

Abstract
Purpose.
Bietti crystalline dystrophy (BCD) is a rare, autosomal recessive, progressive, degenerative eye disease caused by mutations in the CYP4V2 gene, for which no treatments are currently available. Cyp4v3 is the murine ortholog to CYP4V2, and to better understand the molecular pathogenesis of this disease we have established a Cyp4v3-null mouse line.
Methods.
Cyp4v3−/− mice were generated by homologous recombination in embryonic stem cells. Ocular morphologic characteristics were evaluated via fundus imaging, plasma lipid profiling, and histologic analysis via Oil Red O reactivity, hematoxylin and eosin staining, and transmission electron microscopy.
Results.
The Cyp4v3−/− mouse recapitulates the characteristic features of corneoretinal crystal accumulation and systemic dyslipidemia seen in BCD. The Cyp4v3−/− mice behave normally and are viable and fertile when maintained under specific pathogen-free (SPF) housing conditions.
Conclusions.
Cyp4v3−/− mice represent a promising preclinical model that may be used to better understand the disease etiology and to evaluate pharmacotherapies for this devastating condition.
Keywords: Bietti crystalline dystrophy, preclinical model, CYP4V2
Cyp4v3−/− mice were generated as a model for Bietti crystalline dystrophy. Ocular morphologic characteristics were evaluated via fundus imaging, plasma lipid profiling, Oil Red O staining of ocular lipids, and structural analysis by light and electron microscopy.
Introduction
Bietti crystalline dystrophy (BCD) is a rare, degenerative eye disease that was first identified in 1937 by Gian Battista Bietti, an Italian ophthalmologist who treated three patients with progressive night blindness. These patients developed sparkling crystalline deposits in their corneas and posterior pole of their retinas, leading to declining vision that did not respond to traditional vision correction.1–3 Bietti crystalline dystrophy is regarded as a rare disease with a prevalence estimated at 1:67,000, although it may be underdiagnosed.4,5 There is no available treatment, and patients will eventually become completely blind.
The symptoms and clinical features of BCD are similar to those of many forms of retinitis pigmentosa, characterized by gradual constriction of the visual field, night blindness, abnormal retinal electrophysiology, altered visual acuity, and often color blindness.6 Structural findings are also typically similar to those for retinitis pigmentosa, including degeneration of the retinal pigment epithelium (RPE), pigment clumping, and sclerosis of the choroid.6 Importantly, the hallmark characteristic that differentiates BCD from other retinal disorders is formation of yellow-white crystalline deposits on the cornea and retina.
There is a wide variability in phenotype among BCD patients with differing disease severity and rate of progression. Visual acuity is often diminished, but there are reports of patients with best corrected vision within a normal range despite symptoms such as nyctalopia or blurred vision, the presence of characteristic crystalline deposits, and signs of retinal degeneration.7,8 Optical coherence tomography (OCT) images of BCD patients have revealed not only the presence of crystals in retinal and corneal tissue,9 but also the disruption and disorganization of cellular layers of the retina.8,10 Electroretinography (ERG) examination typically shows decreased amplitudes of b-waves from both rods and cones10; however, in one study that evaluated 15 BCD patients, more than 50% of patients had recordable standard ERG, and 2 had readings within normal limits 20 years after initial BCD diagnosis, suggesting that ERG may not always correlate with disease progression.11 In one case report, standard ERG was normal, but multifocal ERG showed depressed central responses consistent with the macular lesion identified by OCT.12 This suggests that the neuroretinal cells were still functioning in this patient despite RPE degeneration. Reports indicate that neuroretinal cell dysfunction in BCD patients is highly variable, and it is reasonable to suggest that the progression of RPE degeneration in BCD may originate from tissues on the basal side of the RPE rather than the neural cell layers.
In 2004, researchers at the National Eye Institute identified a mutation in the “orphan” cytochrome P450 4V2 (CYP4V2) gene in patients with BCD.13 CYP4V2 is one of 57 functional human enzymes in the cytochrome P450 superfamily of heme-containing monooxygenase enzymes. While the enzyme is ubiquitously distributed, expression in the eye is highest within RPE cells, with weaker expression in the cornea.14,15 CYP4V2 is one of 13 human P450 enzymes classified as orphans because their substrate specificity and physiological role are not well characterized.15 To date, over 50 mutations have been identified in BCD patients, with at least one mutation in each of the gene's 11 exons.9–13,16–35 Genotype analysis has shown that the most common pathologic CYP4V2 mutation is c.802-8_810del17insGC, which results in deletion of exon 7 in the mature transcript, though other mutations in each exon are also linked to the disease.16,17,28 Additionally, a relatively common polymorphism in CYP4V2 (rs13146272; Q259K), with a minor allele frequency of 45%, has been associated with deep vein thrombosis.36 The inheritance pattern of BCD is generally considered to be autosomal recessive, though some reports suggest possible haploinsufficiency, in which a heterozygous carrier may display some phenotypic characteristics, though less severe.10
Clinical systemic dyslipidemia in BCD patients has been reported, possibly due to nonfunctional CYP4V2 enzymes. For example, lymphocytes from BCD patients displayed a lack of two fatty acid-binding proteins, involved with fatty acid trafficking, with molecular weights of 32 and 45 kDa, that are expressed in control subjects. Further analysis showed that the 32-kDa protein preferentially bound docosahexaenoic acid (DHA, C22:6), α-linolenic acid (ALA, C18:3), and palmitic acid (C16:0).37 Abnormal fatty acid processing and storage has been identified in lymphocytes and fibroblasts of BCD patients, including decreased conversion of fatty acid precursors into n-3 polyunsaturated fatty acids (PUFAs) and increased incorporation of ALA into triglycerides.38 Further, fatty acid profiling in BCD serum showed an abnormal composition of fatty acids and reduced activity of the Δ-9-desaturase regardless of the CYP4V2 mutation spectrum.3,39 Lymphocytes and fibroblasts from three BCD patients revealed crystalline deposits, some resembling complex lipid deposits, although the crystal composition was not determined.3 Similarly, the composition of ocular crystals that accrue in BCD has not been determined. Elucidation of the chemical nature of these crystals would be an invaluable step toward a biochemical understanding of BCD; however, the availability of this material from human subjects is extremely limited. Analysis is further complicated by adventitious debris from macular degeneration often present in aging patients, along with the observation that crystals are no longer present in end-stage disease.40,41 To understand the biochemical mechanism underlying this progressive blinding disease, we have previously cloned and expressed the human enzyme, demonstrating that, like other CYP4 enzymes, CYP4V2 characteristically catalyzes medium- and long-chain fatty acid ω-hydroxylation reactions despite sharing only 31% to 37% sequence homology to other CYP4 enzymes.42 In addition, the enzyme has ω-hydroxylase activity for the docosanoids eicosapentaenoic acid (EPA, C20:5(n-3)) and DHA, with kinetic parameters comparable to those of CYP4F2.14
The Cyp4v3 gene is the mouse ortholog of human CYP4V2, and the proteins share 82% identity and 92% similarity.43 Therefore, to better understand BCD etiology, we have generated a strain of Cyp4v3−/− mice that are viable and fertile with no overt phenotype at birth. To determine whether mice lacking functional CYP4V3 recapitulate the pathophysiology of BCD, Cyp4v3−/− mouse eyes were screened for the presence of ocular crystalline deposits by fundus examination. In addition, ocular lipid deposition was examined histochemically by Oil Red O staining; retinal structural features were assessed by both light microscopy of hematoxylin and eosin (H&E)-stained sections and transmission electron microscopy (TEM); and plasma PUFA profiles were compared to those of wild-type animals.
