

Abstract
Purpose.
In the pathogenesis of diabetic retinopathy, matrix metalloproteinase (MMP)-9 damages retinal mitochondria, activating the apoptotic machinery. Transcription of MMP-9 is regulated by nuclear factor kappa B (NF-κB), and the activation of NF-κB is modulated by the acetylation of its p65 subunit. Sirtuin 1 (Sirt1), a deacetylase, plays an important role in the acetylation-deacetylation of p65. The goal of this study is to investigate the role of Sirt1 in the activation of MMP-9 in diabetic retinopathy.
Methods.
The effect of hyperglycemia and Sirt1 activator, resveratrol, on acetylation of p65 and its binding at MMP-9 promoter—and mitochondrial damage and apoptosis—was assessed in the retinal endothelial cells. Role of oxidative stress in the regulation of Sirt1 was evaluated in the cells incubated in H2O2. The results were confirmed in the retina from diabetic mice with Sod2 or MMP-9 gene manipulated.
Results.
High glucose decreased Sirt1 activity and increased p65 acetylation, and resveratrol prevented increase in p65 acetylation, binding of p65 at MMP-9 promoter and MMP-9 activation, mitochondria damage, and cell apoptosis. While Sirt1 was decreased by H2O2, MMP-9 was significantly increased. Retina from wild-type diabetic mice presented similar decrease in Sirt1, and diabetic mice with Sod2 overexpression or MMP-9 deletion had normal retinal Sirt1. Retinal microvasculature from human donors with established diabetic retinopathy also had decreased Sirt1.
Conclusions.
Thus, in diabetes, increase in oxidative stress inhibits Sirt1 and p65 is hyperacetylated, increasing the binding of p65 at MMP-9 promoter. Prevention of Sirt1 inhibition, via modulating acetylation of p65, should protect activation of MMP-9 and inhibit the development of diabetic retinopathy.
Keywords: diabetic retinopathy, MMP-9, NF-kB, posttranslational modification, Sirt1
Increased oxidative stress in diabetes inhibits retinal Sirt1. Nuclear factor kappa B is hyperacetylated, and p65 binding with MMP-9 is increased. Activated MMP-9 damages mitochondria and accelerates capillary cell apoptosis. Sirtuin 1 activators should inhibit hyperacetylation of p65 and inhibit diabetic retinopathy.
Introduction
Retinopathy remains one of the most feared complications of diabetes. Clinical and experimental studies have shown a strong relationship between chronic hyperglycemia and the development of diabetic retinopathy.1,2 Many metabolic abnormalities that are triggered in hyperglycemic milieu, including polyol pathway, protein kinase C activation, and oxidative stress3,4—and a number of genes associated with these pathways5—have been implicated in the development of diabetic retinopathy, but the underlying mechanism of how hyperglycemia causes its development remains elusive.
Diabetic environment also stimulates secretion of matrix metalloproteinases (MMPs), a class of approximately 25 zinc-dependent proteinases important in degrading at least one component of the extracellular matrix, and increased levels of MMP-2 and MMP-9 are observed in the vitreous, retina, and retinal capillary cells of diabetic patients and rodents.4,6–11 Matrix metalloproteinases have a wide range of cellular functions, including apoptosis, proliferation, differentiation, and angiogenesis. Among this MMP family, MMP-9 is the largest and the most complex member.12 We have shown that diabetes activates retinal MMP-9 via Ras/Raf/MEK/ERK signaling cascade,8,10 and in the pathogenesis of diabetic retinopathy, activated MMP-9 plays an apoptotic role via damaging retinal mitochondria.9 The regulation of MMP-9 is mediated by number of transcriptional factors (e.g., AP-1, NF-κB, and SP1 are associated with its transcription).13 Our recent study has shown that epigenetic modifications in MMP-9 promoter, brought up by diabetic environment, regulate the interactions of MMP-9 with p65 subunit of NF-κB; due to hypomethylation of lysine 9 of histone 3 at MMP-9 promoter, that lysine 9 is acetylated, and this enables p65 to bind with MMP-9 promoter.11
Activation of NF-κB is regulated by its acetylation-deacetylation; while acetylated form of NF-κB is considered active, removal of acetyl group results in the loss of its transcriptional activity.14 Nicotinamide adenine dinucleotide (NAD)-dependent deacetylase sirtuin-1 (Sirt1) is considered to play an important role in the acetylation of NF-κB.15,16 In addition, in human fibrosarcoma cells, Sirt1 is also implicated in the regulation of MMP-9.17 Sirtuin 1 is present in both the nucleus and cytoplasm with dominant expression in the nucleus, and its levels are regulated by oxidative stress and intracellular redox thiol (GSH/GSSG) pool.18–20 However, how acetylation-deacetylation status by Sirt1 regulates MMP-9 in the retina in diabetes remains to be investigated.
The goal of this study is to investigate the role of Sirt1 in the activation of MMP-9 in the development of diabetic retinopathy. Using retinal endothelial cells, we investigated the effect of high glucose on Sirt1 activity. The role of Sirt1 in the regulation of MMP-9 was evaluated by using Sirt1 activator, resveratrol, on the acetylation of p65, its binding at MMP-9 promoter and mitochondria damage. The results were confirmed in retina from mice with MMP-9 gene manipulated, and the key findings were investigated in the retinal microvasculature from human donors with diabetic retinopathy.
