

Abstract
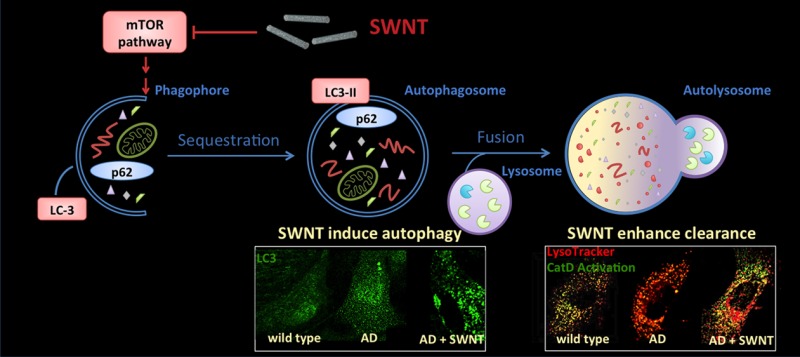
Defective autophagy in Alzheimer’s disease (AD) promotes disease progression in diverse ways. Here, we demonstrate impaired autophagy flux in primary glial cells derived from CRND8 mice that overexpress mutant amyloid precursor protein (APP). Functionalized single-walled carbon nanotubes (SWNT) restored normal autophagy by reversing abnormal activation of mTOR signaling and deficits in lysosomal proteolysis, thereby facilitating elimination of autophagic substrates. These findings suggest SWNT as a novel neuroprotective approach to AD therapy.
Keywords: Single-walled carbon nanotubes, primary glia, autophagy, Alzheimer’s disease
Autophagy, a lysosomal degradative pathway for recycling obsolete cellular constituents and damaged organelles, contributes to the pathogenesis of a range of human disease states.1 Mounting evidence strongly implicates defective autophagy in the development of several major neurodegenerative disorders, especially Alzheimer’s disease (AD).2,3 In this study, we explored a strategy for reversing neural autophagy impairments in a mouse model of AD using a novel nanoparticle strategy. Carbon nanotubes have promising physical properties, including high electrochemically accessible surface area,4 excellent electronic current,5,6 effective thermal conductivity,7 and super mechanical strength,8,9 which recommend them for diverse applications in nanomedicine.10−12 We used single walled carbon nanotubes (SWNT), a form of carbon nanotubes with relatively high water-solubility and dispersibility, conferring enhanced ability to translocate through cell membranes with relatively low cytotoxicity.13,14 Here, we took advantage of recent reports that SWNT can strongly upregulate autophagy.15 In most of these studies, SWNT were used at high concentrations to promote death of cancer cells, although the specific contribution of autophagy induction to the cytotoxicity is unclear.15−18 At lower concentrations, however, SWNT have cytoprotective effects19,20 involving mechanisms of action that have not been carefully explored. We therefore investigated the potential of low nontoxic concentrations of SWNT to upregulate autophagy and to reverse defects in the autophagic turnover of proteins within astroglial cells from the brains of AD model mice.
Although several forms of autophagy are known,21 the major form, macroautophagy, hereafter referred to as autophagy, is induced mainly by a decrease in signaling by the protein kinase mammalian Target of Rapamycin (mTOR) (Figure 1). Down-regulation of mTOR activates a series of macromolecular complexes that coordinate a rearrangement of subcellular membranes and creation of a cup-shaped phagophore that envelops cytoplasmic constituents and closes to form an autophagosome (Figure 1). Autophagosomes ultimately fuse with lysosomes to form acidic autolysosomes, wherein the degradation of proteins, lipids, and other autophagy substrates occurs. Although autophagy is constitutive in most cells, it is induced under conditions of nutrient starvation or cell stress, such as protein aggregate accumulation, and thereby provides energy and enables more adaptive new synthesis from amino acids and other building blocks released from lysosomes during substrate degradation.21−23 The rate of substrate capture and degradation (“autophagy flux”) can be monitored by measuring changes in levels of specific proteins involved in substrate sequestration, such as LC3 and p62, which are subsequently degraded along with other captured substrates.24
Figure 1.

Graphic illustrates the autophagy pathway and its impairment in CRND8 glial cells at two levels: lowered autophagy induction and lysosomal proteolytic dysfunction, which are believed to contribute to AD pathogenesis,6,27 Macroautophagy, hereafter referred to as autophagy, involves induction steps dependent on the inhibition of mTOR, elongation of a membrane structure (phagophore) around a substrate or a region of cytoplasm, closure of this structure to form an autophagosome, fusion of the autophagosome with lysosomes creating an autolysosome within which autophagosomes are degraded by acidic hydrolases, yielding a lysosome. To assess autophagy, we assayed the activation state of mTOR (induction), formation, and translocation of LC3-II to autophagosomes (autophagosome formation), autolysosomal degradation of autophagy substrates, including two autophagy related proteins, LC3-II and p62. Lysosome function is monitored by assay of cathepsin D (CatD) activation (proteolysis), CatD maturation (enzyme activation and lysosomal pH), and lysosomal acidification. The graphic depicts the sites of action of SWNT, internalized by endocytosis, in restoring normal autophagy function by depressing mTOR activity (asterisks) to stimulate autophagy induction and enhancing lysosomal proteolysis by increasing cathepsin activation possibly by restoring normal acidification of lysosomes (asterisks).