Materials and Methods
Chemicals and Reagents
Atropine (1% ophthalmic solution), ketamine (100 mg/mL), xylazine (20 mg/mL), and GenTeal lubricant eye gel were purchased from the University of Washington Drug Services (Seattle, WA, USA). 2,2-d2-Stearic acid was purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA). All chemicals were of the highest purity available.
Production and Characterization of Cyp4v3−/− Mice
Embryonic stem cells (clone ID KO-1055, Cyp4v3_BB5) with targeted disruption of the Cyp4v3 gene were obtained from the Knockout Mouse Project (https://www.komp.org/, University of California, Davis [in the public domain]). The targeting vector design from Velocigene (Regeneron Pharmaceuticals, Inc., Tarrytown, NY, USA) contains the 5′ untranslated region and start codon of exon 1 of Cyp4v3 in the 5′ arm, and the 3′ arm starts after the stop codon of exon 11. Thus, homologous recombination of the targeting vector results in a complete absence of any CYP4V3 coding sequence (Fig. 1). With assistance from the University of Washington Transgenic Core Facility, the embryonic stem (ES) cell clone was expanded on embryonic feeder cells, evaluated for correct gene targeting by polymerase chain reaction (PCR), and then injected into albino C57Bl/6 blastocysts to generate chimeric mice. Mice with high degrees of chimerism were backcrossed with albino C57Bl/6 mice to test for germline transmission. Offspring that inherited the targeted Cyp4v3 allele were interbred as heterozygotes for production of Cyp4v3−/− mice. The Cyp4v3−/− mice behave normally and are viable and fertile when maintained under specific pathogen-free (SPF) housing conditions to exclude the possibility of infectious agents known to cause murine diseases that may confound phenotypes attributable to the gene targeting event.
Figure 1.
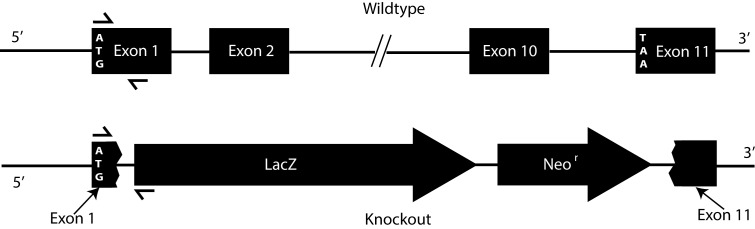
Mouse Cyp4v3 gene structure and targeting vector design. Top: The mouse Cyp4v3 gene is composed of 11 coding exons and maps to chromosomal region 8 B1.1 (syntenic with human CYP4V2, chromosome 4q35.2). Bottom: Arrangement of the Cyp4v3 locus following homologous recombination with the targeting vector. The replacement vector (http://www.velocigene.com/komp/detail/10578 [in the public domain]) deletes exons 2 to 10 and portions of exons 1 and 11 of the Cyp4v3 gene (deletion size 26,040 bp) and inserts a splice acceptor-lacZ reporter and ubC-Neor resistance cassette (ZEN-Ub1). The locations of the primers used for PCR genotyping are indicated by directional arrows over exon 1 and the lacZ reporter gene.
To confirm the null allele in Cyp4v3−/− mice, genotype analysis was performed by PCR. Primers for Cyp4v3, LacZ, and fatty acid binding protein (FABP) were mixed with DNA extracted from Cyp4v3−/− mice, heterozygote mice, and wild-type controls in a master mix containing GemTaq 5× Reaction Buffer and GemTaq polymerase (MGQuest; Lynnwood, WA, USA), deoxyribonucleotide triphosphates (dNTPs; Qiagen, Valencia, CA, USA), 5 M betaine (Sigma-Aldrich Corp.), and dimethyl sulfoxide (DMSO; Sigma-Aldrich Corp.). Polymerase chain reaction amplicons were separated by agarose gel electrophoresis and stained with ethidium bromide (Bio-Rad, Hercules, CA, USA); data were acquired on a UVP Doc-ItLS image acquisition system (Upland, CA, USA).
To detect the nontargeted wild-type allele, primers Cyp4v3-Ex1.for and Cyp4v3-Ex1.rev were used. The forward primer spans nucleotides (nt) 19 to 37 of the Cyp4v3 mRNA (NM_133969.2) and the reverse primer nt 241 to 265, resulting in a 246-bp amplicon. The lacZ.rev primer is located at nt 111 to 132 of the ZEN-Ub1 lacZ-NeoR selection cassette designed by Velocigene for Knockout Mouse Project (KOMP) and, in combination with primer Cyp4v3-Ex1.for, yields a 335-bp amplicon for a correctly targeted allele. The location of the Cyp4v3-Ex1.rev primer is beyond the portion of the Cyp4v3 gene that is deleted in the replacement vector. Thus, as seen in Figure 1, it yields a product only for wild-type or heterozygous mice. To control for the presence of PCR-amplifiable genomic DNA, each sample was subjected to analysis using primers for the single-copy somatic gene (FABP) as previously described.44
To confirm that the Cyp4v3 gene targeting event is truly null, CYP4V3 protein expression was assessed by Western blot analysis. Microsomes were prepared from excised livers from Cyp4v3−/− mice (n = 3) and wild-type control mice (n = 3) as previously described.45 Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) was used to separate proteins in samples containing 15 μg total protein per well. Separated proteins were transferred to nitrocellulose membranes (Invitrogen, Carlsbad, CA, USA) using iBlot (Invitrogen), blocked overnight in Odyssey blocking buffer (Li-Cor, Lincoln, NE, USA), and probed for CYP4V3 using a rabbit polyclonal antibody raised against human CYP4V214; β-actin (Li-Cor) was used as a loading control. Immunoreactivity was detected using Li-Cor secondary antibodies, and images were generated with Odyssey CLX (Li-Cor).
