

The vitamin D receptor (VDR) belongs to the nuclear hormone receptor (NHR) superfamily and has been recognized as the primary mediator of the physiological actions of the active form of vitamin D, 1, alpha, 25 dihydroxy-vitamin D3 [1,25-(OH)2D3] or calcitriol, and its synthetic analogs[1]. Akin to classical nuclear hormone receptors, VDR functions as a ligand-dependent transcription factor that binds to vitamin D response elements (VDREs) within the chromatin as a heterodimer with its obligate partner, retinoid X receptor (RXR)[1]. Activation of VDR via its cognate ligand alters the expression of a network of genes involved in both calcium/phosphorus homeostasis and mineral metabolism[1]. Consistent with the biological role of vitamin D signaling, both VDR knockout and vitamin D-deficient mice have been shown to develop hypocalcaemia, rickets and hyperparathyroidism[1], re-capitulating the phenotypes of these vitamin D deficient human diseases. Thus, both biochemical and genetic evidence not only support VDR as a master regulator of skeletal health but also suggest potential therapeutic effects of VDR ligands in human diseases. Indeed, vitamin D is used therapeutically for the treatment of rickets, secondary hyperparathyroidism and osteoporosis[1]. Clinical interest in VDR ligands as a therapeutic modality has been further spurred by the observation that VDR is expressed in virtually all organs and a diverse range of cell types[1,2]. In this regard, studies over the last two decades have led to an appreciation of the multifaceted roles of VDR in mediating homeostatic effects across non-skeletal tissues and raised the possibility of using VDR ligands to manage a broad spectrum of diseases including, but not limited to, metabolic syndrome, cardiovascular diseases, auto-immune diseases, skin and muscle disorders, and certain types of cancer[2]. However, it is intriguing that the liver, a pivotal organ in many aspects of metabolism and energy homeostasis, appeared devoid of vitamin D/VDR functionality [3,4].
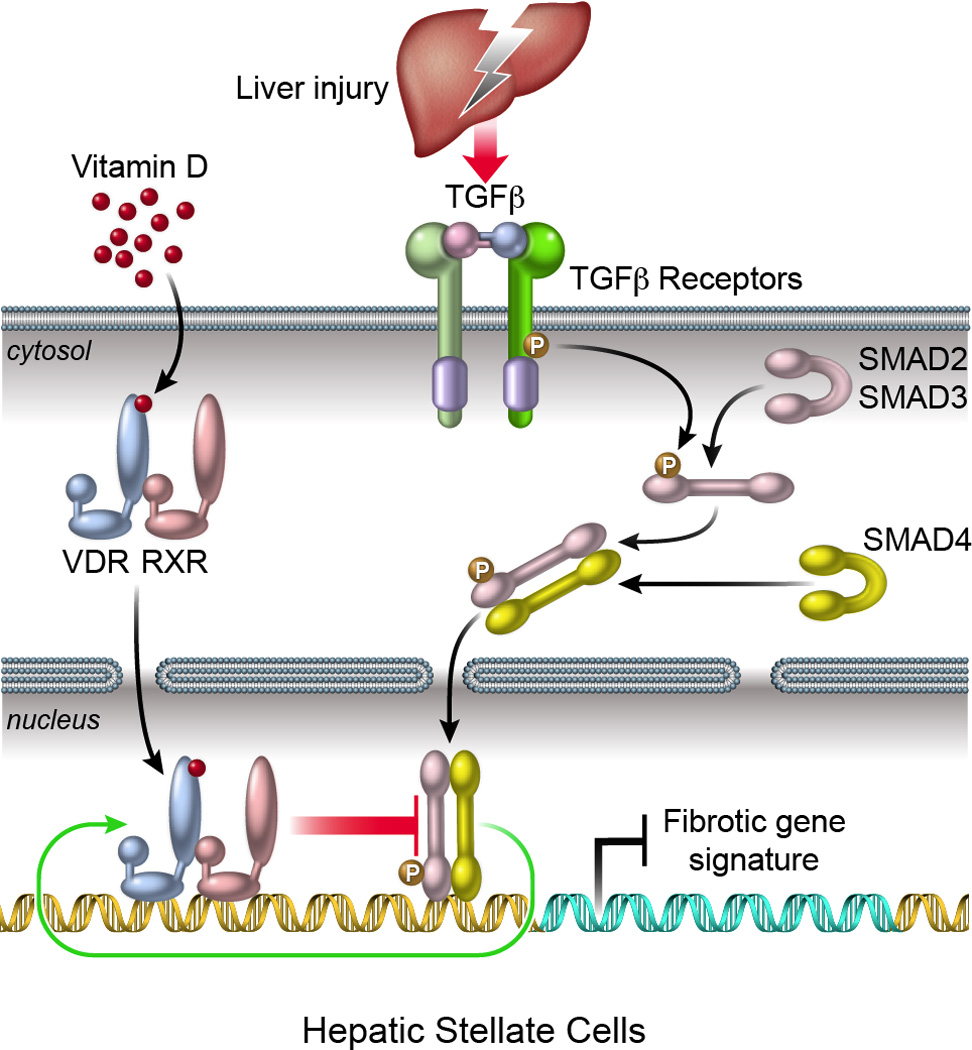
In contrast to hepatocytes, which constitute over 90% of liver mass and exhibit little or no VDR expression, VDR is highly expressed in the non-parenchymal cells of the liver. Kupffer cells (KCs), sinusoidal endothelial cells and especially hepatic stellate cells (HSCs) [3] all express VDR. In regards to chronic liver diseases, HSCs are the major effector cells in the wound healing process leading to liver fibrosis, being the predominant source of both normal and pathological hepatic extracellular matrix[5,6]. This cell-specific expression pattern of VDR suggests that the liver could be vitamin D responsive mediated through its non-parenchymal constituents. In support of this notion, activation of VDR in HSCs has recently been shown to strongly antagonize TGFβ signaling, the most potent pro-fibrogenic pathway in liver, resulting in suppression of a broad range of pro-fibrotic genes including collagens, integrins and tissue inhibitors of