Methods
Retinal endothelial cells were isolated from calf eyes (BRECs), and cells from the fourth to seventh passage (∼80% confluence) were incubated for days either in normal glucose (5 mM) or high glucose (20 mM glucose added to the medium). To investigate the effect of Sirt1 on MMP-9, Sirt1 activity analogue resveratrol (25 μM21; Sigma-Aldrich Corp., St. Louis, MO, USA) was added during 4 days of normal or high glucose incubation. Each experiment included osmotic control where the cells were incubated in 20 mM mannitol instead of 20 mM glucose.8,10,11
To determine the effect of increased oxidative stress on Sirt1, cells were exposed to 250 μM H2O2 for 1 hour,22,23 quickly washed with DMEM, and incubated in 5 mM glucose for 4 additional days. The choice of these conditions is based on our previous study showing activation of a small molecular weight G-protein, H-Ras under similar conditions, and H-Ras is considered to play a major role in the regulation of MMP-9 activation.10,12,22
Mice, wild-type (WT) C57BL/6 J or MMP-knockout (KO; B6.FVB [Cg]-MMP-9tm1Tvu/J) were obtained from Jackson Laboratory (Bar Harbor, ME, USA), and mice overexpressing Sod2 (Sod2-Tg) have been produced in our laboratory.9,23,24 A group of MMP-KO and WT mice, and Sod2-Tg and their WT littermates were made diabetic by streptozotocin injection (55 mg/kg BW) for 5 consecutive days. Mice with blood glucose 250 mg/dL or higher, 3 days after the last injection of streptozotocin, were considered diabetic. Age-matched normal WT and MMP-KO mice were used as controls. Seven months after induction of diabetes, mice were killed by carbon dioxide asphyxiation and the retina was isolated for analysis. The treatment of the animals conformed to the Association for Research in Vision and Ophthalmology Resolution on the Use of Animals in Research and Institutional Guidelines (Wayne State University).9,10,23,25
Human retina was enucleated within 6 to 8 hours after death from eyes obtained from the Midwest Eye Banks (Ann Arbor, MI, USA). Donors with established proliferative retinopathy (aged 54–77 years) had diabetes for 10 to 30 years. Age-matched nondiabetic donors (aged 55–77 years) served as control. A small portion of the freshly isolated retina (∼5 mm2) was incubated in distilled water for 1 hour at 37°C, and after hypotonic shock, the debris was cleaned from the vasculature under a microscope by repetitive inspiration and ejection through Pasteur pipette. Retinal microvessels from these donors have been used by us recently for other parameters, and as shown previously, the microvessels prepared by this method are generally devoid of nonvascular components.10,26
Chromatin immunoprecipitation (ChIP) was carried out using an assay kit (ChIP Assay Kit, catalog #17-295; Millipore Corp., Temecula, CA, USA). Retina and cells were cross-linked and sonicated, and the protein-DNA complex (100–120 μg) was immunoprecipitated with the antibody against p65 (Abcam, Cambridge, MA, USA), or normal rabbit IgG. Fragments of DNA were recovered by phenol:chloroform:isoamyl alcohol extraction, followed by ethanol precipitation, and resuspended in water for polymerase chain reaction (PCR). The extracted DNA for p65-MMP-9 promoter in mouse (−1547 to −1432) and in BRECs (−546 to −719) were quantified by SYBR green-based quantitative PCR (qPCR). The product size was confirmed by analyzing the samples on a 1.2% agarose gel. Normal rabbit IgG was used as negative antibody control and DNA from the input (30-μg protein–DNA complex) as an internal control.11,27,28 Each ChIP measurement was made in four to five samples/group.
Gene expression was determined by SYBR green-based qPCR using a commercial PCR system (7500 Real-Time PCR System; Applied Biosystems, Foster City, CA, USA). Chromatin immunoprecipitation–purified DNA or cDNA was amplified using the primers listed in the Table. The specific products were confirmed by SYBR green single melting curve and a single-correct-size product on a 1.2% agarose gel. Samples were measured in duplicate. Values in each cDNA were normalized to the Ct value from β-actin in the same sample, and values in each ChIP-purified DNA were normalized to the Ct value from the input sample. Relative fold changes were calculated by setting the mean fraction of normal samples (cells in 5 mM glucose or WT-N).
Table.
Primers Used
| Bovine | ||
| MMP-9 | Forward | 5′-AGACGCCACAACACTCCCA-3′ |
| Reverse | 5′-TCCTCTCCCTGCTCCACCTG-3′ | |
| MMP-9 promoter | Forward | 5′-CAGACGCCACAACACTCCCA-3′ |
| Reverse | 5′-TCCTCTCCCTGCTCCACCTG-3′ | |
| Sirt1 | Forward | 5′-ACACGAATGGAAACCGTTGG-3′ |
| Reverse | 5′-GGCTTACAGGGCCTATCCAG-3′ | |
| p65 | Forward | 5′-CAATGGCTCGACAGTAGCG-3′ |
| Reverse | 5′-GCGTACGAAGGGTCAGAAGG-3′ | |
| Cytb | Forward | 5′-CGATACATACACGCAAACGG-3′ |
| Reverse | 5′-AGAATCGGGTAAGGGTTGCT-3′ | |
| β-actin | Forward | 5′-CCTCTATGCCAACACAGTGC-3′ |
| Reverse | 5′-CATCGTACTCCTGCTTGCTG-3′ | |
| Mouse | ||
| MMP-9 | Forward | 5′-GGGGTTTGCCCCATGGAAT-3′ |
| Reverse | 5′-GAGCCCATCCCCACACTGTA-3 | |
| MMP-9 promoter | Forward | 5′-GCCTGCTGGAGCTAGGGGTTTG-3′ |
| Reverse | 5′-GAGTGCAGCCTGGAGCCCATC-3′ | |
| Sirt1 | Forward | 5′-TTGGCACCGATCCTCGAA-3′ |
| Reverse | 5′-CCCAGCTCCAGTCAGAACTAT-3′ | |
| p65 | Forward | 5′-GCGTACACATTCTGGGGAGT-3′ |
| Reverse | 5′-ACCGAAGCAGGAGCTATCAA-3′ | |
| β-actin | Forward | 5′-CCTCTATGCCAACACAGTGC-3′ |
| Reverse | 5′-CATCGTACTCCTGCTTGCTG-3′ | |
| Human | ||
| MMP-9 | Forward | 5′-CACTGTCCACCCCTCAGAGC-3′ |
| Reverse | 5′-GCCACTTGTCGGCGATAAGG-3′ | |
| Sirt1 | Forward | 5′-TGCCGGAAACAATACCTCCACCTG-3′ |
| Reverse | 5′-ACAGACACCCCAGCTCCAGTT-3′ | |
| β-actin | Forward | 5′-AGCCTCGCCTTTGCCGATCCG-3′ |
| Reverse | 5′-TCTCTTGCTCTGGGCCTCGTCG-3′ |
Localization of Sirt1 in the nucleus was performed immunohistochemically. The cells grown on coverslips in 5 or 20 mM glucose for 4 days were fixed with cold methanol for 15 minutes at −20°C, and blocked in 1% BSA for 1 hour. The cells were then incubated with anti-mouse-Sirt1 overnight, and rinsed with PBS. This was followed by incubation with anti-mouse-Texas red (red; Vector Laboratories, Inc., Burlingame, CA, USA) for 1 hour, washing with PBS, and mounting with DAPI containing mounting medium (blue). The slides were imaged with commercial microscopy (Zeiss ApoTome; Carl Zeiss AG, Jena, Germany) using ×40 magnification.9,10,23,29
Sirt1 deacetylase activity was assessed in the nuclear fraction29 using a commercial fluorescence kit (Cayman Chemical Company, Ann Arbor, MI, USA). We incubated 30 to 40 μg of protein with the substrate (coupled to the fluorophore and quencher) and NAD+, and the fluorescence emitted due to deacetylation of the substrate by Sirt1 was measured at 345-nm excitation and 450-nm emission wavelengths.