Fundus Imaging
To screen for changes in ocular appearance, a Micron II small animal retinal imaging system from Phoenix Research Laboratories (Pleasanton, CA, USA) was used. In brief, mouse eyes were first treated with topical atropine (Bausch & Lomb, Tampa, FL, USA) and then the mice were anesthetized with ketamine/xylazine (University of Washington Drug Services) prior to fundus imaging. GenTeal lubricant eye gel (Novartis, East Hanover, NJ, USA) was applied to the eyes of anesthetized mice to keep them moist during imaging. Mice were placed on the stage of the imaging system and placed under the camera to capture retinal images. Animal studies were conducted in accordance with all applicable animal care and use laws, regulations, and guidelines, including adherence to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research guidelines, under a protocol approved by the University of Washington Institutional Animal Care and Use Committee in an Association for Assessment and Accreditation of Laboratory Animal Care–accredited facility.
Plasma PUFA Profiling
Analysis of 20 μL sera from Cyp4v3 knockout and wild-type mice for PUFA content was performed using methanol and methyl-t-butyl ether (MTBE) as described previously.46 Specifically, 1 μg 2,2-d2-stearic acid (internal standard) was added to the biological sample, followed by 0.75 mL methanol (Thermo Fisher Science, Pittsburgh, PA, USA). After shaking for 5 minutes, 1.5 mL MTBE was added to the mixture and shaken for 1 hour at room temperature. Then, 0.63 mL nanopure water was added, and the mixture was centrifuged at 1000g for 10 minutes. The upper organic layer was transferred to a clean centrifuge tube, and the extraction procedure was repeated a second time. The pooled organic layers were washed with small quantities of 0.2 M aqueous potassium chloride and nanopure water. Next, the organic solvent was dried under a stream of N2 gas. The extracted fraction was reconstituted with a small amount of ethyl acetate and derivatized using a molar excess of diazomethane. After derivatization, the sample was dried under a stream of N2 gas. The extracted fraction was reconstituted with 60 μL ethyl acetate and analyzed by gas chromatography and electron ionization mass spectrometry (GC-EI/MS). At this point, all free fatty acids had been derivatized to their methyl ester forms. The following procedure released fatty acids from complex lipids and transesterified them. The derivatized samples in ethyl acetate were transferred to a glass tube and mixed with 200 μL toluene and 1 mL 2% sulfuric acid in methanol (vol/vol). After the tube was sealed and vortexed quickly, the lipids were heated at 100°C in a sand bath for 65 minutes. Then the lipids were cooled on ice, and 1.2 mL nanopure water was added. Lipids were extracted with 0.7 mL hexane, and the extraction was repeated two more times. The combined solvent was dried under a stream of N2 gas, and the dried fraction was reconstituted in 60 μL ethyl acetate. Finally, total fatty acids were analyzed by GC-EI/MS.
Lipid Profiling by GC-EI/MS
The transesterified lipids were analyzed on a Shimadzu QP2010 GCMS (Shimadzu, Kyoto, Japan) fitted with a 60-m fused-silica capillary column (DB-1) using selected ion monitoring. The analytes were injected at a temperature of 100°C. After 2 minutes, the oven temperature was raised at 30°C/min to 130°C, 10°C/min to 180°C, held for 2 minutes, 4°C/min to 210°C, 10°C/min to 235°C, held for 3 minutes, 4°C/min to 255°C, 10°C/min to 310°C, then held for 4 minutes. Under these conditions, the derivatized saturated and unsaturated fatty acids eluted as follows: d2-C18:0 at 21.30 minutes, C14:0 at 13.86 minutes, C16:0 at 17.77 minutes, C16:1 at 17.36 minutes, C18:0 at 21.30 minutes, C18:1 cis-9 at 20.91 minutes, C18:1 cis-11 at 21.01 minutes, C18:2 at 20.76 minutes, C18:3 at 20.83 minutes, C20:4 at 23.66 minutes, C20:5 at 23.57 minutes, and C22:6 at 27.65 minutes. Quantitation was achieved by selected ion monitoring of the characteristic [M-31]+ and [M-43]+ fragment ions found in EI spectra of the majority of fatty acid methyl esters. The PUFAs' most abundant ions appeared at less than 100 mass-to-charge ratio (m/z), unlike other saturated and mono- and diunsaturated fatty acids. Thus 74 m/z is utilized as a characteristic ion for penta- and hexaunsaturated fatty acids as described previously.14,42 Standard curves (r2 > 0.98) for each fatty acid were generated with their authentic chemical standards. Differences between two means within a group were tested with Student's t-test (two-tail, two-sample unequal variance by Student's t-test using GraphPad Prism [GraphPad Software, Inc., La Jolla, CA, USA]).
Histologic Analysis
Whole eyes dissected from mice were immediately submerged in 10% formalin (Fisher Scientific, Hampton, NH, USA) and stored at 4°C to fix the tissue for standard histology processing by the University of Washington Histology and Pathology Specialized Services Core as described previously.47 Briefly, formalin-fixed tissues were dehydrated in graded ethanols and embedded in paraffin. Sections (10 μm) were deparaffinized, rehydrated, and treated with sodium citrate buffer before staining with H&E. Slides were washed thoroughly and fixed, and images were captured on a Nikon Ti-S microscope (Nikon Instruments, Inc., Melville, NY, USA).
