

Abstract
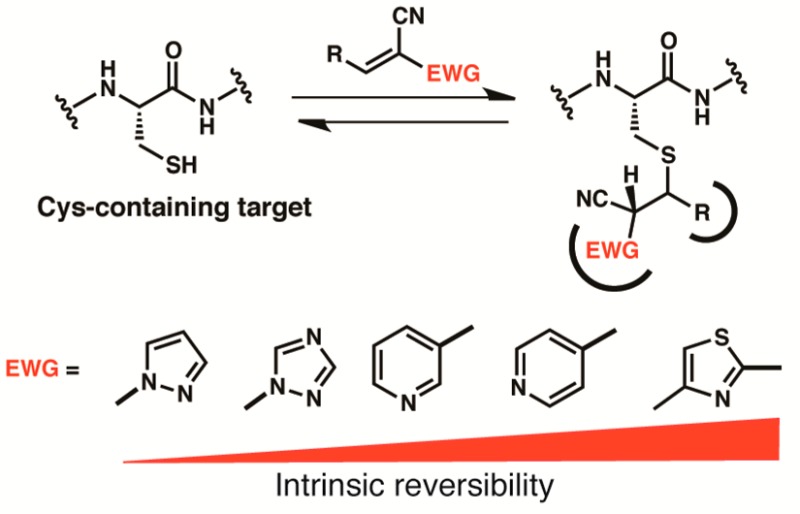
Electrophilic probes that covalently modify a cysteine thiol often show enhanced pharmacological potency and selectivity. Although reversible Michael acceptors have been reported, the structural requirements for reversibility are poorly understood. Here, we report a novel class of acrylonitrile-based Michael acceptors, activated by aryl or heteroaryl electron-withdrawing groups. We demonstrate that thiol adducts of these acrylonitriles undergo β-elimination at rates that span more than 3 orders of magnitude. These rates correlate inversely with the computed proton affinity of the corresponding carbanions, enabling the intrinsic reversibility of the thiol-Michael reaction to be tuned in a predictable manner. We apply these principles to the design of new reversible covalent kinase inhibitors with improved properties. A cocrystal structure of one such inhibitor reveals specific noncovalent interactions between the 1,2,4-triazole activating group and the kinase. Our experimental and computational study enables the design of new Michael acceptors, expanding the palette of reversible, cysteine-targeted electrophiles.
Introduction
Chemical probes that bind covalently to a protein target often have prolonged target-residence times, resulting in increased potency.1−3 Covalent probes can also be highly selective, especially when designed to react with a protein nucleophile that is not essential for enzymatic catalysis and poorly conserved among closely related proteins.4 Examples of covalent kinase inhibitors that target a noncatalytic cysteine thiol include the BTK inhibitor, ibrutinib,5 and the EGFR inhibitor, afatinib,6 recently approved for advanced B cell and lung cancers, respectively. Both of these drugs employ an α,β-unsaturated carboxamide to form an irreversible covalent bond with a poorly conserved cysteine in the kinase active site.7
Despite their advantages, covalent drugs are seldom developed for diseases other than cancer because of the potential for adverse effects caused by irreversible modification of off-target nucleophiles.8−13 Exceptions to this trend include older drugs whose covalent mechanism was initially unknown (e.g., clopidogrel14 and omeprazole15), as well as recently developed protease inhibitors that form reversible covalent bonds with catalytically essential nucleophiles (e.g., telaprevir,16 odanacatib,17 and saxagliptin18). The latter compounds employ weak electrophiles such as ketones or nitriles, which have not found general utility outside the specific context of protease targets.19 A major challenge in the field of covalent inhibitor design is the identification of electrophiles that can form energetically favorable yet reversible covalent bonds with noncatalytic cysteines, which are often less nucleophilic than catalytic cysteines.
Activated acrylonitriles bearing a carboxylic ester or carboxamide α-substituent have been reported to react rapidly and reversibly with thiols.20,21 The thiol-Michael adducts could not be isolated and, upon dilution, underwent β-elimination to form the starting cyanoacrylates/amides. Reversible thiol reactivity appears to be a general property of cyanoacrylamides, and this insight led to the design of reversible, cysteine-targeted kinase inhibitors such as cyanoacrylamide 1 (Figure 1a), which inhibits RSK1/2/4 kinases in the picomolar to low nanomolar range.21 Recently, we took advantage of the intrinsic reversibility of thiol/cyanoacrylamide reactions to develop an electrophilic fragment-based approach to ligand discovery. This strategy led to the first cysteine-targeted inhibitor of the MSK1 C-terminal kinase domain.22 The cyanoacrylamide inhibitors exhibit slow off-rates when bound to the intact, folded kinase domain, yet dissociate rapidly when the protein is unfolded or degraded by proteases. As revealed by the cocrystal structure of cyanoacrylate 2 bound to RSK2, the pyrrolopyrimidine scaffold forms specific noncovalent interactions with the kinase, orienting the electrophile and cooperatively stabilizing the covalent complex. In addition, interactions between the tert-butyl ester group and the glycine-rich loop of RSK2 likely contribute to the slow off-rate.