Matrix metalloproteinase 9 activity was quantified in the homogenate (30–40 μg protein) by fluorescence kit using a specific anti-MMP-9 monoclonal antibody and a fluorogenic substrate (SensoLyte Plus 520 MMP-9 Assay Kit; AnaSpec, Inc., Fremont, CA, USA). The MMP-9 induced cleavage of the fluorogenic peptide was measured at 490-nm excitation and 520-nm emission wavelengths.9,11,30
Acetylation of p65 was determined by coimmunoprecipitation technique.10,31 Protein (120–150 μg) was incubated overnight at 4°C with acetylated lysine antibody (Cell Signaling Technology, Inc., Beverly, MA, USA), followed by 1 hour with 20 μL agarose immunoprecipitation beads reagent (Protein A and G Plus, pre-washed and suspended in lysis buffer; Cell Signaling Technology, Inc.). The beads were washed four times with lysis buffer, and the proteins were separated by SDS-PAGE. The membranes were immunoblotted with p65 antibodies.
Reactive oxygen species (ROS) were quantified by the 2′,7′-dichlorofluorescein diacetate (DCHFDA; Sigma-Aldrich Corp.) method.26,30 Briefly, 5 to 10 μg of protein was incubated in PBS with 2 μM DCHFDA for 10 minutes and fluorescence was measured at 485-nm excitation wavelength and 530-nm emission wavelength.
Mitochondrial damage was evaluated by quantifying the gene transcripts of mtDNA-encoded cytochrome b (Cytb).23,30
Statistics analysis of the data was performed using statistical software (SigmaStat; Systat Software, Inc., San Jose, CA, USA). The results are represented as means ± standard deviation, and P < 0.05 was considered statistically significant. The Shapiro-Wilk test was performed to test for normal distribution, and the results with normal distribution were analyzed by either t-test (two groups) or ANOVA followed by Bonferroni test for variables (>2 groups). The data that did not present normal distribution was analyzed by Mann-Whitney U test (two groups) or by Kruskal-Wallis test followed by Dunn's (>2 groups).
Results
Retinal Endothelial Cells
Sirtuin 1 regulates transactivation potential of the transcription factor by removing acetyl groups from the acetylated lysine residues. Deacetylation of the acetylated lysine in p65 is reported to inhibit the transactivation potential of NF-κB14,15 and in diabetes, the binding of p65 with MMP-9 promoter regulates retinal MMP-9.11 Figure 1a shows that Sirt1 expression (gene and protein) was decreased by ∼50% in the retinal endothelial cells exposed to high glucose. This was accompanied by significant decrease in its deacetylase activity (Fig. 1b), and a decrease in nuclear accumulation (Fig. 1c), compared with the cells incubated in normal glucose. High glucose also increased acetylation of p65 by over 50% compared with the values obtained from cells in normal glucose (Fig. 2a). Consistent with our previous results10 in the same samples, the binding of p65 at MMP-9 promoter, and the activity of MMP-9, were also significantly increased in high glucose medium (Figs. 2b–d).
Figure 1.

Sirtuin 1 and high glucose exposure of retinal endothelial cells. (a) In retinal endothelial cells incubated in normal (5 mM) or high (20 mM) glucose for 4 days, in the presence or absence of 25 μM resveratrol (RV), gene transcript and protein expression of Sirt1 were determined by qPCR and Western blot technique, respectively. We used β-actin as a housekeeping gene or loading protein. (b) Sirtuin 1 deacetylase activity was measured in the nuclear fraction by using a fluorescence kit from Cayman Chemical. (c) The cells grown on coverslips in normal or high glucose for 4 days were used to immunohistochemically localize Sirt1 in the nucleus. Anti-mouse secondary antibody was conjugated with Texas red, and the mounting reagent contained DAPI blue. The images are representative of three or more different experiments. Data are presented as mean ± SD from three to four preparations in each group with the values obtained from cells incubated in 5 mM glucose adjusted to 1 or 100%. *P < 0.05 compared with the values obtained from the cells incubated in 5 mM glucose. Five mM and 20 mM cells in 5 or 20 mM glucose, respectively; 20 + RV, cells incubated in 20 mM glucose in the presence of resveratrol; Mann, cells in 20 mM mannitol instead of 20 mM glucose.
Figure 2.