AD, the most prevalent human neurodegenerative disease, is characterized by intraneuronal aggregates of tau protein, extracellular β-amyloid deposits associated with dystrophic and degenerating neurites, and neuronal loss, ultimately leading to dementia.25 In AD, autophagy-related vesicular compartments, especially autolysosomes, accumulate within the grossly swollen dystrophic neurites, reflecting a markedly defective clearance of autophagic substrates by lysosomes.4 The pathogenic significance of this defect is underscored by the ability of mTOR inhibitors that induce autophagy26,27 or treatments that restore lysosomal proteolysis to prevent diverse neuronal and cognitive deficits in mouse models of AD.28 In several lysosomal storage disorders where neuronal autophagy is markedly impaired,29 glial cells are also critical to clearing abnormally accumulated or misfolded proteins26 and conferring neuroprotection.30 Glial cells may also protect neurons in AD by internalizing Aβ and other potentially toxic proteins via endocytosis and clearing them within lysosomes.31−33
In a well-established transgenic mouse model of AD-related amyloidosis (CRND8), we investigated the therapeutic properties of a SWNT preparation34 that was previously found to have cytoprotective effects in a model of ethanol-induced neurodegeneration (Xue et al. submitted). Notably, ethanol inhibits mTOR, which is an apparent neuroprotective response because a further degree of autophagy induction with the mTOR inhibitor, rapamycin, attenuates ethanol-induced neuronal death while autophagy inhibition exacerbates it.35 Under control of the Prp-promoter, CRND8 mice overexpress a human amyloid-β precursor protein (App) gene carrying two mutations (i.e., “Swedish” and “Indiana”) associated with familial early onset of AD.36 Transgene overexpression leads to progressive β-amyloid deposition and marked deficits of lysosomal proteolysis that impair turnover of autophagy substrates and are associated with increased amyloidogenesis and significant cognitive decline.37,38 For the first time, we establish in this report that autophagy is markedly impaired in primary glia from CRND8 mice and demonstrate that autophagy dysfunction and autophagic substrate clearance are reversed by SWNT. Our results further support the potential therapeutic value of enhancing autophagy in AD and suggest a novel approach to restoring normal autophagy activity when lysosomal function is impaired.
Primary CRND8 Glia as a System for Evaluating Nanotube Effects
In our previous study,34,39 a highly dispersed carboxyl-functionalized SWNT system was sorted by density gradient ultracentrifugation (DGU) in aqueous solution to obtain single-dispersed SWNT. These SWNT fractions were found to have higher yield (50% output), better reproducibility, and less cytotoxicity than previous preparations.19,34 The SWNT fraction was further characterized by atomic force microscope (AFM) (Supporting Information Figure S1A), which revealed that the SWNTs were well dispersed and had an average diameter of 1–2 nm (Supporting Information Figure S1B). This is consistent with our previous study by AFM height measurements with HR-TEM results.34
We investigated the competence of autophagy in purified primary glial cells (Supporting Information Figure S2) from CRND8 mice and wild-type (WT) controls and the influences of SWNT treatment. To mimic extracellular fluid (ECF) circulation in the brain, culture medium was continuously removed and replaced by fresh medium with/without SWNT at a constant rate using a mechanical pump. By also removing APP metabolites secreted into the medium, this procedure eliminated the possibility of superimposed effects of Aβ reinternalized after its release from the cells. To evaluate autophagy and the impact of SWNT exposure, we cultured WT and CRND8 glial cells in medium without or with SWNT at 0.05 μg/mL, a concentration determined to be 10-fold lower than that which induced toxicity assessed by MTT assay (Supporting Information Figure S3). This SWNT concentration was previously found to be neuroprotective in our previous analyses of a neurodegeneration model (Xue et al. submitted). As a positive control condition for autophagy responsiveness, we used the specific mTOR inhibitor, rapamycin (5 nM), an established autophagic inducing agent.40 Culture medium containing either SWNT, rapamycin, or deoxycholate sodium vehicle alone was continuously flowed over glial cells for 10 h, following which the glial cells were cultured with fresh medium without any treatment.
