

Abstract
Brain vascular malformations are resource-intensive to manage effectively, are associated with serious neurological morbidity, lack specific medical therapies, and have no validated biomarkers for disease severity and progression. Investigators have tended to work in “research silos” with suboptimal cross-communication. We present here a paradigm for interdisciplinary collaboration to facilitate rare disease research. The Brain Vascular Malformation Consortium (BVMC) is a multidisciplinary, inter-institutional group of investigators, one of 17 consortia in the Office of Rare Disease Research Rare Disease Clinical Research Network (RDCRN). The diseases under study are: familial Cerebral Cavernous Malformations type 1, common Hispanic mutation (CCM1-CHM); Sturge-Weber Syndrome (SWS); and brain arteriovenous malformation in hereditary hemorrhagic telangiectasia (HHT). Each project is developing biomarkers for disease progression and severity, and has established scalable, relational databases for observational and longitudinal studies that are stored centrally by the RDCRN Data Management and Coordinating Center. Patient Support Organizations (PSOs) are a key RDCRN component in the recruitment and support of participants. The BVMC PSOs include Angioma Alliance, Sturge Weber Foundation, and HHT Foundation International. Our networks of clinical centers of excellence in SWS and HHT, as well as our PSOs, have enhanced BVMC patient recruitment. The BVMC provides unique and valuable resources to the clinical neurovascular community, and recently reported findings are reviewed. Future planned studies will apply successful approaches and insights across the three projects to leverage the combined resources of the BVMC and RDCRN in advancing new biomarkers and treatment strategies for patients with vascular malformations.
Approximately 25 million people in the United States are affected by an estimated 7,000 rare diseases or conditions leading to significant morbidity and mortality. An operational definition of “rare disease” was proposed in an amendment1 to the Orphan Drug Act of 1983:2 “any condition affecting fewer than 200,000 Americans or a disease with a greater prevalence but for which no reasonable expectation exists that the costs of developing or distributing a drug can be recovered from the sale of the drug in the United States.” In addition to the unmet healthcare needs, many rare diseases result from unique single or multiple gene defects that offer insight into normal biologic function. Understanding the pathogenesis of rare diseases may advance our understanding of other more common medical disorders.
The longitudinal natural history has not been sufficiently characterized for many rare diseases, including the neurovascular disorders we are studying: (1) Cerebral Cavernous Malformations type 1, common Hispanic mutation (CCM1-CHM); (2) Sturge-Weber syndrome (SWS); and (3) brain arteriovenous malformation (bAVM) in hereditary hemorrhagic telangiectasia (HHT). Treatment can be equally challenging with many unanswered questions and a lack of evidence-based guidelines concerning clinical management. Rare disease patients are small populations, in most instances dispersed over wide geographic areas. There are significant logistical challenges to systematic study of rare diseases,3 which requires collaboration of scientists from multiple disciplines sharing what are typically scarce research resources and patient samples. The Brain Vascular Malformation Consortium (BVMC), part of the Rare Diseases Research Network (RDCRN),4 addresses these challenges.
History and Organization of the Rare Diseases Clinical Research Network
An NIH Office of Rare Disease Research (ORDR) Special Emphasis Panel made recommendations on the special research and health care issues posed by rare diseases,5 followed by passage of the Rare Diseases Act of 2002 that directed ORDR to support regional centers of excellence for clinical research into, training in, and demonstration of diagnostic, prevention, control, and treatment methods for rare diseases. The ORDR responded with creation of the RDCRN utilizing a U54 (NIH cooperative agreement award) mechanism. The first request for application (RFA) for funding was issued in 2003 and represented a collaboration between ORDR and several NIH Institutes/Centers, including the National Center for Research Resources (NCRR), National Institute of Neurological Disorders and Stroke (NINDS), National Institute of Child Health and Human Development (NICHD), National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), and National Heart Lung and Blood Institute (NHLBI). A similar request for application for funding was then issued by ORDR in 2008, in collaboration with NINDS, NIAMS, NIDDK, NHLBI, the National Institute of Alcohol Abuse and Alcoholism, National Cancer Institute, and National Institute of Dental and Craniofacial Research, to which the BVMC projects responded. The BVMC is currently in Year 4 of the five-year funding cycle.
The RDCRN is a cooperative network composed of 17 Rare Diseases Clinical Research Consortia (RDCRC) and a single Data Management and Coordinating Center (DMCC) to facilitate clinical research in rare diseases carried out by the RDCRCs. A Steering Committee is composed of the Principal Investigators (Program Directors) of each RDCRC, the Principal Investigator (Director) of the DMCC, the RDCRN Program Director from ORDR, NCATS, and program scientists from NIH Institutes. Each RDCRC generally includes a consortium of clinical investigators, institutions, CTSA units (or their equivalent), and relevant organizations, including patient support organizations (PSO), for the study of a group of at least three rare diseases that are relevant to the NIH Institutes. The DMCC provides RDCRN coordination, data management infrastructure, and other administrative support.
Having the BVMC function within the larger context of the RDCRN has created the opportunity to collaborate with other consortia with varying degrees of common interests, and as importantly, the opportunity to interact with more than 95 PSOs, representing over 200 rare disorders. The RDCRN has allowed an economy of scale in terms of providing assistance with the standardization of data collection, both conceptually and in terms of data collection instruments. The DMCC also provides the centers support in protocol and amendments for submission to the NIH, preparation for audits, and assistance in obtaining IRB approval for their protocols. Biannual RDCRN in-person Steering Committee meetings and every-other-year conferences provide a venue for interactions and discussions that directly benefit development of the BVMC. If the individual projects existed in isolation from the BVMC and RDCRN context, these opportunities would be missed, including the ability to learn from what has worked—and not worked—for other rare disease consortia.
Brain Vascular Malformation Consortium (BVMC)
The three projects correspond to the three rare neurovascular disorders (Figure 1). These projects are supported by an Administrative Unit (AU) and a Genetics and Statistical Analysis Core (GSAC). Two additional components are a Training Module that provides partial support for one rare disease research trainee per year and a Pilot/Demonstration Project Program (PPP) that supports one pilot project per year (Table 1).
Figure 1.
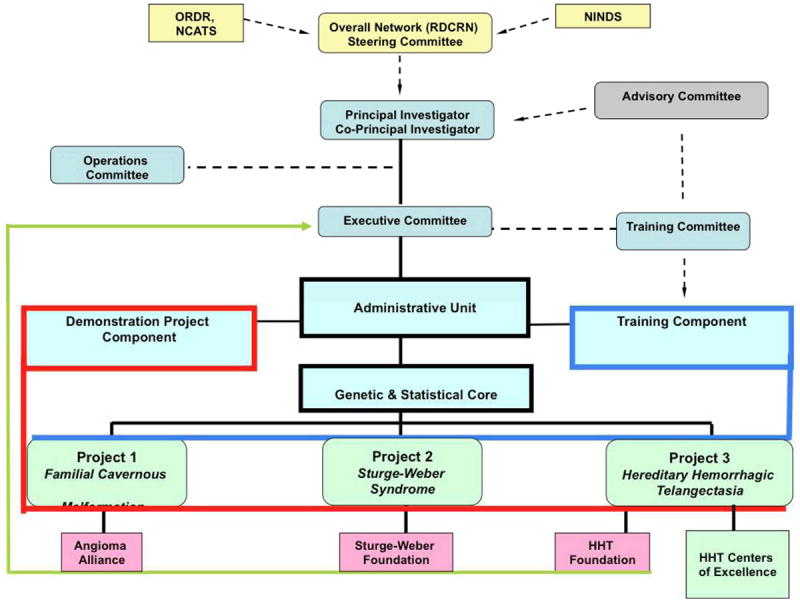
Organizational chart for Rare Diseases Clinical Research Consortium administrative functions (light blue) and experimental service cores serving all projects. NINDS, National Institute of Neurological Disorders and Stroke; ORDR, Office of Rare Diseases Research; NCATS, National Center for Advancing Translational Sciences; RDCRN, Rare Diseases Clinical Research Network
Table 1.