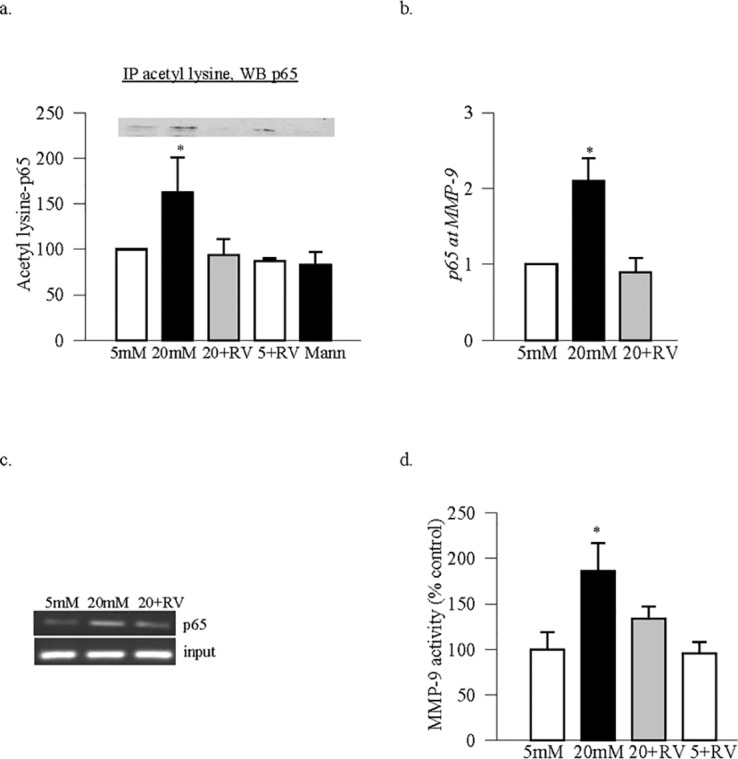
Acetylation of p65 and its binding with MMP-9 promoter: Retinal endothelial cells incubated in the presence or absence of resveratrol were analyzed for (a) acetylation of p65 by coimmunoprecipitation technique. In the acetyl lysine immunoprecipitate, p65 was Western blotted. (b) The binding of p65 at MMP-9 was determined by ChIP technique. The precipitated DNA was amplified for MMP-9 promoter region (−546 to −719) by SYBR green-based qPCR, and (c) the product size was confirmed on a 1.2% agarose gel. (d) Activity of MMP-9 was determined in 30 to 40 μg protein of cell homogenate using a fluorometric assay. *P < 0.05 compared with the values obtained from normal rats. Five + RV and 20 + RV = cells incubated in 5 or 20 mM glucose, respectively in the presence of resveratrol.
Supplementation of high glucose with Sirt1 activator resveratrol, as expected, ameliorated inhibition of Sirt1 activity (Fig. 1b), and prevented increase in the acetylation of p65, binding of p65 with MMP-9 promoter and activation of MMP-9 (Figs. 2a–d). Inclusion of resveratrol in normal glucose medium had no significant effect on p65 acetylation and MMP-9 activity (Figs. 2a–d).
In the pathogenesis of diabetic retinopathy, activation of MMP-9 is implicated in mitochondria damage.9,11 To confirm the role of Sirt1 in the regulation of MMP-9 mediated mitochondrial damage, results in Figure 3 show that resveratrol also attenuated increase in ROS levels, and ameliorated glucose-induced decrease in the transcripts of mtDNA-encoded Cytb.
Figure 3.
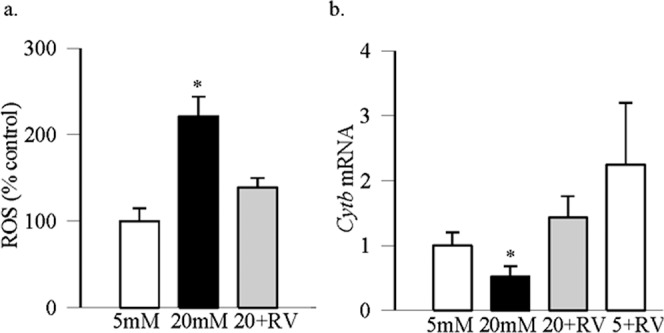
Effect of resveratrol on oxidative stress: (a) Total ROS levels were quantified using 2′,7′-dichlorofluorescein diacetate, and fluorescence was measured at 485-nm excitation wavelength and 530-nm emission wavelength. (b) The transcripts of mtDNA-encoded Cytb were quantified by real time qPCR using β-actin as a housekeeping gene. Values are represented as mean ± SD obtained from three to four cells preparations, and each measurement is made at least in duplicate. *P < 0.05 compared with 5 mM glucose.
Sirtuin 1 is a redox-sensitive deacetylase,18–20 and oxidative stress is increased in the retina in diabetes.25,32 To understand the role of oxidative stress in the regulation of Sirt1, cells treated with H2O2 were analyzed. Consistent with the results from others demonstrating inhibition of Sirt1 activity by H2O2 in human bronchial epithelial cells,33 Figure 4a shows that exposure of retinal endothelial cells to H2O2 also significantly decreased its deacetylase activity. Also, H2O2 increased MMP-9 gene transcripts and enzyme activity by ∼2-fold compared with the cells incubated in normal glucose (Fig. 4b).
Figure 4.
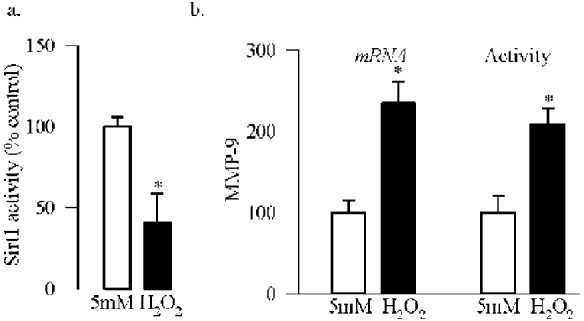
Oxidative stress and inhibition of Sirt1. Cells incubated with 250 μM H2O2 for 1 hour, followed by incubation in normal glucose for 4 additional days were analyzed for (a) deacetylase activity of Sirt1, and (b) MMP-9 mRNA by qPCR and enzyme activity using a fluorogenic substrate, 5-FAM/QXL520 FRET peptide. Each experiment was repeated with three to five different cell preparations. 5 mM, 5 mM glucose; H2O2, cells incubated with H2O2. *P < 0.05 compared with 5 mM glucose.
Mouse Retina.
As with the retinal endothelial cells, deacetylase activity of Sirt1 was also decreased by over 40% in the retina from WT mice diabetic for >7 months compared with the values obtained from age-matched WT-normal mice (Fig. 5a). Consistent with this, acetylation of p65 and the binding of p65 at retinal MMP-9 promoter were also significantly increased in these diabetic mice (Figs. 5b, 5c).
Figure 5.
