

1. Introduction: The Roles of Metals in Biology
From the perspective of a chemist, biology confers a rich variety of roles on a number of metal ions. It is widely agreed that a large fraction of the genomic output of living things contains metal or metalloid ions, although estimates of this fraction vary widely and depend upon which metal ions are considered.1−3 Moreover, recent reports suggest that, at least in some cases, there are many uncharacterized metalloproteins.4 With inclusion of the s-block metals such as Na, K, Mg, and Ca, the proportion likely approaches 100%; recent estimates from the protein data bank indicate that the prevalence of heavier metal ions of atomic number above 20 within proteins is around 22%,5 with Zn2+ proteins alone constituting about 11%. Living organisms have an inherent and very rich physical structure, with relevant length scales ranging from the nanometer scale for subcellular structure to hundreds of micrometers and above for tissue, organ, or organism-level organization. The ability to derive the spatial distribution of elements on this diversity of length scales is a key to understanding their function.
Metals play essential and central roles in the most important and chemically challenging processes required for life, with active site structures and mechanisms that, at the time of their discovery, have usually not yet been duplicated in the chemical laboratory. Furthermore, diseases of metal dysregulation can cause disruption in the distribution of metals.6 For example, Menke’s disease and Occipital Horn Syndrome,7 and Wilson’s disease,8 involve disruption in copper uptake and excretion, respectively, through mutation in the ATP7A and ATP7B Cu transporters.9 The mechanisms of action of toxic elements such as mercury and arsenic are also of interest, as are essential nonmetal trace elements, such as selenium. Likewise, an increasing number of pharmaceuticals include metals or heavier elements; such chemotherapeutic drugs include the platinum derivatives cisplatin and carboplatin,10 some promising new ruthenium drugs,11 and arsenic trioxide, which has been used to treat promyelocytic leukemia.12 Understanding the localization, speciation, and distribution of these at various length scales is of significant interest.
A wide variety of heavier elements can be probed by X-ray spectroscopic methods; these are shown graphically in Figure 1. X-ray fluorescence imaging is a powerful technique that can be used to determine elemental and chemical species distributions at a range of spatial resolutions within samples of biological tissues. Most modern applications require the use of synchrotron radiation as a tunable and high spectral brightness source of X-rays. The method uses a microfocused X-ray beam to excite X-ray fluorescence from specific elements within a sample. Because the method depends upon atomic physics, it is highly specific and enables a wide range of chemical elements to be investigated. A significant advantage over more conventional methods is the ability to measure intact biological samples without significant treatment with exogenous reagents. The technique is capable of determining metal and nonmetal distributions on a variety of length scales, with information on chemical speciation also potentially available. Figure 2 shows examples of rapid-scan X-ray fluorescence imaging at two contrasting length scales: rapid-scan imaging13 of a section of a human brain taken from an individual suffering from multiple sclerosis and showing elemental profiles for Fe, Cu, and Zn;14 and a high-resolution image showing mercury and other elements in a section of retina from a zebrafish larva treated with methylmercury chloride.15 We will discuss both the state of the art in terms of experimental methods and some recent applications of the methods. This Review considers X-ray fluorescence imaging with incident X-ray energies in the hard X-ray regime, which we define as 2 keV and above. We review technologies for producing microfocused X-ray beams and for detecting X-ray fluorescence, as well as methods that confer chemical selectivity or three-dimensional visualization. We discuss applications in key areas with a view to providing examples of how the technique can provide information on biological systems. We also discuss synergy with other methods, which have overlapping or complementary capabilities. Our goal is to provide useful and pertinent information to encourage and enable further use of this powerful method in chemical and biochemical studies of living organisms.
Figure 1.

Periodic table of the elements showing elements of biological interest that can be probed using X-ray fluorescence imaging. Elements are divided into three categories, those that are physiologically important, those that are pharmacologically active, and those that are toxic or of environmental concern. Those elements that can be detected using hard X-ray XFI are shown in bold color, while those that cannot are indicated by a lighter shade of the same color. Some elements will fall into more than one category; for example, selenium is both essential and toxic in excess with a relatively narrow margin separating adequate supply and the onset of toxic symptoms. Similarly, bromine is used pharmacologically but is also a physiologically important element.
Figure 2.

Examples of biological X-ray fluorescence imaging (XFI), illustrating the different accessible length-scales. Panel a shows a coronal section of human brain taken post-mortem from an individual suffering from multiple sclerosis,14 with normalized levels of iron, zinc, and copper indicated as shades of red, green, and blue. Panel b shows a thin section of larval zebrafish retina imaged at high resolution showing preferential accumulation of mercury in the photoreceptor cells.15
Several acronyms are used for X-ray fluorescence imaging; some commonly in use are X-ray fluorescence (XRF), synchrotron X-ray fluorescence (SXRF), X-ray fluorescence imaging (XFI), synchrotron X-ray fluorescence imaging (SXFI), synchrotron radiation-induced X-ray emission (SRIXE), rapid scan X-ray fluorescence (RSXRF), as well as X-ray fluorescence microscopy and X-ray fluorescence mapping (both of which are commonly abbreviated XFM). All of these correspond to essentially the same experiment. Purists might argue that most of what we discuss here is predominantly mapping and not imaging; however, given that we will cover both two-dimensional and three-dimensional applications, the acronym used here is X-ray fluorescence imaging (XFI).
2. Experimental Methods and Strategies
In essence, the technique of X-ray fluorescence imaging uses a small or microfocused X-ray beam incident upon a sample, an energy-dispersive detector to monitor the X-ray fluorescence, and sample translation so that the X-ray beam sequentially interrogates different parts of the sample to develop an image of the sample. Essentially all modern applications of XFI in biological systems employ synchrotron radiation as a high spectral brightness source of X-rays. Synchrotron radiation from a state-of-the-art third-generation source is of many orders of magnitude greater spectral brightness than even the most powerful rotating anode X-ray sources; the low concentrations of metals in biological tissues mean that XFI is only practical on these samples using synchrotron light. A detailed discussion of the origins and properties of synchrotron light is outside the scope of this Review, and has been reviewed by others.16 A schematic of the experimental apparatus is shown in Figure 3. Below we review different aspects of the experiment, with an emphasis on those components that are likely to be selected or adjustable by the experimenter.
Figure 3.

