

Abstract
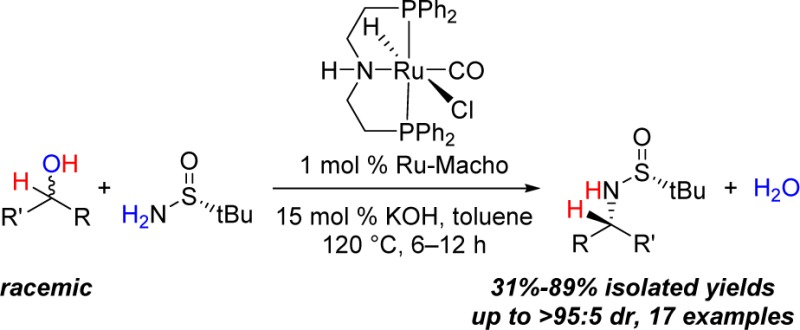
A commercially available ruthenium(II) PNP-type pincer catalyst (Ru-Macho) promotes the formation of α-chiral tert-butanesulfinylamines from racemic secondary alcohols and Ellman’s chiral tert-butanesulfinamide via a hydrogen borrowing strategy. The formation of α-chiral tert-butanesulfinylamines occurs in yields ranging from 31% to 89% with most examples giving >95:5 dr.
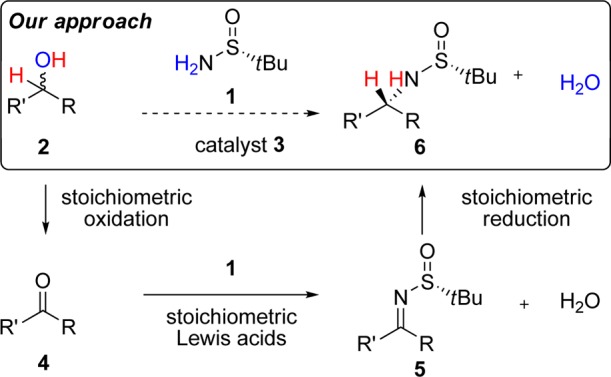
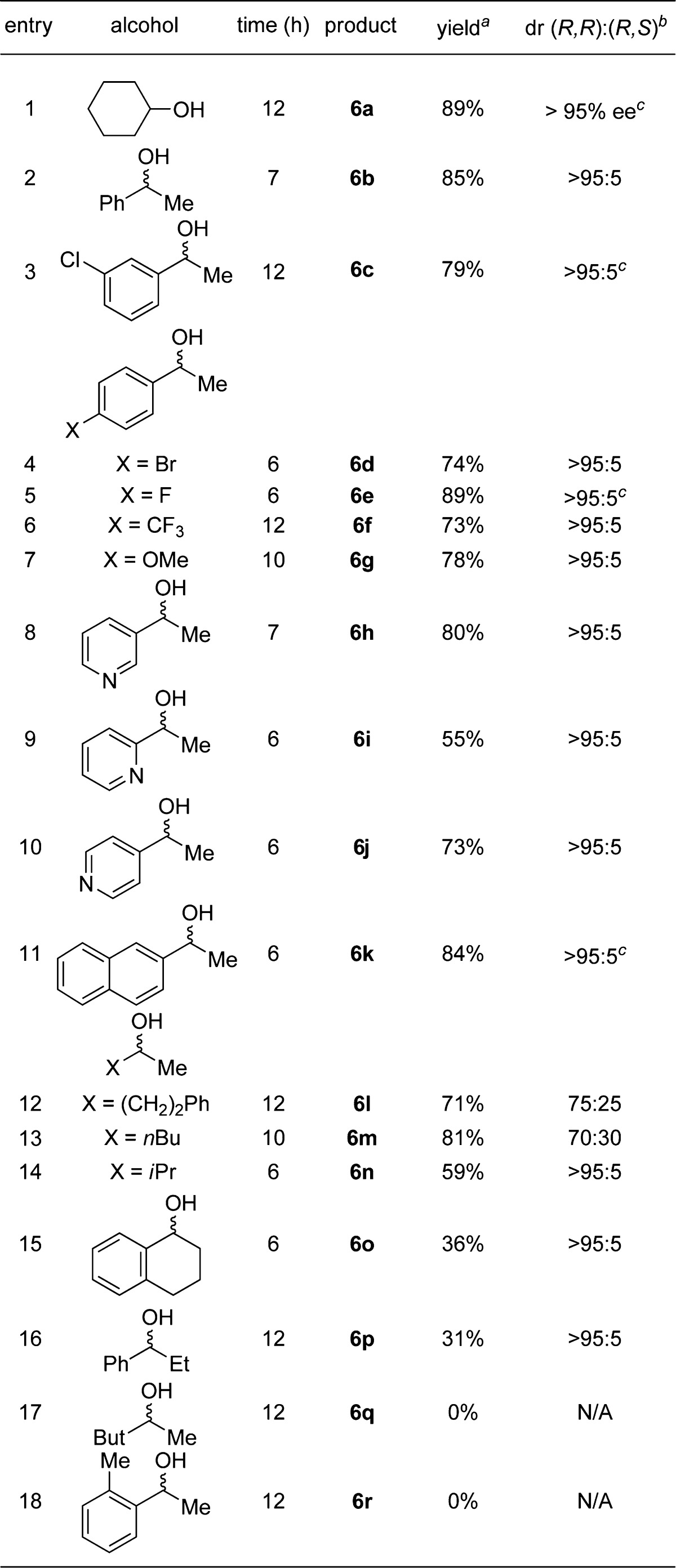
Because of the high value of α-chiral amines in both medicinal and synthetic chemistry, there is a need to develop more efficient methods for the stereoselective construction of C–N bonds.1 Since its discovery in 1997, Ellman’s sulfinamide has become a widely used reagent for the synthesis of α-chiral primary amines, with many industrially relevant applications.2,3 Considering the high practicality of this ammonia equivalent, we sought a catalytic method for the diastereoselective N-alkylation of Ellman’s sulfinamide (1) using readily available secondary alcohols 2 (Scheme 1).4,5 This formal nucleophilic substitution would transform racemic alcohols into enantiopure amines and generate water as the only byproduct. Herein we describe a Ru-catalyzed approach that overcomes the need for stoichiometric reagents and achieves in a single operation what traditionally requires three chemical steps (i.e., oxidation, condensation using Ti(OEt)4, and then reduction), as shown in Scheme 1.
