

Abstract
The level of the p53 transcription factor is negatively regulated by the E3 ubiguitin ligase murine double minute clone 2 (MDM2). The interaction between p53 and MDM2 is essential for the maintenance of genomic integrity for most eukaryotes. Previous structural studies revealed that MDM2 binds to p53 transactivation domain (p53TAD) from residues 17 to 29. The K24N mutation of p53TAD changes a lysine at position 24 to an asparagine. This mutation occurs naturally in the bovine family and is also found in a rare form of human gestational cancer called choriocarcinoma. In this study we have investigated how the K24N mutation affects the affinity, structure, and dynamics of p53TAD binding to MDM2. Nuclear magnetic resonance studies of p53TAD show the K24N mutant is more flexible and has less transient helical secondary structure than the wildtype. Isothermal titration calorimetry measurements demonstrate that these changes in structure and dynamics do not significantly change the binding affinity for p53TAD-MDM2. Finally, free energy perturbation and standard molecular dynamics simulations suggest the negligible affinity change is due to a compensating interaction energy between the K24N mutant and MDM2 when it is bound. Overall, the data suggests that the K24N-MDM2 complex is able to at least partly compensate for an increase in the conformational entropy in unbound K24N with an increase in the bound state electrostatic interaction energy.
Keywords: intrinsically disordered protein, binding mechanism, nuclear magnetic resonance spectroscopy, isothermal titration calorimetry, molecular dynamics
INTRODUCTION
The p53 protein is a transcription factor that regulates the cell cycle.1 P53 is essential for the maintenance of genomic integrity. It is constitutively expressed but in the absence of cellular stress the activity of p53 is suppressed and it is actively targeted for degradation by the proteosome.2 It is stabilized and activated in response to a variety of cellular stresses, such as UV light, osmotic shock and hypoxia, leading to cell cycle arrest and the subsequent transcription of target genes to revive the cell.3 If the damage is irreparable, p53 will initiate apoptotic pathways. If p53 fails to respond to cellular stresses the damaged DNA will not be repaired. This can lead to genome instability, uncontrolled proliferation, and tumorigenesis. P53 is a gatekeeper of the cell cycle and a tumor suppressor and mutated forms of p53 are found in 50% of solid tumors.4
The level of p53 is largely regulated by the E3 ubiquitin ligase, murine double minute clone 2 (MDM2). 3 Normally, MDM2 binds and ubiquinates p53 to trigger the degradation process. 3 However, some p53 proteins escape, thereby enabling the transcription of the MDM2 gene, which maintains the feedback loop. 3 Therefore, the levels of p53 are kept low by the interaction with MDM2 in non-stressed cells. This low level of p53 is able to ensure a rapid response to stresses. Under stressed conditions, the binding between p53 and MDM2 is abrogated by phosphorylation, leading to the activation of p53.
Structural and biochemical studies of the p53-MDM2 complex have identified the regions that are necessary for binding. An ordered N-terminal domain of MDM2 that is comprised of residues 1 to 109 binds to a short, disordered segment of the p53 transactivation domain (p53TAD). 5,6 Residues 15–30 make up the short, disordered segment of p53TAD that binds to MDM2.5–9 This binding site is found within a larger region of disorder that extends from residues 1 to 90.10 The disordered MDM2 binding region of p53TAD undergoes coupled folding and binding with MDM2,8,9,11 that is, the disordered segment of p53TAD folds into an ordered helical structure concomitant with binding to MDM2. There is an entropic cost to fold a disordered protein that can be paid by an increase in the binding enthalpy during the coupled folding and binding process.
Figure 1 presents the crystal structure of MDM2 from residues 25 to 109 in complex with a p53 fragment from residues 17 to 29 from protein data bank (PDB) identifier 1YCR.7 For the current study we will denote this protein fragment in complex as p53TADc. The PDB structure shows that an amphipathic alpha helical structure of p53TADc fits into a hydrophobic cleft on MDM2, 7 indicating MDM2 and p53TADc are held together largely by hydrophobic forces. At the binding interface, residues F19, L22, W23, and P27 of p53 make hydrophobic contacts with MDM2, among which W23 is deeply buried in the hydrophobic cleft. 7 Several other studies also support the conclusion that residues F19, W23, and L26 of p53 are the key residues responsible for the binding with MDM2, and to be functional F19 and W23 cannot be replaced with other amino acids.12–15 The alpha helix structure of p53 is largely held by the hydrogen bonds between residues T18 and D21 7 and stabilized by a salt bridge between two charged amino acids: D21 and K24.7,16
Figure 1.
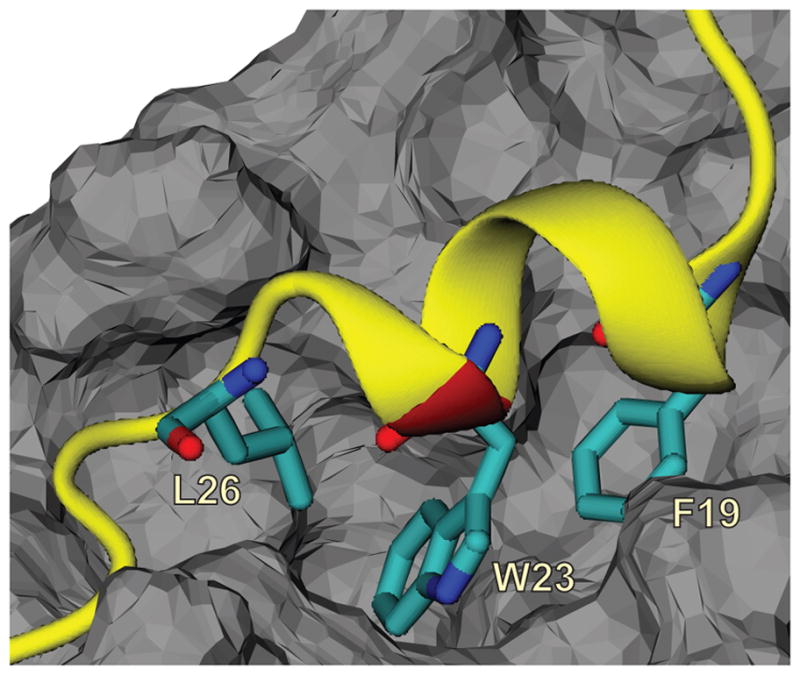
Structure of the p53TADf-MDM2 complex from PDB ID: 1YCR 7 with three critical residues, F19, W23, and L26 shown in sticks. The p53TADf peptide is presented as a ribbon in yellow with the K24N mutation site colored in red, and MDM2 is shown as grey solid surface.