Highly simplified and schematic plan view of the apparatus for conducting X-ray fluorescence imaging experiments. The X-ray source is typically synchrotron light, used in conjunction with upstream X-ray optics (not shown) and in most cases an X-ray double crystal monochromator.
2.1. Micro-Focus Optics
There are five important methods of generating a small or microfocused X-ray beam: simple apertures, Kirkpatrick–Baez (K–B) mirrors, capillary optics, Fresnel zone plates, and compound refractive lenses. Excellent reviews of these methods have been reported relatively recently,17−20 and therefore we will discuss each method only briefly.
2.1.1. Apertures
The simplest method of generating small X-ray beams, apertures, is most suitable when relatively large beams are required. Because they do not focus X-rays, they are often neglected in reviews, but much outstanding work has been conducted using simple apertures. An aperture usually consists of a precision slit assembly or a laser drilled aperture or pinhole of fixed size (e.g., 20–100 μm), usually fabricated from a high atomic number material such as tungsten or tantalum. In general, it is good practice to locate the aperture close to the sample, especially if upstream focusing optics are used, as noncolinearity of the X-ray beams and scattering from the edges of the aperture may result in different size projection, sometimes called beam blow-up, at the sample. These effects, especially the latter, can be reduced by means of a scatter slit or second aperture downstream of the first.
2.1.2. Specular Optics
Two of the common methods of X-ray microfocusing depend upon X-ray reflection: K–B mirrors and capillary optics. Both require that the incident angle θ between the optic surface and the incident X-rays is less than the X-ray critical angle θc. Outside of the total external reflection condition (i.e., θ > θc), the reflectivity of all materials is negligible. The value of θc is dependent both upon the X-ray energy and upon the atomic number and density of the material used for the reflective optics. X-ray mirrors are often made of silicon and coated with a material of high atomic number (such as nickel, platinum, gold, or rhodium), as high atomic number coatings give higher θc values, which for hard X-rays and common coatings is generally between 0.1 and 10 mrad. An alternative approach is to use multilayer mirrors. Here, alternating layers of high and low refractive index materials are used to achieve high reflectivity at angles greater than θc through constructive interference at multiple surface interfaces.
2.1.3. Kirkpatrick–Baez (K–B) Mirrors
Probably the most commonly used method of obtaining microfocused X-ray beams are K–B mirrors.21 These comprise two spherical or cylindrical mirrors in a crossed configuration21 with adjustable bend radii to achieve optimum focus at different wavelengths. A schematic diagram of a K–B pair is shown in Figure 4. In most cases, the beam sizes achievable with K–B mirrors are of the order of 1–5 μm, but in some cases sub-100 nm spot sizes have been achieved.22 The focal length of most modern devices is such that several centimeters are typically available between the physical end of the optic and the sample. Recently, side-by-side K–B mirror configurations, sometimes called Montel mirrors, have been discussed.23,24 These provide a more compact system that is capable of higher demagnification than traditional sequential K–B mirrors, but at the time of writing they are not in routine use.
Figure 4.

Schematic representation of a two-mirror Kirkpatrick–Baez system. The lines indicate the flight path of rays from the source to the sample. The angle of incidence θ is shown on the first mirror. The two mirrors focus in orthogonal directions.
2.1.4. Capillary Optics
In most cases, capillary optics are made of glass, although metal devices have been explored. There are two basic types of capillary optics: polycapillaries and monocapillaries. Both function by total external X-ray reflection from the inner walls of the glass (Figure 5). Polycapillaries have multiple small channels that guide X-rays by multiple internal reflections along the channel’s inner wall, and focal spot sizes in the range 10–50 μm are achievable.25 Polycapillaries have the major advantage of being able to collect larger solid angles than monocapillaries. Monocapillaries can be further divided into condensing capillaries and single bounce ellipsoidal capillaries. The latter focus the X-ray with a single reflection from its inner surface with the advantage of superior efficiency. The focal length of glass devices typically provides several millimeters between the tip of the optic and the sample. Tapered metal capillaries have been explored26 and used in a research setting;27,28 these have the advantage of greater efficiency and superior performance especially at low X-ray energies,29 and can produce beams sizes of around 3–15 μm. The main disadvantage of these devices is that the focal length is very short, so that the sample needs to be located at about 0.1 mm from the beam exit of the capillary. Polycapillaries fitted to detectors have been used in the construction of confocal X-ray fluorescence imaging systems, which we will discuss below (section 2.7).
Figure 5.
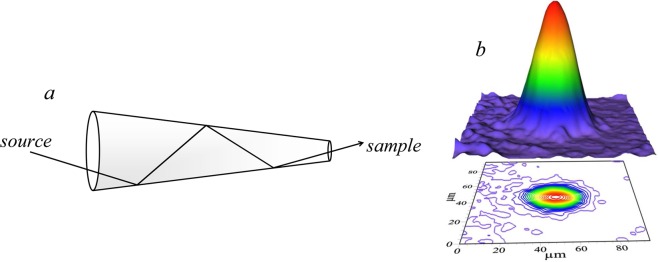
Capillary microfocus optics. (a) Schematic diagram of a multiple-bounce tapered monocapillary microfocus optic. (b) The beam intensity profile of a tapered metal capillary optic,26 measured using a cadmium tungstate scintillator placed at the focal point of the device.
2.1.5. Compound Refractive Lenses
X-ray refractive lenses, analogous to the glass lenses commonly used for visible light, were considered impractical for many years due to the weak relative refraction and strong absorption typical of X-rays. However, lenses fabricated from low atomic number materials such as Be, B, C, and Al have been used to minimize X-ray attenuation due to absorption. Because of the low refractive index, focusing refractive X-ray lenses must have a concave shape. To achieve reasonable focal lengths, many individual lenses can be stacked, forming a compound refractive lens and yielding working distances of the order of 1 m. While they remain a promising alternative, especially for high energy applications,30 at the time of writing compound refractive lenses are little used for microfocus optics in X-ray fluorescence imaging applications, and we will not consider them further.
2.1.6. Fresnel Zone Plates
Effectively lenses for monochromatic X-rays, Fresnel zone plates consist of a series of concentric rings where the rings become narrower at larger radii until the last and narrowest ring is reached (Figure 6). The radii of the zone plate edges r are given by eq 1:
| 1 |
where n is the zone number, λ is the X-ray wavelength, and F is the focal length. We consider here applications in which Fresnel zone plates are used to focus a larger beam onto a small spot. The focusing ability is based upon constructive interference of the X-ray wavefront, modified by passage through the zone plate, either in phase or more commonly by attenuation due to the zones. In most cases, the working distance from the zone plate to the sample is of the order of centimeters, and the spot sizes achievable are defined by both the X-ray energy (wavelength λ) and the outermost zone width Δr = rN – rN–1, where N is the number of zones. To avoid chromatic blurring, N must be less than the inverse of the X-ray wavelength resolution λ/Δλ. As with all grating optics, Fresnel zone plates give different diffraction orders (Figure 6B), with the higher orders having focal points that are closer to the optic, zero order corresponding to a straight-through beam path, and negative orders giving diverging beams. Only the first order beam is used, and the other orders are predominantly rejected by using an order-sorting aperture, which is placed between the focus of the first-order beam (the location of the sample) and the zone plate. Fresnel zone plates were first applied in soft X-ray transmission microscopy31 for which spot sizes below 50 nm can be obtained. In the hard X-ray regime, beam spots of the order of 100–300 nm can often be achieved. Fresnel zone plates differ from the other commonly used micro focus optics considered here (K–B mirrors and capillaries) in that their focal length is dependent on the X-ray energy. This can cause problems when performing μ-XAS experiments in which energy is scanned (section 2.6) because ideally the sample should be translated as a function of energy so as to maintain its position at the X-ray focus; the alignment need only be slightly imperfect for the beam to move across the sample during the course of the μ-XAS scan. If the energy range being scanned in μ-XAS is not large, then it may be better to maintain a stationary sample position to avoid such problems.
Figure 6.