BVMC Rare Disease Research Trainees and Pilot Projects.
| Year | Training Period | Institution | Project No. | Trainee |
|---|---|---|---|---|
| 1 | 9/1/09-6/30/10 | University of New Mexico | 1 | Yasir Khan |
| 2 | 7/1/10-6/30/11 | University of Toronto and Yale University | 3 | Takeo Nishida |
| 3 | 7/1/11-6/30/12 | Kennedy Krieger | 2 | Eboni Lance |
| 4 | 7/1/12-6/30/13 | University of California, SanFrancisco | 1 | Hélène Choquet |
| 5 | 7/1/13-6/30/13 | TBD | TBD | |
| Year | Project Pilot Period | Institution | Project No. | Description |
| 1 | 9/1/09-6/30/10 | Kennedy Krieger | 2 | Establish reliability of quantitive EEG, transcranial Doppler, behavioral outcomes and optical coherence tomography in SWS: a first step toward biomarker development |
| 2 | 7/1/10-6/30/11 | University of New Mexico | 1 | Permeability in MRI in cerebral cavernous Type 1 in New Mexico: effects of statins |
| 3 | 7/1/11-6/30/12 | University of Toronto | 3 | Topical antiangiogenic therapy of telangiectasia in HHT: proof of concept |
| 4 | 7/1/12-6/30/13 | TBD | ||
| 5 | 7/1/13-6/30/14 | TBD |
Abbreviations: EEG, electroencephalogram; HHT, hereditary hemorrhagic telangiectasia; MRI, magnetic resonance imaging; SWS, Sturge-Weber syndrome; TBD, to be determined
In many respects, both basic and clinical researchers in the area of cerebrovascular malformations have tended to work in “research silos” with suboptimal cross-communication among diseases, even those in the same vascular bed. This “silo problem” was one of the motivations for NINDS to hold a workshop on “Biology of Vascular Malformations of the Brain” in March 2008,6 in which most of the BVMC key investigators participated; the workshop was a key event to set the stage for development of the BVMC.
The three disorders were chosen for several reasons. Even though causative genetic defects have been identified for two of the three disorders, they are all poorly understood in terms of biological mechanisms, are resource-intensive to manage effectively, and have high probability of serious neurological morbidity. Each disease is characterized by the development of a distinct set of vascular malformations and a unique spectrum of clinical and phenotypic outcomes, for which biological risk factors are either poorly understood or completely unknown.
All three disorders share a common biological theme: a brain vascular phenotype resulting from failure of the normal physiological mechanisms of blood vessel formation or maintenance. Thus, each of these disease states represents some degree of disordered angiogenesis and vascular remodeling.6-9 Although they presumably proceed through different final pathways, insights from one disease may help illuminate the biological basis of or treatment strategies for the others, as well as related cerebrovascular malformations that are more common.9 There is considerable overlap in the neurological morbidity of these disorders, e.g., intracranial hemorrhage (ICH), seizures and focal deficits. Therefore, therapy aimed at stabilizing the abnormal vessels in one disorder may well be adaptable to more than one disease. Importantly, specific medical therapy is not currently available for any of the three disorders. The establishment of clinical research infrastructure and data registries will facilitate multi-center clinical studies of all types.
Central themes of the BVMC are the development of patient registries (both cross-sectional and longitudinal) and biomarkers; the program structure (Figure 1) serves these themes. The first aim of each project is development of a deidentified relational database. Each of the diseases has a large number of dedicated clinicians and scientists studying basic and clinical aspects of the disease, while providing the best currently available care. Nonetheless, progress has been hampered, and the pace of discovery held back, by the lack of a central repository for standardized clinical information with common data elements. The extant registries for each disease were either center-specific or, in the case of foundation listings, developed for communication purposes and not designed as research-grade relational databases that utilized common data elements. The BVMC infrastructure and the attendant central support from the DMCC have been an historic opportunity that has brought together investigators in each disease area and created large-scale observational databases of affected individuals. The BVMC structure allows for cross-collaboration of experienced researchers between and within projects that includes sharing of resources and knowledge. Monthly conferences with BVMC key personnel, DMCC representatives and NIH Program staff are highly interactive and allow continual external input into the ongoing management and development of the BVMC.
Biomarker development is intended not only for use in risk stratification but also for insights into the genetic and molecular underpinnings of these diseases. The methodological technology for creating and validating biomarkers can be shared across projects, facilitated by the GSAC. Biomarkers for gauging natural history and disease progression risk are particularly important for vascular lesions of the brain: the risk of intervention, e.g., surgical or endovascular therapy, may outweigh the risks of the natural history of minimally-symptomatic disease. This issue has recently gained attention though an international multi-center randomized controlled trial that tests whether patient outcome is less morbid by conservative management than best procedural intervention (endovascular embolization, microsurgical resection or radiotherapy, alone or in combination). This trial, A Randomized Trial of Unruptured Brain Arteriovenous Malformation (ARUBA),10 has sparked considerable controversy as to whether unruptured bAVMs need to be treated because of their relatively benign course (low spontaneous hemorrhage rate and relatively mild sequelae of ICH) and relatively high risk of causing iatrogenic injury in minimally symptomatic patients.11-13 Interventionalists argue that intervention results in a better long-term outcome for patients, based on referral-based retrospective case series.14, 15 What is not controversial, however, is the need for better risk markers that can identify patients at highest risk for spontaneous hemorrhage in order to optimally and expeditiously provide treatment.16 Although ARUBA is concerned with sporadic bAVM, the issues confronting invasive treatment of HHT-related bAVM, and indeed all minimally symptomatic cerebrovascular lesions, are highly relevant to the focus of this BVMC and the clinical neuroscience community.
Patient Support Organizations (PSOs)
A mission-critical aspect of the RDCRN program is the collaborative involvement of PSOs—with their leadership as co-investigators—to provide patient-oriented input, advice on assessing research priorities, assistance with public awareness of the program, and facilitation of patient recruitment. To our knowledge, this is the first incorporation of PSOs into an ongoing scientific project in cerebrovascular disease as active collaborators.
Commonalities Among the 3 Patient Support Organizations
The Patient Support Organizations (PSOs) of the BVMC are international voluntary organizations driven by similar missions to inform and support individuals affected by vascular malformations, and to facilitate improved diagnosis and management through education and research. The PSOs work to support the scientific community by funding direct research through pilot grants, and by providing valuable communication and coordination roles, including organizing the annual BVMC executive committee face-to-face meeting. On a rotational basis, the meeting is planned to precede a CCM, HHT, or SWF Patient and Family Conference. This cooperative planning allows for BVMC investigators to both attend the executive meeting and present at the PSO-sponsored family conference in a cost- and time-effective manner.
The PSOs, in addition to scientists, government, industry and medical professionals, are key stakeholders toward the collaborative efforts working to advance pathobiological understanding and clinical care for brain vascular malformations. Each of the PSOs supports independent disease registries and patient databases. These registries are distinct from the aims of the BVMC, as they were designed for communication, to collect unique datasets, and/or to include data and/or participants beyond the scope of the BVMC projects. The purpose of each of these registries was not as a prospective natural history study. Importantly, however, the PSO registries provided valuable models for the design and development of BVMC natural history databases.
The PSOs also aim to improve quality of life for those impacted by diagnosis. To this end, the PSOs sponsor advocacy and provide information and support for patients and their families. Up-to-date disease information is provided through the individual PSO websites and newsletters. Each website also features a discussion forum, and regional networking is facilitated and encouraged through social media and at Patient and Family Conferences. These activities foster a sense of community and trust between the PSOs and affected individuals. The strength of these community relationships and tireless efforts of the PSOs offer the BVMC both support from the patient community as well as the vital observational data that helped to originally identify commonalities among CCM, HHT and SWS and initiated genesis of the BVMC.
All three PSOs have patient registries, but they primarily are geared for patient identification, whereas the BVMC databases are more granular, use standardized common data elements, and were specifically designed to address individual project aims, including longitudinal follow-up.
The BVMC PSOs include the following three groups
Angioma Alliance (AA)
In addition to the common patient support efforts described above, AA is the host of the “CCM Scientific Meeting.” Since its inauguration in 2005, this annual meeting has brought together an international group of basic, translational and clinical researchers, including investigators from all three BVMC projects, to share pre-publication findings and explore possible collaborations. In an effort to facilitate recruitment for research and clinical studies, AA launched the International Cavernous Angioma (CCM) Patient Registry in 2010. This web-based communication tool is designed to link the patient and research communities to distribute information about studies to potential participants. Importantly, the AA-managed patient registry is inclusive of all forms of CCM, and is intended to facilitate recruitment for a wide range of CCM research studies. In 2006, to further support the CCM research community, AA established an independent CCM DNA & Tissue Bank, including genetic testing facilities and an extensive clinical database. The data collected include information from medical records, imaging, and patient interviews with annual follow-up. These data are intended to mirror and expand upon those collected by the Scottish Intracranial Vascular Malformations Study (SIVMS),17, 18 and were also used as a model for development of the BVMC CHM-CCM research registry to allow for future international collaboration and use of a comprehensive data set including all forms of CCM.