metalloproteinase (TIMPs)[7–9]. Moreover, associated genetic studies in mice reveal that spontaneous liver fibrosis occurs when one or both Vdr alleles are knocked out, with more severe fibrosis observed in Vdr−/− animals[8]. In humans, multiple studies have reported a high prevalence of vitamin D deficiency in patients with chronic liver diseases of diverse etiology[10], though the cause of this remains uncertain. However, studies correlating vitamin D deficiency with progression of chronic liver disease have generated conflicting results. Among patients with advanced liver fibrosis due to chronic hepatitis C infection, vitamin D status did not appear to impact disease progression[10]. In contrast, very low circulating levels of vitamin D were associated with worse liver disease, a higher rate of cirrhosis and increased mortality in patients with alcoholic liver disease[11]. Additionally, vitamin D and VDR polymorphisms appear to impact on the success of anti-viral therapy for HCV (hepatitis C virus) infection [12], while hepatitis B replication measured by serum HBV-DNA is inversely correlated with serum vitamin D[13]. Thus, while preclinical, clinical and epidemiological research all suggest a link between vitamin D and liver disease, the nature of this association is poorly understood.
In order to advance the use of VDR ligands as treatments for chronic liver disease and especially to define if they have a role in attenuating hepatic fibrogenesis, a comprehensive understanding of the mechanistic basis of VDR action in the liver is required. At a molecular level, VDR primarily operates as a ligand-dependent transcription factor to regulate gene expression through three different mechanisms: 1. Direct activation of gene expression by binding to VDREs in proximal promoter regions; 2. Repression of gene expression by binding to negative VDREs; 3. Repression of gene expression by antagonizing the action of other transcription factors[1]. A powerful tool to investigate these mechanisms has recently become available, combining chromatin immunoprecipitation with high-throughput deep-sequencing (ChIP-Seq). Utilizing this technology, we examined genome-wide protein-DNA interactions and discovered that VDR suppresses the fibrotic response in HSCs through antagonism of SMAD3 recruitment, a transcription factor in the TGFβ signaling pathway, to regulatory regions of pro-fibrotic genes[8]. Specifically, TGFβ induces chromatin remodeling, allowing ligand-bound VDR/RXR heterodimers to access genomic sites occupied by SMAD3 and displace SMAD3 from the chromatin[8]. Discovery of this dynamic genomic circuit not only represents a novel gene-regulatory mechanism for VDR but also exemplifies the power of NextGen sequencing technology for predicting biological pathways with potential applications in the clinic. Costs of NextGen sequencing have decreased significantly since 2005[14], increasing both the availability of this technology and its utility in identifying potential therapeutic pathways.