Fresnel zone plate microfocus optics. (A) Simplified schematic diagram of a Fresnel zone plate microfocus optic. The X-rays from the source are depicted as parallel rays, and following the zone plate converge on a focus at the sample. (B) Positioning of order sorting aperture and beam stop in a typical Fresnel zone plate-based XFI experiment. The order sorting aperture serves to reject the light from diffraction orders other than 1, with the sample ideally located at the order 1 beam focus.
The different methods used for generating microscopic X-ray beams in XFI experiments are summarized and compared in Table 1.
Table 1. Comparison of Optics for Generating Micro-Beams for Hard X-ray XFIa.
| apertures | Kirkpatrick–Baez mirrors | capillaries | Fresnel zone plates | |
|---|---|---|---|---|
| max X-ray energy | N/A | 30 keV | 20–40 keV | 30 keV |
| typical resolution | 50–100 μm | 2–20 μm | 1–20 μm | 150–300 nm |
| best resolution | ca. 20 μm | 40 nm | 1 μm | 60 nm |
| focal length | N/A | several cm | 0.1–5 mm | several cm |
| disadvantages | inefficient (low photon flux), poor resolution | short focal length | energy-dependent focal length |
Only commonly used optics are considered; values given are those for an X-ray energy of 10 keV.
2.2. X-ray Fluorescence Detection
When the energy E of an X-ray is above an absorption edge of an element, it can excite core electrons, creating a photoelectron and a core hole. The core hole is rapidly filled by decay of an outer electron with either the loss of an Auger electron or the emission of an X-ray fluorescence photon. The division between these two modes of decay is specified by the fluorescence yield,32 and as it is the latter that concerns us here we will not discuss electron yield further. Figure 7 shows a simplified schematic diagram of the underlying physics together with the fluorescence emission spectra of a selenium-containing sample illuminated with X-rays above and below the Se K-edge.
Figure 7.

Physics of X-ray absorption and X-ray fluorescence. (A) Schematic diagram of the physics. The incident X-ray photon excites a core–electron as a photoelectron creating a core hole in the process. This core hole is filled by dipole-allowed decay of an inner electron with concomitant emission of an X-ray fluorescence photon. (B) Experimental X-ray emission spectra collected with incident X-ray energy above and below the selenium K-absorption edge of a biological sample containing high levels of selenium. The spectrum with the incident X-ray energy below the Se K-edge shows only scattered X-rays, whereas peaks due to the Se Kα and Se Kβ X-ray fluorescence are clearly visible when the incident X-ray energy is above the Se K-edge.
The X-ray fluorescence emission lines of the elements occur at characteristic energies, which are listed in the so-called “orange book”, otherwise known as the X-ray data booklet.33 The specific X-ray fluorescence lines for K and L edges are summarized in Figure 8. They obey the familiar dipole selection rules common to most electronic spectra with Δl = ±1, where l is the azimuthal quantum number. Thus, for a K-edge, the most intense fluorescence lines are the Kα1 and Kα2, which arise from the decay of 2p3/2 and 2p1/2 electrons, respectively, to fill the 1s core hole. Similarly, the lines of secondary intensity, the Kβ1 and Kβ3, arise from decay of 3p3/2 and 3p1/2 electrons, respectively. Because the K-edges of heavy elements such as third row transition elements may be higher in energy than the working range of most XFI beamlines, in these cases the L-edges are used. Here, the most intense fluorescence lines arise from decay of 3d electrons to fill the 2p hole; for example, for LIII edges, the Lα1 and Lα2 arise from 3d5/2 → 2p3/2 and 3d3/2 → 2p3/2 transitions, respectively. Figure 9 shows a typical X-ray fluorescence emission spectrum from a biological sample. In an XFI experiment, conventionally the Kα1 and Kα2 lines cannot be resolved by the detector (section 2.3) and are labeled as an average Kα peak, likewise for the Kβ peak. All of the other, more minor, lines from the K-shell elements can typically be neglected (Figure 9). The Kα is almost always the fluorescence line employed for experimental measurements because it is the most intense; the Kβ may need to be taken into account in cases where it overlaps with the fluorescence of another element. For L-shell excitation with an energy above all three L-edges, the primary fluorescence appears as a characteristic pair of intense peaks, the Lα12 and the Lβ1, the most intense lines from the LIII and LII edges, respectively. In principle, the X-ray fluorescence lines from all elements in a sample with absorption edge energies below that of the incident beam are observable, but in practice experimental factors such as attenuation of lower energy X-rays and low fluorescence yields for the lighter elements mean that only fluorescence from elements heavier than silicon is typically observable, unless the element is present in great abundance. With biological samples the X-ray emission spectrum is often dominated not by X-ray fluorescence but by scattered X-rays. X-ray scattering can be divided into elastic or Rayleigh scattering, and inelastic or Compton scattering. The former occurs at the same energy as the incident X-ray beam, while the latter occurs at lower energies and shows a broad asymmetric energy profile with a low-energy tail that can extend into regions of the emission spectrum where X-ray fluorescence of interest occurs (Figure 9). The Compton scattering energy decreases as (1 – cos ϑ), where ϑ is the observation angle relative to the incident beam, which creates challenges for detectors that accept a large solid angle, such as array detectors.
Figure 8.