Sturge-Weber Foundation (SWF)
To speed the pace of discovery in SWS, the SWF funded a Translational Research Summit and subsequently established Centers of Excellence (COE), to collaborate in gathering clinical and scientific data and establish standards of diagnosis and care. The SWF has co-sponsored two NIH workshops on angiogenesis: the biology of vascular malformations,6 and an NIH meeting specifically focused on research priorities for SWS. The SWF has been instrumental in facilitating the collection of blood, brain, and skin tissue samples for scientific research stored at National Disease Research Interchange. Additionally, the SWF has collected clinical data in a natural history registry of over 4,300 SWS patients that includes up to 25 years of follow-up, including demographic and at least partial clinical data on both the patients and their family members. This database project and data collection is ongoing.
HHT Foundation International
The HHT Foundation is engaged in increasing access to expert HHT care throughout North America, overseeing and supporting the establishment of 16 North American HHT Treatment Centers of Excellence to date. The Foundation brought HHT genetic testing to clinical care in North America, advocating for patients and facilitating negotiations among stakeholders. The Foundation’s grant program has funded 11 Research Grants totaling $2.5M since 2004, as well as 12 Young Researcher Grants. Several conferences have been supported by the HHT Foundation, including an NIH HHT Research Conference in June 2006, the conference for development of the first International HHT Guidelines,19 a National Disease Research Exchange for HHT Tissue research, two NIH workshops on angiogenesis and neurovascular malformations, and the Genetics and Genomics of Vascular Disease Workshop sponsored by the North American Vascular Malformations Research meeting. Since the first International Scientific Conference in 1996, HHT Foundation International has supported 10 biennial international conferences bringing together basic, translational and clinical researchers. In terms of patient, family and physician education, the Foundation has sponsored two hands-on physician training programs, 16 national conferences, four regional conferences in collaboration with HHT Centers of Excellence, and continuing education medical programs for physicians and healthcare professionals. Furthermore, the HHT Foundation supported the first-ever Centers for Disease Control HHT Conference in March 2008. The Foundation has worked closely with the BVMC investigators, actively participating in participant recruitment through communications with their large patient contact registry and social networks. In addition, the Foundation is currently driving an initiative to develop a prospective North American HHT database, including comprehensive data regarding natural history of all aspects of HHT, which will be complementary to the BVMC HHT bAVM database.
Main Disease Projects
Project 1: Modifiers of disease severity in cerebral cavernous malformations
Cerebral Cavernous Malformation (cavernous angioma; cavernoma; cryptic vascular malformation) is a vascular abnormality of the central nervous system (brain and spinal cord) with extra-CNS involvement of skin in some patients. The lesions consist of a cluster of abnormal, dilated capillaries invariably containing extravasated blood products of various ages; lesions are low-flow without shunting of blood, and detected on MRI by the typical patterns of blood breakdown products (Figure 2).20 CCMs are a significant source of neurological morbidity, including intracranial hemorrhage (ICH), seizures, and focal neurological deficits. CCMs can occur in a solitary, sporadic form, or occur in an autosomal dominant condition typically characterized by multiple lesions on brain MRI that appear to increase with age (Figure 3).21
Figure 2.
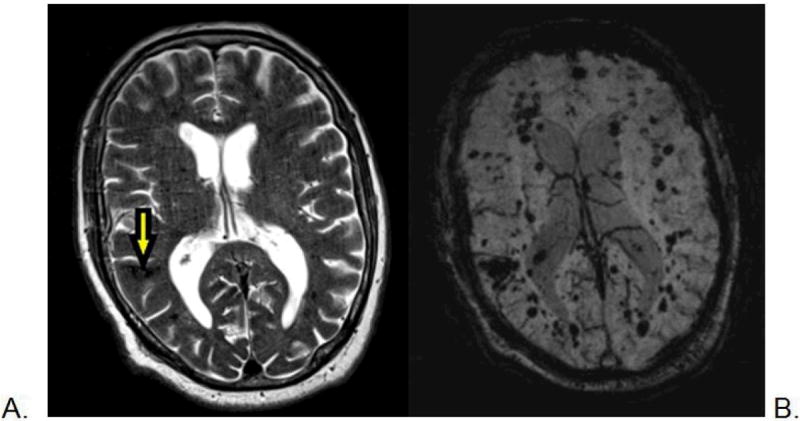
Magnetic resonance imaging scan of a 78-year-old man with CCM1-CHM. Axial T2-weighted image (A) shows a posterior right temporal lobe typical CCM with reticulated, mixed signal internally and peripheral hemosiderin rim (arrow), and a few small additional small foci of low signal. The axial susceptibility-weighted image (B) is more sensitive for blood breakdown products and shows numerous areas of low signal intensity (dark areas on the image) within small CCMs.
Figure 3.
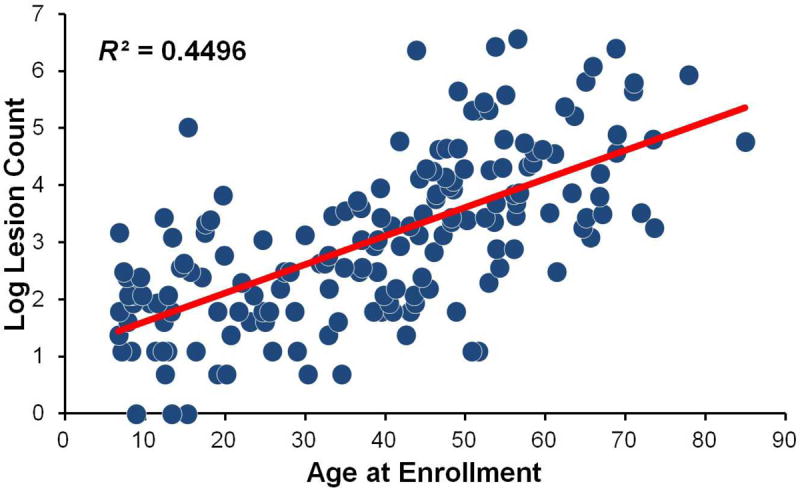
Scatterplot of log-transformed lesion count by age in 176 CCM1-CHM patients showing a significant positive linear relationship (R2=0.4496, P<.001). Linear regression analysis showed a 5% increase in lesion count for every 1-year increase age (95% CI=1.04-1.06, P<.001).
Project 1 studies one of the familial forms of the disease known as CCM1 caused by loss-of-function mutations in the KRIT1 (Krev Interaction Trapped 1) gene. In the Hispanic population of New Mexico, virtually all cases are caused by the same KRIT1 founder mutation (Q248X), termed the Common Hispanic Mutation (CHM).22-24 Genealogy studies conducted in New Mexico CCM families suggest the mutation originated in a common ancestor from Spain who relocated to Mexico in the 1500s, traveled up El Camino Real, and settled in the New Mexico area. Because of this founder mutation, the autosomal dominant transmission of disease, large Hispanic families, and settling in this geographic area, CCM1-CHM is a huge public health burden in the State of New Mexico. The estimated population prevalence of CCM1-CHM worldwide is less than 1/100,000, but in New Mexico the prevalence is much higher at 15/100,000.
The natural history of CCM is not clearly established nor is it known why CCM patients present with a wide range in symptoms and lesions, even among those with the same gene mutation or age. The overall goal of this project is to identify and understand modifiers of disease severity in familial CCM to improve patient care and prognosis. The project will recruit a relatively large cohort (n=500) of familial CCM1 cases, all carriers of the CHM (CCM1-CHM) and geographically clustered in New Mexico, to identify clinical characteristics and modifier genes influencing lesion burden, a marker for disease severity. We are also evaluating the association between lesion burden and risk of hemorrhage, seizures and other neurological sequelae. Results from this genetically homogenous group are likely to yield important findings that may have relevance to the CCM community at large.