A comprehensive replacement analysis by A. Böttger et al. suggested that K24 of p53 can be replaced by 15 other amino acids without a noticeable influence on binding affinity (less than 3-fold).14 However, they did not mention which 15 amino acids can be substituted, except that E should replace K24 in the best binding sequence.14 This is in contrast to the three key residues F19, W23 and L26 that are highly conserved across a wide spectrum of species.17–20
The K24 to N (K24N) mutation on p53 is found to be prevalent in some organisms other than homo sapiens, such as cow21, sheep22, dog23, and cat24. Interestingly, Y. Yaginuma et al. identified the K24N mutation in a human NUC-1 choriocarcinoma cell line.25 Choriocarcinoma is a very malignant and metastatic form of gestational cancer.25 In spite of the importance of the K24N mutation there have not been any studies mapping the K24N mutation to its biological role.
For unbound p53TAD, structural studies confirmed a flexible conformation. According to the nuclear magnetic resonance (NMR) on full length p53TAD, this protein is mostly disordered in the unbound state. 16,26 Based on the sequential dNN NOEs and a helical prediction algorithm AGADIR, p53TAD contains a so called “preformed” helix from residues 18 to 26, and two nascent turns (residues 40–45 and residues 49–54) at 278 K.16,27 This “preformed” helical region is in the binding site for MDM2, and forms a stable helix when bound to MDM2.16 Therefore people have yet to obtain a well-defined structure for the unbound state. An NMR study at 275 K of a p53 fragment corresponding to residues 17 to 29 detected several weak NOEs between amide protons, indicating the presence of two beta turn structures (residues 19 to 22 and residues 22 to 25).28 At room temperature, 300 K, circular dichroism spectrum showed that the peptide p53TAD (residues 9 to 25) exhibits features of random coil. 15
In this report, NMR spectroscopy, isothermal titration calorimetry (ITC) and, molecular dynamics (MD) simulations were used to investigate how the K24N mutation in p53TAD affects the structure and dynamics of p53TAD, MDM2 and the p53TAD-MDM2 complex. It is currently not understood how the K24N mutation affects dynamics and binding with MDM2. The NMR results show the K24N mutant is more flexible and has less transient helical secondary structure than wild type. ITC measurements demonstrate that these changes in structure and dynamics do not significantly change the binding affinity of p53TAD-MDM2 complex. Finally, free energy perturbation (FEP) and standard MD simulations suggest the small affinity change is due to a compensating interaction energy between K24N and MDM2 when bound. Overall, the data suggests that the K24N-MDM2 complex is able to at least partly compensate for this increase in the conformational entropy of unbound K24N with an increase in the bound state electrostatic interaction energy.
MATERIALS AND METHODS
Protein purification
Samples of human p53TAD (residues 1–73) that were uniformly labeled with either 15N or 15N and 13C, were prepared as previously described.26 Samples of the K24N mutant were prepared using this same method.
NMR data collection and analysis
Resonance assignments for human p53TAD were previously reported.26 Experiments on K24N were carried out at 298 K on a Varian VNMRS 600 MHz spectrometer equipped with a triple resonance pulse field Z-axis gradient cold probe. To make the amide 1H and 15N as well as 13Cα, 13Cβ and 13CO resonance assignments, sensitivity enhanced 1H-15N HSQC and three dimensional HNCACB experiments were performed on the uniformly 15N and 13C labeled sample of K24N in 90%H2O/10% D2O, PBS buffer, at a pH of 6.8. For the HNCACB experiment, data were acquired in 1H, 13C and 15N dimensions using 8012.8 (t3) × 12000 (t2) × 2000 (t1) Hz sweep widths, and 512 (t3) × 128 (t2) × 32 (t1) complex data points. The sweep widths and complex points of the HSQC were 8012.8 (t2) × 2000 (t1) Hz and 512 (t2) × 128 (t1), respectively. For K24N, processing and analysis of the HNCACB data resulted in 60 non-proline, amide 1H, 15N, 13Cα and 13Cb resonance assignments plus 12 proline 13Cα and 13Cβ resonance assignments.
All NMR spectra were processed with nmrPipe and analyzed using nmrView software. 29 Apodization was achieved in the 1H, 13C and 15N dimensions using a squared sine bell function shifted by 70°. Apodization was followed by zero filling to twice the number of real data points and linear prediction was used in the 15N dimension of the HNCACB and HNCO.
1H-15N steady-state NOE experiments were recorded in the presence and absence of a 120° off-resonance 1H saturation pulse every 5 ms for a total of 3 s. A total of 512 (t2) × 128 (t1) complex points were recorded with 128 scans per increment. The NHNOE values were determined by taking the quotient of the intensity for resolved resonances in the presence and absence of proton saturation. Three measurements were made on each protein and the values were averaged.
ITC data collection and analysis
ITC experiments were conducted using a GE Microcal VP-ITC. Samples were exchanged into a buffer containing 50 mM Sodium Phosphate, 150 mM sodium chloride, 1 mM EDTA, 0.02% sodium azide, 8 mM beta-mercapto-ethanol at a pH of 6.8. Solutions of wildtype and the K24N variant of p53TAD, at a concentration of 50 μM were loaded into the syringe and injected into the sample cell containing MDM2 at a concentration of 5 μM. 38 7.5 ul injections were used for MDM2 titrations. The sample cell was equilibrated at 298 K. All of the data was analyzed using the Origin 7.0 ITC software (OriginLab, Northampton, MA). The integrated ITC data was best fit with a single-site binding model and the binding stoichiometries were between 0.8 and 1.0. The values listed in Table II are the averages and standard deviations from multiple ITC measurements.
Table II.
Isothermal titration calorimetry (ITC) results for wildtype and K24N mutant.
| p53TAD | K24N | |
|---|---|---|
| ΔG | −8.93 (0.17) | −9.12 (0.13) |
| ΔH | −10.90 (0.83) | −10.50 (0.29) |
| TΔS | −1.96 (0.77) | −1.38 (0.37) |
Values are in kcal/mol. The values for p53TAD are the averages and standard deviations (in parentheses) from two separate protein preparations with three repeats each. The averages and standard deviations (in parentheses) for K24N are based on three ITC measurements performed on the same protein preparations.