Origins and nomenclature of the major and minor X-ray fluorescence emission lines. The bold lines indicate the most intense fluorescence transitions, with the boldest lines indicating those most suitable for X-ray fluorescence imaging experiments.
Figure 9.

Biological X-ray fluorescence emission spectrum. The spectrum shown is that of a sample of human cerebral cortex from an individual poisoned through skin exposure to dimethylmercury, plotted with counts per second (CPS) on the ordinate. Two copies of the trace are shown, one full-scale, and the other (labeled) with the vertical scale expanded by a factor of 20. The spectrum was collected using a Canberra LeGe detector using analog electronics with a Gaussian shaping amplifier employing a shaping time of 2 μs. The incident X-ray energy was 13 200 eV. Data were collected on SSRL 9-3.
To select for specific characteristic fluorescence lines, almost all X-ray fluorescence imaging is achieved using solid-state energy dispersive detectors, which allow the intensity of all photons emanating from the sample to be plotted as a function of the photon energy. Figure 10 shows the results of an X-ray fluorescence imaging measurement on a biological sample, the tip of a leaf from a selenium hyperaccumulating plant, showing the levels of different elements in the sample together with the fluorescence emission spectrum.
Figure 10.

Example of elemental mapping using X-ray fluorescence imaging. The sample imaged is a leaf tip of the selenium hyperaccumulator Astragalus bisulcatus, grown hydroponically with selenate-containing media. The X-ray emission spectrum is shown in the lower part of the figure, with the Kα lines of different elements detected indicated. The Ar fluorescence arises from the air in the experiment. The optical micrograph is shown in opt, plus XFI maps of Ca, Zn, and Se, together with a map for scattered X-rays. Ca can be seen to be concentrated in the leaf hairs (trichomes), Zn in the spongy mesophyll cell layer within the leaf, and the Se is relatively uniformly distributed throughout the leaf. We note the relative intensities of the lower and higher energy fluorescence lines, such as Ca and Se, do not reflect the absolute relative levels of these elements because of air and window attenuation of the low-energy X-rays. Data were collected on SSRL 9-3, using a glass capillary microfocus optic, a silicon drift (Vortex) detector, and a Gaussian shaping amplifier with a shaping time of 0.5 μs.
2.3. Solid-State Energy Dispersive X-ray Detectors
A variety of solid-state detectors34,35 are commercially available, each with their advantages and disadvantages; we will discuss a few different types. The properties sought are good efficiency over a wide energy range, good energy resolution, and high count rate capability. The literature on detectors tends to be specialized, and at the time of writing we know of no reviews that are readily accessible to researchers in chemistry or the life sciences. We therefore briefly summarize below the different types of detector currently available, their modes of operation, and their advantages and drawbacks.
2.3.1. Germanium Detectors
The first of the commonly used energy dispersive detectors that we will consider are the Ge detectors (Figure 11). These essentially consist of large reverse-bias diodes, typically arranged so that X-ray photons enter through a thin ion-implanted p+ contact, with the n+ contact at the opposite end of the device (Figure 11). The passage of an X-ray photon into the detector causes a cloud of electron–hole pairs to form within the diode, the number of which is proportional to the X-ray energy, assuming that the X-ray photon and any scattered or fluorescent daughter photons are all absorbed within the diode. The sweeping voltage causes the electrons and holes, respectively, to drift toward the front and the rear of the detector, giving a current pulse that is proportional to the X-ray energy. Ge detectors give excellent energy resolution and good efficiency at high X-ray energies, although escape peaks can be problematic for incident X-ray energies above the energy of the Ge K-edge [E(Ge K) = 11.1 keV]. Escape peaks arise from X-ray fluorescence from the Ge of the detector itself; if these fluorescence photons are absorbed by the detector, then there will be no effect, but if they “escape” outside the detector, then the energy registered is that of the incident photon displaced to lower energy by the Ge fluorescence energy. The result is that for X-rays incident on the detector with energy Ein > E(Ge K), there is a reproduction of all structures above E(Ge K) displaced to lower energy by the fluorescence energy (primarily the Kα), as illustrated in Figure 12. The presence of escape peaks for incident energies typical of XFI experiments can often obscure the fluorescence lines of lighter elements in the sample; for example, in Figure 12 sulfur fluorescence would be difficult to observe because it would be overlaid by the escape peaks. A small inconvenience of Ge detectors is that they must be cooled to liquid nitrogen temperatures. This is because at room temperature the thermal excitation of the valence electrons can be sufficient to allow them to cross the band gap and reach the conduction band. In an appropriately cooled detector, the charge is transferred to a field-effect transistor (FET) to convert the current into a voltage as part of the preamplifier.
Figure 11.

Germanium detector. (a) Canberra LeGe germanium detector, and (b) a schematic diagram showing the basis of its function. The beryllium window, the preamplifier, and the liquid nitrogen cryostat are indicated on (a).
Figure 12.