Scheme 1. Diastereoselective Amination: Proposed Catalytic Strategy (One Step) versus the Conventional Approach (Three Steps).
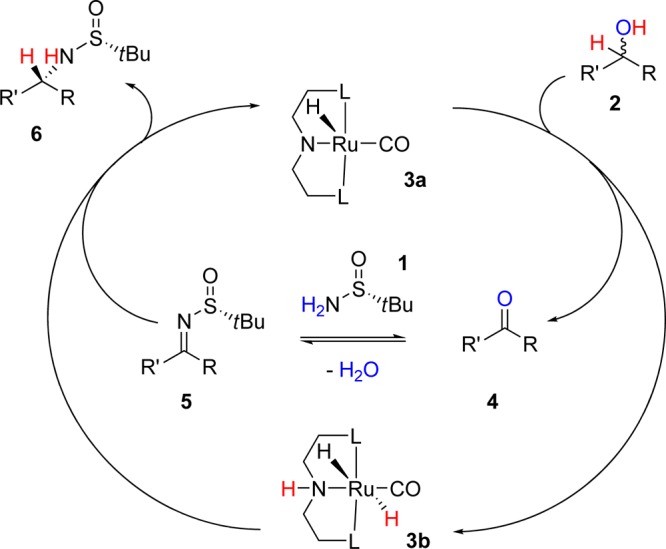
The direct amination of alcohols has been demonstrated with various catalysts,6,7 but only one stereoselective amination has been achieved to date, by Zhao and co-workers.8 However, this Ir-catalyzed method is limited to the synthesis of chiral anilines.8 Guided by previous studies using Ru–pincer complexes,4,9 we proposed that racemic alcohol 2 (Scheme 2) would undergo oxidation by Ru–pincer complex 3a to form ketone 4 and Ru hydride 3b. Ketone 4 would undergo condensation with sulfinamide 1 to form the sulfinylimine 5.10 Hydrogenation of imine 5 via Ru hydride 3b would proceed with high and predictable diastereoselectivity to generate the α-chiral sulfinylamine 6.10 To achieve this novel alkylation, we recognized the challenge of identifying a catalyst that would favor hydrogen transfer over competing pathways (e.g., acceptorless dehydrogenation).11,12 Moreover, the Ru catalyst would need to tolerate H2O as the main byproduct.