Figure 1.
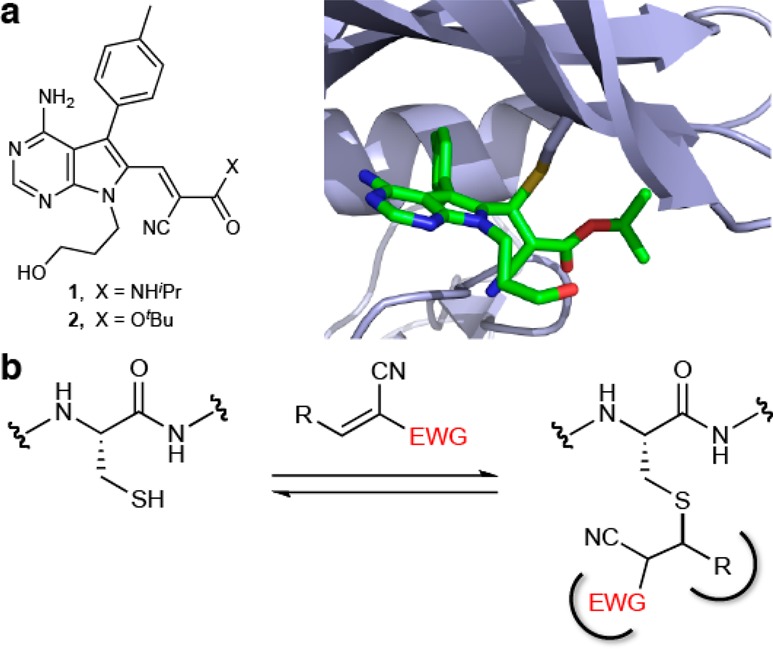
Reversible covalent binding of activated Michael acceptors to cysteine thiols. (a) RSK2 inhibitors, 1 and 2. A cocrystal structure (PDB code: 4D9U) reveals hydrophobic interactions between the tert-butyl group of 2 and the glycine-rich loop of RSK2. (b) Reversible covalent binding of an activated acrylonitrile to a hypothetical cysteine-containing protein. In this example, the electron-withdrawing group (EWG) and the β-substituent (R) both contribute to the free energy of binding by providing favorable noncovalent interactions with the protein.
The latter observation suggested the possibility of exploiting the electron-withdrawing groups not only for their ability to activate the olefin toward conjugate addition but also for their potential to contribute specific noncovalent interactions (Figure 1b). More generally, we recognized that, in many targets, the orientation of the cysteine relative to the binding site would be such that one of the olefin activating groups could serve as the primary noncovalent recognition element. While electrophilic olefins such as α-cyanoenones,23 alkylidene rhodanines,24 and alkylidene thiazolidine diones25 have also been found to form reversible covalent adducts with protein thiols, little is known about the relationship between an activated olefin’s electron-withdrawing substituents and its propensity to react reversibly with thiols.26
Results and Discussion
Kinetic and Computational Studies of Model Thiol/Acrylonitrile Adducts
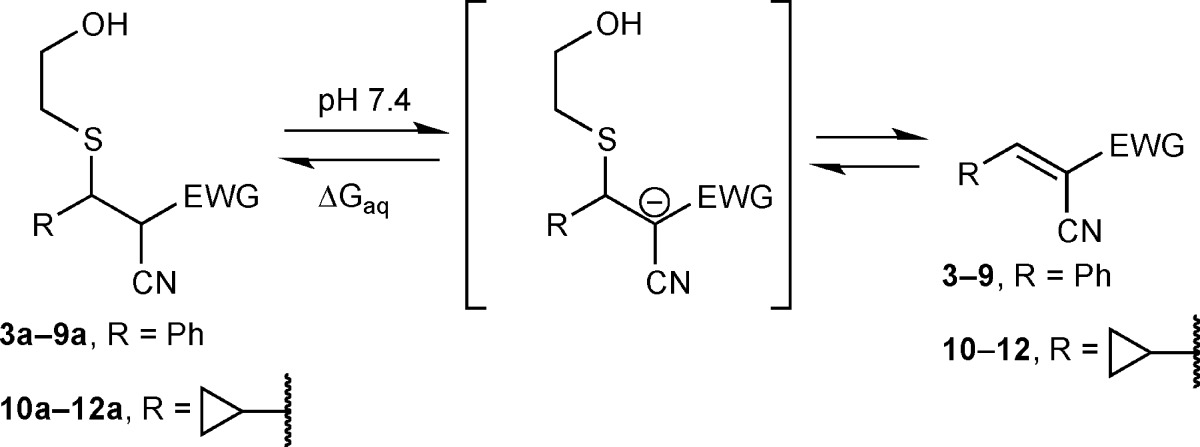
To expand the structural diversity of reversible cysteine-targeted electrophiles, we replaced the carboxamide group of 2-cyanoacrylamides with aryl or heteroaryl activating groups. The nitrile group was retained because of its small size and ability to form both polar and hydrophobic interactions with proteins.27 At the outset, it was not obvious how different aryl, or especially heteroaryl, activating groups would affect the intrinsic reversibility of the thiol-Michael reaction, i.e., the rate of thiol elimination from acrylonitrile-derived Michael adducts. To our knowledge, no systematic studies on the reactivity of such compounds toward thiols have been reported. With the goal of establishing kinetic trends that could be extrapolated to more complex structures, we synthesized a series of model acrylonitriles (3–9) bearing different aryl or heteroaryl activating groups at the α-position and a phenyl group at the β-position (Table 1). We then measured β-elimination rates of the corresponding Michael adducts derived from the simple thiol, β-mercaptoethanol (BME). These rates should reflect the relative propensity of α-activated acrylonitriles to dissociate from cysteines in unstructured regions of proteins (spurious off-target adducts) or from unfolded globular proteins prior to their degradation by cellular proteases.