X-ray fluorescence emission spectrum showing Ge detector escape peaks. The data set is the same as that of Figure 9 but replotted with a wider energy range to show the escape peaks, plotted with counts per second (CPS) on the ordinate. X-rays impinging the detector with energies above the Ge K-edge (indicated) generate additional escape peaks due to loss of Ge X-ray fluorescence from the detector. The escape peaks corresponding to both the Ge Kα and the Kβ fluorescence lines are seen as echoes of the scatter peaks displaced to low energy by the Ge Kα and Kβ fluorescence energies, respectively. Data were collected on SSRL 9-3.
2.3.2. Lithium Drifted Silicon Detectors
Lithium drifted silicon (SiLi) detectors are alternatives to germanium detectors that do not suffer from problems due to escape peaks obscuring low-energy fluorescence lines of interest. These devices also require cooling with liquid nitrogen. They are less efficient at high X-ray energies than the Ge detector, but more efficient than the silicon drift detector, considered below, as the typical diode thickness is 3–5 mm. In general, these devices are not much used for modern XFI experiments as silicon drift detectors are preferred.
2.3.3. Silicon Drift Detectors
Silicon drift detectors offer many of the most desirable features, providing high count rate ability with excellent energy resolution in a system that does not require liquid nitrogen cooling. Reliable and relatively affordable silicon drift detectors have now been available for a number of years, and these systems are increasingly considered the detector of choice in XFI experiments. Silicon drift detectors have a concentric ring structure composed of p+ silicon implanted on one side of an n-type silicon chip. These concentric rings are designed to generate an electric field gradient across the device (Figure 13) with a small centrally positioned contact that serves as the anode of the device. A homogeneous p+ junction implanted on the reverse side of the detector acts as an X-ray entrance window. As with other types of solid-state detector, electron–hole pairs are generated by the incoming X-ray photons, and these drift toward the anode under the influence of the electric field. At the anode the charge is transferred to the FET. Silicon drift detectors do require cooling but not to the liquid nitrogen temperatures needed for Ge detectors, and in the case of single detectors or small arrays of discrete detectors, Peltier cooling is sufficient. Silicon drift detectors do not suffer from escape peak problems because the low energy silicon fluorescence photon is so readily absorbed that it has negligible probability of escaping from the device. Because the overall thickness of the device is usually of the order of 450 μm, the efficiency at high X-ray energies is low, which can be a major disadvantage of such devices. Thicker devices that are more efficient at high X-ray energies have recently been created by stacking individual silicon drift detectors,36 and these are a promising new technology, combining high count rate ability and excellent energy resolution with no escape peak issues. We note in passing that silicon drift detectors are also suitable for specialized experiments such as low temperature X-ray magnetic circular dichroism.37 This is because the Hall effect from the applied magnetic field that will divert the current in a Ge detector will not do so in a drift detector because in the latter case the current flow is transverse to the detector axis.
Figure 13.
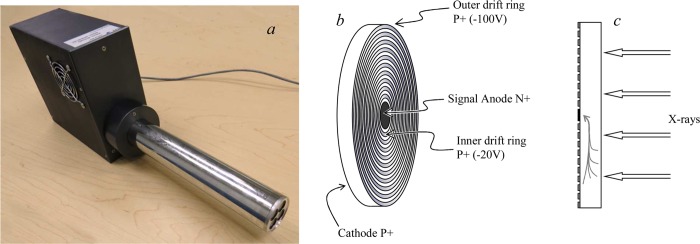
Silicon drift detector. Part (a) shows a SII Vortex 4-element array detector, with the beryllium windows for the four detectors seen on the right side of the photograph, part (b) shows a schematic diagram of a single silicon drift detector element showing the surface of the detector with the drift rings, and part (c) shows a side-on view of a single silicon drift detector element with the current flow indicated.
2.3.4. Solid-State Detector Data Readout and Preprocessing
The data from a solid-state detector are typically processed by analog electronics or by a digital equivalent. With both analog and digital systems, a charge preamplifier with an FET generates a pulse train that feeds the subsequent processing electronics. The preamplifier is generally an integral part of the detector and is not normally adjusted by the user. With analog electronics, the preamplifier pulse train is fed to a shaping amplifier, operating with user-selected shaping time (see discussion in Detector Dead Time Effects), which in turn is used to feed either a set of single channel analyzers adjusted to discriminate voltages corresponding to the fluorescence lines of interest or an analog-to-digital converter (ADC), which would be used to record the whole fluorescence emission spectrum. With the digital alternative, the preamplifier pulse train is fed to a digital signal processing unit containing a high-speed ADC and firmware that analyzes the shape of the pulse train to effectively give the entire fluorescence emission spectrum. The function of processing electronics is summarized in a simplified schematic manner in Figure 14.
Figure 14.