Simulation
The initial structure of the wildtype p53TADc-MDM2 complex was taken from the protein databank (PDB ID: 1YCR)7, that includes residues 17–29 of p53 and residues 25–109 of MDM2. The coordinates for missing light chain atoms in the crystal structure were guessed by VMD psfgen package.30 The unbound fragment p53TADf was also initiated from 1YCR with an absence of MDM2. Since it is known that unbound p53TADf is intrinsically disordered we generated five plausible structures using our broad ensemble generator with re-weighting (BEGR) method.31,32 By examining their local root mean square fluctuation (RMSF) we chose the structure that has the most significant difference in flexibility compared with the wildtype. The increased flexibility of the mutant is consistent with the experimental observation. In this report, we denote p53TADf for unbound p53 fragment simulations initiated from the crystal structure, and p53BEGRf for unbound simulations starting from BEGR structures. The initial structure for the bound/unbound mutant fragment K24Nc/K24Nf was prepared by changing the residue 24 of p53TADc/p53TADf from K to N using the mutator plugin of VMD.30 And similarly, K24Nf is the unbound mutant fragment initiated from the crystal structure and K24NBEGRf represents the one started from BEGR structures.
We calculated the free energy difference for MDM2 binding to p53TAD and K24N fragments by performing alchemical free energy perturbation. 33,34 We used the dual topology approach implemented in NAMD35–37, where wildtype and mutant are present at the same time but do not interact with each other. The wildtype protein was set as the initial state (λ = 0), the final state (λ = 1) was the mutant. During the simulation, we calculated the potential energy function for both solutes as weighted by λ and (1−λ) for the wildtype and the mutant. For both simulations we used values of λ = 0, 0.0001, 0.01, 0.05 – 0.95 with increment 0.05, 0.99, 0.9999, 1.0. FEP simulations starting at λ = 0 are denoted forward, and simulations starting at λ = 1 are denoted backward. A soft-core potential was employed to avoid the so-called “end-point catastrophes” 38 by scaling electrostatic and van der Waals interactions as λ varies.39–41 For each λ window, 2 ps of energy minimization, 100 ps of MD equilibration, and 2 ns of production were performed for the unbound p53 fragments; 2 ps minimization, 40 ps equilibration, 1 ns production were performed for the complexes. The longer simulation times were performed for the unbound p53 fragments since it is known that p53TAD is intrinsically disordered and thus the conformational space is larger than for the complex. All parameters were the same as described above. To calculate the corresponding entropic contribution, we performed FEP simulations at variable temperatures, 270 K, 300 K and 330 K. The initial structures for the FEP simulations were prepared using the mutator plugin of VMD,30 followed by minimization and equilibration with FEP at λ = 0 or 1. In present study, we did FEP simulations on K24N mutation of p53TADf, p53BERGRf and p53TADc-MDM2.
Each of the eight simulations systems (p53TADf, K24Nf, p53BEGRf, K24NBEGRf, p53TADc-MDM2, and K24Nc-MDM2; FEP p53TADf and p53TADc-MDM2) was solvated in a cubic TIP3P water box that extended 14 Å in each direction from the solute. 42 Each system was given a neutral charge by adding 150 mM sodium chloride (NaCl) to achieve neutralization. 30 Molecular dynamics simulations were carried out for 100 ns at 300 K using the CHARMM22 force field 43 using the NAMD software version 2.7b4.37 The long range electrostatic interactions were calculated using Particle Mesh Ewald (PME) 44, and the short range van der Waals interactions were truncated at 12 Å. Periodic boundary conditions were also applied. The vibration of the bonds involving hydrogen atoms were constrained using SHAKE 45, and a time step of 2 fs was used for dynamics. Trajectory snapshots were saved every 2 ps. Each system was initially minimized with the backbone atoms fixed and then with all constraints removed. The minimized structures were gradually heated up to 300 K in 300 ps followed by equilibration at constant pressure and temperature for 100 ps. The production run was performed for each system at constant pressure and temperature. Simulations were repeated three times with independent minimization and equilibration processes for each system. The trajectories were processed using VMD 30 to get an aligned solute without explicit water and counter-ions. The RMSF of the last 50 ns simulation for each system was computed in VMD and plotted using GRACE-5.1.22. 46 Secondary structures based on featured atomic coordinates of protein were computed for each residue using STRIDE 47 and plotted as the evolution of simulation time using the timeline plugin of VMD. 30
Thermodynamic cycle
Figure 2 shows the thermodynamic cycle used to estimate the difference between the mutant and wildtype affinities. Since the free energy is a state function, the change from unbound to bound state is independent of path.48 The quantities and were obtained through two FEP simulations of the K24N mutation in p53TAD fragment, one for the unbound p53TADf peptide and the other for the p53TADc-MDM2 complex. The horizontal arrows correspond to the binding free energy of wiltype and the mutant . Adding these quantities in the directions of arrows, we will get zero and thus:
and,
Figure 2.
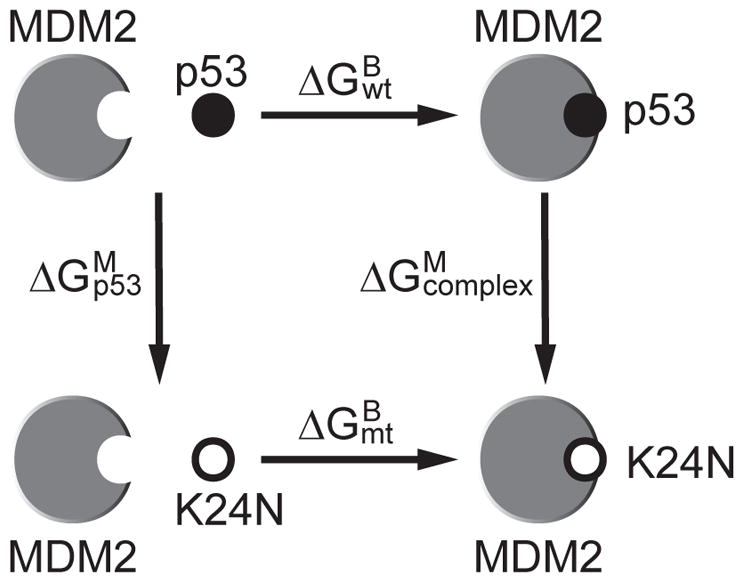
Thermodynamic cycle used to estimate the relative binding affinity between wildtype and mutant p53 for MDM2. The vertical arrows correspond to the K24N mutation in the p53 fragment and in the complex. The horizontal arrows correspond to the binding of p53 fragments to MDM2.
where is the binding affinity difference between the wildtype and the mutant. For simplicity, we will ignore the superscript and subscript, and denote the binding affinity difference as ΔΔG.