Table 1. β-Elimination Rates and Calculated Proton Affinities of BME/Acrylonitrile Adducts 3a–9a.
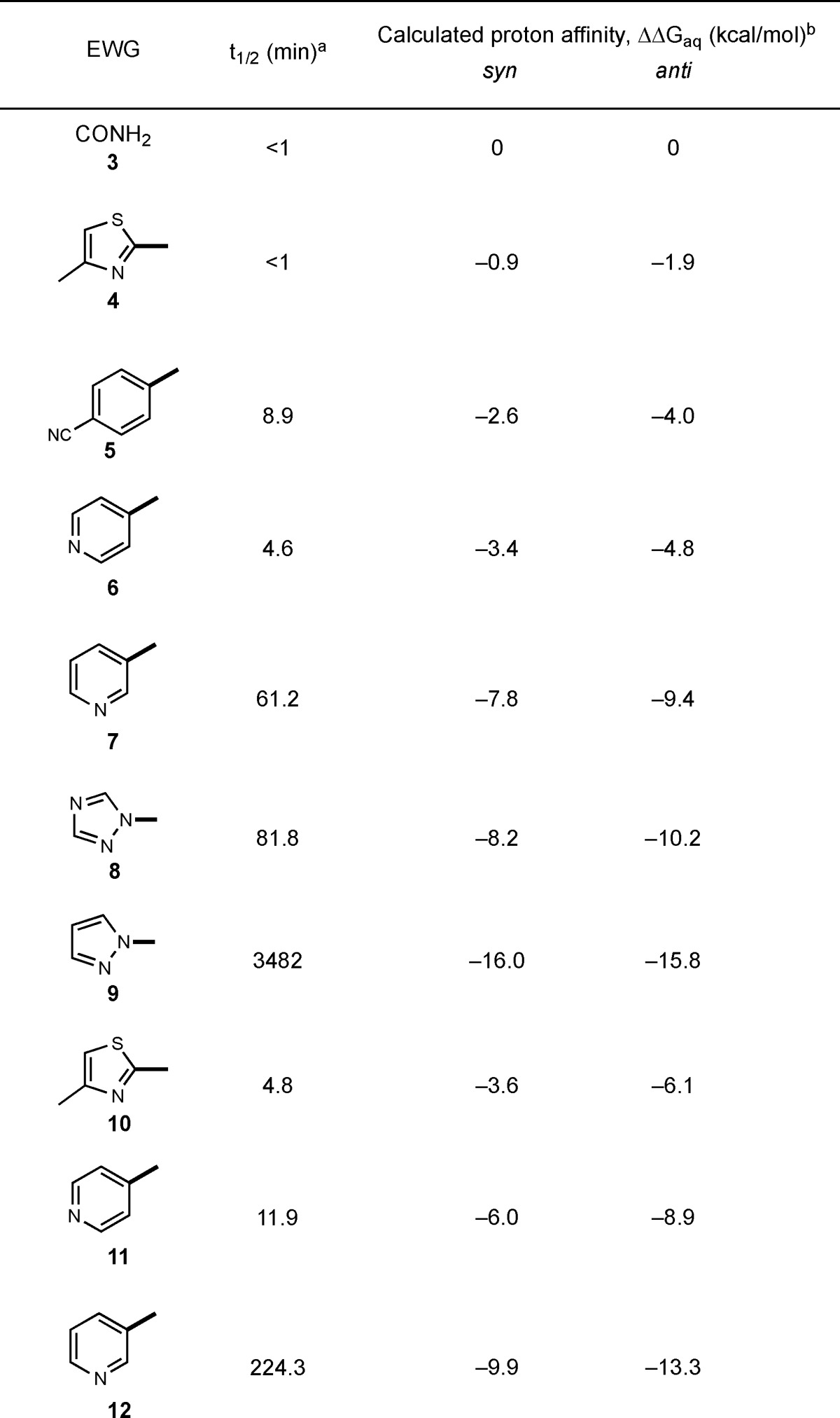
Determined by 1H NMR for adducts 3a–6a and 10a–11a after dilution into DMSO-d6/PBS-d (3:1 v/v) and by LC-MS for adducts 7a–9a and 12a after dilution into PBS. Diastereomeric ratios of 3a–6a, 8a, and 10a–11a remained constant after dilution, suggesting rapid interconversion. The diastereomers of 7a and 12a were not separable during LC-MS analysis. For 9a, a 1:1 ratio of diastereomers was subjected to the β-elimination reaction.