To characterize autophagy, we conducted immunoblot analyses using a panel of autophagy pathway markers that allowed us to interrogate multiple steps in the autophagy pathway (Figure 1), including mTOR activation state (i.e., p70 S6 kinase phosphorylation state), autophagosome formation (i.e., LC3-positive vesicles), and lysosome function (i.e., LC3 and p62 turnover, cathepsin D (CatD) maturation, lysosome acidification). Comparisons of untreated WT versus CRND8 glial cells in Figure 2 established two autophagy defects, an abnormal activation of mTOR signaling, reflecting suppression of autophagy induction, and an impairment of lysosome function in CRND8 glia (Figure 3).
Figure 2.

Effects of SWNT on autophagic induction. (A) Formation of LC3-positive vesicular “puncta” (autophagosomes) in CRND8 glial cells treated with SWNT and rapamycin detected by immunofluorescence using LC3 antibody. (B) Western blot analysis showing LC3 levels with rapamycin (5 nM, 24 h) as a positive control and actin as a loading control. *p < 0.05, **p < 0.01, ***p < 0.001 versus CRND8 control group (CTRD8-Ctrl); #p < 0.05, ##p < 0.01, ###p < 0.001 versus the WT-Ctrl group; n = 3. (C) Western blot analysis of LC3-II levels in the absence and presence of leupeptin, a cysteine protease inhibitor, which blocks lysosomal degradation of LC3-II. The difference in LC-II levels under these two conditions is a measure of autophagosome formation.42 Data are presented as means ± SD *p < 0.05, **p < 0.01, ***p < 0.001 versus CRND8 control group (CTRD8-Ctrl); #p < 0.05, ##p < 0.01, ###p < 0.001 versus the WT-Ctrl group; n = 3. (D) Autophagic flux detection showing by Western blots with antiphospho-mTOR (p-mTOR), antitotal-mTOR (t-mTOR), antiphospho-p70S6k (p-p70S6k), antitotal-p70S6k (t-p70S6k),antiphospho-ULK1 (p-ULK1), and antitotal-ULK1 (t-ULK1) antibodies after treatment with SWNT 10 h following fresh medium instead for 14 h and rapamycin for 24 h. The ratio of p-mTOR/t-mTOR (E), p-p70S6k/t-p70S6k (F), p-ULK1/t-ULK1 (G) are quantified. Data are presented as means ± SD *p < 0.05, **p < 0.01, ***p < 0.001 versus CRND8 control group (CTRD8-Ctrl); #p < 0.05, ##p < 0.01, ###p < 0.001 versus the WT-Ctrl group; n = 3.
Figure 3.

Effects of SWNTs on lysosomal function. (A) CatD immunoblots showing the mature form (matu-CatD, 32 kDa) and their pro-form (pro-CatD, 49 kDa) in both WT and CRND8 glial cells. The proportions of mature-CatD and pro-CatD are measured. Data are presented as means ± SD *p < 0.05, **p < 0.01, ***p < 0.001 versus CRND8 control group (CRND8-Ctrl); #p < 0.05, ##p < 0.01, ###p < 0.001 versus the WT-Ctrl group; n = 3. (B) CatD activity assays are assessed with LysoTracker and Bodipy-FL-pepstatin A, which binds to active CatD. The scale bar represents 10 μm. (C) Quantitative analysis of the intensity of Bodipy-FL-pepstatin A-positive compartments. Data are presented as means ± SEM n = 40–50 each, randomly; ***p < 0.001 versus CRND8 control group (CRND8-Ctrl); ###p < 0.001 versus the WT-Ctrl group. (D) Immunofluorescence images showing double labeling with Lamp2-positive lysosome (green) and CatD-postitive lysosomal enzyme (red) in representative WT and CRND8 glial cells with high-magnification images shown in inset. CatD/Lamp2-positive vesicles were counted with ImageJ, presenting their relative size (E) and relative number (F) of lysosomes. Data are presented as means ± SEM n = 40–50 each, randomly; ***p < 0.001 versus CRND8 control group (CRND8-Ctrl); ###p < 0.001 versus the WT-Ctrl group. (G) Western blot analysis of p62 levels in the absence and presence of leupeptin. Data are presented as means ± SD *p < 0.05, **p < 0.01, ***p < 0.001 versus CRND8 control group (CTRD8-Ctrl); n = 3.