For histochemical lipid analysis, whole eyes were harvested from mice, immediately frozen at −80°C in Tissue-Tek optimum temperature cutting compound (Electron Microscopy Sciences, Fort Washington, PA, USA), and submitted for histologic processing and Oil Red O staining for lipid by the University of Washington Histology and Pathology Specialized Services Core. Briefly, frozen sections were sliced 8 to 10 μm thick and mounted on slides. The slides were dehydrated in propylene glycol and then stained in Oil Red O solution for 8 to 10 minutes at 60°C. Slides were then placed in 85% propylene glycol solution for 2 to 5 minutes for differentiation, rinsed, and counterstained in hematoxylin solution for 30 seconds. Slides were washed thoroughly and fixed, and images were captured on a Nikon Ti-S microscope (Nikon Instruments, Inc.).
For analysis of retinas by TEM, whole eyes harvested from mice were cut open to facilitate fixative penetration and then submerged in 1/2× Karnovsky's fixative (2.5% glutaraldehyde with 2% formaldehyde in 0.1 M buffer). The tissue was postfixed in osmium tetroxide and processed, sectioned, and examined by TEM according to standard protocols as described previously.47
Results
To confirm successful knockout of Cyp4v3 alleles and to verify the absence of CYP4V3 protein in Cyp4v3−/− mice, we performed PCR genotyping analysis and Western blot analysis. The Cyp4v3 amplicon of 246 bp in length is present in both wild-type and heterozygous mice, but absent in mice with the null allele. The LacZ gene is a marker in the knockout construct and appears as a band at 335 bp in knockout and heterozygote mice. Fatty acid binding protein is a control to verify genomic DNA integrity and should be present in all samples tested. Figure 2a shows the results of PCR genotyping for Cyp4v3−/−, Cyp4v3+/−, and wild-type control mice. Lanes 1, 6, and 11 contain molecular weight markers in 100-bp increments; lanes 2, 7, and 12 contain DNA samples from wild-type control mice; lanes 3, 8, and 13 contain DNA from heterozygote mice; lanes 4, 9, and 14 contain DNA from Cyp4v3−/− mice; and lanes 5, 10, and 15 contain a nontemplate control. As expected, the wild-type control has bands positive for Cyp4v3 and FABP, but is negative for LacZ that was part of the knockout construct. The heterozygote has bands for all three markers, while the Cyp4v3−/− mouse is negative for Cyp4v3 but positive for LacZ and FABP. This confirms correct gene targeting in Cyp4v3−/− mice.
Figure 2.

Confirmation of genotype and protein expression of Cyp4v3−/− mice compared to wild-type controls. (a) PCR of wild-type (lanes 2, 7, and 12), heterozygote (lanes 3, 8, and 13), and Cyp4v3−/− mice (lanes 4, 9, and 14) shows that wild-type mice have positive bands for Cyp4v3 (lane 2), negative bands for the LacZ reporter construct (lane 7), and positive bands for FABP to verify the presence of genomic DNA (lane 12). Heterozygotes show bands for all three genes, and Cyp4v3−/− mice are negative for Cyp4v3 (lane 4), positive for the LacZ reporter construct (lane 9), and positive for FABP (lane 14). This confirms that Cyp4v3−/− mice lack functional Cyp4v3, and the targeting vector was successfully incorporated into the genome. (b) Western blot analysis of Cyp4v3−/− mice (lanes 2, 3, and 4), wild-type mice (lanes 5, 6, and 7), and 0.18 pmol human purified CYP4V2 as a validation control. Lower bands are β-actin, used to verify the presence of equivalent total protein in each lane, and upper bands show the presence (wild-type) and absence (Cyp4v3−/−) of CYP4V3 protein expression, as expected. This confirms that mice without functional Cyp4v3 do not produce the corresponding protein.
Western blotting was performed to verify that Cyp4v3−/− mice lack CYP4V3 protein. Microsomes were prepared from mouse livers according to previously published protocols and proteins separated by SDS-PAGE electrophoresis.45 CYP4V3 has a molecular weight of approximately 60 kDa and appears as the upper band in Figure 2b. The lower band at approximately 42 kDa is β-actin and confirms that equivalent amounts of protein were loaded in each lane of the gel. Lane 1 is the molecular weight marker; lanes 2, 3, and 4 contain liver microsomes (15 μg total protein) from Cyp4v3−/− mice; lanes 5, 6, and 7 contain liver microsomes (15 μg total protein) from wild-type control mice; and lane 8 contains 0.18 pmol purified human CYP4V2.
As crystalline deposits are a hallmark of BCD, we performed fundus imaging on Cyp4v3−/− mice (Fig. 3). At 6 months of age (n = 4 each wild-type and Cyp4v3−/−), sparse crystals (10–20 μm in diameter) were visible on retinal exam, and by 12 months of age (n = 6 wild-type and n = 8 Cyp4v3−/−) the crystalline pattern became stark and approached confluence. Microscope measurements indicated that each deposit was approximately 10 to 20 μm in diameter, on average. These are similar in absolute size to those observed in BCD patients,4 and appear larger in mice due to the much smaller relative globe size. From a species comparison, a 6-month-old mouse approximates humans in their 20s, corresponding to the approximate age of BCD diagnosis. A 12-month-old mouse approximates humans in their 50s, corresponding to the approximate age of legal blindness in BCD.3,48 Thus, the gross ocular pathology of Cyp4v3−/− mice recapitulates the clinical timeline of BCD pathogenesis, and crystal morphometry mimics the human disease.7
Figure 3.

Fundus images of Cyp4v3−/− mice and a Cyp4v3+/+ control. (a) Normal retina of a 12-month-old Cyp4v3+/+ mouse; (b) retina of a 6-month-old Cyp4v3−/− mouse with sparse, reflective crystalline deposits (arrowheads); (c) retina of 12-month-old and (d) 18-month-old Cyp4v3−/− mice with highly confluent crystalline deposits (arrowheads).
To gain more detailed knowledge of the ocular pathology of Cyp4v3−/− mice, we evaluated histologic sections from wild-type and Cyp4v3−/− mice (Fig. 4). All mice were older than 18 months and of similar age. In Figure 4, the top row (Figs. 4a, 4b) shows histologic sections of two individual wild-type mice. The bottom row (Figs. 4c, 4d) shows histologic sections of two individual Cyp4v3−/− mice. The sections clearly delineate the layers of the retina, and there is no visible difference in the neural retinal tissue between wild-type and Cyp4v3−/− mice. Of particular interest, however, is a distinct linear array of optically clear vacuoles (arrowheads, Figs. 4c, 4d) contained deep within the photoreceptor outer segment and adjacent to the pigment epithelium. Contents of the vacuoles were lost during tissue processing; however, their size and location are consistent with our knowledge of the crystalline deposits observed in BCD patients and in our Cyp4v3−/− mice as shown in Figure 3.