Scheme 2. Mechanistic Proposal Featuring Hydrogen Borrowing via a Ruthenium(II) Pincer Complex.
To test our hypothesis, we investigated the coupling of sulfinamide 1 and racemic alcohol 1-phenylethanol (2b) with a number of Ru–pincer complexes (Table 1). A few structurally related pincer complexes, including Milstein’s catalyst 3c (entry 1) as well as catalysts 3d and 3f (entries 2 and 3), showed no desired reactivity, even though these catalysts are known to activate both C–N and C–O bonds.11,13 In a previous study,14 we discovered that Ru-Macho (3f)15 acts as an efficient catalyst for the dehydrogenative synthesis of amides and imines from alcohols and amines. We envisioned that this complex could potentially act as a hydrogen-transfer catalyst to afford the desired product. Indeed, Ru-Macho afforded α-chiral sulfinylamine 6 in 71% conversion (entry 4). By further tuning the reaction parameters, we identified the optimal conditions to be the use of toluene as the solvent and KOH as the base at 120 °C (89% conversion; entry 10). The transformation occurs efficiently using only 1 mol % catalyst.
Table 1. Optimization of Reaction Conditions for the Formation of N-tert-Butanesulfinylamines.
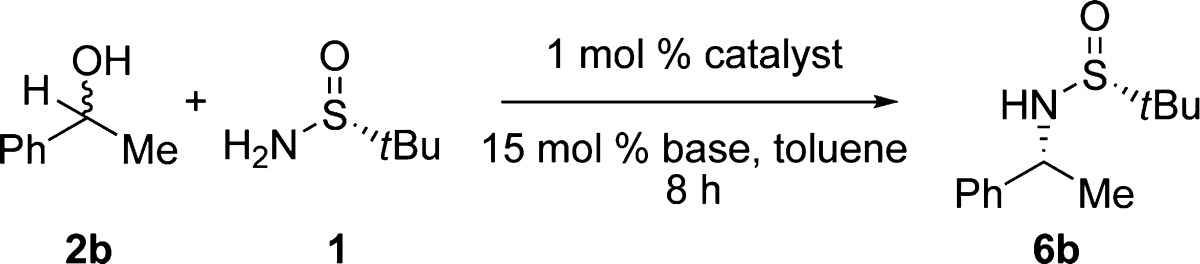
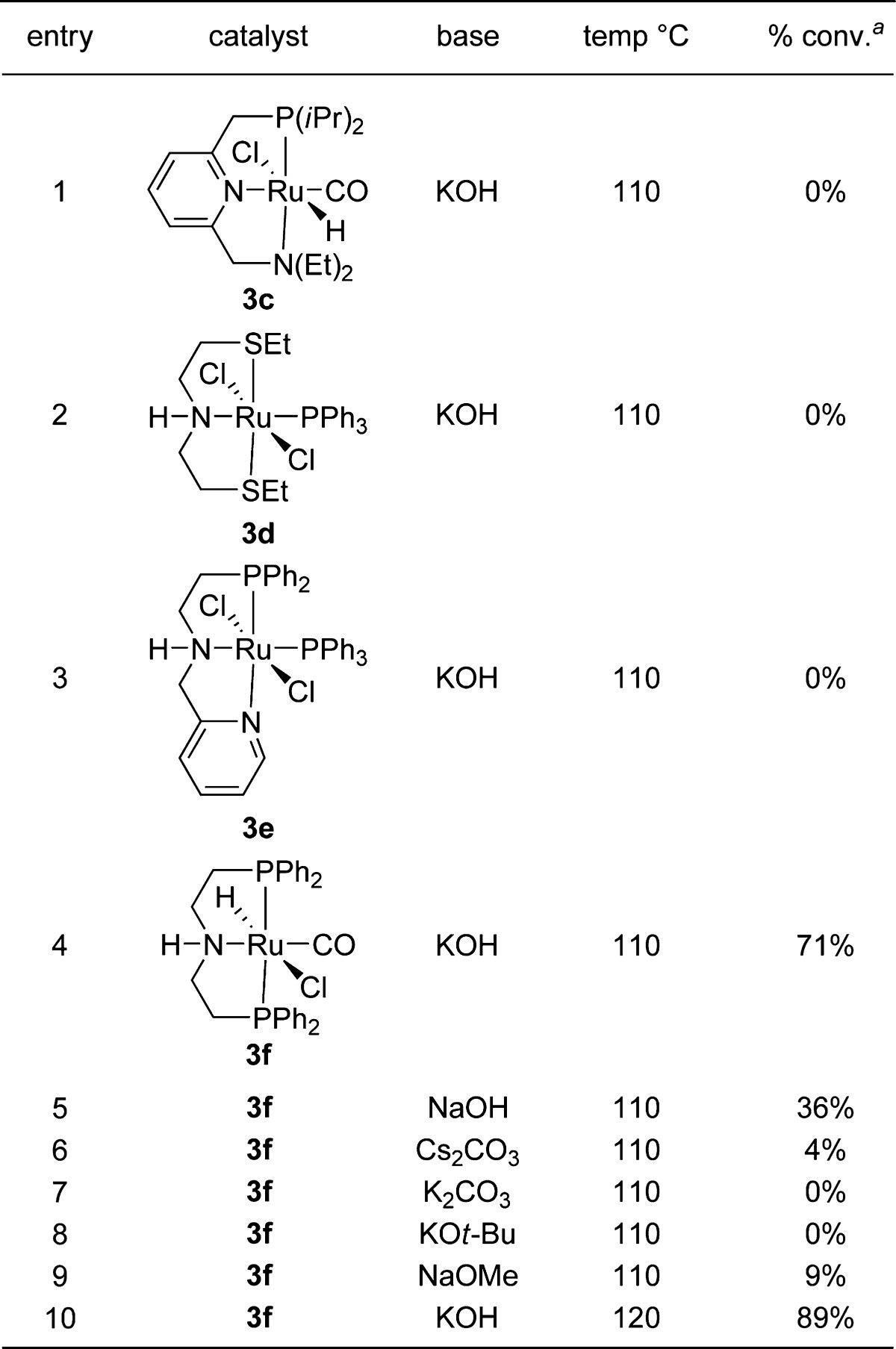
Reaction conditions: 0.5 mmol of 2b, 0.5 mmol of 1, 1 mol % 3, 15 mol % base, 0.5 mL of toluene, 110 °C, 8 h.
Conversion measured by GC with a known amount of dodecane standard.