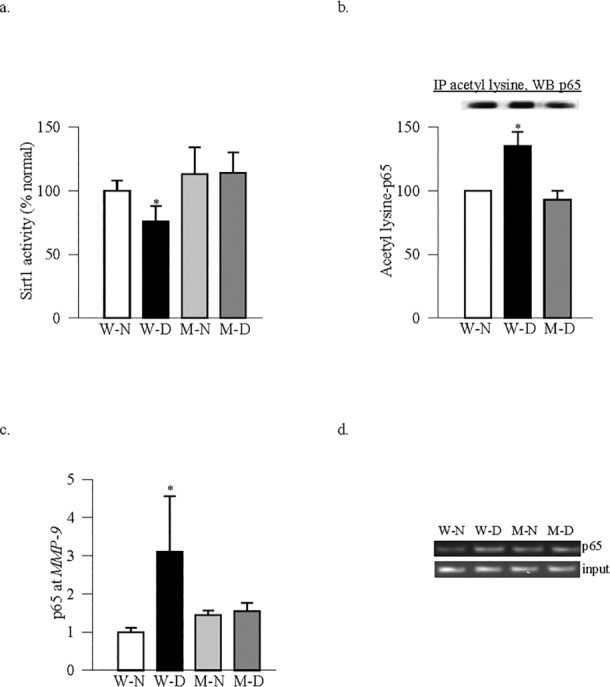
Genetic regulation of MMP-9 and Sirt1. Retina from MMP-9–KO and WT mice, diabetic for 7 months, and their age-matched normal control mice were analyzed for (a) deacetylase activity of Sirt1, (b) acetylation of p65 by coimmunoprecipitation, and (c) p65 binding at MMP-9 promoter by ChIP technique. (d) The product size was confirmed on an agarose gel. Measurements were made in five to seven animals in each group, and the results are represented as mean ± SD. W-N and M-N, wild-type and MMP-KO normal mice; W-D and M-D, wild-type and MMP-KO diabetic mice. *P < 0.05 compared with W-N or M-N.
Abrogation of MMP-9 gene in mice, in addition to inhibiting diabetes-induced activation of retinal MMP-9, also prevents mitochondrial damage and the development of retinopathy.8 Figure 5a shows that deletion of MMP-9 also prevents diabetes-induced decrease in Sirt1 activity. The values obtained from MMP-9–KO diabetic or nondiabetic mice were similar, and these values were not different from those obtained from WT-normal mice. Retina from the same diabetic MMP-9–KO mice had decreased acetylation of p65 and reduced binding of p65 at MMP-9 promoter compared with the values obtained from WT-diabetic mice (Figs. 5b, 5c).
Regulation of oxidative stress by overexpression of Sod2 in mice prevented diabetes-induced decrease in Sirt1 and increase in p65 (Fig. 6a), and the values obtained from diabetic Sod2-Tg mice were not different from those obtained from nondiabetic WT or Sod2-Tg mice. In the same diabetic animals, an increase in MMP-9 gene expression and activity were also protected by overexpression of Sod2 (Fig. 6b).
Figure 6.
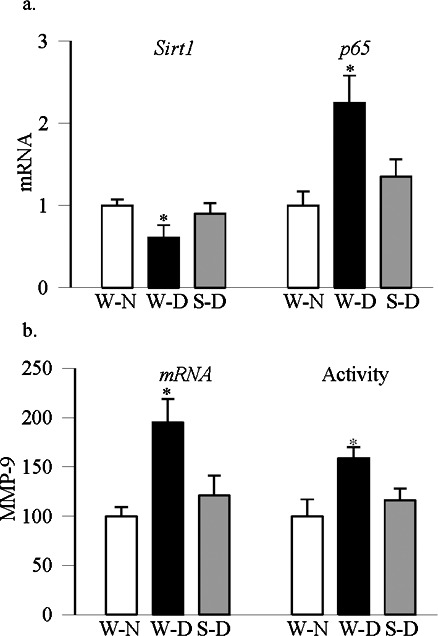
Diabetic mice overexpressing Sod2 are protected from decrease in retinal Sirt1 and increase in MMP-9. Gene transcripts of (a) Sirt1 and p65 were quantified in the retina by SYBR green-based qPCR using a sequence detection system (Applied Biosystems). The data were normalized to β-actin expression. (b) MMP-9 mRNA was quantified by qPCR, and its activity using an assay kit (AnaSpec, Inc.). Results are from six to eight mice in each group and are represented as mean ± SD. W-N, wild-type normal mice; W-D and S-D, wild-type and Sod2-Tg diabetic mice. *P < 0.05 compared with W-N.
Human Retinal Microvessels.
In order to further confirm the role of Sirt1 in the regulation of MMP-9 in diabetic retinopathy, Sirt1 was quantified in the retinal microvessels, the site of histopathology, from donors with established diabetic retinopathy. Retinal microvessels from donors with diabetic retinopathy had ∼50% decreased Sirt1 gene expression and activity compared with their age-matched nondiabetic donors (Fig. 7a). In agreement with the decrease in Sirt1, the same microvessel preparations had over 70% higher MMP-9 mRNA levels and enzyme activity (Fig. 7b).
Figure 7.
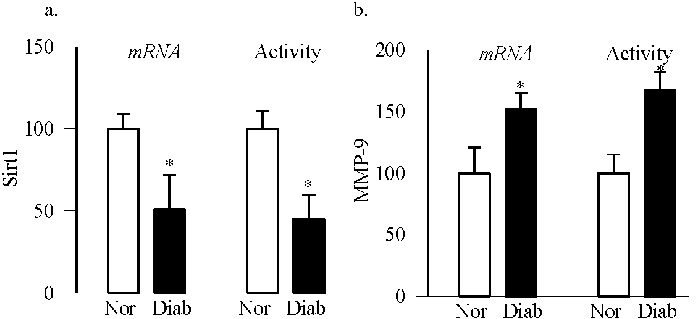
Sirtuin 1 in the retinal microvasculature from human donors with diabetic retinopathy: Microvessels from retina of diabetic subjects with documented retinopathy (Diab) and age-matched nondiabetic subjects (Norm) were analyzed for (a) Sirt1 gene expression and deacetylase activity, and (b) MMP-9 gene expression and its activity (using a specific anti-MMP-9 monoclonal antibody and MMP fluorogenic substrate). Each measurement was performed in duplicate in the retinal microvessels from four to five nondiabetic and diabetic donors. *P < 0.05 compared with nondiabetic donors.