Figure 4.

Histologic sections of murine retinas at 20× magnification. (a, b) Images from two individual wild-type mice and (c, d) from two individual Cyp4v3−/− mice. The tissue structures appear normal in all four mice. However, in the Cyp4v3−/− mouse (c, d), circular vacuoles are present at the base of the photoreceptor layer just anterior to the RPE cells (arrowheads). These vacuoles are consistent in size, location, and composition with the crystalline deposits observed by fundus imaging in BCD patients and Cyp4v3−/− mice. Ch, choroid; G, ganglion cell layer; I, inner nuclear layer; O, outer nuclear layer; Ph, photoreceptor cell layer; RPE, retinal pigmented epithelium.
Images gathered from TEM of retinal sections from wild-type and Cyp4v3−/− mice are shown in Figure 5. Again, all mice were older than 18 months and of similar age. Images in Figures 5a and 5b are from two different wild-type mice, and Figures 5c and 5d are from two separate Cyp4v3−/− mice. Osmification of tissues for examination by EM results in a marked increase in the electron density of lipid-containing structures. Consequently, on electron microscopy (EM), the lipid-filled vacuoles represented as optically clear spaces in Figure 4 appear as either scattered solitary foci or confluent black globular structures arranged in various patterns in Figure 5 due to the number and close adjacency of vacuoles. In one sample from a Cyp4v3−/− albino mouse (Fig. 5d), the pigment normally present within the pigment epithelium is missing, but the other structures are still present.
Figure 5.

Transmission electron microscopy images of murine retinas. (a, b) Retinas of two different wild-type control mice. (c, d) Retinas of two Cyp4v3−/− mice. The presence of osmium increases the electron density of lipid-containing structures, so lipid-filled vacuoles appear as scattered solitary foci or confluent black globular structures. In one sample from a Cyp4v3−/− albino mouse, the pigment normally present within the pigment epithelium was missing, but other structures were the same. Ch, choroid; M, melanin pigment granule; N, nucleus; Ph, photoreceptors; RPE, retinal pigmented epithelium.
Biochemical analysis of murine plasma revealed changes in several fatty acids in knockout mice. At 12 months of age, C18:0 levels were significantly lower, while C18:2 and C18:3 levels were significantly higher in knockout mice versus wild-type controls. A previous study in BCD patients also showed a difference in C18:0 levels of similar magnitude, but in the opposite direction to our results in mice. Specifically, BCD patients exhibited a significantly higher level of C18:0 compared to healthy controls (P = 0.007). The n-3 PUFA DHA level was numerically higher in knockout versus wild-type mice, but just failed to reach statistical significance (P = 0.07; Fig. 6). Levels of DHA showed a similar trend toward elevated levels in BCD patients that, as in Cyp4v3−/− mice, approached but did not reach statistical significance (P = 0.12).39 These results suggest that systemic dyslipidemia is a common component in Cyp4v3−/− mice and BCD patients, but the mechanism and clinical relevance remain unclear.
Figure 6.

Serum fatty acid concentrations in Cyp4v3−/− mice compared with age-matched Cyp4v3+/+ controls.
In addition to plasma lipid analysis, we examined the eyes of Cyp4v3−/− mice using the lysochrome diazo dye Oil Red O. While 6-month-old mice appeared normal (data not shown), 12-month-old mice exhibited significant focal accumulation of Oil Red O lipid droplets within the choroid layer (Fig. 7). This suggests that lipid processing in ocular tissue is abnormal in Cyp4v3−/− mice and has an age-related progression. Indeed, ocular lipid processing becomes much less efficient with advanced age, and it has been observed that lipids accumulate in the Bruch's membrane over time even in normal, healthy eyes. This accumulation has been suggested to influence the pathological formation of lesions in age-related macular degeneration.49,50
Figure 7.
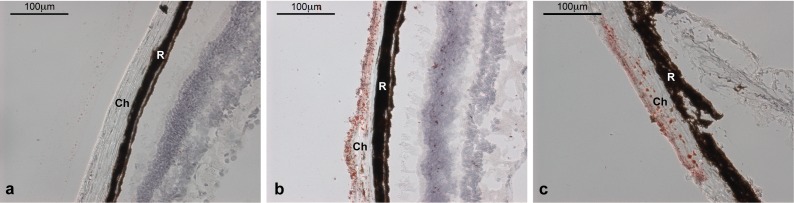
Oil Red O staining of cross-sectional eye tissue samples from 12-month-old Cyp4v3−/− mice and an age-matched Cyp4v3+/+ control. (a) Normal Cyp4v3+/+; (b, c) two individual Cyp4v3−/− mice with red-stained lipid droplets visible throughout. Ch, choroid; R, retinal pigment epithelium.
Discussion
Here we describe generation and characterization of a Cyp4v3−/− mouse developed as a preclinical model for BCD. This model displays age-related progression of the disease that mimics the human pathology in ocular tissue, and also exhibits systemic dyslipidemia as reported in human patients. Human CYP4V2 and the murine ortholog CYP4V3 are likely involved in lipid homeostasis in that the human enzyme is known to catalyze preferential ω-hydroxylase activity on medium-chain fatty acids.42 Of particular interest is the role of CYP4V2 in metabolism of DHA both systemically and in ocular tissue, because DHA is an essential component of retinal rod outer segment membranes, and is also a known precursor to resolvins and protectins (essential components in innate anti-inflammatory modulation).51–54 Pathology from nonfunctional CYP4V2 may be related to inflammation in ocular tissue. This would follow other age-related retinal degenerative diseases such as macular degeneration, in which inflammatory response has an important role.55
Recycling of rod outer segment membranes is a very active process, with as many as 10% of disc membranes shed each day in a normal eye.56 Dysfunction in CYP4V2 may disrupt membrane processing in the RPE, leading to localized dyslipidemia and photoreceptor degradation. Given the slow, age-related progression of BCD, however, CYP4V2 may not be critical for recycling rod outer segments, as significant disruption of the high turnover would imply more rapidly progressing pathology.