With this protocol in hand, we transformed a variety of racemic alcohols into optically active amines. (Table 2).16 The reaction conditions do not epimerize the chiral sulfinamide, and the expected enantiomer was produced after coupling with an achiral alcohol (entry 1). Both electron-donating and electron-withdrawing groups on the β-phenyl group relative to the amine could be incorporated (73–89% yield, >95:5 dr; entries 3–7). Fluoro-containing amines were obtained in good yields (73–89%) with high diastereocontrol (>95:5 dr) (entries 5 and 6). Next, we found that chiral pyridyl derivatives (6h–j) could be generated in 55–80% isolated yield with high diastereoselectivity (>95:5 dr) (entries 8–10). A racemic alcohol bearing a larger aromatic substituent, such as the 2-naphthyl group (entry 11) underwent coupling in 84% yield with >95:5 dr. When the aryl group was substituted for saturated hydrocarbon chains (entries 12 and 13), a drop in diastereoselectivity (75:25 and 70:30 dr, respectively) was observed, but the reaction yield remained between 71 and 81% (6l and 6m, respectively). Our observations are in agreement with those of previous studies by Ellman and others, who found that reduction of sulfinylimines proceeds with lowered diastereocontrol when the β-substituents are similar in size.2,17 We propose that the lower diastereocontrol in entries 12 and 13 is due to the similar steric bulk of a methyl group and an n-butyl or −(CH2)2Ph group.18 In contrast, an alcohol with a isopropyl substituent (entry 14) was shown to form the corresponding sulfinylamine with higher diastereoselectivity (>95:5) as a result of the greater steric difference in the corresponding substituents (6n).19
Table 2. Variation of Secondary Alcohols Used in Diastereoselective Amination.