Discussion
Diabetes activates MMP-9 in the retina and its capillary cells, and the enzyme continues to be activated at duration of diabetes when capillary cell apoptosis and histopathology of diabetic retinopathy can be observed. Activated MMP-9 damages the mitochondria accelerating the apoptosis of capillary cells. Diabetic MMP-9–KO mice have normal retinal mitochondria homeostasis, and these mice are protected from the development of retinopathy.4,8–11,30 Transcription of MMP-9 is controlled by transcriptional factors including AP-1, NF-κB and SP1.10,13,34 Our recent study has shown that epigenetic modifications at MMP-9 promoter play an important role in the increased recruitment of NF-κB.11 Activation of NF-κB is closely related with MMP-9 activation,13,35 and while acetylated form of NF-κB is considered active, removal of acetyl group results in the loss of its transcriptional activity.15 Here, we show that Sirt1, a deacetylase, is inhibited in the retina and its capillary cells in hyperglycemic milieu, and p65 is hyperacetylated with increased binding of p65 with MMP-9. Activation of Sirt1 by resveratrol, in addition to inhibiting glucose-induced increase in acetylation of p65, also inhibits MMP-9, and protects the mitochondria damage. Oxidative stress appears to mediate Sirt1 activation, and mice protected from diabetes-induced increase in retinal superoxide radicals, are also protected from decrease in deacetylase activity of Sirt1. Together, these results strongly suggest that increased oxidative stress, induced by diabetes, inhibits retinal Sirt1, and Sirt1, via modulating acetylation status of p65, regulates the activation of MMP-9.
Sirtuin 1, mainly a nuclear protein, removes acetyl groups from proteins by transferring the acetyl group to NAD+. By deacetylating histones and nonhistone proteins and transcription factors, Sirt1 regulates various metabolic pathways, including inflammation and apoptosis.15,36 The activity of this enzyme depends on the availability of cellular NAD+, which can be controlled by various factors including cellular redox status and NAD+ synthesis.37,38 Sirtuin 1 regulates transcription activity of NF-κB,14–16,39 and also regulates mitochondria homeostasis.40 Sirtuin 1 is localized in various ocular tissues, including cornea, lens, and retina. Also, decreased levels of Sirt1 are observed in chronic ocular diseases, such as cataract, retinal degeneration, and optic neuritis.41,42 Here, we show that in diabetic retinopathy, another chronic ocular disease, which is associated with dysfunctional mitochondria and impaired mitochondria biogenesis,23,43 Sirt1 expression and its deacetylase activity are also decreased in the retina and its capillary cells.
Activated retinal MMP-9 in diabetes damages the mitochondria, which initiates the apoptotic machinery, and increased apoptosis of retinal capillary cells is implicated in the histopathology, including increased degenerative capillaries and pericyte loss.9,44,45 Transcription of MMP-9 is regulated by NF-κB, and in diabetes, epigenetic modifications of MMP-9 facilitate the binding of p65 and activate MMP-9. Furthermore, diabetes also activates the transcriptional activity of retinal NF-κB, and activated NF-κB acts as a proapoptotic factor.46,47 Results presented here show that the acetylation of p65 subunit of NF-κB is increased in the retina in diabetes, and resveratrol ameliorates acetylation of p65, confirming the role of Sirt1 in p65 acetylation in diabetic retinopathy. In support, recent studies have suggested a functional interrelationship between Sirt1 and NF-κB, where deacetylation of p65 by Sirt1 inhibits the transactivation potential of NF-κB.39,48,49 The role of Sirt1 in the activation of retinal MMP-9 in diabetes is further confirmed by the beneficial effect of resveratrol on glucose-induced increased p65 binding at MMP-9 promoter, and inhibition of MMP-9 activation. In accordance with our results, resveratrol is shown to inhibit MMP-9 in fibrosarcoma cells, and the mechanism appears to be via its regulation of Sirt1.17
Increase in retinal MMP-9 in diabetes damages the mitochondria and increases oxidative stress.9 Others have shown that the regulation of Sirt1 in retinal cells increases their sensitivity to hyperglycemic stress, and regulates mitochondrial ROS.50 Overexpression of Sirt1 is shown to prevent increase in oxidative stress markers.51 Here, we show that regulation of Sirt1 by resveratrol inhibits glucose-induced increase in ROS and also prevents mitochondria damage, as evidenced by significantly higher expression of mtDNA-encoded Cytb in glucose-treated cells incubated with resveratrol compared with the cells without resveratrol. Consistent with our results, Sirt1 has been shown to regulate mitochondria homeostasis.40
Our results show that incubation of retinal endothelial cells with H2O2 inhibits the deacetylase activity of Sirt1 by over 50%, and this is accompanied by increased MMP-9 transcripts and activity. Sirtuin 1 is a redox-sensitive enzyme that is regulated by oxidative stress and intracellular redox thiol (GSH/GSSG) pool, and increased oxidative stress downregulates the enzyme.18–20,33 Exposure of H2O2 exposure of human bronchial epithelial cells has been shown to inhibit Sirt1 activity without altering its protein expression, suggesting that posttranslational modifications of Sirt1 could be playing a role in its decreased enzyme activity.33 In the pathogenesis of diabetic retinopathy, oxidative stress is increased in the retina, GSH levels are decreased26,32,52,53 and proteins are posttranslationally modified,11,28,31 and the role of posttranslational modifications in inhibiting retinal Sirt1 activity in diabetes cannot be ruled out. Oxidative stress–mediated Sirt1 inhibition in the regulation NF-κB–MMP-9 is further confirmed by our in vivo model; retina from diabetic mice in which oxidative stress is regulated by overexpression of Sod2 is protected from decrease in Sirt1 and increase in p65. The retina of these Sod2-Tg mice also has normal MMP-9 activity, and we have shown previously that these mice are also protected from mitochondria damage and the development of diabetic retinopathy.23,54,55 Thus, our in vitro and in vivo results clearly show that the conditions that favor oxidative stress also inhibit Sirt1 activity and activate MMP-9.
The results presented here showing a decrease in Sirt1 in diabetes in the retina and its capillary cells are further supported by decreased Sirt1 and increased MMP-9 in the retinal microvasculature of human donors with established diabetic retinopathy compared with age-matched nondiabetic human donors. Consistent with these, signaling mechanism for activation of MMP-9 and the members of NADPH oxidase-2 subunits are increased in these microvessels,10,26 further strengthening the role of Sirt1 in the regulation of MMP-9 in the development of diabetic retinopathy.
In conclusion, our study clearly demonstrates that in diabetes, increase in oxidative stress inhibits Sirt1 resulting in hyperacetylated p65, and the binding of p65 with MMP-9 promoter is increased. Prevention of Sirt1 inhibition, via modulating acetylation of p65, has potential to protect activation of MMP-9 and mitochondria damage. Thus, the regulation of Sirt1 by pharmacological means could serve as a potential target to prevent/delay the development of diabetic retinopathy. Optimistically, efforts are being put into testing activators of Sirt1 for the treatment of other chronic diseases, and our results provide strong background for their use to prevent the development of diabetic retinopathy.