Uncertainties for the ΔG values were estimated by computing the standard deviation over at least three independent pairs of forward and backward FEP simulations. Independent trials were generated using different starting configurations. Then the uncertainties of ΔΔG were calculated as , where σmt and σwt are standard deviations of ΔGmt and ΔGwt separately.
Estimation of entropic and enthalpic contribution
The entropic contribution to the relative binding affinity ΔΔG was determined by the derivative of the free energy with respect to the system temperature T using the following relation, 49,50
The above derivative was estimated by computing ΔΔG at three temperatures 270 K, 300 K and 330 K and then using a three-point finite difference approximation.
The enthalpic contribution ΔΔH was then estimated by,
The uncertainties of TΔΔS and ΔΔH were derived from the standard deviations of ΔΔG using the same method for computing the uncertainties of ΔΔG.
RESULTS AND DISCUSSION
For the current study, both experiments and molecular dynamics computer simulations were used to determine the effects of the K24N mutation on the flexibility of p53TAD and the thermodynamic properties of p53TAD binding to MDM2. The nomenclature and sequence information for this study is shown in Table I.
Table I.
Protein sequences derived from the transactivation domain of human p53 (p53TAD) used in this study.
| Peptide name | Derived from | Others |
|---|---|---|
| p53TAD | Human p53 residues 1 to 73 | |
| K24N | K24N mutant of human p53 residues 1 to 73 | Prepared by gene expression 26 |
| p53TADf | Human p53 residues 17 to 29 at unbound state | From crystal structure PDB ID: 1YCR 7 |
| p53TADc | Human p53 residues 17 to 29 bound with MDM2 | From crystal structure PDB ID: 1YCR 7 |
| K24Nf | K24N mutant of human p53 residues 17 to 29 at unbound state | Mutate K24 to N24 in p53TADf using the Mutator plugin of VMD 30 |
| K24Nc | K24N mutant of human p53 residues 17 to 29 bound with MDM2 | Mutate K24 to N24 in p53TADf using the Mutator plugin of VMD 30 |
| p53BEGRf | Human p53 residues 17 to 29 | Generated by the software BEGR 31,32 |
| K24NBEGRf | K24N mutant of human p53 residues 17 to 29 | Mutate K24 to N24 in p53BEGRf using the Mutator plugin of VMD 30 |
| Primary sequence of p53TAD (GenBank accession no. BAC16799.1) | ||
| MEEPQSDPSVEPPLSQETFSDLWKLLPENNVLSPLPSQAMDDLMLSPDDIEQWFTEDPGPDEAPRMPEAAPRV | ||
The names, sequences, and other information of proteins used in this study are shown in the upper panel. The primary sequence of p53TAD is shown in the lower panel. The underlined segment was used for simulations and the bold residue is the site of the K24N mutation.
I. Effects of K24N mutation on the structure and dynamics of p53TAD and MDM2
The K24N mutant has reduced helicity compared to wildtype
The NMR alpha carbon secondary chemical shifts (CA Δδ) for p53TAD and K24N are shown in Figure 3(a) and (b) with solid and clear bars, respectively. Backbone resonance assignments and dynamics measurements on K24N were performed as previously described for p53TAD. 26 The CA Δδ values were calculated using the new neighbor corrected random coil chemical shift library developed by Mulder and colleagues.51 The CA Δδ values provide a sensitive measure of secondary structure at single residue resolution. For CA atoms, a positive value indicates the presence of helical secondary structure and a negative value indicates the presence of beta or extended structures.52–55 For IDPs it is typical to see Δδ values close to zero in addition to adjacent values that switch between negative and positive over the length of the protein. The CA Δδ values are plotted in parts per million on the y-axis and residue position is plotted on the x-axis. The MDM2 binding region of p53TAD includes residues 17–29. Several groups have previously shown that this region contains transient helical structure and this finding is confirmed in Figure 3(a). 16,26,56
Figure 3.
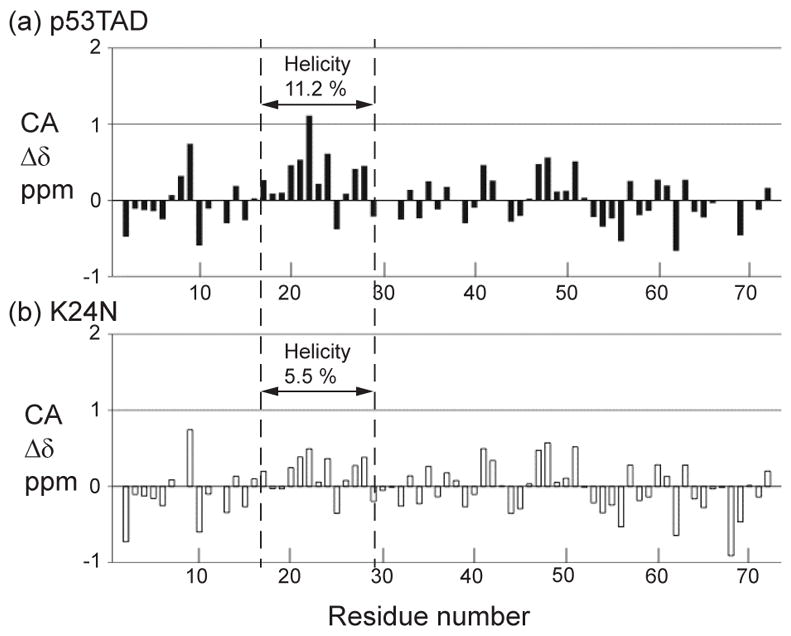
Alpha carbon secondary chemical shifts for (a) p53TAD and (b) K24N with the helicity calculated on residues 17–29. The dashed lines indicate the MDM2 binding site.
Figure 3 indicates that changing a single amino acid from K to N reduces the fractional helicity of the unbound p53 fragment by a factor of two. The fractional helicity was calculated using a relationship developed by Wright and Dyson based on the secondary chemical shifts. 57 When this calculation is performed on residues 17–29 of p53TAD a value of 11.2% is obtained. By contrast, the fractional helicity of the K24N mutant is around 5.5%.