CRND8 Glia Exhibit Reduced mTOR-Dependent Autophagy Induction and Autophagosome Formation, Which Are Reversed by SWNT
To assess autophagosome formation, we monitored LC3, which shifts from the cytosolic LC3-I form to LC3-II, its lipidated vesicle-associated form, when LC3 is recruited to membranes to form new autophagosomes.41 LC3-II is then degraded when autophagosomes are cleared by lysosomes; therefore, steady-state levels of this protein reflect its formation and degradation. These two processes can be further differentiated experimentally by blocking LC3-II degradation by inhibiting lysosomal cysteine proteases with the inhibitor leupeptin.42 At baseline, LC3-II positive vesicles, marking the presence of autophagosomes, were uncommon in both CRND8 and WT cells, suggesting a low rate of formation (Figure 2A). Immunoblot analysis of LC3-II steady-state levels, however, was increased in CRND8 glia (Figure 2B). Together, these results suggested a low rate of autophagosome formation but impaired LC3-II degradation in CRND8 cells. To further substantiate a low rate of autophagosome formation in CRND8 cells, we evaluated changes in LC3-II in the presence or absence of leupeptin. When LC3-II degradation is blocked by leupeptin, its accumulation reflects only the formation of autophagosomes. This analysis revealed 88% less LC3-II build-up in CRND8 glia than in WT cells, establishing that autophagosome formation is substantially reduced and that reduced lysosomal degradation is the cause of the elevated LC3-II level (Figure 2C).
To explain the lowered autophagosome formation, we analyzed upstream mTOR signaling that controls the induction of autophagosome production. In CRND8 glial cells, mTOR, a constitutive suppressor of autophagy induction, exhibited increased activation, as reflected in its higher phosphorylation state by Western blot analysis (Figure 2D,E), paralleling an increased phosphorylation state of one of its substrates, p70S6k (Figure 2D,F). In addition, the phosphorylation state of another mTOR substrate UNC051 kinase 1 (ULK1) (at Ser757), which is directly linked to the process of autophagosome formation was also increased (Figure 2D,G). These findings confirmed increased mTOR signaling, which is associated with a suppression of autophagy induction and autophagosome formation.
SWNT treatment reversed abnormally suppressed autophagy induction in CRND8 glia but, notably, had minimal effect on WT glia. The ratios of phospho-variant to total forms of mTOR and p70S6k in CRND8 glial were both lowered by SWNT treatment back to WT levels (Figure 2D–F). In addition, the phosphorylation state of p-ULK1 was also reduced in CRND8 glia to a level similar to that in WT glia (Figure 2D,G). Interestingly, the autophagy inducer rapamycin had similar effects to SWNT in CRND8 glia (Figure 2D–G), but unlike SWNT, rapamycin also induced autophagy in WT cells, as expected. Thus, SWNT had relatively selective effect in reversing APP-driven pathological activation of mTOR signaling in CRND8 glia without affecting the normal baseline signaling in WT cells. These selective effects of SWNT in reducing mTOR signaling and inducing autophagy in CRND8 glia are supported by further immunocytochemical studies showing markedly increased numbers of LC3-positive puncta in SWNT treated CRND8 cells (Figure 2A).
CRND8 Glia Exhibit Lysosomal Dysfunction, Which Is Reversed by SWNT
All forms of autophagy share the common cardinal feature of autophagic substrate digestion within lysosomes.3 In addition to having impaired autophagy induction, CRND8 glia exhibited significant lysosomal dysfunction. CatD maturation was markedly inhibited as evidenced by greatly lowered levels of the mature (most active) 32 kDa form and an upward shift in the apparent molecular size of full-length pro-forms of the enzyme (Figure 3A), suggesting a more immature glycosylation state. Consistent with these alterations of active CatD in CRND8 glia, we observed markedly decreased in situ lysosomal active CatD, reflected by reduced fluorescence signal of the affinity ligand Bodipy-FL-pepstatin, which binds only to the active state of CatD (Figure 3B,C).43 Moreover, the sizes of lysosomes were significantly enlarged as determined by morphometric analysis after immunocytochemical detection of lysosome-associated membrane protein 2 (Lamp2) and the lysosomal protease cathepsin D (CatD) (Figure 3D,E). Enlargement was accompanied by a decrease in lysosome number in CRND8 glia (59%, ###p < 0.001) (Figure 3F), suggesting a possible impairment in lysosomal reformation.44
SWNT treatment reversed these CatD maturation abnormalities (Figure 3A) and the markedly increased levels of CatD activity in CRND8 glia (51%, ***p < 0.001) assessed by Bodipy-FL-pepstatin A labeling, restoring these levels to levels those comparable to vehicle-treated WT cells (Figure 3B,C). Although total lysosomal protein, reflected by levels of Lamp2, was not changed (Supporting Information Figure S4), the abnormally marked enlargement of Lamp2- and CatD-positive lysosomes seen in CRND8 cells was also corrected by SWNT (Figure 3D,E). The 31% reduction (***p < 0.001) in average size of Lamp2/CatD-positive lysosomes after SWNT treatment compared to the vehicle-treated CRND8 glia represented a full restoration to WT average lysosome size (Figure 3E). Additionally, ImageJ analysis revealed significantly fewer lysosomes in CRND8 glia (59%, ###p < 0.001) compared to WT glia. SWNT treatment partially rescued this abnormality (67% recovery, ***p < 0.001) (Figure 3F). Finally, abnormally high levels of p62 (SQSTM1/sequestome1), an established autophagy substrate, were substantially lowered in CRND8 glial cells treated with SWNT (Figure 3G). Importantly, SWNT treatment of WT glia had negligible effects on these parameters.