Aim 1 is to establish a database of CCM1-CHM cases recruited through the University of New Mexico in collaboration with the RDCRN DMCC and the Angioma Alliance PSO. Thus far, we have recruited 218 patients towards our goal of 500 familial CCM1-CHM patients by 2014. We are collecting detailed clinical characteristics, neurological exams, imaging features from MRI and susceptibility-weighted imaging, and blood specimens at baseline; we are also performing annual follow-up phone calls to obtain outcome information. We will identify clinical characteristics associated with lesion burden (i.e., number of lesions) and define specific clinical and imaging features unique to this large cohort, paying particular attention to how the disease features cluster within families.
Table 2 summarizes characteristics for eligible participants enrolled in our study as of December 2012: 190 subjects from 43 families with 2 or more members and 42 singletons had partial or complete information entered from data collected at baseline visits. At study enrollment, most participants were symptomatic (88%) with a mean age of 39.1 ± 19.7 years; the mean age at diagnosis was younger (31.8 ± 19.3 years). Approximately 24% presented with a hemorrhage, and 26% had surgical resection for a CCM lesion. As expected, lesion count was highly variable and the distribution was right skewed. The mean number of lesions was 59.5 ± 114.9 and the median was 18 (IQR: 6 – 50.5). Three subjects had no lesions and five harbored a single lesion (Figure 3), reflecting our ascertainment strategy of recruiting family members based on CCM1-CHM carrier status and not phenotype. This strategy ensures that we see a broad spectrum of phenotypes associated with the disease. Of interest, two of the cases with a single lesion were in their third decade of life. CCM1-CHM cases also had a high prevalence of co-morbid conditions. Clinically, the majority of cases were neurologically normal (93%) and had little to no significant disability (88% had modified Rankin score of <3).
Table 2.
Baseline Demographic and Clinical Characteristics of Patients with Familial CCM1-CHM Enrolled in Project 1.
| Baseline Characteristics | Value |
|---|---|
|
| |
| Female gender, No. (%) | 123 (65) |
|
| |
| Age at enrollment, mean (SD), year | 39.1 (19.7) |
|
| |
| Age at diagnosis mean (SD), year | 31.8 (19.3) |
|
| |
| Symptomatic at presentation, No. (%) | 122 (64) |
| Hemorrhage | 46 (24) |
| Seizure | 46 (24) |
| NH-FND | 7 (4) |
| Headache | 41 (22%) |
|
| |
| Ever symptomatic, No. (%) | 167 (88) |
| Hemorrhage | 63 (33) |
| Seizure | 59 (31%) |
| NH-FND | 9 (5) |
| Headache | 101 (53) |
|
| |
| Comorbidities | |
| Obesity | 68 (38) |
| Hypertension | 56 (29) |
| Hyperlipidemia | 98 (52) |
| Depression/Anxiety | 34 (18) |
| Diabetes | 18 (9) |
| Arthritis | 22 (12) |
| Hypothyroidism | 13 (7) |
Abbreviations: CCM1-CHM, cerebral cavernous malformations type 1, common Hispanic mutation, NH-FND, Nonhemorrhagic Focal Neurological Deficit
Aim 2 is discovery-driven, based on the overarching hypothesis that there is a distinct set of genetic modifiers acting in concert with CCM1-CHM to result in higher lesion burden. We will perform a cross-sectional genome-wide association study (GWAS) to identify modifier genes influencing CCM lesion burden. We have already genotyped 176 CCM1-CHM cases using the Affymetrix Axiom Latino array at the University of California, San Francisco, and will test for association with lesion burden when we achieve our target enrollment of 500. Our primary GWAS analysis will adjust for potential confounders, including age, gender, and Hispanic ancestry, and other important clinical covariates identified in Aim 1, followed by additional fine-mapping and haplotype analysis. Secondary analysis will examine copy number variants (CNV).
Aim 3 is to perform longitudinal analysis of lesion growth (number and size) over the study observation period. The first 100 patients enrolled in Year 1 of the study will be brought back for a follow-up MRI at Year 5 to assess change in lesion growth. Exploratory outcomes will include changes in modified Rankin scores and SF-36 quality of life scores.
Project 1 utilizes a unique practice setting at the University of New Mexico (UNM) and patient population, along with statistical and genetics expertise at UCSF from the GSAC, to identify biomarkers for CCM disease progression, using both cross-sectional and longitudinal designs. The longitudinal part of the design will be especially valuable as it will allow estimates for risk of hemorrhage (and clinical sequelae), development of seizures or epilepsy, and headaches over the period of study. Rare disease research is challenging; however we have benefitted greatly from the RDCRN infrastructure, including the RDCRN registry, and the AA for helping support educational family meetings, recruitment of study participants, and for identifying additional subjects for the study.
Project 2: Innovative approaches to gauge progression of Sturge-Weber Syndrome
Sturge-Weber Syndrome (SWS) is a rare congenital vascular disorder characterized by brain, ocular and cutaneous vascular anomalies. Clinically, SWS is characterized by a facial port-wine birthmark, glaucoma and choroidal angioma, leptomeningeal angioma, epilepsy, stroke-like episodes, headache, and cognitive impairment. The characteristic extracranial manifestations are facial port-wine stain and glaucoma. The population prevalence is estimated to be 5/100,000.
The severity of neurologic symptoms varies greatly, but can include blindness, paralysis, severe mental retardation, or sudden unexplained death in epilepsy. The cellular and molecular etiology of SWS remains unknown. However, the disease is characterized by variable degrees of vascular overgrowth, and histopathological studies of SWS lesion tissue show abnormal expression of vascular phenotypes and angiogenic factors. These observations suggest that the primary defect in SWS is abnormal angiogenesis (vascular development). The hypothesis of aberrant angiogenesis forms one arm of our central hypothesis and suggests that longitudinal analysis of angiogenesis biomarkers may have clinical relevance in the prediction of disease severity and outcome.
The vascular malformation of the brain in SWS consists of enlarged and tortuous leptomeningeal vessels and dilated deep venous vessels, most often involving the occipital cortex. Impaired venous drainage from the involved brain regions results in reduced arterial perfusion to these regions.29 SPECT (Single Photon Emission Computed Tomography) studies in young infants with SWS have shown that cerebral perfusion goes from being generous in the very young infant to being deficient in the involved cortical region by the end of the first year.30
The vessels of leptomeningeal angiomas are thin-walled venous structures, many of which are hugely dilated. The vessels are innervated only by noradrenergic sympathetic nerve fibers31 and show increased endothelin-1 expression,32 suggesting that there may be increased vasoconstrictive tone in SWS. The impaired venous drainage through these vessels results in reduced microcirculation and hypoxia in the surrounding brain tissue. Microscopically, the cortical tissue underlying the angioma shows neuronal loss, calcium deposition, hypoplastic blood vessels, breakdown of the blood-brain barrier and gliosis–reviewed in Comati et al.8 Changes are also seen in the underlying white matter: there is an early phase of hypermyelination, which is then followed by white matter loss.33
The brain lesions in SWS are progressive, suggesting that there is ongoing angiogenesis in these lesions. Consistent with this idea, increased levels of endothelial proliferation and apoptosis were seen in leptomeningeal vessels from SWS patients relative to those of controls,8 as well as over-expression of fibronectin.34 The vessels also showed increased levels of expression of VEGF, VEGF receptors 1 and 2, neuropilin, Tie-2, and HIF-1α and HIF-2α.8 Thus, leptomeningeal angiomas in SWS appear not to be static lesions, but rather show evidence of ongoing vascular remodeling.
The second arm of our central hypothesis stems from clinical observations and relates to the role of early somatic mosaicism (somatic mutation) in neurocutaneous mosaic phenotypes. Rudolf Happle first proposed the concept of mutation of autosomal lethal genes occurring early in development and surviving only in a mosaic state, to explain the genetic basis of several syndromes characterized by the following characteristics: (almost always) sporadic occurrence, distribution of lesions in a scattered or asymmetrical pattern, variable extent of involvement, lack of diffuse involvement of entire organs, and equal sex ratio.35 This phenotypic pattern was termed paradominant inheritance. Significantly, SWS was one of the phenotypes Happle used to formulate his theory.