Proton affinities in water (ΔGaq) were calculated for syn- and anti-diastereomers of thioethers 3a–12a using the B3LYP/6-311+G(d), IEFPCM method. ΔΔGaq values relative to 3a are shown.
After reacting the acrylonitriles (3–9) with excess BME, we diluted the isolated adducts (7a–9a), or equilibrium mixtures in the case of nonisolable adducts (3a–6a), into pH 7.4 buffer (with DMSO cosolvent) to promote thiol elimination. β-elimination rates were determined by monitoring the disappearance of the adducts and reappearance of the acrylonitriles by NMR or LC/MS (Supplementary Figures S1–S10). Depending on the activating group, the half-times for thiol elimination varied from less than 1 min to more than 2 days (Table 1 and Supplementary Table S1). Among the aryl/heteroaryl activating groups tested, the methylthiazole (4a) exhibited the greatest intrinsic reversibility (fastest elimination rate, t1/2 < 1 min); the 3-pyridyl (7a) and 1,2,4-triazol-1-yl (8a) adducts reverted to the starting acrylonitriles with intermediate rates, whereas the pyrazol-1-yl adduct (9a) was essentially irreversible (t1/2 > 58 h).
To explore the potential for structural variation at the electrophilic β-position, we tested three heteroaryl-activated acrylonitriles (10–12) bearing a cyclopropyl in place of a phenyl group (Table 1). Although elimination from the cyclopropyl-substituted BME adducts was 2–3 times slower than the corresponding phenyl-substituted adducts, the results demonstrate that an aromatic substituent in the β-position is not essential for reversibility of thiol-Michael reactions with activated acrylonitriles. A smaller cyclopropyl group might be preferred in reversible acrylonitrile-based ligands in which the β-substituent is solvent-exposed and the α-activating group serves as the primary noncovalent recognition element.
We used a computational approach to gain insight into the structure/reactivity trends (Table 1), reasoning that increased acidity of the α-carbon bearing the nitrile and the second electron-withdrawing group would lead to faster thiol elimination via an E1cB mechanism.28−31 Using density functional theory, we calculated the proton affinities32 in aqueous solution for the conjugate base (α-carbanion) of each BME/acrylonitrile adduct (expressed as ΔΔGaq, relative to the cyanoacrylamide-derived adduct 3a, Table 1). Similar trends were observed using calculated proton affinities for either the syn- or anti-diastereomers of the BME adducts. A Brønsted-type plot33 revealed a linear correlation between the calculated proton affinities and a three-log range of experimental rate constants for thiol β-elimination (R2 = 0.96, Figure 2 and Supplementary Figure S11). This combined computational/empirical analysis suggests the possibility of predicting the approximate rate of thiol elimination from adducts bearing novel, as-yet-uncharacterized activating groups (Supplementary Table S2), simply by calculating the proton affinity of the corresponding α-carbanion (ΔGaq). To test this, we prepared a new β-phenyl-substituted acrylonitrile bearing a pyrazine activating group. The calculated ΔΔGaq for the derived BME adduct is −3.4 kcal/mol; fitting this value to the Brønsted relationship shown in Figure 2 led to a predicted β-elimination t1/2 of 6.1 min. Using NMR spectroscopy, we determined the t1/2 for BME β-elimination to be 7.2 min (k = 0.096 min–1), close to the predicted value (Supplementary Figure S12).
Figure 2.

Brønsted-type plot of computed proton affinity (ΔΔGaq for syn-diastereomers) vs β-elimination rate (log k, min–1) for BME/acrylonitrile adducts 5a–12a (Table 1). Adducts 3a and 4a are not shown, as only the upper limit of their t1/2 values (<1 min) could be determined. BME/acrylonitrile adducts with higher proton affinity (more negative ΔΔGaq) undergo β-elimination at slower rates. Plotting ΔΔGaq for the anti-diastereomers affords a similar correlation (Supplementary Figure S11).