Schematic diagram of energy dispersive solid-state detector signal processing. The preamplifier pulse train shows sharp step-like voltage increases corresponding to the arrival of photons at the detector, superimposed upon a slower exponential decay. The size of the preamplifier voltage steps is proportional to the energy of the photons. The preamplifier pulse train can then be analyzed either by older analog electronics, using a shaping amplifier (a typical oscilloscope type display is depicted, triggering on the voltage rise), the output of which is sent to an analog to digital converter, or directly by modern digital signal processing. The latter involves a fast analog to digital converter, and real-time shape analysis of the preamplifier pulse train.
With an analog system, the output of the shaping amplifier can be passed to an ADC and to a multichannel analyzer (MCA) in which pulses of different voltages are accumulated as a histogram of the emission spectrum of the samples, which is commonly referred to as the MCA spectrum. If a sufficiently fast ADC is available, then the entire MCA spectrum can be recorded to computer for each data point. Alternatively, a number of single channel analyzers (SCAs) can be used to register shaping amplifier pulses within voltage ranges corresponding to fluorescence lines of interest. The MCA is, in any case, useful to determine the SCA voltage limits, and is often used in conjunction with a delay amplifier and coincident and anticoincident gating of the ADC to visualize this process, which is commonly called windowing.
In general, the digital systems have the significant advantage of providing the complete emission spectrum (or MCA spectrum) much more readily than the analog systems. In the recent past, analog systems could still operate at higher count rates for a given energy resolution than the digital alternative, but recently the gap between the digital and analog systems has closed as the digital systems have increased in speed. The Maia array detector38−40 is discussed below (in Array Detectors), but we will briefly consider the electronics of this system here. This large array detector system has very impressive performance with real-time low-latency pulse processing employing high-density application-specific integrated circuits. The data processing pipeline is implemented using a large field programmable gate array (FPGA) processing subsystem employing fine-grained parallelism.39,40 Another advantage of the new digital systems is also exemplified by the Maia electronics, which is the compact nature of the hardware. Thus, analog electronics for Maia would require close to 10 full height racks, with considerable quantities of cable while the Maia digital processing system is attached to the detector, with an overall system size of only 140 × 310 × 42 mm3.39,40 Some of the newer commercial digital processing systems such as the Xspress 3 system from Quantum Detectors (Harwell, Oxford, UK) or the Falcon X (XIA, Hayward, CA) can now easily exceed analog systems in performance. At the time of writing, and in most cases, the optimum readout electronics for XFI are considered to be the digital option.
2.3.5. Detector Dead Time Effects
All solid-state detectors suffer from electronic dead time effects. If the time taken to process a photon is τ, the detector will not register the arrival of another photon within τ. Moreover, the dead time is extended by a further τ following the arrival of the second photon. This gives rise to what is known as a paralyzing or extending dead time,34,41 for which the measured count rate r is related to the incident count rate ρ by
| 2 |
where τ̅ is the average value of τ over the entire spectrum. The effects of electronic dead time are illustrated in Figure 15. While dead time correction is often possible,42 it is generally considered desirable to operate solid-state detectors at count rates that are sufficiently low that the exponential term approximates to unity, in what is called the pseudolinear regime. In general, there is a trade-off between dead time effects and energy resolution. At shorter pulse shaping times (or the digital equivalent, often called the peaking time), the detector will be able to operate faster (at higher count-rates) but with a poorer energy resolution. With longer shaping times, the detector resolution can approach the theoretical optimum as discussed below, but in this case count rates are limited.
Figure 15.
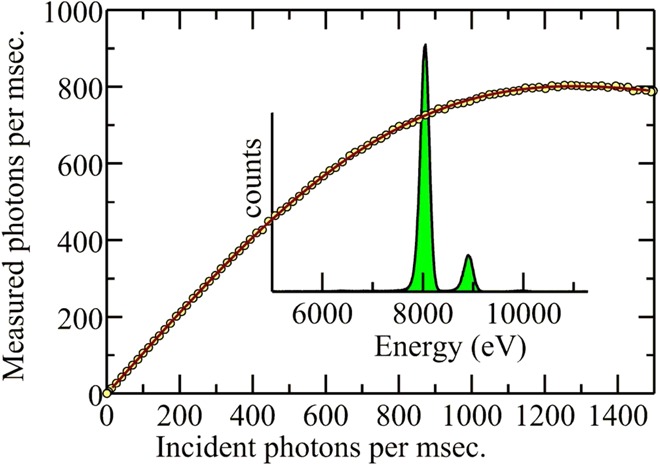
Solid-state detector electronic dead time effects. The detector employed for the measurements was a Canberra LeGe with a spectroscopy amplifier Gaussian shaping time of 0.125 μs. The sample was 1 wt % cupric oxide in a polyvinylpolypyrrolidone pressed disk with incident count rates adjusted by driving a slit blade through the beam incident on the sample. Data were collected on SSRL 2-3.
2.3.6. Pile-Up
Pile-up events occur when pulses originating from two photons arrive closer in time than the pulse resolution of the system.34 There are two types of pile-up that are of interest in XFI detection. The first, more common type of pile-up, is where the second pulse arrives on the tail of the first, potentially resulting in incorrect pulse amplitude determination and broadening or distorting the energy peak in the output spectrum. The second type of pile-up happens when photons arrive sufficiently close in time that the two pulses appear as a single pulse with amplitude the sum of the two individual amplitudes. This is registered just as if it were arising from a single photon with the sum of the energy of the two photons, and gives rise to an apparent high energy peak called a pile-up peak. The process by which pile-up events are discriminated is called pile-up rejection or detection, and a variety of electronic34 and digital43 means are employed. In most cases, these methods are sufficiently effective that users of modern spectrometer systems do not need to be concerned about their effects. With analog detector electronics, pile-up events are rejected with loss of signal, but modern digital signal processing electronics can in principle provide effective estimation of nearly overlapping pulses resulting in fewer rejected events.
2.3.7. Energy Resolution
As discussed above, the energy resolution of a solid-state detector is a function of the selected operating conditions. Under conditions where dead time effects are not prevalent, the measured peak shape is usually best approximated as a Gaussian, with detector energy resolution usually expressed as a full width at half-maximum (fwhm) or half-width at half-maximum (hwhm), relating to the width of the peak at half of the maximal measured intensity. The energy resolution is a function of the amplifier pulse shaping time or the digital peaking time (which are essentially functionally equivalent but numerically different), the operating count-rate, and the level of detector saturation (dead time). Because the use of a short shaping time will not include all of the preamplifier pulse, and the use of a very long shaping time will include too much signal noise, careful matching of the preamplifier output is required to obtain optimum resolution. For a well-optimized detector system such as a modern germanium or silicon drift detector, operating at around 10 keV, an energy resolution close to the theoretical best possible resolution of about 150 eV fwhm should be achievable at count rates with negligible dead time. Thus, for the Ge detector example shown in Figure 9, a relatively long amplifier shaping time of 2 μs was used with count rates limited to nonsaturating values, resulting in an energy resolution of about 160 eV fwhm. In most cases, however, the selected amplifier shaping times will be shorter to allow collection of high count-rate data. Thus, a 0.125 μs shaping time will allow high count rates (Figure 15) with energy resolutions in the range of 350–400 eV fwhm and count-rates in the vicinity of 100 000 counts per second. An intermediate example is shown in Figure 10, in which a 0.5 μs shaping time provides an energy resolution of 200 eV fwhm with a silicon drift detector. Using the most modern digital signal processing hardware, considerably better energy resolution at high count-rates is possible, with little degradation in resolution even at the highest count rates.
2.3.8. Array Detectors
One solution to the dead time problem of limited count rates is the use of array detectors.42 A number of individual detectors are combined, each with similar (ideally) independent electronic dead time to multiply both the overall count rate and the solid angle of fluorescence detected by the number of detectors within the array. Arrays can be composed of monolithic devices or arrays of discrete detectors packed into a single housing. In general, the larger arrays are found on beamlines dedicated to X-ray absorption spectroscopy, as these have larger X-ray beam sizes and can typically provide much greater photon fluxes at the sample, leading to correspondingly greater detector dead time problems. Nevertheless, it is common to find small arrays such as the four-element silicon drift detector array Vortex-ME4 (Hitachi High-Technology Science America, Inc., Northridge, CA), which has an active area of 170 mm2. Monolithic detectors are also available, typically germanium devices, which can accept good solid angles with little dead area between the pixels. The Maia detector38−40 is a sizable monolithic array that is currently in use at the X-ray fluorescence microscopy beamline at the Australian Synchrotron and most recently at the Cornell High Energy Synchrotron Source. The system consists of a 20 × 20 pixel silicon monolith detector, with the middle square of 4 × 4 pixels removed to make a central hole, yielding 384 potentially active pixels in the monolithic array. The illuminating X-ray beam is projected through the central hole and onto the sample, which must be about 10 mm away. The chief advantage of this geometry is that the detector accepts more solid angle, and the chief disadvantage is that it must also accept substantial scattered radiation. Siddons et al.40 have pointed out that this is not overwhelming, but can be useful in providing an estimate of sample density. Because each pixel is 1 × 1 mm2, the system accepts a very large solid angle and has an integrated pipelined, parallel processor with embedded data analysis. The individual array pixels have a maximum count rate of about 30 000 counts per second, and the resolution is worse than competing systems, requiring sophisticated data analysis methods; nevertheless, the system is very impressive, with high sensitivity and rapid scan ability.
2.3.9. Other Detector Systems
Although what follows does not relate to solid-state detectors, we briefly discuss them here as they present alternative strategies that may have applications for specific systems. Considerably higher energy resolutions are available through the use of crystal optics, with commercial detectors including a log-spiral bent Laue detector,44 available from FMB Oxford, UK. These systems use crystal optics to provide high energy resolution for fluorescence in a narrow range, and must be used with another detector (e.g., a nondispersive detector) employed to register the X-rays. The major advantage of these systems is improved discrimination of nearby fluorescence lines and superior background rejection, both of which arise from the high energy resolution. Moreover, in principle there are no inherent count-rate limitations due to detector dead time effects. The disadvantage is that fluorescence from only one element at a time is registered, and while these systems could be used in combination with a solid-state detector, they will likely be restricted to niche applications. Similar arguments apply to commercial crystal-based electron microscopy wavelength dispersive detectors (e.g., Oxford FMB INCAWave spectrometers) that can be found on a number of beamlines capable of XFI measurements. These systems typically have a variety of different crystals and use a Johannson geometry Rowland circle affording excellent energy resolution, corresponding to about 30 eV fwhm, but probing a low solid angle.
2.4. Sample Scanning Systems and Experimental Strategies
With synchrotron light sources the X-ray beams are static, and the sample must be moved relative to the incident light. The stage system selected will depend upon the required spatial resolution and speed of movement, and can include several types. Stepper motor driven stages can have a large range of travel, be scanned reasonably rapidly, and move with a typical precision of the order of 1 μm. So-called DC-servo stages combine a direct current motor plus gear reducer to a coupled position sensor in a closed feedback loop. They have the advantage of being able to move very rapidly and precisely and have excellent ranges. Piezoelectric stages are capable of the most precise movement, depending upon deformation of piezoelectric materials under the influence of an applied electric field; their main disadvantage is that the range of motion is typically quite small.
Irrespective of the type of stage used, two different types of data collection strategy are employed in modern systems: point-by-point and continuous or fly scan. With the point-by-point data acquisition strategy, the sample is moved to a location, and, possibly following a settling time, the data are acquired on a stationary sample. The time taken to scan samples with this strategy is often considerable; for example, a typical midscale resolution X-ray fluorescence mapping data set might be 250 pixels ×250 pixels, with a collection time of 0.2 s per point, with an additional 0.1 s per point required to move the stage and perhaps allow for settling. A single energy image collected in this way would take more than 5 h. Continuous scan is a much faster strategy; here, the sample is moved continuously with data being collected while the sample is in motion. The simplest such strategy is to scan the sample in one direction and then rapidly return to the start of the next raster, but more efficient still is a bidirectional mode in which left-to-right and right-to-left raster scans are interlaced. Effective dwell times can be reduced to the milliseconds range, and a scan that would take many hours in point-by-point mode can be rapidly completed in a fraction of the time, albeit with reduced signal-to-noise corresponding to shorter effective count times. Continuous scan mode leads to some inevitable motion blurring of the detail in the image, and is most effective for samples in which signal counts are not statistically limited by dilution of the elements of interest. In dilute cases, the point-by-point mode may be the most effective because accumulation of sufficient counts for the fluorescence signal of interest can require averaging. Point-by-point also potentially yields the best spatial resolution as no motion blurring should be present. Very large samples can be imaged using continuous scanning, with examples including paintings,45 ancient manuscripts,46 and large fossils.47,48
In some experiments, individual images of samples at a number of different X-ray excitation energies are used to determine chemical information. This method is discussed below in section 2.6. In this case, we effectively scan in three dimensions, one being the X-ray energy and the other two orthogonal axes of sample motion. In many XFI setups, the X-ray monochromator will take considerably longer to complete an energy move than the sample stage does to move between pixels. In this case, the most effective scanning method is generally to scan an individual raster at successive X-ray energies, and then go to the next raster on completion of the sequence of energies.29 This method, summarized in Figure 16, minimizes the time difference between energies, and allows accumulation of a complete fractional map at all energies in case of equipment or software failure or sample deterioration during the experiment.
Figure 16.