The central hypothesis brings these two seminal concepts together and proposes that SWS is caused by an early somatic mutation in a gene involved in angiogenesis. We propose that a somatic mutation occurs in a cell derived from the primordial vascular plexus. As predicted by the paradominant inheritance hypothesis, we propose that this gene is essential to survival due to its critical importance in angiogenesis, such that only somatic mutations in this gene are compatible with life. The extent of the phenotype associated with SWS would depend on the time during development when the somatic mutation occurred. Angiogenesis biomarkers might therefore correlate with disease severity. Our work has investigated our central hypothesis with clinical, biochemical, and molecular analyses organized under the following aims.
Aim 1 creates a standardized SWS registry and clinical database to comprehensively define the phenotype of SWS brain involvement (Figure 4) and foster future clinical research. The registry is a resource for future clinical and molecular investigations of SWS. This database is being created through the collaborative efforts of the lead clinical site at the Kennedy Krieger Institute, the DMCC, the SWF, and the other recruiting SWF Centers of Excellence. It has also utilized the statistical resources of the BVMC. The contributing centers are currently working together to analyze the current database (see Table 3) and are being solicited for ideas for future research utilizing it.
Figure 4.
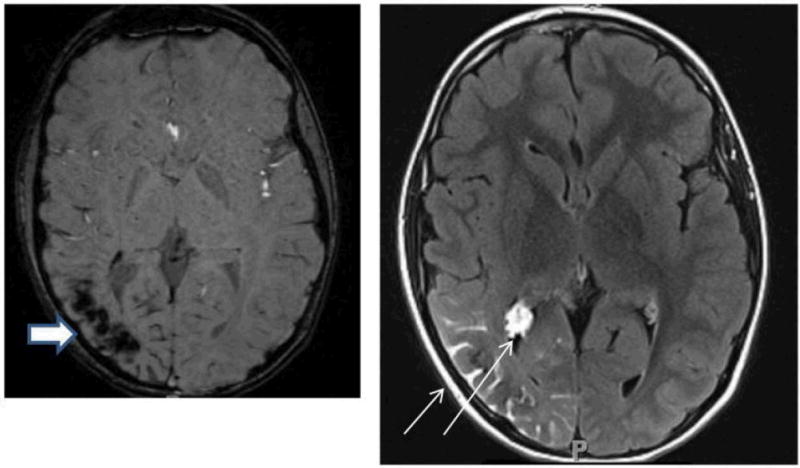
Axial MRI images from the same individual with Sturge-Weber syndrome showing typical imaging findings of loss of signal on susceptibility-weighted imaging in the right parietal-occipital region, indicating calcification (thick arrow, left panel) and leptomeningeal and choroid plexus enhancement (thin arrow, right panel).
Table 3.
Characteristics of Patients with Sturge-Weber Syndrome, Obtained from Deidentified Research Questionnaire.
| Characteristic | Study Population, No. (%) |
|---|---|
|
| |
| Gender (N=108) | |
|
| |
| Male | 46 (42.6) |
|
| |
| Female | 62 (57.4) |
|
| |
| Brain involvement (N=108) | |
|
| |
| Unilateral | 94 (87.0) |
|
| |
| Bilateral | 14 (13.0) |
|
| |
| Skin involvement (N=108) | |
|
| |
| Unilateral | 55 (50.9) |
|
| |
| Bilateral | 41 (38.0) |
|
| |
| None | 12 (11.1) |
|
| |
| Eye involvement (N=108) | |
|
| |
| Unilateral | 43 (39.8) |
|
| |
| Bilateral | 10 (9.3) |
|
| |
| None | 53 (49.1) |
|
| |
| Unknown | 2 (1.9) |
|
| |
| Epilepsy (N=108) | |
|
| |
| Yes | 97 (89.8) |
|
| |
| No | 8 (7.4) |
|
| |
| Unknown/Unattainable | 3 (2.8) |
|
| |
| Age of Diagnosis (N=101) | Mean=19.8 mo. |
| Range=0-624 mo. | |
Aim 2 evaluates the hypothesis that use of urine levels of angiogenesis biomarkers will correlate with and predict severity of neurologic progression in SWS. The study protocol collects data at baseline and then at 1 and 2 years’ follow-up. Neurological status is estimated using the Sturge-Weber neurologic clinical score.36 This biomarker study is a collaboration between the lead clinical site and the laboratory of Dr. Marsha Moses at Boston Children’s Hospital.
Aim 3 is the search for somatic mutations causing SWS. In collaboration with the University of Maryland Brain Bank, NDRI and the Sturge-Weber Foundation, we have banked autopsy or biopsy vascular tissue and matched control tissue from SWS patients. We will use a variety of whole genome approaches to search for somatic mutations in affected SWS tissue. Aim 3 is a collaborative effort between laboratories at the Kennedy Krieger Institute (Pevsner and Comi) and the Marchuk laboratory at Duke University, with the support of the SWF aiding the donation of tissue to brain and tissue banks. Identifying a causative somatic mutation(s) in SWS will uncover the underlying disease mechanism, and this could, in turn, lead to a definitive biomarker for SWS and the development of novel therapeutic strategies.
This work has resulted in two original clinical research manuscripts,37, 38 a case report,39 and a review of SWS.40 A trainee project with funding from the BVMC consortium summarized the results of treating children with SWS with low-dose aspirin, describing the low-rate of complications, and quantifying outcome through the use of the SWS neurologic clinical score applied prospectively.37 This article adds significantly to the growing literature suggesting that low-dose aspirin is safe, reduces stroke-like episodes, helps to control seizures, and may improve outcome in SWS. Our work has also clarified the approach to the diagnosis and treatment of central hypothyroidism in patients with SWS.38 Pilot funding from the BVMC has aided the development of additional biomarkers relevant to SWS, including quantitative EEG, medical rehabilitation scales, transcranial Doppler, and optical coherence tomography; this has resulted in a submitted manuscript and additional in-progress manuscripts. Significant progress has been made on all three of the main aims above such that other manuscripts directly relating to the aims are currently in submission or progress. The collaborative efforts of the BVMC have provided essential resources and a framework by which the work described above was made possible.
Project 3: Cerebral hemorrhage risk in Hereditary Hemorrhagic-Telangiectasia
Hereditary Hemorrhagic Telangiectasia (HHT, Osler-Weber-Rendu Syndrome) is a disorder of muco-cutaneous fragility and vascular dysplasia. The most common presentation is epistaxis, followed by gastrointestinal bleeding. Further, patients harbor arteriovenous malformations in the pulmonary, hepatic and cerebral circulations; the latter is associated with ICH and attendant risk of neurological morbidity. HHT is an autosomal dominant disorder with a prevalence of at least 10/100,000. The prevalence of bAVM in the general population is ≈ 1/100,000, of which less than 5% are due to HHT.
The disease is an autosomal dominant condition characterized by vascular malformations, specifically developing muco-cutaneous telangiectasia and solid organ AVMs (primarily brain, lung and liver) (Figure 5). The systematic study of the epidemiology of bAVM lesions in HHT is difficult due to the relative rarity of bAVM lesions in HHT (pulmonary AVMs are much more common in HHT). Risk factors for spontaneous ICH are a major management consideration. However, while the clinical behavior of sporadic bAVM has been studied in some depth, comparable studies for HHT are limited to small series of 20-30 cases each. Our project represents the first large-scale collaboration of a number of HHT Centers of Excellence in North America to perform a study of ICH in HHT patients harboring bAVM (Table 4).
Figure 5.