BME has a pKa of 9.6,34 toward the high end of the range for surface cysteines (from a recent study of surface cysteines: mean pKa 9.3, range 8.2–9.9).35 To test the effect of decreasing the pKa of the thiolate leaving group, we purified and characterized the cysteamine (pKa 8.4)36 adducts of triazole 8 and pyrazole 9. Upon dilution into pH 7.4 buffer, we found that the β-elimination rates were 11.2- and 15.8-fold faster, respectively, than the corresponding BME adducts (Supplementary Figures S13 and S14). These results imply that more intrinsically reactive surface cysteines (or cysteines in unfolded regions of proteins), i.e., those with a lower pKa, are less likely to form long-lived adducts with heteroaryl-acrylonitriles and more likely to dissociate upon dilution or clearance of the inhibitor. It is important to note, however, that we do not anticipate any correlation between the elimination rates of model thiol/acrylonitrile adducts (Table 1 and Figure 2) and the potency of inhibitors bearing these electrophiles. This is because potency will be affected by noncovalent interactions (and clashes) between the acrylonitrile activating group and the protein target, in addition to the rates of covalent bond formation and dissociation with the active-site cysteine.
Application of Heteroaryl-Activated Acrylonitriles to Covalent Kinase Inhibitors
We next sought to employ these novel Michael acceptors in the design of reversible, cysteine-targeted kinase inhibitors. We synthesized acrylonitriles 13–18, all of which bear an aryl/heteroaryl activating group, analogous to the model compounds in Table 1. A pyrrolopyrimidine moiety attached to the electrophilic β-carbon serves as the primary noncovalent recognition element (Figure 3a), analogous to the cyanoacrylamide RSK2 inhibitors reported previously.21 In addition to the activating groups studied above (Table 1), we tested pyrazine 14 and pyridine 15 (Figure 3a), as they were both predicted to react reversibly with thiols on the basis of their computed proton affinity (Supplementary Table S2 and Figure S12).
Figure 3.

Targeting RSK2 kinase with aryl/heteroaryl-activated acrylonitriles. (a) Half-maximal inhibitory concentration (IC50, mean ± s. d.) of acrylonitriles 13–18 in kinase assays with wild-type RSK2 (WT) and the Cys436Val mutant (C436V). (b) Binding of acrylonitriles 13–18 (0.1 and 1.0 μM) and FMK (1.0 μM, positive control) to endogenous RSK1 and RSK2 in MDA-MB-231 breast cancer cells, assayed by competitive labeling with the irreversible fluorescent probe, BODIPY-FMK. Cells were incubated with acrylonitriles 13–18 for 2 h prior to treatment with BODIPY-FMK for 1 h. Cells were lysed and proteins were analyzed by gel electrophoresis, followed by fluorescence scanning and immunoblotting for RSK1/2. (c) Dose response for RSK1/2 occupancy in MDA-MB-231 cells by 13 and 14 (RSK1/2 immunoblot: green and red respectively).
In kinase assays with the RSK2 C-terminal kinase domain, pyrazine 14 was the most potent inhibitor (IC50 = 12 nM), whereas the p-cyanophenyl compound 18 was the least potent (IC50 = 770 nM). All of the inhibitors showed reduced potency toward the Cys436 to Val mutant, consistent with covalent bond formation with Cys436. We speculate that noncovalent interactions contribute to the differential potency of compounds 13–18, with thiazole 13 and pyrazine 14 having the best steric and electrostatic complementarity to the RSK2 active site. To test whether acrylonitriles 13–18 bind RSK1/2 in cells, we used a competitive labeling assay with the fluorescent affinity probe, BODIPY-FMK.21 Labeling of RSK1/2 was significantly reduced in cells that were pretreated with compounds 13–16, whereas compounds 17 and 18 were less effective (Figure 3b). Thiazole 13 and pyrazine 14 were especially potent, blocking endogenous RSK1/2 at low nanomolar concentrations (EC50 ≈ 5 nM, Figure 3c).
A desirable attribute of our previously described cyanoacrylamide inhibitors is their ability to dissociate from Cys436 (and presumably any off-target thiols) after unfolding or proteolysis of the intact RSK2 kinase domain.21,22 Our model studies with activated acrylonitriles and BME (Table 1) suggested that inhibitors 13–18 would share this property. To test this, we incubated each inhibitor (1.6 μM) with excess RSK2 (3.2 μM), resulting in quantitative formation of the complex based on the kinase activity assays (Figure 3a). RSK2/inhibitor complexes were then treated with 3 M guanidine (pH 7.4) to unfold the kinase domain, and the released inhibitor was quantified by LC-MS. Similar to our previously reported cyanoacrylamide inhibitors, acrylonitriles 13–18 were recovered in 84–93% yields after guanidine-mediated unfolding of the inhibitor-bound RSK2 kinase domain (Supplementary Table S3).
We recently developed an electrophilic fragment-based approach to ligand discovery, and we used this method to develop potent inhibitors of the MSK1 C-terminal kinase domain.22 These compounds also inhibit RSK2 and are thus useful for interrogating signaling events downstream of the closely related MSK/RSK subfamily of kinases in cellular assays of short duration (<2 h). However, unlike our previously developed pyrrolopyrimidine-based cyanoacrylamides,21 the indazole-based cyanoacrylamides (e.g., compound 19, Figure 4) lose activity over a period of 12–24 h and are degraded in cell culture by unknown mechanisms (unpublished results).
