

Abstract
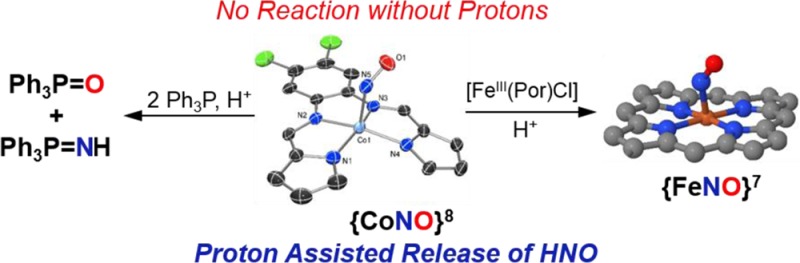
Research on the one-electron reduced analogue of NO, namely nitroxyl (HNO/NO–), has revealed distinguishing properties regarding its utility as a therapeutic. However, the fleeting nature of HNO requires the design of donor molecules. Metal nitrosyl (MNO) complexes could serve as potential HNO donors. The synthesis, spectroscopic/structural characterization, and HNO donor properties of a {CoNO}8 complex in a pyrrole/imine ligand frame are reported. The {CoNO}8 complex [Co(LN4PhCl)(NO)] (1) does not react with established HNO targets such as FeIII hemes or Ph3P. However, in the presence of stoichiometric H+1 behaves as an HNO donor. Complex 1 readily reacts with [Fe(TPP)Cl] or Ph3P to afford the {FeNO}7 porphyrin or Ph3P=O/Ph3P=NH, respectively. In the absence of an HNO target, the {Co(NO)2}10 dinitrosyl (3) is the end product. Complex 1 also reacts with O2 to yield the corresponding CoIII-η1-ONO2 (2) nitrato analogue. This report is the first to suggest an HNO donor role for {CoNO}8 with biotargets such as FeIII-porphyrins.
Among the many biochemical and physiological roles of NO•,1 its redox chemistry to reduced analogues such as nitroxyl (HNO/NO–, pKa = 11.6)2 has not been extensively studied. This is primarily due to the rapid dimerization and dehydration of HNO to N2O and H2O (k = 106 M–1 s–1)3 that makes this particular nitrogen oxide difficult to study. Despite its analogous structure, nitroxyl has demonstrated pharmacological and therapeutic advantages distinct from NO•.4 Such properties include specific targeting of biological thiols and FeIII heme proteins, increasing the plasma concentration of calcitonin gene-related peptide (CGRP, a small neuropeptide involved in vasodilation), and its positive cardiac inotrope effect (heart muscle contraction). These properties have driven the field to better understand the chemical biology of HNO through the design and synthesis of controllable donor molecules.5 The most common donors include diazeniumdiolates such as Na2N2O3 (Angeli’s salt)6 and N-hydroxysulfonamide derivatives (R-SO2-NH-OH) such as Piloty’s acid.5b,7 While reliable as HNO donors, these compounds are limited as they release other potentially reactive species or function at pH > 9.5 Clearly, the development of better HNO donors is needed.
Metal nitrosyls (MNO) could serve as an alternative platform for HNO/NO– delivery. Indeed, the heme enzyme responsible for NO synthesis (NO synthase) has been shown to release HNO via an Fe-bound N-hydroxy-l-arginine intermediate in the absence of its biopterin cofactor.8 Due to the variable redox states of NO when coordinated to metals (i.e., NO+, NO•, NO–), descriptions of bonding typically resort to the Enemark–Feltham (EF) notation as a result of extensive electron delocalization within the MNO moiety.9 We reported the synthesis and properties of nonheme {FeNO}8 complexes (a rare EF notation),10 one which demonstrates nitroxyl-like reactivity with equine skeletal metmyoglobin under pseudo-physiological conditions.10a Due to the inherent reactivity of the {FeNO}8 systems, we have now synthesized isoelectronic {CoNO}8 complexes with more electron-deficient supporting ligands as the next logical step in our goal of customizing an HNO/NO– delivery vehicle with more controllable properties. Herein we describe the synthesis and properties of a non-porphyrin-based {CoNO}8 complex, the H+-induced formation of the fleeting HNO donor intermediate, and its nitroxyl-like reaction with an FeIII-heme model complex and Ph3P.