Stimulating autophagy induction has previously shown promise for multiple neurodegenerative diseases by virtue of its potential to preserve neuronal survival by lowering levels of toxic protein aggregates and recycling nonessential constituents for energy and adaptive protein synthesis under conditions of cellular stress.1 The selective mTOR complex 1 (mTORC1) inhibitor rapamycin, used as a positive control in this study, was previously shown to attenuate neuropathological and functional deficits in multiple AD mouse models.45,46 Recent evidence shows that autophagy can be induced by nanomaterials, such as fullerene C60,47 quantum dots,48 and nanocrystals.49,50 Among these nanomaterials, SWNT hold significant therapeutic promise as neuroprotective agents that enhance neural growth, differentiation, and maturity.51−53 However, few studies have addressed mechanisms underlying these neuroprotective effects, including the potential of SWNT to induce autophagy and enhance lysosomal clearance of autophagic substrates in neurodegenerative diseases, which could shed light on possible therapeutic applications.
We show in this study for the first time that primary glial cells from the CRND8 transgenic mouse model of AD recapitulate the lysosomal dysfunction demonstrated in CRND8 brain, which underlies deficits in autophagic protein turnover and cognition.37,54 We also provide the first evidence that impaired autophagic clearance of substrates in this model is also accompanied by a significant activation of mTOR signaling leading to the suppression of autophagy induction. This combination of impairments would be expected to significantly reduce autophagy substrate turnover and the capacity of neural cells to respond to cellular stress and eliminate potentially toxic materials internalized from the extracellular space or generated intracellularly, such as Aβ peptide. The design of this cell culture system, which incorporated a mechanism to continually remove from the medium metabolites, such as Aβ, that are released from the cells, allows us to conclude that these adverse effects on autophagy are due not to endocytosis of released Aβ, which is potentially toxic to lysosomes, but rather to other factors initiated by the overexpression of mutant APP, which may include intracellular generation and accumulation of Aβ and other APP metabolites or to altered signaling functions of APP.
We further observed remarkable effects of SWNT in reversing both the deficits in autophagy induction caused by abnormal mTOR signaling and the impairments of lysosomal proteolysis leading to accumulation of autophagic substrates and lysosome swelling. Improved lysosome function may also be related to robust up-regulation of mTOR-dependent autophagy induction, which is known to activate transcription factors, including TFEB, that promotes transcription of most genes controlling lysosome biogenesis and hydrolytic function and additional genes regulating autophagy.55,56 The ability of SWNT to enhance autophagic flux represents one potential mechanism to account for previously observed neuroprotective effects of SWNT in several pathological settings. In studies by us (under review) and others, SWNT showed neuroprotective effects at low nontoxic concentrations in several neuropathological states in vitro57 and in vivo,20 including experimental stroke, in one case by triggering a neurotrophic pathway via the stimulation of the Tropomyosin-receptor kinase (Trk) family and their downstream protein molecules. These studies support our current evidence that SWNT could have therapeutic actions in Alzheimer’s disease.
Lysosomal dysfunction is a pathogenic factor in AD, PD, and other neurodegenerative diseases and a potential therapeutic target.23,58,59 To mimic possible in vivo conditions for therapeutic delivery of SWNT, we provided a continuous flow of SWNT over glial cells at very low concentrations that exerted no evident cytotoxicity and are comparable to concentrations used by others without mechanical pump.15,60 Interestingly, concentrations of SWNT that dramatically reversed autophagy deficits in CRND8 glia negligibly altered autophagic/lysosome activation in WT cells. Although it is tempting to speculate that SWNT may have selective effects on the specific pathological processes leading to autophagy deficits in the CRND8 model, it is alternatively possible that healthier WT glial cells internalize and/or accumulate less SWNT or are more resistant to an autophagy-triggering cellular stress induced by SWNT.