Figure 4.

Potent, selective, and durable inhibition of RSK2 by a 1,2,4-triazole-activated acrylonitrile, 20. (a) Dose–response curves for 20 in kinase assays with RSK2, NEK2, and PLK1 (IC50 values for compound 19 reproduced from ref (22)). (b) RSK1/2 occupancy was determined 2 and 24 h after treating cells with 19 and 20 (1 μM) by competitive labeling with BODIPY-FMK (RSK1/2 immunoblot: red and green, respectively). (c) Overlay of 20/RSK2 cocrystal structure (this work) with NEK2 (red, PDB code: 2WQO), highlighting Leu546 of RSK2 and the structurally homologous residue Phe148 of NEK2. The side chain of NEK2 Phe148 would clash with the 1,2,4-triazole.
Remarkably, we were able to solve this problem by replacing the carboxamide with a 1,2,4-triazole as the acrylonitrile activating group. Although triazole 20 is somewhat less potent than 19 in RSK2 kinase assays (IC50 = 47 vs 3 nM, respectively), 20 has similar or better selectivity over NEK2 and PLK1, unrelated kinases with a homologous cysteine in the ATP binding site (Figure 4a). Moreover, the cellular efficacy of 20 was dramatically enhanced, as shown by sustained RSK1/2 occupancy and the absence of compound degradation during a 24 h experiment. By contrast, RSK1/2 occupancy by cyanoacrylamide 19 was lost after 24 h (Figure 4b), correlating with its disappearance from the cell culture medium. Similar to 19, triazole 20 potently inhibited MSK1 autophosphorylation in cells (Supplementary Figure S15) and had no significant effect when tested against a panel of 18 additional kinases, 10 of which contain an active-site cysteine (Supplementary Table S4). Despite the selectivity observed with this panel of cysteine-containing kinases, we cannot rule out binding of 20 to other cellular targets besides MSK/RSK. As expected on the basis of our BME reactivity studies, binding of triazole 20 to RSK2 was reversible upon unfolding the kinase domain (t1/2 ≈ 1 h at room temperature), albeit with slower kinetics relative to 19. By contrast, the corresponding pyrazole-acrylonitrile S3 was essentially irreversible in this assay, showing ∼2% dissociation after 4 h (Supplementary Figure S16).
A cocrystal structure of triazole 20 bound to RSK2 (PDB: 4M8T) confirmed the covalent bond with Cys436 and revealed specific noncovalent interactions between the trimethoxyphenyl indazole scaffold and the kinase active site (Figure 4c). The 1,2,4-triazole projects from the newly formed sp3-carbon toward the floor of the ATP binding site, forming close contacts (4–5 Å) with the methyl groups of Leu546. The analogous region of both PLK1 and NEK2 is occupied by a larger phenylalanine residue, which would likely clash with the triazole. Thus, despite sharing a homologous cysteine, PLK1 and NEK2 are apparently unable to accommodate the triazole upon nucleophilic attack and protonation of the acrylonitrile. The 1,2,4-triazole substituent in 20 serves three critical functions: (1) it activates the acrylonitrile toward reversible nucleophilic attack by Cys436; (2) it enhances selectivity via polar and hydrophobic interactions with RSK2 and a steric clash with PLK1 and NEK2; and (3) it increases cellular stability and potency.
Conclusions and Perspective
In this study, we have characterized a series of aryl- and heteroaryl-activated acrylonitriles, with the ultimate goal of applying these novel electrophiles to the design of reversible, cysteine-targeted probes. We focused our attention on the kinetic stability of the corresponding thiol-Michael adducts, reasoning that electrophiles with greater intrinsic reversibility (i.e., giving rise to kinetically less stable thiol adducts) are less likely to form permanent covalent adducts with off-target cysteines.9 As with irreversible covalent inhibitors, the maximum residence time of reversible covalent inhibitors is limited by the turnover rate of the target. Hence, the reversible covalent strategy would have a limited pharmacodynamic advantage when targeted against a protein with a short half-life, although the selectivity advantage gained by targeting a noncatalytic cysteine would still apply. High intrinsic reversibility may also be advantageous in electrophilic fragment-based screens, although a recently published screen of irreversible acrylate-based fragments suggests that reversible electrophiles may not be required.37 With intrinsically reversible electrophiles, fragment binding would be under thermodynamic control and would require specific noncovalent interactions to cooperatively stabilize a high-affinity covalent interaction with the target.22 Our DFT calculations on thiol/acrylonitrile adducts and their derived carbanions revealed a strong correlation between computed proton affinities and experimental β-elimination rates. Based on this analysis, we can now estimate the relative reversibility of thiol-Michael adducts derived from novel acrylonitriles before synthesizing and testing them. Acrylonitriles activated by 1,2,4- or 1,3,4-oxadiazoles, for example, are predicted to form rapidly reversible thiol adducts (Supplementary Table S2), motivating the synthesis of electrophilic fragment libraries bearing these and similarly activating heteroaryl substituents for future cysteine-targeting applications.