Figure 4 shows the secondary structure as a function of simulation time for p53TADf and K24Nf using the timeline plugin in VMD.30 Standard MD simulations were performed on unbound p53TADf using the bound structure from PDB ID: 1YCR as the starting structure. 7 The K24Nf mutant was created using the VMD mutator plugin. 30 Three independent simulations of 100 ns were performed for both p53TADf and K24Nf and used to estimate the secondary structure for each residue as a function of simulation time.47 As illustrated in Figure 4, from residues 18 to 24, the feature of alpha helix is mostly preserved for both p53TADf and K24Nf in the initial ~20 ns simulation, but the longer simulation times show larger turn and coil propensities. Consistent with experiment (Figure 3), this result shows reduced helicity at L25 of K24Nf. The reduced helicity is like due to the loss of a salt bridge between residues D21 and K24 that is believed to stabilize the transient helical structure of the unbound p53TADf.7,16
Figure 4.

Secondary structure of unbound p53 fragments from six independent 100 ns molecular dynamics simulations as determined by STRIDE.47 Left column is for p53TADf and right for K24Nf. Color key: alpha helix = purple; 3–10 helix = blue; pi-helix = red; turn = aqua; extended configuration = yellow; isolated bridge = khaki; random coil = white.
The K24N mutant is more flexible than wildtype when unbound but not when bound
If the K24N mutant reduces the fractional helicity of p53TAD in the region of the MDM2 binding site then one would expect an increase in the backbone flexibility of this region. This is indeed what was observed when the heteronuclear 1H-15N NOE between the amide nitrogen and proton (NHNOE) was compared for the mutant and wild type. The NHNOE provides a residue specific measure of backbone rotational motions on the nanosecond to picoseconds timescale.58,59 For IDPs, small positive NHNOE’s are often observed in regions that contain transient secondary structure and negative NHNOE’s are observed in more flexible regions. Figure 5(a) and (b) show the NHNOE values for p53TAD and K24N, respectively. As predicted, the K24N mutation induces faster rotational motions on the nanosecond timescale for the MDM2 binding region (residues 17–29) as indicated by the reduction in positive NHNOE values for this region. Taken together the CA Δδ and NHNOE values for K24N indicate the MDM2 binding region is more flexible than p53TAD.
Figure 5.
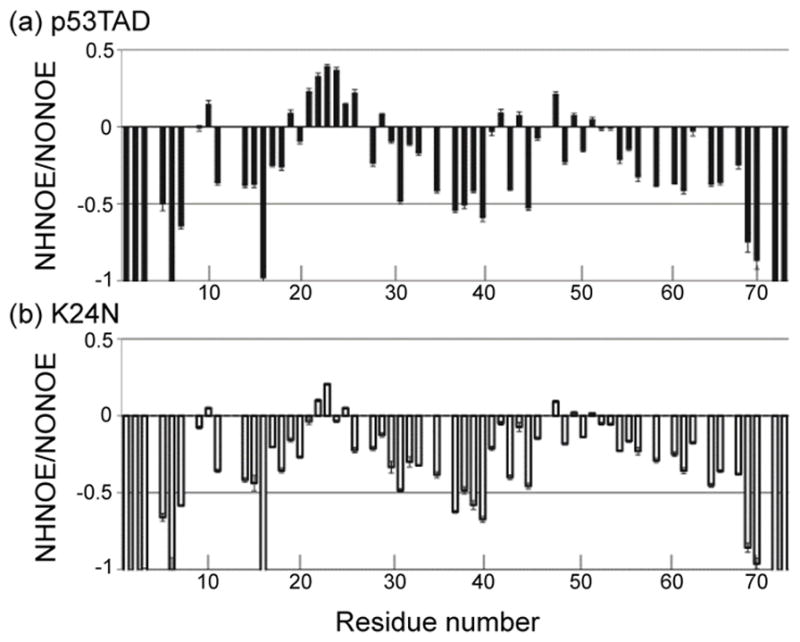
1H-15N steady-state NOE values (NHNOE) for (a) p53TAD and (b) K24N
Figure 6 shows simulation results that also indicate unbound K24Nf is more flexible than p53TADf. Three independent MD simulations for both unbound and bound p53TADf and K24Nf were each run for 100 ns. Averages of root mean square fluctuations (RMSF) were calculated for alpha carbons based on trajectories from the last 50 ns of the simulation. RMSF is a metric for how much the position of an atom deviates from the averaged coordinate during the molecular dynamics simulation and thus is a direct measure of flexibility. The RMSF values computed for K24Nf were mostly larger than p53TADf when the p53 fragment is not bound (Figure 6(a)). However, the bound fragment K24Nc has slightly lower level of flexibility compared with p53TADc. The result indicates K24Nfis more flexible than p53TADf in the unbound state but not in the bound state.
Figure 6.

Alpha carbon root mean square fluctuations (RMSF) in units of Å for (a) p53 fragments and (b) MDM2 at both bound and unbound states. Each curve is an average obtained from three independent 100ns molecular dynamics simulations. The last 50ns of each simulation was used in the analysis. p53TADf and K24Nf are unbound wildtype and mutant p53 fragments; p53TADc and K24Nc are p53 fragments in complexes; MDM2f is at unbound state; MDM2wt and MDM2mt are MDM2 bound to p53TADc and K24Nc respectively.
K24Nc exhibits stronger interaction with MDM2
To further understand the effect of the K24N mutation, we compared the flexibility of MDM2 when bound to either K24Nc or p53TADc using MD simulations. Figure 6(b) shows the comparison of RMSF curves for MDM2 in the bound and unbound states. Both MDM2wt (bound with p53TADc) and MDM2mt (bound with K24Nc) show reductions in RMSF for residues 50 to 65 (α2) and 95 to 104 (α2′) compared with MDM2f (unbound state). These two regions reside at the binding interface with p53TAD. The major differences between MDM2wt and MDM2mt comes from residues 69 and 85 in which the former is in the linker region between β3 and β1′ of MDM2, and the latter is in the α1′ region. However taking the uncertainty into account the difference between MDM2wt and MDM2mt is negligible. This indicates that the K24N mutant has similar binding behavior with the wildtype.
Figure 7 shows the interaction energies between either p53TADc or K24Nc and MDM2 calculated from simulations. Each value is an average obtained from three independent 100ns molecular dynamics simulations. The last 50ns of each simulation was used in the analysis. The errors were estimated as the standard deviation of the three independent simulations. Van der Waals interaction shows negligible difference between the wildtype and the mutant. The primary difference is in the electrostatic interaction energy. This is probably because the net charge of MDM2 is +5e, the charge of p53TADc is −2e, and the charge of K24Nc is −3e. The increased electrostatic interaction between K24N and MDM2 probably accounts for the reduced flexibility of K24N in the bound state, as compared to p53TAD.