While the mechanisms responsible for autophagy rescue by SWNT require further investigation, our current findings provide proof-of-concept evidence for a nanoparticle-based neuroprotective approach for AD and other neurodegenerative diseases in which autophagy impairment is a pathogenic factor. The future success of such biocompatible nanomaterials will depend on optimization of pharmacokinetic/pharmacodynamic properties61 and finding a balance between maximal therapeutic efficacy and minimal adverse reactivity. To date, several studies have demonstrated therapeutic effects of SWNT in brain ischemia by local administration,20,62 illustrating the potential for clinical translation of nanomaterials.
Acknowledgments
This work was supported by the Chinese Natural Science Foundation project (81171455), National Distinguished Young Scholars Grant (31225009) from the National Natural Science Foundation of China, the National Key Basic Research Program of China (2009CB930200), the Chinese Academy of Sciences (CAS) “Hundred Talents Program” (07165111ZX), the CAS Knowledge Innovation Program and the State High-Tech Development Plan (2012AA020804 and SS2014AA020708). The authors also appreciate the support by the “Strategic Priority Research Program” of the Chinese Academy of Sciences, Grant XDA09030301, and support by the U.S. National Institute on Aging (P01AG017617) (RAN) and Takeda Pharmaceuticals, Ltd (RAN).
Supporting Information Available
Materials and methods to characterize SWNT, including AFM and TEM images, animal dissection and established glial primary culture, Western blot and confocal imaging; additional figures and tables. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
(Y.S.) Center for iPS Cell Research and Application (CiRA), Kyoto University, Kyoto 606-8507, Japan.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Rubinsztein D. C.; Codogno P.; Levine B. Nat. Rev. Drug Discovery 2012, 119709–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochfeld W. E.; Lee S.; Rubinsztein D. C. Acta Pharmacol. Sin. 2013, 345600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon R. A. Nat. Med. 2013, 198983–997. [DOI] [PubMed] [Google Scholar]
- Keefer E. W.; Botterman B. R.; Romero M. I.; Rossi A. F.; Gross G. W. Nat. Nanotechnol. 2008, 37434–439. [DOI] [PubMed] [Google Scholar]
- Skakalova V.; Kaiser A. B.; Dettlaff-Weglikowska U.; Hrncarikova K.; Roth S. J. Phys. Chem. B 2005, 109157174–7181. [DOI] [PubMed] [Google Scholar]
- Randeniya L. K.; Bendavid A.; Martin P. J.; Tran C. D. Small 2010, 6161806–1811. [DOI] [PubMed] [Google Scholar]
- Kim P.; Shi L.; Majumdar A.; McEuen P. L. Phys. Rev. Lett. 2001, 8721215502. [DOI] [PubMed] [Google Scholar]
- Treacy M. M.; Krishnan A.; Yianilos P. N. Microsc. Microanal. 2000, 64317–323. [DOI] [PubMed] [Google Scholar]
- Nardelli M. B.; Y B. I.; Bernholc J. Phys. Rev. B 1997, 57, 4277–4280. [Google Scholar]
- Kyle S.; Saha S. Adv. Healthcare Mater. 2014, 10.1002/adhm.201400009. [DOI] [Google Scholar]
- Akhter S.; Ahmad I.; Ahmad M. Z.; Ramazani F.; Singh A.; Rahman Z.; Ahmad F. J.; Storm G.; Kok R. J. Curr. Cancer Drug Targets 2013, 134362–378. [DOI] [PubMed] [Google Scholar]
- Bokara K. K.; Kim J. Y.; Lee Y. I.; Yun K.; Webster T. J.; Lee J. E. Anat. Cell Biol. 2013, 46285–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uo M.; Akasaka T.; Watari F.; Sato Y.; Tohji K. Dent. Mater. J. 2011, 303245–263. [DOI] [PubMed] [Google Scholar]
- Vardharajula S.; Ali S. Z.; Tiwari P. M.; Eroglu E.; Vig K.; Dennis V. A.; Singh S. R. Int. J. Nanomed. 2012, 7, 5361–5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. L.; Zhang Y. L.; Yang N.; Zhang Y. X.; Liu X. Q.; Li C. G.; Zhao Y.; Wang Y. G.; Zhang G. G.; Yang P.; et al. Cell Death Dis. 2011, 2, e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E. J.; Zahari N. E.; Lee E. W.; Song J.; Lee J. H.; Cho M. H.; Kim J. H. Toxicol. In Vitro 2014, 10.1016/j.tiv.2013.12.012. [DOI] [PubMed] [Google Scholar]