Acknowledgments
This work was supported by grants from the U.S. National Institutes of Health (NIH) (GM071434 to J.T.), the California Tobacco Related Disease Research Program (19FT-0091 to S.K.), and the National University of Ireland-Galway (B.T.). We acknowledge the University of California—San Francisco (UCSF) Mass Spectrometry Facility (supported by NIH Grant P41RR001614), the SFI/HEA Irish Centre for High-End Computing (ICHEC), and Prof. Leif A. Eriksson (University of Gothenburg) for helpful discussions.
Supporting Information Available
Detailed experimental procedures, synthesis and spectral characterization of compounds, kinetic measurements, computational methods, crystallographic statistics, and collection parameters. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Potashman M. H.; Duggan M. E. J. Med. Chem. 2009, 52, 1231–1246. [DOI] [PubMed] [Google Scholar]
- Smith A. J. T.; Zhang X. Y.; Leach A. G.; Houk K. N. J. Med. Chem. 2009, 52, 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A.; Pompliano D. L.; Meek T. D. Nat. Rev. Drug Discovery 2006, 5, 730–739. [DOI] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. Nat. Rev. Drug Discovery 2011, 10, 307–317. [DOI] [PubMed] [Google Scholar]
- Byrd J. C.; Furman R. R.; Coutre S. E.; Flinn I. W.; Burger J. A.; Blum K. A.; Grant B.; Sharman J. P.; Coleman M.; Wierda W. G.; Jones J. A.; Zhao W.; Heerema N. A.; Johnson A. J.; Sukbuntherng J.; Chang B. Y.; Clow F.; Hedrick E.; Buggy J. J.; James D. F.; O’Brien S. New Engl. J. Med. 2013, 369, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sequist L. V.; Yang J. C.-H.; Yamamoto N.; O’Byrne K.; Hirsh V.; Mok T.; Geater S. L.; Orlov S.; Tsai C.-M.; Boyer M.; Su W.-C.; Bennouna J.; Kato T.; Gorbunova V.; Lee K. H.; Shah R.; Massey D.; Zazulina V.; Shahidi M.; Schuler M. J. Clin. Oncol. 2013, 31, 3327–3334. [DOI] [PubMed] [Google Scholar]
- a Liu Q.; Sabnis Y.; Zhao Z.; Zhang T.; Buhrlage S. J.; Jones L. H.; Gray N. S. Chem. Biol. 2013, 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schwartz P. A.; Kuzmic P.; Solowiej J.; Bergqvist S.; Bolanos B.; Almaden C.; Nagata A.; Ryan K.; Feng J.; Dalvie D.; Kath J. C.; Xu M.; Wani R.; Murray B. W. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park B. K.; Boobis A.; Clarke S.; Goldring C. E. P.; Jones D.; Kenna J. G.; Lambert C.; Laverty H. G.; Naisbitt D. J.; Nelson S.; Nicoll-Griffith D. A.; Obach R. S.; Routledge P.; Smith D. A.; Tweedie D. J.; Vermeulen N.; Williams D. P.; Wilson I. D.; Baillie T. A. Nat. Rev. Drug Discovery 2011, 10, 292–306. [DOI] [PubMed] [Google Scholar]
- Lin D.; Saleh S.; Liebler D. C. Chem. Res. Toxicol. 2008, 21, 2361–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebler D. C. Chem. Res. Toxicol. 2008, 21, 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetrecht J. Chem. Res. Toxicol. 2008, 21, 84–92. [DOI] [PubMed] [Google Scholar]
- Guengerich F. P.; MacDonald J. S. Chem. Res. Toxicol. 2007, 20, 344–369. [DOI] [PubMed] [Google Scholar]
- a Evans D. C.; Watt A. P.; Nicoll-Griffith D. A.; Baillie T. A. Chem. Res. Toxicol. 2004, 17, 3–16. [DOI] [PubMed] [Google Scholar]; b Evans D. C.; Watt A. P.; Nicoll-Griffith D. A.; Baillie T. A. Chem. Res. Toxicol. 2005, 18, 1777. [DOI] [PubMed] [Google Scholar]
- Pereillo J. M.; Maftouh M.; Andrieu A.; Uzabiaga M. F.; Fedeli O.; Savi P.; Pascal M.; Herbert J. M.; Maffrand J. P.; Picard C. Drug Metab. Dispos. 2002, 30, 1288–1295. [DOI] [PubMed] [Google Scholar]