The intrinsic anti-fibrotic properties of VDR strongly suggest VDR ligands as a novel class of therapeutic agents in the treatment of patients with chronic liver disease, especially those likely to progress to significant liver fibrosis or cirrhosis. Accordingly, preclinical studies have demonstrated that systemic administration of the endogenous VDR natural ligand, calcitrol, or a synthetic ligand such as calcipotriol, effectively inhibited early hepatic fibrogenesis in standard rodent models of liver injury [8,9].
Despite the experimentally-supported rationale for targeting VDR to treat liver diseases, several major challenges remain. Firstly, it is not clear if it is sufficient to replenish vitamin D by administering oral vitamin D3 (cholecalciferol,) or if potent direct VDR ligands, such as 1,25-(OH)2D3 or its synthetic analogues are necessary? Secondly, if the natural ligands prove most effective, strategies are required to minimize hypercalcaemia, due to the impact of VDR ligands on calcium absorption and transport in the intestine and kidney[1]. In this respect, at least two options are available to mitigate this side effect: administration of less calcemic or noncalcemic VDR ligands or targeted delivery of VDR ligands to the liver. As so-called “less-calcemic” VDR ligands have not achieved desirable deprivation of calcemic action and “non-calcemic” VDR ligands are still in development, targeted delivery of VDR ligands appears the most promising therapeutic strategy to maximize their efficacy while simultaneously reducing their side effects.
For example, vitamin A-coupled liposomes could be a powerful delivery approach for targeting HSCs given the unique role of quiescent HSCs for storing retinoids in cytoplasmic lipid droplets[15]. An additional issue is that the VDR-inducible cytochrome P450 24-Hydroxylase (24-OHase, CYP24A1) efficiently catalyzes the inactivation of VDR ligands with a secosteroid backbone (e.g. calcitriol, calcipotriol)[16], which constitutes a self-adjusting regulatory loop by diminishing both the availability and therapeutic activity of VDR ligands at target sites. To maximize the efficacy of VDR ligands, CYP24A1 activity needs to be suppressed by cytochrome P450 or CYP24A1-selective inhibitors[17,18].
Another facet of VDR action relevant to chronic liver disease is its anti-inflammatory activity[1,2]. In most forms of liver disease, inflammation underlies the activation of HSCs, which in turn leads to liver fibrosis. Activation of toll-like receptors (TLRs) expressed in HSCs and KCs is a key aspect of many forms of liver inflammation leading to activation of nuclear factor-kappa B (NF-κB) and transcription factor AP-1 resulting in the induction of numerous pro-inflammatory cytokine and chemokine genes[19]. The contribution of TLR-driven inflammation in fibrosis is evidenced by a recent study where experimental liver fibrosis was suppressed in TLR4-mutant mice[20]. Thus, VDR ligands have the potential to converge anti-inflammatory and anti-fibrotic actions into a single therapeutic agent to provide a much needed treatment option for chronic liver diseases where the underlying cause cannot be removed or significantly abrogated.
In summary, it is now apparent that VDR ligands targeting non-parenchymal cells in liver, as exemplified by HSCs, have the potential to ameliorate one of the most troubling aspects of chronic liver disease, namely the wound healing response that gives rise to liver fibrosis and in severe cases, to cirrhosis. It may not be enough to render patients vitamin D replete through supplementation, necessitating the development of new drugs or the reformulation of existing compounds. Further basic and translational studies, particularly around the development of potent VDR ligands that target HSCs without inducing hypercalcemia, are required before trials can be carried out to evaluate VDR ligands in the clinic. Nevertheless, with increasing interest in VDR biology in the liver, both benchside and bedside studies could move VDR-targeted therapies to the center stage of hepatology and pave the way for the development of a new pipeline of VDR ligands with therapeutic promise for a wide range of liver disorders.
Figure 1.
ACKNOWLEDGEMENTS
We thank C. Brondos and E. Ong for administrative assistance. N.D. was supported by a postdoctoral fellowship from Genentech Foundation. This work was funded by grants from NIH (DK057978, HL105278, DK090962, HL088093, ES010337 and CA014195) and National Health and Medical Research Council of Australia Project Grants 512354 and 632886 (C.L. and M.D.), as well as the Helmsley Charitable Trust, Samuel Waxman Cancer Research Foundation and Ipsen/Biomeasure. R.M.E. and M.D. are supported in part by a Stand Up to Cancer Dream Team Translational Cancer Research Grant, a Program of the Entertainment Industry Foundation (SU2C-AACR-DT0509). R.M.E is an investigator of the Howard Hughes Medical Institute and March of Dimes Chair in Molecular and Developmental Biology at the Salk Institute.