Acknowledgments
The authors thank Doug Putt and Mangayarkarasi ThandampallayamAjjeya for technical help.
Supported in part by grants from the National Institutes of Health, the Juvenile Diabetes Research Foundation, the Thomas Foundation, Research to Prevent Blindness, and Midwest Eye Banks.
Disclosure: R.A. Kowluru, None; J.M. Santos, None; Q. Zhong, None
References
- 1. Engerman RL, Kern TS. Hyperglycemia as a cause of diabetic retinopathy. Metabolism. 1986; 35: 20–23 [DOI] [PubMed] [Google Scholar]
- 2. Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993; 329: 977–986 [DOI] [PubMed] [Google Scholar]
- 3. Frank RN. Diabetic retinopathy. N Engl J Med. 2004; 350: 48–58 [DOI] [PubMed] [Google Scholar]
- 4. Kowluru RA, Zhong Q, Santos JM. Matrix metalloproteinases in diabetic retinopathy: Potential role of MMP-9. Expert Opin Investig Drugs. 2012; 21: 797–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Petrovic D. Candidate genes for proliferative diabetic retinopathy. Biomed Res Int. 2013; 2013: 540416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Giebel SJ, Menicucci G, McGuire PG, Das A. Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood-retinal barrier. Lab Invest. 2005; 85: 567–607 [DOI] [PubMed] [Google Scholar]
- 7. Grant MB, Caballero S, Tarnuzzer RW, et al. Matrix metalloproteinase expression in human retinal microvascular cells. Diabetes. 1998; 47: 1311–1317 [DOI] [PubMed] [Google Scholar]
- 8. Kowluru RA. Role of matrix metalloproteinase-9 in the development of diabetic retinopathy and its regulation by H-Ras. Invest Ophthalmol Vis Sci. 2010; 51: 4320–4326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kowluru RA, Mohammad G, Dos Santos JM, Zhong Q. Abrogation of MMP9 gene protects against the development of retinopathy in diabetic mice by preventing mitochondrial damage. Diabetes. 2011; 60: 3023–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mohammad G, Kowluru RA. Diabetic retinopathy and signaling mechanism for activation of matrix metalloproteinase-9. J Cell Physiol. 2012; 227: 1052–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhong Q, Kowluru RA. Regulation of matrix metalloproteinase-9 by epigenetic modifications and the development of diabetic retinopathy. Diabetes. 2013; 62: 2559–2568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tang CH, Tsai CC. Ccl2 increases MMP-9 expression and cell motility in human chondrosarcoma cells via the Ras/Raf/MEK/ERK/NF-κB signaling pathway. Biochem Pharmacol. 2012; 83: 335–344 [DOI] [PubMed] [Google Scholar]
- 13. Robert I, Aussems M, Keutgens A, et al. Metalloproteinase-9 gene induction by a truncated oncogenic NF-Kappab2 protein involves the recruitment of Mll1 and Mll2 H3K4 histone methyltransferase complexes. Oncogene. 2009; 28: 1626–1628 [DOI] [PubMed] [Google Scholar]
- 14. Liu P, Wilson MJ. miR-520c and miR-373 upregulate MMP9 expression by targeting mTOR and SIRT1, and activate the Ras/Raf/MEK/Erk signaling pathway and NF-kappaB factor in human fibrosarcoma cells. J Cell Physiol. 2012; 227: 867–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jung YJ, Lee JE, Lee AS, et al. Sirt1 overexpression decreases cisplatin-induced acetylation of NF-Kappab p65 subunit and cytotoxicity in renal proximal tubule cells. Biochem Biophys Res Commun. 2012; 419: 206–210 [DOI] [PubMed] [Google Scholar]
- 16. Pavlova S, Klucska K, Vasicek D, et al. The involvement of Sirt1 and transcription factor NF-Kappab (p50/p65) in regulation of porcine ovarian cell function. Anim Reprod Sci. 2013; 140: 180–188 [DOI] [PubMed] [Google Scholar]
- 17. Lee SJ, Kim MM. Resveratrol with antioxidant activity inhibits matrix metalloproteinase via modulation of Sirt1 in human fibrosarcoma cells. Life Sci. 2011; 88: 465–472 [DOI] [PubMed] [Google Scholar]
- 18. Sundar IK, Caito S, Yao H, Rahman I. Oxidative stress, thiol redox signaling methods in epigenetics. Methods Enzymol. 2010; 474: 213–244 [DOI] [PubMed] [Google Scholar]
- 19. Zee RS, Yoo CB, Pimentel DR, et al. Redox regulation of sirtuin-1 by s-glutathiolation. Antioxid Redox Signal. 2010; 13: 1023–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hwang JW, Yao H, Caito S, Sundar IK, Rahman I. Redox regulation of Sirt1 in inflammation and cellular senescence. Free Radic Biol Med. 2013; 61C: 95–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen F, Qian LH, Deng B, Liu ZM, Zhao Y, Le YY. Resveratrol protects vascular endothelial cells from high glucose-induced apoptosis through inhibition of NADPH oxidase activation-driven oxidative stress. CNS Neurosci Ther. 2013; 19: 675–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kowluru V, Kowluru RA. Increased oxidative stress in diabetes regulates activation of a small molecular weight g-protein, H-Ras, in the retina. Mol Vis. 2007; 13: 602–610 [PMC free article] [PubMed] [Google Scholar]
- 23. Santos JM, Tewari S, Goldberg AFX, Kowluru RA. Mitochondria biogenesis and the development of diabetic retinopathy. Free Rad Biol Med. 2011; 51: 1849–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kowluru RA, Kowluru V, Ho YS, Xiong Y. Overexpression of mitochondrial superoxide dismutase in mice protects the retina from diabetes-induced oxidative stress. Free Rad Biol Med. 2006; 41: 1191–1196 [DOI] [PubMed] [Google Scholar]