The {CoNO}8 complex [Co(LN4PhCl)(NO)] (1) (Scheme 1) was synthesized by purging NO(g) into an MeCN solution of in situ prepared (Et4N)2[Co(LN4PhCl)Cl2] at 60 °C. The resulting dark-brown microcrystalline product precipitated from the reaction mixture and was isolated as analytically pure material in 72% yield. Complex 1 is stable to air, moisture, and vacuum in the solid-state. Solution samples (THF, MeCN) of 1 also demonstrate similar stability; however, slow air exposure (1 week) to an MeCN/THF solution of 1 afforded the corresponding nitrato complex [Co(LN4PhCl)(MeCN)(η1-ONO2)] (2) as single crystals (Scheme 1). This result parallels what is seen with other {CoNO}8/O2 reactions11 and highlights the nucleophilic nature of the NO ligand in 1 which is suggestive of its NO– character.
Scheme 1. Reactivity of 1.

HNO-derived products highlighted.
Recrystallization of the {CoNO}8 complex from 2-MeTHF/Et2O afforded X-ray quality crystals of 1, which revealed a square pyramidal Co center originating from the diimine-dipyrrolide N4 ligand (basal plane) with NO in the apical position (Figure 1). Relevant X-ray data and metric parameters are provided in Tables S1 and S2. The near-perfect square pyramidal geometry in 1 is indicated by its low trigonal distortion parameter (τ = 0.08)12 and is due to the extreme planarity of the N4 ligand with Co displaced by 0.264 Å out of the N4 plane and toward NO. The Co–Nimine (avg: 1.895 Å) and Co–Npyrrole (avg: 1.919 Å) distances are comparable to Co–N bonds in other non-porphyrin {CoNO}8 complexes13 and also in CoIII complexes without NO ligands such as complex 2 (avg Co–Nimine: 1.893 Å; avg Co–Npyrrole: 1.926 Å; SI, Figure 1). This comparison suggests a CoIII oxidation state for 1, which was further verified by X-ray absorption spectroscopic (XAS) measurements (vide infra). The Co–N(O) (1.798(3) Å) and bent Co–N–O angle (124.4(3)°) are also representative metrics of five-coordinate {CoNO}8 complexes.13d The bent nature of the Co–N–O unit in solution is further supported by the large downfield shift of the NO nitrogen in the 15N NMR spectrum (δ: 688 ppm vs CH3NO2 in THF-d8, Figure S2).14 Additionally, the N–O bond distance (1.172(4) Å) is in between that reported for 1HNO (1.21 Å)15 and NO (1.15 Å), but more like the latter.13d Consistent with this observation is the solid-state FTIR spectrum of 1, which exhibits νNO at 1667 cm–1 that shifts to 1638 cm–1 (ΔνNO: 29 cm–1) after isolating [Co(LN4PhCl)(15NO)] (1-15NO) using 15NO(g) in the synthesis (Figure S3). The νNO and diamagnetic ground state (see 1H NMR, Figure S1) is typical for this class of Co nitrosyls.13d,16
Figure 1.
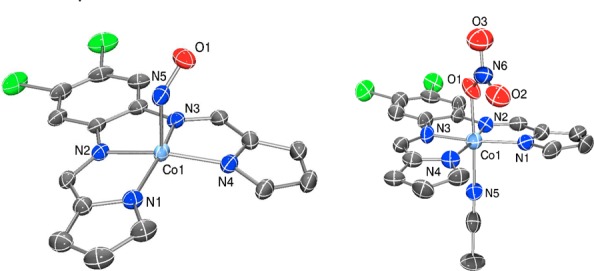
X-ray structures of [Co(LN4PhCl)(NO)] (1) (left) and [Co(LN4PhCl)(MeCN)(η1-ONO2)] (2) (right) (50% probability level). H atoms and distorted THF solvent of crystallization for 1 have been omitted for clarity.