Sample scanning strategy for a multienergy image. Individual rasters are scanned at each energy, building up a complete multienergy map with as little time separation between the individual rasters as possible. In the example, four energies (E1–E4) are in the process of being recorded, with the active raster at E3.
During oversampling, the sample is scanned with a step-size that is finer than the beam-size, which can yield a small amount of additional detail when compared to using a step-size that is the same as or coarser than the beam-size. The beam-size is usually expressed as a full-width half-maximum, determined by scanning a wire or slit blade across the beam, and can be thought of as having a distorted Gaussian-type profile (e.g., Figure 5), sometimes broadened in one dimension. Assuming a Gaussian beam profile, sharp features within an image that are spatially separated by the full-width half-maximum of the beam spot should in principal be resolvable by using oversampling.
2.4.1. Sample Geometry
The geometry of an experimental setup is also an important factor. The two common sample geometries relate to the angle of the surface of a flat sample with respect to the incident beam. Probably the most common is the 45° geometry (Figure 17) in which the sample is inclined at 45° to the incident X-ray beam. The fluorescence detector is oriented at 90° to the incident beam in the horizontal plane, at which angle the X-ray scatter is minimized, allowing a degree of background suppression while minimizing detector saturation. The disadvantage of this geometry is that the non-normal incident X-ray beam is spread out horizontally on the sample by a factor of 21/2 with potential parallax problems. In addition, for imperfectly flat samples, more prominent “edge effects” may result in which physical edges are either preferentially illuminated or cast into shadow, depending upon the orientation with respect to the beam. An alternative geometry is normal incidence in which the sample is oriented at 90° to the incident beam (Figure 17). This has the advantages of being conceptually simpler with optimal horizontal beam size. Edge artifacts and parallax problems may be less prominent, but may still be present as the detector will be at an angle relative to the sample. The major disadvantage of this geometry is that the detector is usually positioned at an angle to the beam at which more scattered X-rays are sampled and the Compton scatter is shifted to lower energy. In some cases, the choice of setup geometry is constrained by the experiment or physical limitations of the equipment, such as with the Maia detector, which is designed to be used only at normal incidence. Each of these experimental configurations has its own advantages and limitations, and ideally the experimenter should be able to choose between them. However, changing configurations is not always convenient and can consume precious beamtime from the assigned allotment so that experimenters are not often presented with this choice.
Figure 17.