- Yu K. N.; Kim J. E.; Seo H. W.; Chae C.; Cho M. H. J. Toxicol. Environ. Health, Part A 2013, 76231282–1292. [DOI] [PubMed] [Google Scholar]
- Tsukahara T.; Matsuda Y.; Usui Y.; Haniu H. Biochem. Biophys. Res. Commun. 2013, 4402348–353. [DOI] [PubMed] [Google Scholar]
- Bari S.; Chu P. P.; Lim A.; Fan X.; Gay F. P.; Bunte R. M.; Lim T. K.; Li S.; Chiu G. N.; Hwang W. Y. Nanomedicine 2013, 981304–1316. [DOI] [PubMed] [Google Scholar]
- Lee H. J.; Park J.; Yoon O. J.; Kim H. W.; Lee do Y.; Kim do H.; Lee W. B.; Lee N. E.; Bonventre J. V.; Kim S. S. Nat. Nanotechnol 2011, 62121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P.; Reggiori F.; Codogno P. Nat. Cell Biol. 2013, 157713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu M.; Thompson C. B. Annu. Rev. Pathol. 2008, 3, 427–455. [DOI] [PubMed] [Google Scholar]
- Shacka J. J.; Roth K. A.; Zhang J. Front Biosci. 2008, 13, 718–736. [DOI] [PubMed] [Google Scholar]
- Klionsky D. J.; Abeliovich H.; Agostinis P.; Agrawal D. K.; Aliev G.; Askew D. S.; Baba M.; Baehrecke E. H.; Bahr B. A.; Ballabio A.; et al. Autophagy 2008, 42151–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman D. M.; Morris J. C.; Goate A. M. Sci. Transl. Med. 2011, 37777sr71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z.; Yan J.; Jiang W.; Yao X. G.; Chen J.; Chen L.; Li C.; Hu L.; Jiang H.; Shen X. J. Neurosci. 2013, 333213138–13149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P.; Zhao H.; Davies P. J. Biol. Chem. 2005, 2804537377–37382. [DOI] [PubMed] [Google Scholar]
- Rubinsztein D. C.; Marino G.; Kroemer G. Cell 2011, 1465682–695. [DOI] [PubMed] [Google Scholar]
- Di Malta C.; Fryer J. D.; Settembre C.; Ballabio A. Autophagy 2012, 8121871–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bernhardi R. Neurotoxic. Res. 2007, 124215–232. [DOI] [PubMed] [Google Scholar]
- Mandrekar S.; Jiang Q.; Lee C. Y.; Koenigsknecht-Talboo J.; Holtzman D. M.; Landreth G. E. J. Neurosci. 2009, 29134252–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolmont T.; Haiss F.; Eicke D.; Radde R.; Mathis C. A.; Klunk W. E.; Kohsaka S.; Jucker M.; Calhoun M. E. J. Neurosci. 2008, 28164283–4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft A. W.; Hu X.; Yoon H.; Yan P.; Xiao Q.; Wang Y.; Gil S. C.; Brown J.; Wilhelmsson U.; Restivo J. L.; et al. FASEB J. 2013, 271187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. R.; Xue X.; Hu X. M.; Wei M. Y.; Zhang C. Q.; Ge G. L.; Liang X. J. Small 2014, 10.1002/smll.201303342. [DOI] [PubMed] [Google Scholar]
- Chen G.; Ke Z.; Xu M.; Liao M.; Wang X.; Qi Y.; Zhang T.; Frank J. A.; Bower K. A.; Shi X.; et al. Autophagy 2012, 8111577–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chishti M. A.; Yang D. S.; Janus C.; Phinney A. L.; Horne P.; Pearson J.; Strome R.; Zuker N.; Loukides J.; French J.; et al. J. Biol. Chem. 2001, 2762421562–21570. [DOI] [PubMed] [Google Scholar]
- Yang D. S.; Stavrides P.; Mohan P. S.; Kaushik S.; Kumar A.; Ohno M.; Schmidt S. D.; Wesson D.; Bandyopadhyay U.; Jiang Y.; et al. Brain 2011, 134Pt 1258–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde L. A.; Kazdoba T. M.; Grilli M.; Lozza G.; Brusa R.; Zhang Q.; Wong G. T.; McCool M. F.; Zhang L.; Parker E. M.; et al. Behav. Brain Res. 2005, 1602344–355. [DOI] [PubMed] [Google Scholar]
- Wang L.; Zhang L.; Xue X.; Ge G.; Liang X. Nanoscale 2012, 4133983–3989. [DOI] [PubMed] [Google Scholar]
- Berger Z.; Ravikumar B.; Menzies F. M.; Oroz L. G.; Underwood B. R.; Pangalos M. N.; Schmitt I.; Wullner U.; Evert B. O.; O’Kane C. J.; et al. Hum. Mol. Genet. 2006, 153433–442. [DOI] [PubMed] [Google Scholar]