XAS measurements support the crystallographic results obtained for 1. The X-ray absorption near edge spectrum (XANES) shows a large pre-edge feature centered at 7710.6 eV, characteristic of CoIII 1s → 3d electronic transitions; the measured peak area (0.387 eV) confirms the metal is coordinated in a non-centrosymmetric ligand environment (Figure 2). The first inflection energy in the XANES spectrum occurs at 7721.3 eV, as in other XAS-characterized CoIII compounds.17 Simulations of the extended X-ray absorption fine structure spectral region are consistent with CoIII in a five-coordinate ligand environment constructed of five O/N ligands at an average distance of 1.88 Å (Figure S4). These results compare favorably to distances obtained from X-ray diffraction (avg: 1.89 Å). Taken together, these results suggest the MNO unit in 1 is best described as low-spin CoIII (S = 0) bound to singlet nitroxyl anion (1NO–; S = 0).
Figure 2.

XANES spectrum for {CoNO}81. Inset: The baseline subtracted pre-edge features for 1.
Despite the formal assignment of the NO ligand as 1NO– in other {CoNO}8 systems,18 we were surprised that their chemical reactivity had not been explored in detail given their potential as HNO donors. Indeed, complexes with the {CoNO}8 notation are generally designated as low-spin CoIII coordinated to 1NO– to yield Stot = 0.18 Perhaps this paucity is due to the well-known kinetic inertness of low-spin d6 metals such as CoIII.19 One common and sensitive test for an HNO donor involves its reaction with FeIII-porphyrins20 or FeIII-heme proteins21 to afford the corresponding {FeNO}7 derivatives. Thus, the capability of 1 to reductively nitrosylate the FeIII-heme model [Fe(TPP)Cl] (TPP = dianion of tetraphenylporphyrin) was performed. When a stoichiometric amount of 1 was added to the FeIII heme in THF at 298 K, no reaction was observed even after 24 h (Scheme 1). Similarly, increasing the temperature to 310 K and monitoring over the same 24 h time period resulted in no reaction. However, when the reaction was repeated in the presence of 1 mol·equiv of an organic-soluble H+ donor (HBF4·Et2O), an entirely different reaction resulted (Scheme 1). In this case, [Fe(TPP)Cl] was almost immediately consumed within 4 min of H+ addition (monitored by UV–vis, FTIR) to afford the corresponding {FeNO}7 heme analogue [Fe(TPP)NO] (νNO: 1698 cm–1 in KBr). Bulk reaction studies further confirmed the near stoichiometric NO transfer between 1 and [Fe(TPP)Cl] (88% yield; Figure S7 for FTIR/EPR of the [Fe(TPP)NO] product). Using isotopically labeled 1-15NO resulted in the corresponding shift of νNO in the IR spectrum of the [Fe(TPP)15NO] product (νNO: 1667 cm–1 in KBr, ΔνNO: 31 cm–1) as well as the expected triplet-to-doublet hyperfine structural change in its X-band EPR spectrum (Figure S7).
Further insight regarding the mechanism of the reductive nitrosylation of [Fe(TPP)Cl] with 1 and HBF4·Et2O come from electrochemical measurements. In THF, 1 displays a quasi-reversible {CoNO}9/{CoNO}8 wave at E1/2 = −1.46 V vs Fc+/Fc in THF (Figure S5). One possible route to [Fe(TPP)NO] is via reduction of [Fe(TPP)Cl] from 1 to yield a {CoNO}7 complex that would release NO• to the FeII-heme model. An irreversible Eox peak at 0.75 V is observed suggesting the {CoNO}7 complex is unstable and likely results in loss of NO•. Since the FeIII/II couple of [Fe(TPP)Cl] is −0.90 V in THF (vs Fc+/Fc),22 it is doubtful that 1 can reduce FeIII to FeII in this reaction. It is possible that HNO transfer is taking place upon H+ addition to 1. Support for this hypothesis comes from reactions performed with DNICs (mononuclear tetrahedral dinitrosyl iron complexes) and FeIII-porphyrins by Darensbourg and co-workers.23 In these studies, addition of H+ greatly enhances the rate of HNO transfer to form the corresponding {FeNO}7 analogues (72 h without H+; 9 h with 0.25 equiv of H+). This rate enhancement of HNO transfer/release from the DNICs has been attributed to the “carrier effect” of H+ for NO– as the neutral HNO should be more mobile in nonpolar aprotic solvents such as CH2Cl2 or THF.