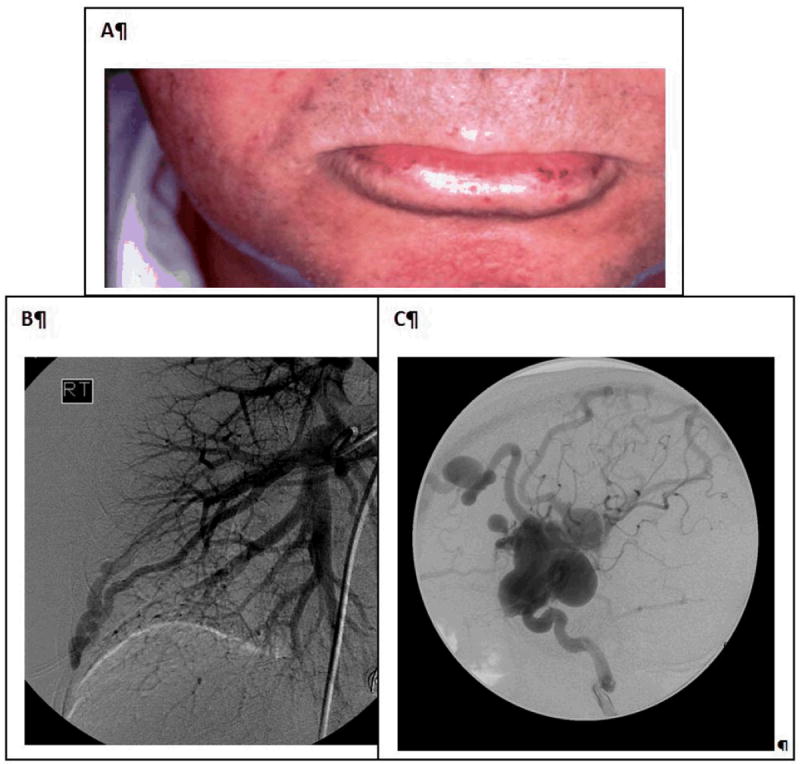
Hereditary hemorrhagic telangiectasia is characterized by the presence of (A) mucocutaneous telangiectasia and (B and C) organ arteriovenous malformations (AVMs): (B) lung AVMs demonstrated on pulmonary angiogram, and (C) two brain AVMs on catheter angiogram in a 4-year-old boy.
Table 4.
HHT Centers of Excellence Across North America Recruiting for BVMC Study Project 3.
| Participating Site Name | Principal Investigator |
|---|---|
|
| |
| Georgia Regents University | James R. Gossage |
|
| |
| Johns Hopkins University School of Medicine | Doris Lin |
|
| |
| Mayo Clinic | Karen Swanson |
|
| |
| University of California, Los Angeles | Justin McWilliams |
|
| |
| University of California, San Francisco | William L. Young |
|
| |
| University of Utah | Jamie McDonald |
|
| |
| Washington University School of Medicine | Murali Chakinala |
| Andrew White | |
|
| |
| Yale University | Robert White Jr. |
|
| |
| University of Alberta | Dilini Vethanayagam |
|
| |
| University of British Columbia / St. Paul’s Hospital | Pearce Wilcox |
|
| |
| University of Toronto / Hospital for Sick Children | Felix Ratjen |
|
| |
| University of Toronto / St. Michael’s Hospital | Marie Faughnan |
Aim 1 is the construction of an HHT clinical research database. Data include demographics, symptomatology, cerebral angioarchitecture, and information about other organ involvement. Approximately half of the HHT cases have mutational analysis (causative gene) information available.
The database has been successfully created with comprehensive data collected for 551 recruited HHT patients to date, 129 of whom have bAVMs. The population characteristics are reported in Table 5 and are similar to those reported in the literature, suggesting that this is a representative population of HHT patients. The database is used to serve the other aims but will also serve as a platform to foster further HHT research, and the centers of excellence that comprise the network have been solicited to submit projects that will utilize the database.
Table 5.
Recruited HHT Population for Project 3.
| Characteristic | Study Population, No. (%)a (N=551) | Published Adult/Mixed Series,19 %b |
|---|---|---|
| Female | 338 (61) | 60 |
| Age mean (range), year | 46.1 (1-89)c | 40-45 |
| Epistaxis | 437 (79) | 50-96 |
| Telangiectasia | 421 (76) | 80-90 |
| Brain AVM | 129 (23)d | 5-23 |
| Pulmonary AVM | 235 (43) | 15-50 |
| HHT GI bleeding | 75 (14) | 25-30 |
| Symptomatic Liver VM | 22 (4) | 8 |
| Anemia | 213 (39) | N/A |
Abbreviations: AVM, arteriovenous malformation; GI, gastrointestinal; NA, not available; VM, venous malformation
All values are shown as number (%) except for age
All values are shown as percentages except for age
Twenty-four children (aged <18 years) at registration
Targeted recruitment proportion of 25%
Aim 2 determines risk factors for hemorrhage in HHT patients with bAVM. Although well known for sporadic AVMs, the risk factors for ICH in HHT bAVM are poorly characterized, and it is unknown whether the risk is similar to that of sporadic cases. This information is essential for rationale planning of potentially high-risk interventional therapy (surgery, embolization, radiation). At the completion of data collection, we will perform a cross-sectional analysis of ICH risk factors in HHT patients. We hypothesize that bAVM HHT lesions will have a lower incidence of hemorrhagic presentation and comprise mostly low-risk AVMs (Spetzler-Martin Grade I or II).41 We predict that, although the fraction of HHT bAVM cases with ICH at presentation will be lower than for sporadic bAVM, adjusting for the effects of demographic, clinical and angioarchitectural risk factors will render HHT bAVMs comparable to sporadic cases. Second, we will compare new ICH rate between HHT patients with bAVM to an already collected cohort of sporadic bAVM after diagnosis but before any intervention (natural history). We will compare ICH rate in this HHT cohort to that in a combined database of sporadic bAVM that has been created consisting both of UCSF and University of Toronto cases. This will be a longitudinal study, analogous to the various analyses that have been carried out for sporadic AVMs.42-45
While recruitment and data collection for this aim continue in twelve centers across North America, we have capitalized on our collaborative network and published new clinically relevant observations for HHT bAVMs. We quantified the association between bAVM multiplicity and HHT, compared with sporadic bAVMs, demonstrating that the presence of bAVM multiplicity is highly predictive of the diagnosis of HHT.46 In addition, we reported gene-phenotype correlations among HHT patients with bAVMs, the first and largest series of its kind with 161 patients, from BVMC participants and patients from collaborating centers.47 The clinically relevant observations were that bAVMs can occur in HHT patients of all HHT genotypes and that ICH can occur in all HHT genotypes and at any age, from infancy onwards. Presently we are combining data from BVMC with databases from BVMC centers to study hemorrhage-free survival in patients with HHT bAVMs versus sporadic bAVMs.
Aim 3 is an exploratory investigation to identify common genetic markers for increased ICH risk utilizing a candidate gene approach. Polymorphic variation in several genes, e.g., single nucleotide polymorphisms (SNP), mostly inflammatory, is associated with ICH in sporadic bAVM patients.48-53 Although probably not causal, certain alleles are associated with increased risk of ICH at presentation or new ICH after diagnosis but before any intervention. Therefore, such SNPs could be developed as biomarkers for clinical management and risk stratification. The primary analysis will be cross-sectional using genotype as the predictor and the outcome as ICH presentation vs. other presentations, but genotype associations with longitudinal risk of ICH will also be assessed.
While recruitment and sample collection for this aim continues, we are currently analyzing 12 candidate SNPs that have been reported to be associated with sporadic bAVMs and ICH presentation.54 The current analysis will compare HHT bAVMs and sporadic bAVMs, and will be the first evaluation of the influence of genotype on ICH risk in HHT bAVM patients.
In summary, the combined Project 3 aims will provide much-needed information about the natural history of bAVMs in HHT, quantifying the risk of ICH and describing risk factors for this morbid outcome.
Summary and Future Directions
The BVMC has provided critical infrastructure and research project funding for three important rare neurovascular disorders. The cohesiveness within and among research groups in the BVMC is a model for interdisciplinary and translational collaboration, which will certainly lead to further synergistic and parallel project development and funding. Key advances that have been accomplished or will be accomplished by the BVMC include but are not limited to: (1) understanding genetic modifiers of disease severity for familial CCM (Project 1); (2) the promise of identifying somatic mutations in a gene (or genes) that causes SWS (Project 2); and (3) improved understanding of underlying genotype (mutation)-phenotype correlations for HHT, as well as modifier genes for disease severity (Project 3). Further, for both SWS and HHT, a high-quality relational database drawing from a novel North American network of centers of clinical excellence has been accomplished. Current cross-project collaborations include evaluating urine angiogenesis biomarkers developed for disease severity and progression in SWS (P1) as markers for diseases severity in CCM1-CHM and HHT. Further, SNPs associated with ICH in sporadic bAVM being tested for HHT are being evaluated for their association with disease severity and progression in CCM1-CHM and SWS. The ORDR initiative to fund RDCRCs has been an historic and highly successful experiment to form a productive, effective and durable alliance among a dedicated cadre of rare disease investigators, with the strongest possible support from three well-recognized and proactive PSOs.