Figure 7.
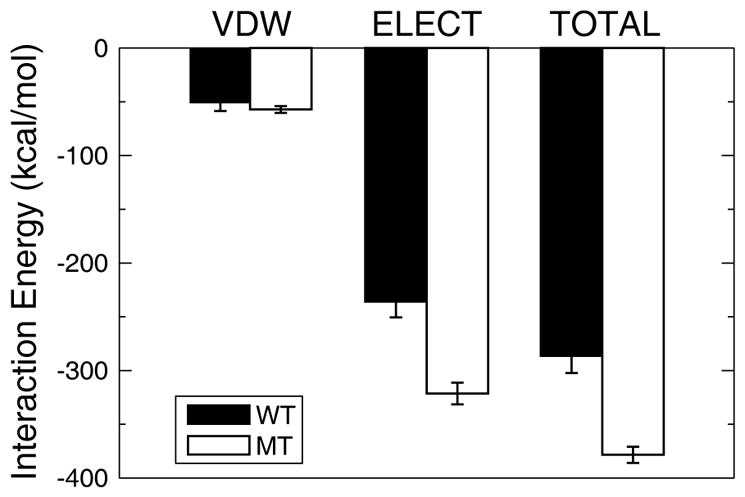
Interaction energy between p53TADc/K24Nc and MDM2. Each bar is an average obtained from three independent 100ns molecular dynamics simulations. The last 50ns of each simulation was used in the analysis. The error bars are the standard deviation of the three independent simulations. Note that the net charge of MDM2 is +5e, the charge of p53TADc is −2e, and the charge of K24Nc is −3e. VDW: Van der Waals interaction; ELECT: Electrostatic interaction; WT: interaction with wildtype (p53TADc); MT: interaction with mutant (K24Nc).
II. Effects of K24N mutation on the thermodynamics of binding
The K24N mutant exhibits similar binding affinity, entropy and enthalpy with wildtype
Our results show the binding free energy, entropy and enthalpy are statistically indistinguishable between p53TAD and K24N from both experimental measurements using isothermal titration calorimetry (ITC) and calculations from computer simulations.
Table II shows that both p53TAD and K24N bind to MDM2 with very similar values for the binding affinity (ΔG), enthalpy (ΔH), and entropy (TΔS). The data also suggest that the binding affinities of p53TAD and K24N are almost exclusively determined by the value of enthalpy. The binding process is mainly driven by enthalpy and thus the enthalpy is the driving force for the coupled folding and binding reaction, and is also responsible for the loss of system entropy.
Table III shows the computed binding free energy results from free energy perturbation (FEP) simulations. The results show that at 300 K the relative affinity (ΔΔG) is about 1 kcal/mol, suggesting that p53TADc binds slightly stronger with MDM2 than K24Nc at room temperature. However, the ITC measurement gave a negligible decreased value, −0.19 (0.21) kcal/mol for ΔΔG. The discrepancy between the simulation and the experiment falls within the expected accuracy of MD simulation (1–2 kcal/mol). 60 To test the effect of the starting structure on the results we also used a p53 fragment (p53BEGRf) generated via BEGR.31,32 BEGR structures are selected from a large pool of candidate structures such that they best fit experimental chemical shift data from NMR. Although the affinity results are very similar for p53BEGRf compared to p53TADf the entropic (TΔΔS) and enthalpic (ΔΔH) contributions are quite different. The uncertainties for TΔΔS and ΔΔH are also quite large. These results highlight the difficulty in estimating the entropic and enthalpic contributions for binding of disordered proteins. The p53TADf simulations incorrectly estimate TΔΔS because the simulations are trapped in a local minima corresponding to the helical structure. Similarly, the p53BEGRf simulations also incorrectly estimate TΔΔS because the unfolded conformational space is too large to thoroughly explore during the timescale of an ordinary MD simulation. Thus, the correct TΔΔS value is expected to lie between the two values; which is true for our results.
Table III.
Thermodynamic properties of p53 binding with MDM2 upon K24N mutation.
| Temperature | FEP (p53TADf) | FEP (p53BEGRf) | ITC | |
|---|---|---|---|---|
| ΔΔG | 270 K | 1.03 (0.71) | 0.54 (0.65) | - |
| 300 K | 0.98 (0.56) | 0.93 (0.58) | −0.19 (0.21) | |
| 330 K | 0.39 (0.67) | 1.01 (0.68) | - | |
| TΔΔS | 300 K | 3.23 (4.60) | −2.32 (4.45) | 0.58 (0.86) |
| ΔΔH | 300 K | 4.21 (4.63) | −1.39 (4.49) | 0.40 (0.88) |
All energy values are evaluated computationally (FEP) and experimentally (ITC), and shown in units of kcal/mol. Errors are in parentheses. The details of the calculation of ΔΔG, ΔΔH, and TΔΔS, and corresponding uncertainties are described in the main text.
CONCLUSIONS
In conclusion, our findings show that the K24N mutation reduces the helicity and increases the flexibility of p53TAD, but has no appreciable effect on the binding affinity, enthalpy, and entropy changes for the binding process. Even though the K24N mutant is more flexible than the wildtype while unbound, the K24N-MDM2 complex is able to at least partly compensate for this increase in the conformational entropy with an increase in the bound state electrostatic interaction energy, thus contributing to the similar binding affinity with the wildtype p53TAD-MDM2 complex. The similar affinity was probably necessary since the mutation was selected by evolution. This is an important observation because it is known that many IDPs, including p53TAD, have a higher rate of amino acid substitutions than ordered proteins.17 Substitutions occurring at a protein-protein interface could be deleterious unless the effect is offset by a mechanism like the one shown in this study. It will be interesting to determine if other interactions between IDPs and their ordered binding partners show a similar behavior.
Acknowledgments
The authors would like to thank for the funding support from various sources. Y.A.Z. and F.M.Y. were supported by the funding from Idaho INBRE and the National Institutes of Health (R21GM83827). G.W.D. was supported by the American Cancer Society (RSG-07-289-01-GMC), the National Science Foundation (MCB-0939014), and the National Institutes of Health (R21GM83827).