Additional proof that 1 behaves as an HNO donor in the presence of stoichiometric H+ stem from its reaction with Ph3P (Scheme 1). King established that triarylphosphines (R3P) react selectively with HNO donors to yield the corresponding phosphine oxide (R3P=O) and aza-ylide (R3P=NH): HNO + 2R3P → R3P=O + R3P=NH.24 These molecules represent unique and quantifiable products as markers of HNO, which is measured by the amount of R3P=O formed (R3P=NH is reactive and hydrolyzes to R3P=O). Accordingly, addition of H+ (1.3 equiv) to the red-brown 1/Ph3P (1/2) mixture resulted in a lightening of this solution to a yellow-tinted transparent brown after 24 h. 31P NMR analysis of this mixture clearly indicated the presence of Ph3P=O (δ: 26.3 ppm), Ph3P=NH (δ: 20.6 ppm), and unreacted Ph3P (−6.0 ppm) (Figure S16). Additional support for Ph3P=NH derives from using 1-15NO, which afforded identical 31P NMR features except for the doublet splitting of the 20.6 ppm peak arising from the 15N nucleus (1JP–N: 135 Hz). This result is definitive proof that 1 in the presence of H+ is an HNO donor. Control experiments with Ph3P and 1 indicated no reaction. To evaluate the extent to which 1/H+ donates HNO, the amount of Ph3P was quantified by HPLC (SI), which was determined to be 33% (2 equiv Ph3P) after 24 h. Utilizing excess (10 equiv) Ph3P improved the yield to 37% (Figure S19). Other peaks in the chromatogram were identified as 1 and free ligand (SI), which may account for the ∼40% yield of Ph3P=O. R3P-based reactions with established HNO donors have also been less than stoichiometric.24b Collectively, these results offer strong support that 1 and H+ generate a compound that is capable of releasing HNO.
In an effort to verify the actual species responsible for the HNO donor property, we attempted in situ characterization of intermediates utilizing high-resolution MS. For example, the MS of the 1 and HBF4 (1/1.3; 30 min) mixture generate a major peak at m/z: 418.974(1) that is assigned to a reduced and doubly protonated cation of general formula [Co(LN4PhClH2)(NO)]+ (Figure 3).25 This peak remains even after 24 h mixing. Fragmenting this peak afforded one signal at m/z: 388.976(1) corresponding to loss of NO (30 amu) and formation of [Co(LN4PhClH2)]+. Experiments with 1-15NO provided analogous results with loss of 31 amu (15NO) (Figure S14). In contrast, 1 reveals no significant peaks in the MS, consistent with its neutral charge. FTIR and 1H NMR measurements reveal additional benchmarks that may be assigned to this intermediate (isotope-sensitive νNO: 1544 cm–1; δNH: 11.5 ppm, Figures S8–S13). A {[Co(LN4PhCl)(HNO)] + H}+ {CoHNO}9 formulation is consistent with the MS; however, the absence of 15N coupling (I = 1/2) to the 11.5 ppm peak in the NMR (using 1-15NO) suggest this signal to arise from protonated ligand. Overall, these observations implicate a three-coordinate, protonated pyrrole-NH and imine-N bound {CoNO}8 (4) as the first intermediate formed in the 1/H+ reaction (Scheme 2). After 24 h mixing, a new species corresponding to [Co(LN4PhClH2)(NO)2]BF4 (3) is observed. Spectroscopic analysis of the reaction mixture confirms this assignment with 15N-sensitive νNO (1869, 1793 cm–1) and m/z: 448.972 (Figures S9, S15). These parameters are consistent with independently synthesized and characterized 3 and other neutral N-bound {Co(NO)2}10 complexes (Figures S20–S23).26 A significant portion of 1 and 4 also remain and implicate the equilibrium depicted in Scheme 2.27
Figure 3.