Acknowledgments
The Brain Vascular Malformation Consortium (BVMC; U54NS065705) is a part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through collaboration between the NIH Office of Rare Diseases Research (ORDR) at the National Center for Advancing Translational Science (NCATS), and the National Institute of Neurological Disorders and Stroke (NINDS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors would like to acknowledge Claudia S. Moy, PhD (NIH/NINDS), for her contribution to this article.
Appendix
Brain Vascular Malformation Consortium Collaborators
Project 1: Beth Baca, Blaine Hart; Project 2: Jonathan Pevsner, Kira Lanier, Charles Swindell, Cathy Bachur, Csaba Juhasz, Warren Lo, Daniel Miles, Angus Wilfong, Rebecca Schultz, Alex Levin, Joshua Ewen, Stacey Suskauer, Henry Jampel, Robert Dejong; Project 3: Myra Slutsky, Nerissa Ko, Timo Krings, Karel TerBrugge, James R. Gossage, Doris Lin, Karen Swanson, Justin McWilliams, Jamie McDonald, Murali Chakinala, Andrew White, Robert White Jr., Dilini Vethanayagam, Pearce Wilcox, Felix Ratjen; DMCC: Callyn Hall; UCSF Core: Phillip Evans
References
- 1.Health Promotion and Disease Prevention Amendments of 1984. 1984 Oct 30; Pub. L. 98-551, 98 Stat. 2815. [Google Scholar]
- 2.Orphan Drug Act of 1983. 1983 Jan 4; Pub. L. No. 97-414, 96 Stat. 2049. [Google Scholar]
- 3.Gupta S, Bayoumi AM, Faughnan ME. Rare lung disease research: strategies for improving identification and recruitment of research participants. Chest. 2011;140:1123–1129. doi: 10.1378/chest.11-1094. [DOI] [PubMed] [Google Scholar]
- 4.Rare Diseases Clinical Research Network. BVMC: Brain Vascular Malformation Consortium. [July 1, 2012]; [Internet]. http://rarediseasesnetwork.epi.usf.edu/BVMC/index.htm.
- 5.Office of Rare Diseases. Department of Health and Human Services, National Institutes of Health; 2001. [July 1, 2012]. Report on Steps to Coordinate Rare Diseases Research Programs: Analysis by and Recommendations of the Special Emphasis Panel of the National Institutes of Health on the Coordination of Rare Diseases Research. http://rarediseases.info.nih.gov/news-reports/fy99annual/SEP.html. [Google Scholar]
- 6.Leblanc GG, Golanov E, Awad IA, et al. Biology of vascular malformations of the brain. Stroke. 2009;40:e694–702. doi: 10.1161/STROKEAHA.109.563692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marchuk DA, Srinivasan S, Squire TL, et al. Vascular morphogenesis: tales of two syndromes. Hum Mol Genet. 2003;12:R97–R112. doi: 10.1093/hmg/ddg103. [DOI] [PubMed] [Google Scholar]
- 8.Comati A, Beck H, Halliday W, et al. Upregulation of hypoxia-inducible factor(HIF)-1alpha and HIF-2alpha in leptomeningeal vascular malformations of Sturge-Weber syndrome. J Neuropathol Exp Neurol. 2007;66:86–97. doi: 10.1097/nen.0b013e31802d9011. [DOI] [PubMed] [Google Scholar]
- 9.Kim H, Pawlikowska L, Young WL. Genetics and vascular biology of brain vascular malformations. In: Mohr JP, Wolf PA, Grotta JC, et al., editors. Stroke: Pathophysiology, Diagnosis, and Management. 5. Chapter 12. Philadelphia: Churchill Livingstone Elsevier; 2011. pp. 169–186. [Google Scholar]
- 10.NINDS. A Randomized Trial of Unruptured Brain AVMs (ARUBA), NCT00389181. [Nov 18, 2011];2011 ClinicalTrials.gov. http://clinicaltrials.gov/ct/show/NCT00389181.
- 11.Stapf C, Mohr JP, Choi JH, et al. Invasive treatment of unruptured brain arteriovenous malformations is experimental therapy. Curr Opin Neurol. 2006;19:63–68. doi: 10.1097/01.wco.0000200546.14668.78. [DOI] [PubMed] [Google Scholar]
- 12.Stapf C, Mohr JP. Unruptured brain arteriovenous malformations should be treated conservatively: yes. Stroke. 2007;38:3308–3309. doi: 10.1161/STROKEAHA.107.504605. [DOI] [PubMed] [Google Scholar]
- 13.Lawton MT, Du R, Tran M, et al. Effect of presenting hemorrhage on outcome after microsurgical resection of brain arteriovenous malformations. Neurosurgery. 2005;56:485–493. doi: 10.1227/01.neu.0000153924.67360.ea. [DOI] [PubMed] [Google Scholar]
- 14.Mohr JP, Moskowitz AJ, Parides M, et al. Hull down on the horizon: A Randomized Trial of Unruptured Brain Arteriovenous Malformations (ARUBA) Trial. Stroke. 2012;43:1744–1745. doi: 10.1161/STROKEAHA.112.653584. [DOI] [PubMed] [Google Scholar]
- 15.Cockroft KM, Jayaraman MV, Amin-Hanjani S, et al. A perfect storm: how a randomized trial of unruptured brain arteriovenous malformations’ (ARUBA’s) trial design challenges notions of external validity. Stroke. 2012;43:1979–1981. doi: 10.1161/STROKEAHA.112.652032. [DOI] [PubMed] [Google Scholar]
- 16.Guo Y, Saunders T, Su H, et al. Silent intralesional microhemorrhage as a risk factor for brain arteriovenous malformation rupture. Stroke. 2012;43:1240–1246. doi: 10.1161/STROKEAHA.111.647263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Al-Shahi R, Bhattacharya JJ, Currie DG, et al. Scottish Intracranial Vascular Malformation Study (SIVMS): evaluation of methods, ICD-10 coding, and potential sources of bias in a prospective, population-based cohort. Stroke. 2003;34:1156–1162. doi: 10.1161/01.STR.0000069012.23858.69. [DOI] [PubMed] [Google Scholar]
- 18.Al-Shahi R, Bhattacharya JJ, Currie DG, et al. Prospective, population-based detection of intracranial vascular malformations in adults: the Scottish Intracranial Vascular Malformation Study (SIVMS) Stroke. 2003;34:1163–1169. doi: 10.1161/01.STR.0000069018.90456.C9. [DOI] [PubMed] [Google Scholar]
- 19.Faughnan ME, Palda VA, Garcia-Tsao G, et al. International guidelines for the diagnosis and management of hereditary hemorrhagic telangiectasia. J Med Genet. 2011;48:73–87. doi: 10.1136/jmg.2009.069013. [DOI] [PubMed] [Google Scholar]
- 20.Rigamonti D, Drayer BP, Johnson PC, et al. The MRI appearance of cavernous malformations (angiomas) J Neurosurg. 1987;67:518–524. doi: 10.3171/jns.1987.67.4.0518. [DOI] [PubMed] [Google Scholar]
- 21.Kattapong VJ, Hart BL, Davis LE. Familial cerebral cavernous angiomas: clinical and radiologic studies. Neurology. 1995;45:492–497. doi: 10.1212/wnl.45.3.492. [DOI] [PubMed] [Google Scholar]
- 22.Gunel M, Awad IA, Finberg K, et al. A founder mutation as a cause of cerebral cavernous malformation in Hispanic Americans. N Engl J Med. 1996;334:946–951. doi: 10.1056/NEJM199604113341503. [DOI] [PubMed] [Google Scholar]
- 23.Sahoo T, Johnson EW, Thomas JW, et al. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1) Hum Mol Genet. 1999;8:2325–2333. doi: 10.1093/hmg/8.12.2325. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J, Clatterbuck RE, Rigamonti D, et al. Mutations in KRIT1 in familial cerebral cavernous malformations. Neurosurgery. 2000;46:1272–1277. doi: 10.1097/00006123-200005000-00064. [DOI] [PubMed] [Google Scholar]
- 25.Petersen TA, Morrison LA, Schrader RM, et al. Familial versus sporadic cavernous malformations: differences in developmental venous anomaly association and lesion phenotype. AJNR Am J Neuroradiol. 2010;31:377–382. doi: 10.3174/ajnr.A1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morrison L, Akers A. Cerebral cavernous malformation, familial. In: Pagon RA, Bird TD, Dolan CR, et al., editors. GeneReviews. Seattle: University of Washington; 2003. [October 10, 2012]. (Updated 2011 May 31) http://www.genetests.org. [Google Scholar]