- Tanida I.; Minematsu-Ikeguchi N.; Ueno T.; Kominami E. Autophagy 2005, 1284–91. [DOI] [PubMed] [Google Scholar]
- Klionsky D. J.; Abdalla F. C.; Abeliovich H.; Abraham R. T.; Acevedo-Arozena A.; Adeli K.; Agholme L.; Agnello M.; Agostinis P.; Aguirre-Ghiso J. A.; et al. Autophagy 2012, 84445–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe D. M.; Lee J. H.; Kumar A.; Lee S.; Orenstein S. J.; Nixon R. A. Eur. J. Neurosci 2013, 37121949–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Y.; Liu M.; Ma L.; Du W.; Zhang H.; Tian Y.; Cao Z.; Li Y.; Ren H.; Zhang C.; et al. Nat. Cell Biol. 2012, 149924–934. [DOI] [PubMed] [Google Scholar]
- Rubinsztein D. C.; Nixon R. A. Proc. Natl. Acad. Sci. U.S.A. 2010, 10749E181.author reply E182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A.; Majumder S.; Richardson A.; Strong R.; Oddo S. J. Biol. Chem. 2010, 2851713107–13120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. M.; Huang S. T.; Huang S. H.; Lin H. W.; Tsai H. P.; Wu J. Y.; Lin C. M.; Chen C. T. Nanomedicine 2011, 71107–114. [DOI] [PubMed] [Google Scholar]
- Seleverstov O.; Zabirnyk O.; Zscharnack M.; Bulavina L.; Nowicki M.; Heinrich J. M.; Yezhelyev M.; Emmrich F.; O’Regan R.; Bader A. Nano Lett. 2006, 6122826–2832. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Zheng F.; Yang T.; Zhou W.; Liu Y.; Man N.; Zhang L.; Jin N.; Dou Q.; Zhang Y.; et al. Nat. Mater. 2012, 119817–826. [DOI] [PubMed] [Google Scholar]
- Yu L.; Lu Y.; Man N.; Yu S. H.; Wen L. P. Small 2009, 5242784–2787. [DOI] [PubMed] [Google Scholar]
- Fabbro A.; Villari A.; Laishram J.; Scaini D.; Toma F. M.; Turco A.; Prato M.; Ballerini L. ACS Nano 2012, 632041–2055. [DOI] [PubMed] [Google Scholar]
- Cellot G.; Cilia E.; Cipollone S.; Rancic V.; Sucapane A.; Giordani S.; Gambazzi L.; Markram H.; Grandolfo M.; Scaini D.; et al. Nat. Nanotechnol 2009, 42126–133. [DOI] [PubMed] [Google Scholar]
- Fan L.; Feng C.; Zhao W.; Qian L.; Wang Y.; Li Y. Nano Lett. 2012, 1273668–3673. [DOI] [PubMed] [Google Scholar]
- Yang D. S.; Stavrides P.; Mohan P. S.; Kaushik S.; Kumar A.; Ohno M.; Schmidt S. D.; Wesson D. W.; Bandyopadhyay U.; Jiang Y.; et al. Autophagy 2011, 77788–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M.; Palmieri M.; di Ronza A.; Medina D. L.; Valenza M.; Gennarino V. A.; Di Malta C.; Donaudy F.; Embrione V.; Polishchuk R. S.; et al. Science 2009, 3255939473–477. [DOI] [PubMed] [Google Scholar]
- Settembre C.; Di Malta C.; Polito V. A.; Garcia Arencibia M.; Vetrini F.; Erdin S.; Erdin S. U.; Huynh T.; Medina D.; Colella P.; et al. Science 2011, 33260361429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L.; Chen R.; Jiang A.; Wang L.; Wang P.; Li C. Z.; Bai R.; Zhao Y.; Autrup H.; Chen C. Small 2013, 99–101786–1798. [DOI] [PubMed] [Google Scholar]
- Lee J. H.; Yu W. H.; Kumar A.; Lee S.; Mohan P. S.; Peterhoff C. M.; Wolfe D. M.; Martinez-Vicente M.; Massey A. C.; Sovak G.; et al. Cell 2010, 14171146–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon R. A.; Yang D. S. Cold Spring Harbor Perspect. Biol. 2012, 4, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvedova A. A.; Kapralov A. A.; Feng W. H.; Kisin E. R.; Murray A. R.; Mercer R. R.; St Croix C. M.; Lang M. A.; Watkins S. C.; Konduru N. V.; et al. PLoS One 2012, 73e30923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali-Boucetta H.; Kostarelos K. Adv. Drug Delivery Rev. 2013, 65152111–2119. [DOI] [PubMed] [Google Scholar]
- Al-Jamal K. T.; Gherardini L.; Bardi G.; Nunes A.; Guo C.; Bussy C.; Herrero M. A.; Bianco A.; Prato M.; Kostarelos K.; et al. Proc. Natl. Acad. Sci. U.S.A. 2011, 1082710952–10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.