References
- 1.El-Deiry W. Regulation of p53 downstream genes. Semin Cancer Biol. 1998;8:345–357. doi: 10.1006/scbi.1998.0097. [DOI] [PubMed] [Google Scholar]
- 2.May P, May E. Twenty years of p53 research: Structural and functional aspects of the p53 protein. Oncogene. 1999;18:7621–7636. doi: 10.1038/sj.onc.1203285. [DOI] [PubMed] [Google Scholar]
- 3.Moman J, Wu H, Dasgupta G. MDM2-Master regulator of the p53 tumor suppressor protein. Genes. 2000;242:15–29. doi: 10.1016/s0378-1119(99)00487-4. [DOI] [PubMed] [Google Scholar]
- 4.Hollstein M, Sidransky D, Vogelstein B, Harris C. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 5.Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362:857–860. doi: 10.1038/362857a0. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Marechal V, Levine AJ. Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol. 1993;13:4107–4114. doi: 10.1128/mcb.13.7.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 Oncoprotein Bound to the p53 Tumor Suppressor Transactivation Domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 8.Bell S, Klein C, Müller L, Hansen S, Buchner J. p53 Contains Large Unstructured Regions in its Native State. Journal of Molecular Biology. 2002;322(5):917–927. doi: 10.1016/s0022-2836(02)00848-3. [DOI] [PubMed] [Google Scholar]
- 9.Ayed A, Mulder FAA, Yi G-S, Lu Y, Kay LE, Arrowsmith CH. Latent and active p53 are identical in conformation. Nature Structural Biology. 2001;8(9):756–760. doi: 10.1038/nsb0901-756. [DOI] [PubMed] [Google Scholar]
- 10.Fields S, Jang SK. Presence of a Potent Transcription Activating Sequence in the p53 Protein. Science. 1990;249:1046–1049. doi: 10.1126/science.2144363. [DOI] [PubMed] [Google Scholar]
- 11.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nature Reviews Molecular Cell Biology. 2005;6(3):197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 12.Zondlo SC, Lee AE, Zondlo NJ. Determinants of Specificity of MDM2 for the Activation Domains of p53 and p65: Proline27 Disrupts the MDM2-Binding Motif of p53. Biochemistry. 2006;45:11945–11957. doi: 10.1021/bi060309g. [DOI] [PubMed] [Google Scholar]
- 13.Massova I, Kollman PA. Computational Alanine Scanning To Probe Protein-Protein Interactions: A Novel Approach To Evaluate Binding Free Energies. J Am Chem Soc. 1999;121:8133–8143. [Google Scholar]
- 14.Böttger A, Böttger V, Garcia-Echeverria C, Chène P, Hochkeppel H-K, Sampson W, Ang K, Howard SF, Picksley SM, Lane DP. Molecular characterization of the hdm2-p53 interaction. Journal of Molecular Biology. 1997;269:744–756. doi: 10.1006/jmbi.1997.1078. [DOI] [PubMed] [Google Scholar]
- 15.Uesugi M, Verdine GL. The α-helical FXXFF motif in p53: TAF interaction and discrimination by MDM2. Proc Natil Acad Sci USA. 1999;96:14801–14806. doi: 10.1073/pnas.96.26.14801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee H, Mok K, Muhandiram R, Park K, Suk J, Kim D, Chang J, Sung Y, Choi K, Han K. Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J Biol Chem. 2000;275:29426–29432. doi: 10.1074/jbc.M003107200. [DOI] [PubMed] [Google Scholar]
- 17.De Laurenzi V, Melino G. Evolution of functions within the p53/p63/p73 family. Ann N Y Acad Sci. 2000;926:90–100. doi: 10.1111/j.1749-6632.2000.tb05602.x. [DOI] [PubMed] [Google Scholar]
- 18.Liu W, Midgley C, Stephen C, Saville M, Lane D. Biological significance of a small highly conserved region in the N terminus of the p53 tumour suppressor protein. J Mol Biol. 2001;313:711–731. doi: 10.1006/jmbi.2001.5082. [DOI] [PubMed] [Google Scholar]
- 19.Lane DP, Cheok CF, Brown CJ, Madhumalar A, Ghahessy FJ, Verma C. Mdm2 and p53 are highly conserved from placozoans to man. Cell Cycle. 2010;9:540–547. doi: 10.4161/cc.9.3.10516. [DOI] [PubMed] [Google Scholar]
- 20.Lane DP, Cheok CF, Brown CJ, Madhumalar A, Ghadessy FJ, Verma C. The Mdm2 and p53 genes are conserved in the Arachnids. Cell Cycle. 2010;9:748–754. doi: 10.4161/cc.9.4.10616. [DOI] [PubMed] [Google Scholar]
- 21.Dequiedt F, Willems L, Burny A, Kettmann R. Nucleotide sequence of the bovine P53 tumor-suppressor cDNA. DNA Sequence:the journal of DNA sequencing and mapping. 1995;5(4):261–264. doi: 10.3109/10425179509030977. [DOI] [PubMed] [Google Scholar]
- 22.Dequiedt F, Kettmann R, Burny A, Willems L. Nucleotide sequence of the ovine P53 tumor-suppressor cDNA and its genomic organization. DNA Sequence:the journal of DNA sequencing and mapping. 1995;5(4):255–259. doi: 10.3109/10425179509030976. [DOI] [PubMed] [Google Scholar]
- 23.Sanchez-Servin A, Martinez S, Cordova-Alarcon E, Fajardo R. TP53 Polymorphisms allow for genetic sub-grouping of the canine transmissible venereal tumor. J Vet Sci. 2009;10:353–355. doi: 10.4142/jvs.2009.10.4.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okuda M, Umeda A, Sakai T, Ohashi T, Momoi Y, Youn H, Watari T, Goitsuka R, Tsujimoto H, Hasegawa A. Cloning of feline p53 tumor-suppressor gene and its aberration in hematopoietic tumors. Int J Cancer. 1994;58:602–607. doi: 10.1002/ijc.2910580425. [DOI] [PubMed] [Google Scholar]
- 25.Yaginuma Y, Yamashita T, Takuma N, Katayama H, Ishikawa M. Analysis of the p53 gene in human choriocarcinoma cell lines. Br J Cancer. 1995;71:9–12. doi: 10.1038/bjc.1995.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vise PD, Baral B, Latos AJ, Daughdril GW. NMR chemical shift and relaxation measurements provide evidence for the coupled folding and binding of the p53 transactivation domain. Nucleic Acids Research. 2005;33:2061–2077. doi: 10.1093/nar/gki336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chi S, Lee S, Kim D, Ahn M, Kim J, Woo J, Torizanwa T, Kainosho M, Han K. Structural details on mdm2-p53 interaction. J Biol Chem. 2005;280:38795–38802. doi: 10.1074/jbc.M508578200. [DOI] [PubMed] [Google Scholar]
- 28.Botuyan MVE, Momand J, Chen Y. Solution conformation of an essential region of the p53. Folding & Design. 1997;2:331–342. doi: 10.1016/S1359-0278(97)00047-3. [DOI] [PubMed] [Google Scholar]