Top: High-resolution ESI-MS (positive mode) of the reaction of 1 + HBF4·Et2O. Bottom: Theoretical isotopic distribution.
Scheme 2. Reactivity of 1 with H+.

The results obtained provide insight into the potential paths traversed with 1/H+. In the absence of HNO targets, 4 appears as the first CoNO intermediate after H+ addition, providing strong support that this species is likely responsible for the observed reaction chemistry based on IR, MS, and NMR. While this proposal does not completely eliminate a transient Co-HNO, none of the current evidence supports its formation. Indeed, the lack of definitive proof for Co-HNO is not too surprising as the isolation and characterization of first-row M-HNO adducts have met with limited success due to the reactive nature of the nitroxyl ligand.28 Attempts to isolate 4 either from the reaction mixture or directly have not been successful.29,30 One hypothesis for the reductive nitrosylation of [Fe(TPP)Cl] is the generation of a bridging {Co(μ-NO)(μ-Cl)Fe} intermediate where trans-nitroxylation occurs to yield the {FeNO}7 heme, a route suggested for the isoelectronic {FeNO}8 complex.10b H+ addition simply enhances the rate of nitroxyl transfer as suggested for DNICs.23 Such bridging intermediates have also been proposed in the dismutation of {CoNO}8 species to {Co(NO)2}10 and CoIII indicative of formal NO– transfer.31 This result is analogous to the fate of 1 to give 3 in the absence of HNO-reactive molecules (Scheme 2). The reaction with Ph3P may take place from 4 (via tautomerization) or a transient Co-HNO intermediate structurally similar to 1. Both free and coordinated Ph3P represent viable possibilities as well. More detailed studies were precluded by the complexity of the process and the difficulty in isolating and detecting every reaction product. Regardless, the observed results are consistent with an HNO donor property for 1/H+.
In conclusion, we report the synthesis of {CoNO}81 and its reaction with H+ to afford an HNO donating intermediate. In the absence of an HNO target, reaction of 1 and H+ ultimately leads to the Co-dinitrosyl complex 3 (via intermediate 4). To our knowledge the reaction of 1 and H+ is the first example of a {CoNO}8 complex that specifically behaves as an HNO donor to known HNO targets (FeIII-heme and Ph3P). Proton-induced formation of other signaling molecules such as H2S has been observed with synthetic 2Fe-2S clusters and excess NO.34 In contrast, 1 alone does not exhibit any reactivity from temperatures ranging 298–310 K, emphasizing the importance of HNO versus NO– and the underexplored potential of metal nitrosyls as HNO donors. Furthermore, these results confirm that {CoNO}8 complexes are not inert and function as sources of HNO under specific pH conditions, thereby opening up a new frontier in HNO donor research.
Acknowledgments
T.C.H. thanks the NSF (CHE-0953102) and UGA for financial support. T.L.S. and A.V.R acknowledge the NIH and the American Heart Association/Friedreich’s Ataxia Research Alliance awards DK068139 and 12PRE11720005, respectively. XAS was collected at the National Synchrotron Radiation Laboratory (NSLS) on beamline X3b. NSLS is supported by the U.S. DOE, Division of Materials Sciences and Division of Chemical Sciences (contract no. DEAC02-98CH10886). J.L.U. and R.J.U. thank the Office of the Vice President for Research (OVPR) and the Office of the Provost at UGA for financial support. We wish to thank Dr. Dennis R. Phillips, Dr. Chau-Wen Chou, and Prof. I. Jonathan Amster for their assistance with high-resolution MS experiments. We acknowledge Henry Niedermaier for assistance with NMR.