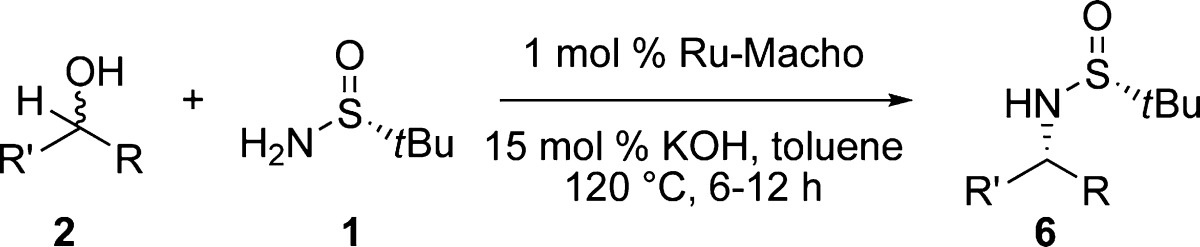
Reaction conditions: 0.5 mmol of 1, 0.5 mmol of 2, 1 mol % Ru-Macho, 15 mol % KOH, 0.5 mL of toluene, 120 °C.
Isolated yields.
Determined by 1H NMR analysis.
Determined by polarimetry.
The synthesis of α-chiral amines bearing a β-methyl group is challenging to achieve because of the low diastereocontrol in the conventional addition of MeLi to N-tert-butylsulfinylaldimine.2,3 In contrast, our method is most effective for the construction of amines bearing a β-methyl group (Table 2, entries 2–14). Ru-Macho appears to be sensitive to the steric bulk at the β-position on the sulfinylimine intermediate during hydrogen transfer (5 in Scheme 2). For example, lower isolated yields are encountered when bulkier groups were placed at the β-position on both sides of the alcohol (31–36%; entries 15 and 16), but the diastereoselectivities remain high (>95:5 dr) (6o and 6p). We found that more bulky substrates (e.g., a tert-butyl-substituted alcohol or ortho-arylated alcohol) show no reactivity under these conditions (entries 17 and 18).
In summary, this study provides an effective method for chiral amine synthesis by combining the power of the widely used Ellman’s sulfinamide auxiliary with the practicality of the inexpensive and industrially relevant Ru-Macho catalyst.15 By using a hydrogen borrowing strategy, α-chiral amines can be accessed directly from racemic alcohols in one step.6 In addition to fewer required steps, this catalytic approach overcomes the need to use stoichiometric Lewis acids.2,3 Moreover, the resulting diastereomeric products can be easily separated.2,3 Importantly, this methodology enables access to methyl-substituted chiral amines that are challenging to obtain with methyl organometallic reagents.2,3 Ongoing studies to expand substrate scope via catalyst design are underway.
Acknowledgments
Z.G. is thankful for the financial support received from the U.S. National Institutes of Health (DK098446) and the U.S. National Science Foundation (CHE-1012422). V.M.D. is thankful for the financial support received from the U.S. National Institutes of Health (GM105938).