- 29.Johnson BA, Blevins RA. NMRView: A computer program for the visualization and analysis of NMR data. Journal of Biomolecular NMR. 1994;4(5):603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 30.Humphrey W, Dalke A, Schulten K. VMD - Visual Molecular Dynamics. Journal of Molecular Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 31.Daughdrill GW, Kashtanov S, Stancik A, Hill SE, Helms G, Mushol M, Receveur-Bréchot V, Ytreberg FM. Understanding the structural ensembles of a highly extended disordered protein. Molecular BioSystems. 2012;8:308–319. doi: 10.1039/c1mb05243h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kashtanov S, Borcherds W, Wu H, Daughdrill GW, Ytreberg FM. Methods in Molecular Biology. New York: Humana Press Inc; 2012. Intrinsically Disordered Proteins: Volume I. Experimental Techniques. [DOI] [PubMed] [Google Scholar]
- 33.Dixit SB, Chipot C. Can absolute free energies of association be estimated from molecular. J Phys Chem A. 2001;105:9795–9799. [Google Scholar]
- 34.Hénin Jeo, Gumbart J, Harrison C, Chipot C. Theoretical biophysics group. University of Illinois and Beckman Institute; Urbana, IL: 2010. In silico alchemy: A tutorial for alchemical free-energy perturbation calculations with NAMD. [Google Scholar]
- 35.Gao J, Kuczera K, Tidor B, Karplus M. Hidden thermodynamics of mutant proteins: A molecular dynamics analysis. Science. 1989;244:1069–1072. doi: 10.1126/science.2727695. [DOI] [PubMed] [Google Scholar]
- 36.Pearlman DA. A comparison of alternative approaches to free energy calculations. J Phys Chem. 1994;98:1487–1493. [Google Scholar]
- 37.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. Journal of Computational Chemistry. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chipot C, Pohorille A. Theory and applications in chemistry and biology. Springer Verlag; 2007. Free energy calculations. [Google Scholar]
- 39.Zacharias M, Straatsma TP, McCammon JA. Separation-shifted scaling, a new scaling method for Lennard-Jones interactions in thermodynamic integration. J Chem Phys. 1994;100:9025–9031. [Google Scholar]
- 40.Beutler TC, Mark AE, van Schaik RC, Gerber PR, van Gunsteren WF. Avoiding singularities and neumerical instabilities in free energy calculations based on molecular simulations. Chem Phys Lett. 1994;222:529–539. [Google Scholar]
- 41.Pitera JW, van Gunsteren WF. A comparison of non–bonded scaling approaches for free energy calculations. Mol Sim. 2002;28:45–65. [Google Scholar]
- 42.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 43.MacKerell J, AD, Bashford D, Bellott M, Dunbrack J, RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuchnir K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher I, WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 44.Darden T, York D, Pedersen L. Particle mesh Ewald: an N. log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 45.van Gunsteren WF, Berendsen HJC. Algorithms for macromolecular dynamics and constraint dynamics. Mol Phys. 1977;34:1311–1327. [Google Scholar]
- 46.Turner Paul J. Grace Development Team. http://plasma-gate.weizmann.ac.il/Grace/
- 47.Frishman D, Argos P. Knowledge-based protein secondary structure assignment. Proteins: structure, function and genetics. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 48.Kollman PA. Free energy calculations: Applications to chemical and biochemical phenomena. Chem Review. 1993;93:2395–2417. [Google Scholar]
- 49.Ghosh T, García AE, Garde S. Enthalpy and entropy contributions to the pressure dependence of hydrophobic interactions. J Chem Phys. 2002;116:2480–2486. [Google Scholar]
- 50.Choudhury N, Pettitt BM. Enthalpy-Entropy Contributions to the Potential of Mean Force of Nanoscopic Hydrophobic Solutes. J Phys Chem B. 2006;110:8459–8463. doi: 10.1021/jp056909r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tamiola K, Acar B, Mulder FA. Sequence-specific random coil chemical shifts of intrinsically disordered proteins. J Am Chem Soc. 2010;132:18000–18003. doi: 10.1021/ja105656t. [DOI] [PubMed] [Google Scholar]
- 52.Wishart DS, Case DA. Use of chemical shifts in macromolecular structure determination. Methods Enzymol. 2001;338:3–34. doi: 10.1016/s0076-6879(02)38214-4. [DOI] [PubMed] [Google Scholar]
- 53.Wishart DS, Sykes BD. Chemical shifts as a tool for structure determination. Methods Enzymol. 1994;239:363–392. doi: 10.1016/s0076-6879(94)39014-2. [DOI] [PubMed] [Google Scholar]
- 54.Wishart DS, Sykes BD, Richards FM. The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry. 1992;32:1647–1651. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- 55.Wishart DS, Sykes BD, Richards FM. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J Mol Biol. 1991;222:311–333. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- 56.Wells MH, Tidow H, Rutherford TJ, Markwick P, Jensen MR, Myloans E, Svergun DI, Blackledge M, Fersht AR. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc Natl Acad Sci U S A. 2008;105:5762–5767. doi: 10.1073/pnas.0801353105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dyson HJ, Wright PE. Insights into the structure and dynamics of unfolded proteins from nuclear magnetic resonance. Adv Protein Chem. 2002;62:311–340. doi: 10.1016/s0065-3233(02)62012-1. [DOI] [PubMed] [Google Scholar]
- 58.Kay LE. Protein dynamics from NMR. Biochem Cell Biol. 1998;76:145–152. doi: 10.1139/bcb-76-2-3-145. [DOI] [PubMed] [Google Scholar]
- 59.Kay LE, Torchia DA. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 60.Chodera JD, Mobley D, Shirts MR, Dixon RW, Branson K, Pande VS. Alchemical Free Energy Methods for Drug Discovery: Progress and Challenges. Current Opinion in Structural Biology. 2011;21(2):150–160. doi: 10.1016/j.sbi.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]