- 27.Morrison L. Familial cavernous malformations: a historical survey. In: Rigamonti D, editor. Cavernous Malformations of the Nervous System. Chapter 3. Cambridge: Cambridge University Press; 2011. pp. 15–20. [Google Scholar]
- 28.Morrison L. Genetic counseling. In: Rigamonti D, editor. Cavernous Malformations of the Nervous System. Chapter 18. Cambridge: Cambridge University Press; 2011. pp. 181–184. [Google Scholar]
- 29.Lin DD, Barker PB, Hatfield LA, et al. Dynamic MR perfusion and proton MR spectroscopic imaging in Sturge-Weber syndrome: correlation with neurological symptoms. J Magn Reson Imaging. 2006;24:274–281. doi: 10.1002/jmri.20627. [DOI] [PubMed] [Google Scholar]
- 30.Adamsbaum C, Pinton F, Rolland Y, et al. Accelerated myelination in early Sturge-Weber syndrome: MRI-SPECT correlations. Pediatr Radiol. 1996;26:759–762. doi: 10.1007/BF01396195. [DOI] [PubMed] [Google Scholar]
- 31.Cunha e Sá M, Barroso CP, Caldas MC, et al. Innervation pattern of malformative cortical vessels in Sturge-Weber disease: an histochemical, immunohistochemical, and ultrastructural study. Neurosurgery. 1997;41:872–876. doi: 10.1097/00006123-199710000-00020. [DOI] [PubMed] [Google Scholar]
- 32.Rhoten RL, Comair YG, Shedid D, et al. Specific repression of the preproendothelin-1 gene in intracranial arteriovenous malformations. J Neurosurg. 1997;86:101–108. doi: 10.3171/jns.1997.86.1.0101. [DOI] [PubMed] [Google Scholar]
- 33.Juhasz C, Lai C, Behen ME, et al. White matter volume as a major predictor of cognitive function in Sturge-Weber syndrome. Arch Neurol. 2007;64:1169–1174. doi: 10.1001/archneur.64.8.1169. [DOI] [PubMed] [Google Scholar]
- 34.Comi AM, Weisz CJ, Highet BH, et al. Sturge-Weber syndrome: altered blood vessel fibronectin expression and morphology. J Child Neurol. 2005;20:572–577. doi: 10.1177/08830738050200070601. [DOI] [PubMed] [Google Scholar]
- 35.Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol. 1987;16:899–906. doi: 10.1016/s0190-9622(87)80249-9. [DOI] [PubMed] [Google Scholar]
- 36.Hatfield LA, Crone NE, Kossoff EH, et al. Quantitative EEG asymmetry correlates with clinical severity in unilateral Sturge-Weber syndrome. Epilepsia. 2007;48:191–195. doi: 10.1111/j.1528-1167.2006.00630.x. [DOI] [PubMed] [Google Scholar]
- 37.Lance EI, Sreenivasan AK, Zabel TA, et al. Aspirin use in Sturge-Weber Syndrome: side effects and clinical outcomes. J Child Neurol. 2013;28:213–218. doi: 10.1177/0883073812463607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siddique L, Sreenivasan A, Comi AM, et al. Importance of utilizing a sensitive free T4 assay in Sturge-Weber Syndrome. J Child Neurol. 2013;28:269–274. doi: 10.1177/0883073812463606. [DOI] [PubMed] [Google Scholar]
- 39.Lopez J, Yeom KW, Comi A, et al. Case report of subdural hematoma in a patient with Sturge-Weber Syndrome and literature review: questions and implications for therapy. J Child Neurol. 2013;28:672–675. doi: 10.1177/0883073812449514. [DOI] [PubMed] [Google Scholar]
- 40.Lo W, Marchuk DA, Ball KL, et al. Updates and future horizons on the understanding, diagnosis, and treatment of Sturge-Weber syndrome brain involvement. Dev Med Child Neurol. 2012;54:214–223. doi: 10.1111/j.1469-8749.2011.04169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Willemse RB, Mager JJ, Westermann CJ, et al. Bleeding risk of cerebrovascular malformations in hereditary hemorrhagic telangiectasia. J Neurosurg. 2000;92:779–784. doi: 10.3171/jns.2000.92.5.0779. [DOI] [PubMed] [Google Scholar]
- 42.Mast H, Young WL, Koennecke HC, et al. Risk of spontaneous haemorrhage after diagnosis of cerebral arteriovenous malformation. Lancet. 1997;350:1065–1068. doi: 10.1016/s0140-6736(97)05390-7. [DOI] [PubMed] [Google Scholar]
- 43.Halim AX, Johnston SC, Singh V, et al. Longitudinal risk of intracranial hemorrhage in patients with arteriovenous malformation of the brain within a defined population. Stroke. 2004;35:1697–1702. doi: 10.1161/01.STR.0000130988.44824.29. [DOI] [PubMed] [Google Scholar]
- 44.Stapf C, Mast H, Sciacca RR, et al. Predictors of hemorrhage in patients with untreated brain arteriovenous malformation. Neurology. 2006;66:1350–1355. doi: 10.1212/01.wnl.0000210524.68507.87. [DOI] [PubMed] [Google Scholar]
- 45.Kim H, Sidney S, McCulloch CE, et al. Racial/ethnic differences in longitudinal risk of intracranial hemorrhage in brain arteriovenous malformation patients. Stroke. 2007;38:2430–2437. doi: 10.1161/STROKEAHA.107.485573. [DOI] [PubMed] [Google Scholar]
- 46.Bharatha A, Faughnan ME, Kim H, et al. Brain arteriovenous malformation multiplicity predicts the diagnosis of hereditary hemorrhagic telangiectasia: quantitative assessment. Stroke. 2012;43:72–78. doi: 10.1161/STROKEAHA.111.629865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishida T, Faughnan ME, Krings T, et al. Brain arteriovenous malformations associated with hereditary hemorrhagic telangiectasia: Genotype-phenotype correlations. Am J Med Genet A. 2012;158(A):2829–2834. doi: 10.1002/ajmg.a.35622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pawlikowska L, Tran MN, Achrol AS, et al. Polymorphisms in genes involved in inflammatory and angiogenic pathways and the risk of hemorrhagic presentation of brain arteriovenous malformations. Stroke. 2004;35:2294–2300. doi: 10.1161/01.STR.0000141932.44613.b1. [DOI] [PubMed] [Google Scholar]
- 49.Pawlikowska L, Tran MN, Achrol AS, et al. Polymorphisms in transforming growth factor-B-related genes ALK1 and ENG are associated with sporadic brain arteriovenous malformations. Stroke. 2005;36:2278–2280. doi: 10.1161/01.STR.0000182253.91167.fa. [DOI] [PubMed] [Google Scholar]
- 50.Achrol AS, Pawlikowska L, McCulloch CE, et al. Tumor necrosis factor-alpha-238G>A promoter polymorphism is associated with increased risk of new hemorrhage in the natural course of patients with brain arteriovenous malformations. Stroke. 2006;37:231–234. doi: 10.1161/01.STR.0000195133.98378.4b. [DOI] [PubMed] [Google Scholar]
- 51.Chen Y, Pawlikowska L, Yao JS, et al. Interleukin-6 involvement in brain arteriovenous malformations. Ann Neurol. 2006;59:72–80. doi: 10.1002/ana.20697. [DOI] [PubMed] [Google Scholar]
- 52.Kim H, Hysi PG, Pawlikowska L, et al. Common variants in interleukin-1-beta gene are associated with intracranial hemorrhage and susceptibility to brain arteriovenous malformation. Cerebrovasc Dis. 2009;27:176–182. doi: 10.1159/000185609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weinsheimer S, Kim H, Pawlikowska L, et al. EPHB4 gene polymorphisms and risk of intracranial hemorrhage in patients with brain arteriovenous malformations. Circ Cardiovasc Genet. 2009;2:476–482. doi: 10.1161/CIRCGENETICS.109.883595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sturiale CL, Puca A, Sebastiani P, et al. Single nucleotide polymorphisms associated with sporadic brain arteriovenous malformations: where do we stand? Brain. 2013;136 (Pt 2):665–681. doi: 10.1093/brain/aws180. [DOI] [PubMed] [Google Scholar]