- Olbe L.; Carlsson E.; Lindberg P. Nat. Rev. Drug Discovery 2003, 2, 132–139. [DOI] [PubMed] [Google Scholar]
- Lin C.; Kwong A. D.; Perni R. B. Infect. Disord. Drug Targets 2006, 6, 3–16. [DOI] [PubMed] [Google Scholar]
- Gauthier J. Y.; Chauret N.; Cromlish W.; Desmarais S.; Duong L. T.; Falgueyret J.-P.; Kimmel D. B.; Lamontagne S.; Léger S.; LeRiche T.; Li C. S.; Massé F.; McKay D. J.; Nicoll-Griffith D. A.; Oballa M. R.; Palmer J. T.; Percival M. D.; Riendeau D.; Robichaud J.; Rodan G. A.; Rodan S. B.; Seto C.; Thérien M.; Truong V.-L.; Venuti M. C.; Wesolowski G.; Young R. N.; Zamboni R.; Black W. C. Bioorg. Med. Chem. Lett. 2008, 18, 923–928. [DOI] [PubMed] [Google Scholar]
- Metzler W. J.; Yanchunas J.; Weigelt C.; Kish K.; Klei H. E.; Xie D.; Zhang Y.; Corbett M.; Tamura J. K.; He B.; Hamann L. G.; Kirby M. S.; Marcinkeviciene J. Protein Sci. 2008, 17, 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frizler M.; Stirnberg M.; Sisay M. T.; Gütschow M. Curr. Top. Med. Chem. 2010, 10, 294–322. [DOI] [PubMed] [Google Scholar]
- Pritchard R. B.; Lough C. E.; Currie D. J.; Holmes H. L. Can. J. Chem. 1968, 46, 775–781. [Google Scholar]
- Serafimova I. M.; Pufall M. A.; Krishnan S.; Duda K.; Cohen M. S.; Maglathlin R. L.; McFarland J. M.; Miller R. M.; Frödin M.; Taunton J. Nat. Chem. Biol. 2012, 8, 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R. M.; Paavilainen V. O.; Krishnan S.; Serafimova I. M.; Taunton J. J. Am. Chem. Soc. 2013, 135, 5298–5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch R. D.; Browning R. G.; Honda T.; Gribble G. W.; Wright D. L.; Sporn M. B.; Anderson A. C. Bioorg. Med. Chem. Lett. 2005, 15, 2215–2219. [DOI] [PubMed] [Google Scholar]
- Powers J. P.; Piper D. E.; Li Y.; Mayorga V.; Anzola J.; Chen J. M.; Jaen J. C.; Lee G.; Liu J.; Peterson M. G.; Tonn G. R.; Ye Q.; Walker N. P. C.; Wang Z. J. Med. Chem. 2006, 49, 1034–1046. [DOI] [PubMed] [Google Scholar]
- Patch R. J.; Searle L. L.; Kim A. J.; De D.; Zhu X.; Askari H. B.; O’Neill J. C.; Abad M. C.; Rentzeperis D.; Liu J.; Kemmerer M.; Lin L.; Kasturi J.; Geisler J. G.; Lenhard J. M.; Player M. R.; Gaul M. D. J. Med. Chem. 2011, 54, 788–808. [DOI] [PubMed] [Google Scholar]
- For a review of reversible Michael acceptors in biologically active small molecules, see:Johansson M. H. Mini-Rev. Med. Chem. 2012, 12, 1330–1334. [DOI] [PubMed] [Google Scholar]
- Fleming F. F.; Yao L.; Ravikumar P. C.; Funk L.; Shook B. C. J. Med. Chem. 2010, 53, 7902–7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbein J. C.; Jencks W. P. J. Am. Chem. Soc. 1988, 110, 5075–5086. [Google Scholar]
- Fishbein J. C.; Jencks W. P. J. Am. Chem. Soc. 1988, 110, 5087–5095. [Google Scholar]
- Bordwell F. G. Acc. Chem. Res. 1972, 5, 374–381. [Google Scholar]
- Heo C. M. K.; Bunting J. W. J. Org. Chem. 1992, 57, 3570–3578. [Google Scholar]
- Fu Y.; Liu L.; Li R.-Q.; Liu R.; Guo Q.-X. J. Am. Chem. Soc. 2004, 126, 814–822. [DOI] [PubMed] [Google Scholar]
- Anslyn E. V.; Dougherty D. A.. Modern Physical Organic Chemistry; University Science Books; Sausalito, CA, 2006; pp 464–471. [Google Scholar]
- Jencks W. P.; Salvesen K. J. Am. Chem. Soc. 1971, 93, 4433–4436. [Google Scholar]
- Jensen K. S.; Pedersen J. T.; Winther J. R.; Teilum K. Biochemistry 2014, 53, 2533–2540. [DOI] [PubMed] [Google Scholar]
- Connett P. H.; Wetterhahn K. E. J. Am. Chem. Soc. 1986, 108, 1842–1847. [Google Scholar]
- Kathman S. G.; Xu Z.; Statsyuk A. V. J. Med. Chem. 2014, 57, 4969–4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.