Supporting Information Available
Details of the syntheses, reactivity, spectroscopic, and crystallographic (CIF) results. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Moncada S.; Higgs E. A. Br. J. Pharmacol. 2006, 147, S193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartberger M. D.; Liu W.; Ford E.; Miranda K. M.; Switzer C.; Fukuto J. M.; Farmer P. J.; Wink D. A.; Houk K. N. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 10958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafirovich V.; Lymar S. V. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 7340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine J. C.; Ritchie R. H.; Favaloro J. L.; Andrews K. L.; Widdop R. E.; Kemp-Harper B. K. Trends Pharmacol. Sci. 2008, 29, 601. [DOI] [PubMed] [Google Scholar]
- a Miranda K. M. Coord. Chem. Rev. 2005, 249, 433. [Google Scholar]; b Paolocci N.; Jackson M. I.; Lopez B. E.; Miranda K.; Tocchetti C. G.; Wink D. A.; Hobbs A. J.; Fukuto J. M. Pharmacol. Ther. 2007, 113, 442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeli A.; Angelico A.; Scurti F. Chem. Zentralbl. 1902, 73, 691. [Google Scholar]
- Bonner F. T.; Ko Y. Inorg. Chem. 1992, 31, 2514. [Google Scholar]
- a Adak S.; Wang Q.; Stuehr D. J. J. Biol. Chem. 2000, 275, 33554. [DOI] [PubMed] [Google Scholar]; b Rusche K. M.; Spiering M. M.; Marletta M. A. Biochemistry 1998, 37, 15503. [DOI] [PubMed] [Google Scholar]
- Enemark J. H.; Feltham R. D. Coord. Chem. Rev. 1974, 13, 339. [Google Scholar]
- a Patra A. K.; Dube K. S.; Sanders B. C.; Papaefthymiou G. C.; Conradie J.; Ghosh A.; Harrop T. C. Chem. Sci. 2012, 3, 364. [Google Scholar]; b Sanders B. C.; Patra A. K.; Harrop T. C. J. Inorg. Biochem. 2013, 118, 115. [DOI] [PubMed] [Google Scholar]
- Trogler W. C.; Marzilli L. G. Inorg. Chem. 1974, 13, 1008. [Google Scholar]
- Addison A. W.; Rao T. N.; Reedijk J.; van Rijn J.; Verschoor G. C. J. Chem. Soc., Dalton Trans. 1984, 1349. [Google Scholar]
- a Franz K. J.; Doerrer L. H.; Spingler B.; Lippard S. J. Inorg. Chem. 2001, 40, 3774. [DOI] [PubMed] [Google Scholar]; b Hess J. L.; Conder H. L.; Green K. N.; Darensbourg M. Y. Inorg. Chem. 2008, 47, 2056. [DOI] [PubMed] [Google Scholar]; c Kozhukh J.; Lippard S. J. J. Am. Chem. Soc. 2012, 134, 11120. [DOI] [PubMed] [Google Scholar]; d McCleverty J. A. Chem. Rev. 2004, 104, 403. [DOI] [PubMed] [Google Scholar]
- Mason J.; Larkworthy L. F.; Moore E. A. Chem. Rev. 2002, 102, 913. [DOI] [PubMed] [Google Scholar]
- Dalby F. W. Can. J. Phys. 1958, 36, 1336. [Google Scholar]
- Wyllie G. R. A.; Scheidt W. R. Chem. Rev. 2002, 102, 1067. [DOI] [PubMed] [Google Scholar]
- a Padden K. M.; Krebs J. F.; MacBeth C. E.; Scarrow R. C.; Borovik A. S. J. Am. Chem. Soc. 2001, 123, 1072. [DOI] [PubMed] [Google Scholar]; b Padden K. M.; Krebs J. F.; Trafford K. T.; Yap G. P. A.; Rheingold A. H.; Borovik A. S.; Scarrow R. C. Chem. Mater. 2001, 13, 4305. [Google Scholar]