- 25. Madsen-Bouterse SA, Mohammad G, Kanwar M, Kowluru RA. Role of mitochondrial DNA damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxid Redox Signal. 2010; 13: 797–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kowluru RA, Kowluru A, Veluthakal R, et al. Tiam1-Rac1 signaling axis mediated activation of NADPH oxidase-2 initiates mitochondrial damage in the development of diabetic retinopathy. Diabetologia. 2014; 57: 1047–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhong Q, Kowluru RA. Epigenetic changes in mitochondrial superoxide dismutase in the retina and the development of diabetic retinopathy. Diabetes. 2011; 60: 1304–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhong Q, Kowluru RA. Epigenetic modification of Sod2 in the development of diabetic retinopathy and in the metabolic memory: role of histone methylation. Invest Ophthalmol Vis Sci. 2013; 54: 244–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhong Q, Mishra M, Kowluru RA. Transcription factor Nrf2-mediated antioxidant defense system in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2013; 54: 3941–3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Santos JM, Tewari S, Lin JY, Kowluru RA. Interrelationship between activation of matrix metalloproteinases and mitochondrial dysfunction in the development of diabetic retinopathy. Biochem Biophys Res Commun. 2013; 438: 760–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Santos JM, Mishra M, Kowluru RA. Posttranslational modification of mitochondrial transcription factor A in impaired mitochondria biogenesis: implications in diabetic retinopathy and metabolic memory phenomenon. Exp Eye Res. 2014; 121: 168–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kowluru RA, Tang J, Kern TS. Abnormalities of retinal metabolism in diabetes and experimental galactosemia. VII. Effect of long-term administration of antioxidants on the development of retinopathy. Diabetes. 2001; 50: 1938–1942 [DOI] [PubMed] [Google Scholar]
- 33. Caito S, Rajendrasozhan S, Cook S, et al. Sirt1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J. 2010; 24: 3145–3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sato H, Seiki M. Regulatory mechanism of 92 Kda type IV collagenase gene expression which is associated with invasiveness of tumor cells. Oncogene. 1993; 8: 395–405 [PubMed] [Google Scholar]
- 35. Xu B, Chen H, Xu W, et al. Molecular mechanisms of MMP9 overexpression and its role in emphysema pathogenesis of SMAD3-deficient mice. Am J Physiol Lung Cell Mol Physiol. 2012; 303: L89–L96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brooks CL, Gu W. How does sirt1 affect metabolism, senescence and cancer? Nat Rev Cancer. 2009; 9: 123–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chung S, Yao H, Caito S, Hwang JW, Arunachalam G, Rahman I. Regulation of Sirt1 in cellular functions: role of polyphenols. Arch Biochem Biophys. 2010; 501: 79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yu J, Auwerx J. Protein deacetylation by sirt1: An emerging key post-translational modification in metabolic regulation. Pharmacol Res. 2010; 62: 35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-Kappab and Sirt1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013; 25: 1939–1948 [DOI] [PubMed] [Google Scholar]
- 40. Menzies KJ, Hood DA. The role of Sirt1 in muscle mitochondrial turnover. Mitochondrion. 2012; 12: 5–13 [DOI] [PubMed] [Google Scholar]
- 41. Jaliffa C, Ameqrane I, Dansault A, et al. Sirt1 involvement in rd10 mouse retinal degeneration. Invest Ophthalmol Vis Sci. 2009; 50: 3562–3572 [DOI] [PubMed] [Google Scholar]
- 42. Mimura T, Kaji Y, Noma H, Funatsu H, Okamoto S. The role of Sirt1 in ocular aging. Exp Eye Res. 2013; 116: 17–26 [DOI] [PubMed] [Google Scholar]
- 43. Kowluru RA, Abbas SN. Diabetes-induced mitochondrial dysfunction in the retina. Invest Ophthalmol Vis Sci. 2003; 44: 5327–5334 [DOI] [PubMed] [Google Scholar]
- 44. Mizutani M, Kern TS, Lorenzi M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest. 1996; 97: 2883–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kern TS, Tang J, Mizutani M, Kowluru R, Nagraj R, Lorenzi M. Response of capillary cell death to aminoguanidine predicts the development of retinopathy: comparison of diabetes and galactosemia. Invest Ophthalmol Vis Sci. 2000; 41: 3972–3978 [PubMed] [Google Scholar]
- 46. Romeo G, Liu WH, Asnaghi V, Kern TS, Lorenzi M. Activation of nuclear factor-kappab induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes. 2002; 51: 2241–2248 [DOI] [PubMed] [Google Scholar]
- 47. Kowluru RA, Koppolu P, Chakrabarti S, Chen S. Diabetes-induced activation of nuclear transcriptional factor in the retina, and its inhibition by antioxidants. Free Radic Research. 2003; 37: 1169–1180 [DOI] [PubMed] [Google Scholar]
- 48. Yeung F, Hoberg JE, Ramsey CS, et al. Modulation of NF-Kappab-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004; 23: 2369–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang XD, Tajkhorshid E, Chen LF. Functional interplay between acetylation and methylation of the RelA subunit of NF-kappaB. Mol Cell Biol. 2010; 30: 2170–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zheng Z, Chen H, Li J, et al. Sirtuin 1-mediated cellular metabolic memory of high glucose via the Ikb1/AMPK/ROS pathway and therapeutic effects of metformin. Diabetes. 2012; 61: 217–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen H, Wan Y, Zhou S, et al. Endothelium-specific sirt1 overexpression inhibits hyperglycemia-induced upregulation of vascular cell senescence. Sci China Life Sci. 2012; 55: 467–473 [DOI] [PubMed] [Google Scholar]
- 52. Kern TS, Kowluru R, Engerman RL. Abnormalities of retinal metabolism in diabetes or galactosemia. ATPases and glutathione. Invest Ophthalmol Vis Sci. 1994; 35: 2962–2967 [PubMed] [Google Scholar]
- 53. Kanwar M, Kowluru RA. Role of glyceraldehyde 3-phosphate dehydrogenase in the development and progression of diabetic retinopathy. Diabetes. 2009; 58: 227–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kanwar M, Chan PS, Kern TS, Kowluru RA. Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Invest Ophthalmol Vis Sci. 2007; 48: 3805–3811 [DOI] [PubMed] [Google Scholar]
- 55. Tewari S, Santos JM, Kowluru RA. Damaged mitochondrial DNA replication system and the development of diabetic retinopathy. Antioxid Redox Signal. 2012; 17: 492–504 [DOI] [PMC free article] [PubMed] [Google Scholar]