, 116.16, 115.76, 111.89, 111.49, 56.23, 21.32. LCMS retention time: 3.154 min. LCMS purity at 214 nm: 100%. HRMS: m/z calcd for C22H20N2O3 (M + H+) 361.1474, found 361.1557.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.