Because the onset of BCD is unlike early-onset, aggressive ocular diseases (e.g., Leber's congenital amaurosis or primary congenital glaucoma), the hypothesis that CYP4V2 is critical to recycling of essential PUFA components of rod outer segments seems unlikely given the rapid turnover of these organelles. An attractive alternative hypothesis is that CYP4V2 plays a role in attenuating the signaling pathway mediated by resolvins/protectins, potent anti-inflammatory mediators.53 We have previously demonstrated that CYP4V2 metabolizes DHA and EPA to their ω-hydroxy products. It is possible that ω-hydroxy docosanoids are not substrates for the lipoxygenase enzymes responsible for generating resolvins/protectins from DHA/EPA, thus limiting their production. Alternatively, the resolvin/protectin molecules themselves may be substrates for CYP4V2. This would be an interesting counterbalance to another CYP4 enzyme (CYP4F3A), which is also involved in modulating inflammatory signaling pathways. In the case of CYP4F3A, this neutrophil-specific splice variant enzyme plays a key role in mitigating leukotriene B4 signaling by ω-hydroxylation, thus inactivating this arachidonic acid–derived n-6 proinflammatory lipid molecule.57 Considering that resolvins/protectins are anti-inflammatory lipid signaling molecules, CYP4V2 could be playing a role in terminating the resolution phase of the inflammatory process. Importantly, evaluation of ARPE-19 cells revealed that CYP4V2 is the only known ω-hydroxylase enzyme expressed in RPE cells.14 In such a scenario, it is plausible that acute loss of CYP4V2 is not detrimental, and that it is only with time and other confounding factors (e.g., environmental or dietary) that BCD progresses to a clinically diagnosed state. More work is required to test these admittedly speculative scenarios.
There are a number of limitations to this study. First, as this was a preliminary investigation into the phenotype of a newly established Cyp4v3−/− mouse line, the number of mice evaluated was limited, although the fundus pathology with crystalline deposits occurred consistently across all individuals examined. Second, although there is documented dyslipidemia in BCD patients, the clinical relevance has not yet been determined. While Oil Red O staining of the eye shows a clear difference in the knockout mouse, it does not provide information on specific lipid composition. Oil Red O is a lysochrome diazo dye that reacts with neutral lipids and cholesterol esters, but it is relatively nonspecific and does not distinguish different lipid species.58 Light microscopy and TEM analyses further suggest that the crystalline deposits are composed of lipids, but we still have no indication regarding the identity of the lipids present. Therefore, it will be critical to profile the ocular lipid composition of the knockout mice in future studies, utilizing more advanced methodology such as matrix-assisted laser desorption ionization (MALDI) imaging. In summary, Cyp4v3−/− mice exhibit phenotypic characteristics that correspond well to BCD in humans, in terms of both progressive crystal deposition in the eye and the systemic nature of the lipid abnormalities. To validate this knockout mouse as a model of BCD, further detailed characterization is warranted, including electrophysiological changes (ERG), spectral-domain OCT, and optokinetic measurements of visual acuity. Finally, the availability of a mouse model of BCD will allow detailed investigations into the molecular and biochemical changes that occur as a result of genetic defects in CYP4V enzyme activity.
Acknowledgments
For their invaluable input and guidance, the authors thank Richard G. Weleber, MD, from the Casey Eye Institute at Oregon Health Sciences University; Russell van Gelder, MD, PhD, professor and chair of the Department of Ophthalmology at the University of Washington; and J. Fielding Hejtmancik, MD, PhD, from the National Eye Institute. They also thank the University of Washington Drug Metabolism, Transport and Pharmacogenomic Research program; the University of Washington School of Pharmacy Mass Spectrometry Center; Warren Ladiges, DVM, MS, director of the Department of Veterinary Medicine, at the University of Washington; Kelly Hudkins, MS, for histology and pathology support; H. Denny Liggitt, DVM, PhD, for his veterinary pathology expertise; and Scott Greenwald, PhD, for assistance with fundus imaging.
Supported by the University of Washington Drug Metabolism, Transport and Pharmacogenomic Research Program and National Institutes of Health Grant R01 GM49054.
Disclosure: C.M. Lockhart, None; M. Nakano, None; A.E. Rettie, None; E.J. Kelly, None
References
- 1. Bietti G. Ueber familiaeres vorkommen von “retinitis punctata albescens” (verbunden mit “dystrophia marginalis cristallinea corneae”), glitzern des glaskoerpers und anderen degenerativen augenveraenderungen. Klin Monbl Augenheilkd. 1937; 99: 21 [Google Scholar]
- 2. National Eye Institute, National Institutes of Health. Facts about Bietti's crystalline dystrophy. Available at: http://www.nei.nih.gov/Health/biettis/index.asp Accessed August 7, 2014. [Google Scholar]
- 3. Kaiser-Kupfer MI, Chan CC, Markello TC, et al. Clinical biochemical and pathologic correlations in Bietti's crystalline dystrophy. Am J Ophthalmol. 1994; 118: 569–582 [DOI] [PubMed] [Google Scholar]
- 4. Mataftsi A, Zografos L, Milla E, Secretan M, Munier FL. Bietti's crystalline corneoretinal dystrophy: a cross-sectional study. Retina. 2004; 24: 416–426 [DOI] [PubMed] [Google Scholar]
- 5. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006; 368: 1795–1809 [DOI] [PubMed] [Google Scholar]