- a McCleverty J. A. Chem. Rev. 1979, 79, 53. [Google Scholar]; b Duffin P. A.; Larkworthy L. F.; Mason J.; Stephens A. N.; Thompson R. M. Inorg. Chem. 1987, 26, 2034. [Google Scholar]
- Huheey J. E.; Keiter E. A.; Keiter R. L.. Inorganic Chemistry: Principles of Structure and Reactivity, 4th ed.; HarperCollins College Publishers: New York, 1993. [Google Scholar]
- a Bari S. E.; Martí M. A.; Amorebieta V. T.; Estrin D. A.; Doctorovich F. J. Am. Chem. Soc. 2003, 125, 15272. [DOI] [PubMed] [Google Scholar]; b Martí M. A.; Bari S. E.; Estrin D. A.; Doctorovich F. J. Am. Chem. Soc. 2005, 127, 4680. [DOI] [PubMed] [Google Scholar]
- a Bazylinski D. A.; Goretski J.; Hollocher T. C. J. Am. Chem. Soc. 1985, 107, 7986. [Google Scholar]; b Bazylinski D. A.; Hollocher T. C. J. Am. Chem. Soc. 1985, 107, 7982. [Google Scholar]; c Doyle M. P.; Mahapatro S. N.; Broene R. D.; Guy J. K. J. Am. Chem. Soc. 1988, 110, 593. [Google Scholar]; d Miranda K. M.; Nims R. W.; Thomas D. D.; Espey M. G.; Citrin D.; Bartberger M. D.; Paolocci N.; Fukuto J. M.; Feelisch M.; Wink D. A. J. Inorg. Biochem. 2003, 93, 52. [DOI] [PubMed] [Google Scholar]
- Bottomley L. A.; Kadish K. M. Inorg. Chem. 1981, 20, 1348. [Google Scholar]
- Chiang C.-Y.; Darensbourg M. Y. J. Biol. Inorg. Chem. 2006, 11, 359. [DOI] [PubMed] [Google Scholar]
- a Reisz J. A.; Klorig E. B.; Wright M. W.; King S. B. Org. Lett. 2009, 11, 2719. [DOI] [PubMed] [Google Scholar]; b Reisz J. A.; Zink C. N.; King S. B. J. Am. Chem. Soc. 2011, 133, 11675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Oxidation–reduction reactions have been previously shown to occur in coordination complexes when examined by ESI-MS; b Henderson W.; McIndoe J. S.. The ESI MS Behaviour of Coordination Complexes. In Mass Spectrometry of Inorganic and Organometallic Compounds; John Wiley & Sons Ltd: Chichester, 2005. [Google Scholar]
- Gwost D.; Caulton K. G. Inorg. Chem. 1973, 12, 2095. [Google Scholar]
- According to peak integrations, 3 makes up ∼20% of the reaction mixture (Figure S12). We attribute the low amount of 3 due to its poor solubility and the large amount of unreacted 1.
- a Berto T. C.; Speelman A. L.; Zheng S.; Lehnert N. Coord. Chem. Rev. 2013, 257, 244. [Google Scholar]; b Farmer P. J.; Sulc F. J. Inorg. Biochem. 2005, 99, 166. [DOI] [PubMed] [Google Scholar]; c Pellegrino J.; Bari S. E.; Bikiel D. E.; Doctorovich F. J. Am. Chem. Soc. 2010, 132, 989. [DOI] [PubMed] [Google Scholar]; d Sanders B. C.; Rhine M. A.; Harrop T. C. Struct. Bonding (Berlin) 2014, 160, 57. [Google Scholar]
- Addition of NO(g) to MeOH solutions containing LN4PhClH2 and Co(II) afford a mixture of species. A medium intensity peak at ∼2200 cm–1 may be from a coordinated N2O ligand, which is a known disproportionation product of coordinated NO: 3NO → N2O + NO2. See ref (30).
- Franz K. J.; Lippard S. J. J. Am. Chem. Soc. 1999, 121, 10504. [Google Scholar]
- Del Zotto A.; Mezzetti A.; Rigo P. Inorg. Chim. Acta 1990, 171, 61. [Google Scholar]
- Tran C. T.; Kim E. Inorg. Chem. 2012, 51, 10086. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.