References
- 1.Bouillon R, Carmeliet G, Verlinden L, et al. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr Rev. 2008;29(6):726–776. doi: 10.1210/er.2008-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosen CJ, Adams JS, Bikle DD, et al. The nonskeletal effects of vitamin D: an Endocrine Society scientific statement. Endocr Rev. 2012;33(3):456–492. doi: 10.1210/er.2012-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gascon-Barre M, Demers C, Mirshahi A, Neron S, Zalzal S, Nanci A. The normal liver harbors the vitamin D nuclear receptor in nonparenchymal and biliary epithelial cells. Hepatology. 2003;37(5):1034–1042. doi: 10.1053/jhep.2003.50176. [DOI] [PubMed] [Google Scholar]
- 4.Han S, Li T, Ellis E, Strom S, Chiang JY. A novel bile acid-activated vitamin D receptor signaling in human hepatocytes. Mol Endocrinol. 2010;24(6):1151–1164. doi: 10.1210/me.2009-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88(1):125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115(2):209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Potter JJ, Liu X, Koteish A, Mezey E. 1,25-dihydroxyvitamin D3 and its nuclear receptor repress human alpha1 (I) collagen expression and type I collagen formation. Liver Int. 2013;33(5):677–686. doi: 10.1111/liv.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding N, Yu RT, Subramaniam N, et al. A Vitamin D Receptor/SMAD Genomic Circuit Gates Hepatic Fibrotic Response. Cell. 2013;153(3):601–613. doi: 10.1016/j.cell.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abramovitch S, Dahan-Bachar L, Sharvit E, et al. Vitamin D inhibits proliferation and profibrotic marker expression in hepatic stellate cells and decreases thioacetamide-induced liver fibrosis in rats. Gut. 2011;60(12):1728–1737. doi: 10.1136/gut.2010.234666. [DOI] [PubMed] [Google Scholar]
- 10.Song BJ, Rockey DC. Status of research on vitamin D supplementation in treating or preventing liver fibrosis. Liver Int. 2013;33(5):653–655. doi: 10.1111/liv.12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trepo E, Ouziel R, Pradat P, et al. Marked 25-hydroxyvitamin D deficiency is associated with poor prognosis in patients with alcoholic liver disease. J Hepatol. 2013;8278(13) doi: 10.1016/j.jhep.2013.03.024. 00203-00201. [DOI] [PubMed] [Google Scholar]
- 12.Rahman AH, Branch AD. Vitamin D for your patients with chronic hepatitis C? J Hepatol. 2013;58(1):184–189. doi: 10.1016/j.jhep.2012.07.026. [DOI] [PubMed] [Google Scholar]
- 13.Farnik H, Bojunga J, Berger A, et al. Low vitamin D serum concentration is associated with high levels of hepatitis B virus (HBV) replication in chronically infected patients. Hepatology. 2013;2013(22):26488. doi: 10.1002/hep.26488. [DOI] [PubMed] [Google Scholar]
- 14.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26(10):1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 15.Sato Y, Murase K, Kato J, et al. Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat Biotechnol. 2008;26(4):431–442. doi: 10.1038/nbt1396. Epub 2008 Mar 1030. [DOI] [PubMed] [Google Scholar]
- 16.Lohnes D, Jones G. Further metabolism of 1 alpha,25-dihydroxyvitamin D3 in target cells. J Nutr Sci Vitaminol (Tokyo) 1992:75–78. [PubMed] [Google Scholar]
- 17.Schuster I, Egger H, Bikle D, et al. Selective inhibition of vitamin D hydroxylases in human keratinocytes. Steroids. 2001;66(3–5):409–422. doi: 10.1016/s0039-128x(00)00159-8. [DOI] [PubMed] [Google Scholar]
- 18.Kan PB, Hirst MA, Feldman D. Inhibition of steroidogenic cytochrome P-450 enzymes in rat testis by ketoconazole and related imidazole anti-fungal drugs. J Steroid Biochem. 1985;23(6A):1023–1029. doi: 10.1016/0022-4731(85)90062-7. [DOI] [PubMed] [Google Scholar]
- 19.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 20.Seki E, De Minicis S, Osterreicher CH, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13(11):1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]