- 6. Li A, Jiao X, Munier FL, et al. Bietti crystalline corneoretinal dystrophy is caused by mutations in the novel gene CYP4V2. Am J Hum Genet. 2004; 74: 817–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fong AM, Koh A, Lee K, Ang CL. Bietti's crystalline dystrophy in Asians: clinical, angiographic and electrophysiological characteristics. Int Ophthalmol. 2008; 29: 459–470 [DOI] [PubMed] [Google Scholar]
- 8. Ayata A, Tathpinar S, Unal M, Ersanli D, Bilge AH. Autofluorescence and OCT features of Bietti's crystalline dystrophy. Br J Ophthalmol. 2008; 92: 718–720 [DOI] [PubMed] [Google Scholar]
- 9. Chung JK, Shin JH, Jeon BR, Ki CS, Park TK. Optical coherence tomographic findings of crystal deposits in the lens and cornea in Bietti crystalline corneoretinopathy associated with mutation in the CYP4V2 gene. Jpn J Ophthalmol. 2013; 57: 447–450 [DOI] [PubMed] [Google Scholar]
- 10. Garcia-Garcia G-P, Lopez-Garrido M-P, Martinez-Rubio M, Moya-Moya M-A, Belmonte-Martinez J, Escribano J. Genotype-phenotype analysis of Bietti crystalline dystrophy in a family with the CYP4V2 Ile111Thr mutation. Cornea. 2013; 32: 1002–1008 [DOI] [PubMed] [Google Scholar]
- 11. Rossi S, Testa F, Li A, et al. Clinical and genetic features in Italian Bietti crystalline dystrophy patients. Br J Ophthalmol. 2012; 97: 174–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rossi S, Testa F, Li A, et al. An atypical form of Bietti crystalline dystrophy. Ophthalmic Genet. 2011; 32: 118–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li A, Jiao X, Munier FL, et al. Bietti crystalline corneoretinal dystrophy is caused by mutations in the novel gene CYP4V2. Am J Hum Genet. 2004; 74: 817–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakano M, Kelly EJ, Wiek C, Hanenberg H, Rettie AE. CYP4V2 in Bietti's crystalline dystrophy: ocular localization, metabolism of omega-3-polyunsaturated fatty acids, and functional deficit of the p.H331P variant. Mol Pharmacol. 2012; 82: 679–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stark K, Guengerich FP. Characterization of orphan human cytochromes P450. Drug Metab Rev. 2007; 39: 627–637 [DOI] [PubMed] [Google Scholar]
- 16. Kelly EJ, Nakano M, Rohatgi P, Yarov-Yarovoy V, Rettie AE. Finding homes for orphan cytochrome P450s: CYP4V2 and CYP4F22 in disease states. Mol Interv. 2011; 11: 124–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xiao X, Mai G, Li S, Guo X, Zhang Q. Identification of CYP4V2 mutation in 21 families and overview of mutation spectrum in Bietti crystalline corneoretinal dystrophy. Biochem Biophys Res Commun. 2011; 409: 181–186 [DOI] [PubMed] [Google Scholar]
- 18. Haddad NMN, Waked N, Bejjani R, et al. Clinical and molecular findings in three Lebanese families with Bietti crystalline dystrophy: report on a novel mutation. Mol Vis. 2012; 18: 1182–1188 [PMC free article] [PubMed] [Google Scholar]
- 19. Jin Z-B, Ito S, Saito Y, Inoue Y, Yanagi Y, Nao-i N. Clinical and molecular findings in three Japanese patients with crystalline retinopathy. Jpn J Ophthalmol. 2006; 50: 426–431 [DOI] [PubMed] [Google Scholar]
- 20. Lai TYY, Ng TK, Tam POS, et al. Genotype phenotype analysis of Bietti's crystalline dystrophy in patients with CYP4V2 mutations. Invest Ophthalmol Vis Sci. 2007; 48: 5212–5220 [DOI] [PubMed] [Google Scholar]
- 21. Lee KY, Koh AH, Aung T, et al. Characterization of Bietti crystalline dystrophy patients with CYP4V2 mutations. Invest Ophthalmol Vis Sci. 2005; 46: 3812–3816 [DOI] [PubMed] [Google Scholar]
- 22. Lin J, Nishiguchi KM, Nakamura M, Dryja TP, Berson EL, Miyake Y. Recessive mutations in the CYP4V2 gene in East Asian and Middle Eastern patients with Bietti crystalline corneoretinal dystrophy. J Med Genet. 2005; 42: e38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu DN, Liu Y, Meng XH, Yin ZQ. The characterization of functional disturbances in Chinese patients with Bietti's crystalline dystrophy at different fundus stages. Graefes Arch Clin Exp Ophthalmol. 2011; 250: 191–200 [DOI] [PubMed] [Google Scholar]
- 24. Mamatha G, Umashankar V, Kasinathan N, et al. Molecular screening of the CYP4V2 gene in Bietti crystalline dystrophy that is associated with choroidal neovascularization. Mol Vis. 2011; 17: 1970–1977 [PMC free article] [PubMed] [Google Scholar]
- 25. Parravano M, Sciamanna M, Giorno P, Boninfante A, Varano M. Bietti crystalline dystrophy: a morpho-functional evaluation. Doc Ophthalmol. 2011; 124: 73–77 [DOI] [PubMed] [Google Scholar]
- 26. Porter LF, Urquhart JE, O'Donoghue E, et al. Identification of a novel locus for autosomal dominant primary open angle glaucoma on 4q35.1-q35.2. Invest Ophthalmol Vis Sci. 2011; 52: 7859–7865 [DOI] [PubMed] [Google Scholar]
- 27. Shan M, Dong B, Zhao X, et al. Novel mutations in the CYP4V2 gene associated with Bietti crystalline corneoretinal dystrophy. Mol Vis. 2005; 11: 738–743 [PubMed] [Google Scholar]
- 28. Wada Y, Itabashi T, Sato H, Kawamura M, Tada A, Tamai M. Screening for mutations in CYP4V2 gene in Japanese patients with Bietti's crystalline corneoretinal dystrophy. Am J Ophthalmol. 2005; 139: 894–899 [DOI] [PubMed] [Google Scholar]
- 29. Wang Y, Guo L, Cai S-P, et al. Exome sequencing identifies compound heterozygous mutations in CYP4V2 in a pedigree with retinitis pigmentosa. PLoS One. 2012; 7: e33673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yokoi Y, Sato K, Aoyagi H, Takahashi Y, Yamagami M, Nakazawa MA. Novel compound heterozygous mutation in the CYP4V2 gene in a Japanese patient with Bietti's crystalline corneoretinal dystrophy. Case Rep Ophthalmol. 2011; 2: 296–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yokoi Y, Nakazawa M, Mizukoshi S, Sato K, Usui T, Takeuchi K. Crystal deposits on the lens capsules in Bietti crystalline corneoretinal dystrophy associated with a mutation in the CYP4V2 gene. Acta Ophthalmol. 2009; 88: 607–609 [DOI] [PubMed] [Google Scholar]