Supporting Information Available
Substrate preparation and 1H and 13C NMR spectra and characterization data for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Nugent T. C.Chiral Amine Synthesis: Methods, Developments and Applications; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]; Nugent T. C.; El-Shazly M. Adv. Synth. Catal. 2011, 353. [Google Scholar]
- Robak M. T.; Herbage M. A.; Ellman J. A. Chem. Rev. 2010, 110, 3600. [DOI] [PubMed] [Google Scholar]
- Liu G. C.; Cogan D. A.; Ellman J. A. J. Am. Chem. Soc. 1997, 119, 9913. [Google Scholar]
- Gunanathan C.; Milstein D. Angew. Chem., Int. Ed. 2008, 47, 8661. [DOI] [PubMed] [Google Scholar]
- Klinkenberg J. L.; Hartwig J. F. Angew. Chem., Int. Ed. 2011, 50, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]; Pingen D.; Muller C.; Vogt D. Angew. Chem., Int. Ed. 2010, 49, 8130. [DOI] [PubMed] [Google Scholar]
- Bahn S.; Imm S.; Neubert L.; Zhang M.; Neumann H.; Beller M. ChemCatChem 2011, 3, 1853. [Google Scholar]; Watson A. J. A.; Williams J. M. J. Science 2010, 329, 635. [DOI] [PubMed] [Google Scholar]; Hamid M. H. S. A.; Slatford P. A.; Williams J. M. J. Adv. Synth. Catal. 2007, 349, 1555. [Google Scholar]; Guillena G.; Ramón D. J.; Yus M. Chem. Rev. 2010, 110, 1611. [DOI] [PubMed] [Google Scholar]; Dobereiner G. E.; Crabtree R. H. Chem. Rev. 2010, 110, 681. [DOI] [PubMed] [Google Scholar]
- Imm S.; Bahn S.; Neubert L.; Neumann H.; Beller M. Angew. Chem., Int. Ed. 2010, 49, 8126. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Lim C. S.; Sim D. S. B.; Pan H. J.; Zhao Y. Angew. Chem., Int. Ed. 2014, 53, 1399. [DOI] [PubMed] [Google Scholar]
- Monrad R. N.; Madsen R. Org. Biomol. Chem. 2011, 9, 610. [DOI] [PubMed] [Google Scholar]; Yamaguchi K.; He J. L.; Oishi T.; Mizuno N. Chem.—Eur. J. 2010, 16, 7199. [DOI] [PubMed] [Google Scholar]; Watson A. J. A.; Maxwell A. C.; Williams J. M. J. Org. Lett. 2009, 11, 2667. [DOI] [PubMed] [Google Scholar]; Hamid M. H. S. A.; Allen C. L.; Lamb G. W.; Maxwell A. C.; Maytum H. C.; Watson A. J. A.; Williams J. M. J. J. Am. Chem. Soc. 2009, 131, 1766. [DOI] [PubMed] [Google Scholar]; Zhang J.; Balaraman E.; Leitus G.; Milstein D. Organometallics 2011, 30, 5716. [Google Scholar]; Tillack A.; Hollmann D.; Mevius K.; Michalik D.; Bahn S.; Beller M. Eur. J. Org. Chem. 2008, 4745. [Google Scholar]; Hollmann D.; Tillack A.; Michalik D.; Jackstell R.; Beller M. Chem.—Asian J. 2007, 2, 403. [DOI] [PubMed] [Google Scholar]
- Xiao X.; Wang H. W.; Huang Z. Y.; Yang J.; Bian X. X.; Qin Y. Org. Lett. 2006, 8, 139. [DOI] [PubMed] [Google Scholar]; Pablo O.; Guijarro D.; Kovacs G.; Lledos A.; Ujaque G.; Yus M. Chem.—Eur. J. 2012, 18, 1969. [DOI] [PubMed] [Google Scholar]; Guijarro D.; Pablo O.; Yus M. J. Org. Chem. 2010, 75, 5265. [DOI] [PubMed] [Google Scholar]
- Gunanathan C.; Ben-David Y.; Milstein D. Science 2007, 317, 790. [DOI] [PubMed] [Google Scholar]