- 32. Zenteno JC, Ayala-Ramirez R, Graue-Wiechers F. Novel CYP4V2 gene mutation in a Mexican patient with Bietti's crystalline corneoretinal dystrophy. Curr Eye Res. 2008; 33: 313–318 [DOI] [PubMed] [Google Scholar]
- 33. Song Y, Mo G, Yin G. A novel mutation in the CYP4V2 gene in a Chinese patient with Bietti's crystalline dystrophy. Int Ophthalmol. 2013; 33: 269–276 [DOI] [PubMed] [Google Scholar]
- 34. Yin H, Jin C, Fang X, et al. Molecular analysis and phenotypic study in 14 Chinese families with Bietti crystalline dystrophy. PLoS One. 2014; 9: e94960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Halford S, Liew G, Mackay DS, et al. Detailed phenotypic and genotypic characterization of Bietti crystalline dystrophy. Ophthalmology. 2014; 1174–1184 [DOI] [PubMed] [Google Scholar]
- 36. Bezemer ID, Bare LA, Doggen CJ, et al. Gene variants associated with deep vein thrombosis. JAMA. 2008; 299: 1306–1314 [DOI] [PubMed] [Google Scholar]
- 37. Lee J, Jiao X, Hejtmancik JF, Kaiser-Kupfer M, Chader GJ. Identification, isolation, and characterization of a 32-kDa fatty acid-binding protein missing from lymphocytes in humans with Bietti crystalline dystrophy (BCD). Mol Genet Metab. 1998; 65: 143–154 [DOI] [PubMed] [Google Scholar]
- 38. Lee J, Jiao X, Hejtmancik JF, et al. The metabolism of fatty acids in human Bietti crystalline dystrophy. Invest Ophthalmol Vis Sci. 2001; 42: 1707–1714 [PubMed] [Google Scholar]
- 39. Lai TY, Chu KO, Chan KP, et al. Alterations in serum fatty acid concentrations and desaturase activities in Bietti crystalline dystrophy unaffected by CYP4V2 genotypes. Invest Ophthalmol Vis Sci. 2010; 51: 1092–1097 [DOI] [PubMed] [Google Scholar]
- 40. Bernauer W, Daicker B. Bietti's corneal-retinal dystrophy: a 16-year progression. Retina. 1992; 12: 18–20 [PubMed] [Google Scholar]
- 41. Furusato E, Cameron JD, Chan C-C. Evolution of cellular inclusions in Bietti's crystalline dystrophy. Ophthalmol Eye Dis. 2010; 2010: 9–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nakano M, Kelly EJ, Rettie AE. Expression and characterization of CYP4V2 as a fatty acid-hydroxylase. Drug Metab Dispos. 2009; 37: 2119–2122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mackay DS, Halford S. Focus on molecules: cytochrome P450 family 4, subfamily V, polypeptide 2 (CYP4V2). Exp Eye Res. 2011; 102: 111–112 [DOI] [PubMed] [Google Scholar]
- 44. Crouthamel MH, Kelly EJ, Ho RJ. Development and characterization of transgenic mouse models for conditional gene knockout in the blood-brain and blood-CSF barriers. Transgenic Res. 2012; 21: 113–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Parkinson OT, Liggitt HD, Rettie AE, Kelly EJ. Generation and characterization of a Cyp4b1 null mouse and the role of CYP4B1 in the activation and toxicity of Ipomeanol. Toxicol Sci. 2013; 134: 243–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Matyash V, Liebisch G, Kurzchalia TV, Shevchenko A, Schwudke D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J Lipid Res. 2008; 49: 1137–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Alpers CE, Hudkins KL, Pritzl P, Johnson RJ. Mechanisms of clearance of immune complexes from peritubular capillaries in the rat. Am J Pathol. 1991; 139: 855–867 [PMC free article] [PubMed] [Google Scholar]
- 48. Flurkey K, Currer JM, Harrison DE. The mouse in aging research. In: Fox JG. ed The Mouse in Biomedical Research. 2nd ed. Burlington, MA: American College Laboratory Animal Medicine (Elsevier); 2007: 637–372 [Google Scholar]
- 49. Curcio CA, Johnson M, Rudolf M, Huang JD. The oil spill in ageing Bruch membrane. Br J Ophthalmol. 2011; 95: 1638–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Curcio CA, Johnson M, Huang JD, Rudolf M. Apolipoprotein B-containing lipoproteins in retinal aging and age-related macular degeneration. J Lipid Res. 2010; 51: 451–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fliesler SJ, Anderson RE. Chemistry and metabolism of lipids in the vertebrate retina. Prog Lipid Res. 1983; 22: 79–131 [DOI] [PubMed] [Google Scholar]
- 52. Weylandt KH, Chiu CY, Gomolka B, Waechter SF, Wiedenmann B. Omega-3 fatty acids and their lipid mediators: towards an understanding of resolvin and protectin formation. Prostaglandins Other Lipid Mediat. 2012; 97: 73–82 [DOI] [PubMed] [Google Scholar]
- 53. Spite M, Claria J, Serhan CN. Resolvins, specialized proresolving lipid mediators, and their potential roles in metabolic diseases. Cell Metab. 2014; 19: 21–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Duffield JS, Hong S, Vaidya VS, et al. Resolvin D series and Protectin D1 mitigate acute kidney injury. J Immunol. 2006; 177: 5902–5911 [DOI] [PubMed] [Google Scholar]
- 55. Wang Y, Wang VM, Chan CC. The role of anti-inflammatory agents in age-related macular degeneration (AMD) treatment. Eye. 2011; 25: 127–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Young RW. Shedding of discs from rod outer segments in the rhesus monkey. J Ultrastruct Res. 1971; 34: 190–203 [DOI] [PubMed] [Google Scholar]
- 57. Christmas P, Ursino SR, Fox JW, Soberman RJ. Expression of the CYP4F3 gene. Tissue-specific splicing and alternative promoters generate high and low K(m) forms of leukotriene B(4) omega-hydroxylase. J Biol Chem. 1999; 274: 21191–21199 [DOI] [PubMed] [Google Scholar]
- 58. Mehlem A, Hagberg CE, Muhl L, Eriksson U, Falkevall A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat Protoc. 2013; 8: 1149–1154 [DOI] [PubMed] [Google Scholar]