- Schley N. D.; Dobereiner G. E.; Crabtree R. H. Organometallics 2011, 30, 4174. [Google Scholar]; Gnanaprakasam B.; Milstein D. J. Am. Chem. Soc. 2011, 133, 1682. [DOI] [PubMed] [Google Scholar]; Nordstrom L. U.; Vogt H.; Madsen R. J. Am. Chem. Soc. 2008, 130, 17672. [DOI] [PubMed] [Google Scholar]; Chen C.; Zhang Y.; Hong S. H. J. Org. Chem. 2011, 76, 10005. [DOI] [PubMed] [Google Scholar]; Muthaiah S.; Ghosh S. C.; Jee J. E.; Chen C.; Zhang J.; Hong S. H. J. Org. Chem. 2010, 75, 3002. [DOI] [PubMed] [Google Scholar]; Dam J. H.; Osztrovszky G.; Nordstrom L. U.; Madsen R. Chem.—Eur. J. 2010, 16, 6820. [DOI] [PubMed] [Google Scholar]; Shimizu K.; Ohshima K.; Satsuma A. Chem.—Eur. J. 2009, 15, 9977. [DOI] [PubMed] [Google Scholar]; Zweifel T.; Naubron J. V.; Grutzmacher H. Angew. Chem., Int. Ed. 2009, 48, 559. [DOI] [PubMed] [Google Scholar]; Srimani D.; Balaraman E.; Hu P.; Ben-David Y.; Milstein D. Adv. Synth. Catal. 2013, 355, 2525. [Google Scholar]; Rigoli J. W.; Moyer S. A.; Pearce S. D.; Schomaker J. M. Org. Biomol. Chem. 2012, 10, 1746. [DOI] [PubMed] [Google Scholar]; Naota T.; Murahashi S. I. Synlett 1991, 693. [Google Scholar]
- Bertoli M.; Choualeb A.; Lough A. J.; Moore B.; Spasyuk D.; Gusev D. G. Organometallics 2011, 30, 3479. [Google Scholar]; Spasyuk D.; Smith S.; Gusev D. G. Angew. Chem., Int. Ed. 2012, 51, 2772. [DOI] [PubMed] [Google Scholar]; Spasyuk D.; Smith S.; Gusev D. G. Angew. Chem., Int. Ed. 2013, 52, 2538. [DOI] [PubMed] [Google Scholar]
- Oldenhuis N. J.; Dong V. M.; Guan Z. Tetrahedron 2014, 70, 4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyama W.; Matsumoto T.; Ogata O.; Ino Y.; Aoki K.; Tanaka S.; Ishida K.; Kobayashi T.; Sayo N.; Saito T. Org. Process Res. Dev. 2012, 16, 166. [Google Scholar]
- Paul C. E.; Rodríguez-Mata M.; Busto E.; Lavandera I.; Gotor-Fernández V.; Gotor V.; García-Cerrada S.; Mendiola J.; de Frutos Ó.; Collado I. Org. Process Res. Dev. 2014, 18, 788. [Google Scholar]
- Colyer J. T.; Andersen N. G.; Tedrow J. S.; Soukup T. S.; Faul M. M. J. Org. Chem. 2006, 71, 6859. [DOI] [PubMed] [Google Scholar]; Chelucci G.; Baldino S.; Chessa S.; Pinna G. A.; Soccolini F. Tetrahedron: Asymmetry 2006, 17, 3163. [Google Scholar]; Tanuwidjaja J.; Peltier H. M.; Ellman J. A. J. Org. Chem. 2007, 72, 626. [DOI] [PubMed] [Google Scholar]
- See the supporting information for a proposed transition-state structure.
- Hirsch J. A. Top. Stereochem. 1967, 1, 199. [Google Scholar]; Jensen F. R.; Bushweller C. H. Adv. Alicyclic Chem. 1971, 3, 139. [Google Scholar]; Eliel E. L.; Wilen S. H.; Stereochemistry of Organic Compounds; Wiley: New York, 1993; p 696. [Google Scholar]; Eliel E. L.; Wilen S. H.; Doyle M. P.. Basic Organic Stereochemistry; Wiley: New York, 2001; p 443. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.