Plan view schematic diagrams of different sample geometries for X-ray fluorescence imaging. The geometry shown in (a) has the plane of the sample oriented at 45° to the incident beam, while that shown in (b) has the sample plane oriented at normal incidence to the illuminating X-ray beam.
2.4.2. Microscope
Another component common to nearly all XFI experiments is an optical microscope (Figure 3, Figure 17). This is used to check alignment of samples with respect to the beam prior to scanning the sample, and some experimental setups have more than one microscope, each suited to a particular experimental aspect. In the 90° orientation (Figure 17), a microscope downstream of the sample can be used, but care must be taken that X-rays do not strike the lens both because commercial optical glass contains many elements of interest (e.g., high levels of zinc) and because the X-rays will damage the lens by induction of color centers. In some cases, a mirrored pellicle made of a low X-ray cross-section material (such as silicon with a thin aluminum coating on one side) can be used to obtain a straight-on view of the front of the sample with no parallax errors (Figure 17). At many beamlines, the sample is fiducialized at an off-line microscope before the sample is brought to the beamline; this enables the regions of interest to be chosen ahead of time.
2.5. Quantitative Analysis of Two-Dimensional Images
Quantitative two-dimensional maps of flat samples such as tissue sections are often shown in terms of what is sometimes called areal density, typically with the somewhat unorthodox units of μg/cm2, representing the total amount of an element of interest (in μg) within the sample expressed as a function of surface area, and neglecting thickness. Calibration of the raw X-ray fluorescence data to quantitative units of this type is done by using measurements of standards of known composition and relating the measured fluorescence peak areas to the peak areas of the standard. Quantification of fluorescence data is often done by simple binning of counts, within an electronic window, as shown in Figure 10. This may be done if the fluorescence emission spectrum is not available digitally; for example, if the beamline employs only analog electronics, then single channel analyzers can be used to discriminate amplifier pulses within a specific voltage range. In many cases, this binning method can produce reasonably accurate results, although if the energy dispersive data are available, then a peak-fitting approach is usually more accurate.
Well-established methods for peak-fitting are available with sophisticated background corrections;49 these approaches have major advantages over binning if partly overlapping fluorescence peaks are present in the spectrum. In many cases, the distribution in intensities of peaks (e.g., Figure 9) is sufficiently broad that the results are best displayed on a logarithmic scale. Figure 18 shows an example of peak fitting and illustrates the potential benefits of this method. For the case of dilute samples in which the fluorescence energy of primary interest is reasonably close in energy to the incident X-ray energy, the low-energy tail of the inelastic scattering peak can provide a substantial background. This occurs, for example, in measuring selenium in tissue samples using an energy just below the bromine K-edge (e.g., 13 450 eV) (see section 2.13). In such cases, and when a full energy dispersive spectrum is not available on a per-pixel basis, adequate background removal can be achieved by careful measurement of both scatter and fluorescence signal and estimating and subtracting the overlap.50 In some cases, standards are available that contain known quantities of the element or elements of interest, but in other cases elements with nearby fluorescence lines have been used. In the quantification of Hg, for example, certified standards for gold and thallium, the elements adjacent to Hg in the periodic table, were used because standards containing elemental mercury showed a gradual decrease in signal over time, presumably due to loss of elemental mercury vapor.51 In the case of very thin samples, such as tissue sections, which may be 0.2–10 μm thick, there is usually no need for correction for thickness in the hard X-ray regime. In the case of thicker samples, such as whole small organisms, a correction for thickness should be applied. Correcting for thickness is simple in principle, and depends upon the X-ray attenuation by both sample and experiment (e.g., by windows, flight paths, etc.) of incident and fluorescent X-rays. For an X-ray beam penetrating into a sample of depth t, with incident X-ray intensity I0, the X-ray intensity at t is I, given by
| 3 |
where μ is known as the X-ray absorption coefficient. Neglecting the effects of absorption edges, μ varies with X-ray energy E approximately as 1/E3, or more specifically for an element μ ≈ ρZ4/ME3, where ρ is density, Z is the atomic number, and M is the atomic mass. The X-ray absorption coefficient is the product of the X-ray cross-section σ and the density ρ and is related to the absorbance A by
| 4 |
McMaster has tabulated the X-ray cross-section for most of the periodic table, from atomic number Z = 1 to 94, with only a handful of exceptions, all of which are heavy elements (i.e., Z = 84, 85, 87, 88, 89, 91, and 93).52 For an X-ray fluorescence imaging experiment, the attenuation of both the incident X-ray beam at energy E and the fluorescent X-ray beam at energy Ef should be considered.
Figure 18.
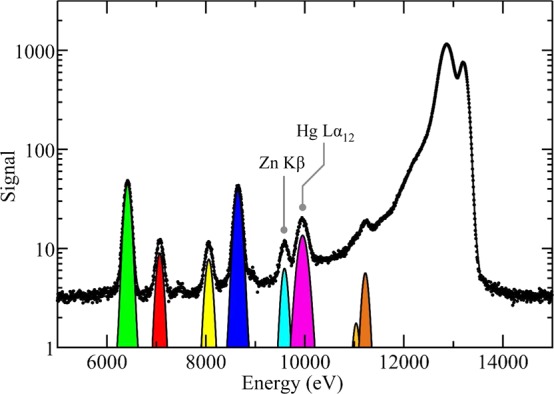
Example of quantification of X-ray fluorescence data using peak fitting. The data used in this example are those shown in Figure 9; here, the points represent experimental data, while the continuous (black) line shows the best fit. Selected individual fluorescence contributions are shown as filled colored areas. Partly overlapping fluorescence lines such as the Zn Kβ and Hg Lα (indicated in the figure) would be difficult to properly quantify without peak-fitting, especially under lower energy resolution–higher count rate conditions. The elastic scatter, inelastic scatter, and background contributions to the fit are not shown, and some of the lower intensity fluorescence peaks are below the bottom of the plot due to the log scale.
Pickering et al.50 have described a simple method that can be used for samples of irregular thickness, such as plant tissues. This uses the measured transmittance of the sample and assumes that the absorption of the plant material can be approximated as water (i.e., using σ and ρ for water). Thus, if Ae is the experimentally measured absorbance, and A and Ab are the absorbances of the sample and the background (from X-ray windows, flight paths, etc.), respectively, then the sample absorbance can be obtained by the difference A = Ae – Ab = σρt, and the sample thickness t easily be obtained as
| 5 |
In many biological cases, approximating the sample as being composed essentially of water is either an excellent approximation, or the materials present are composed of other abundant light elements (C, N, O, H) in which case they will have X-ray absorption characteristics similar to those of water. Calculation of molar quantities and concentrations from the areal densities and the per-pixel sample thickness t is then a relatively simple matter.
Even with a constant incident X-ray energy E, different elements will have different fluorescence energies Ef, and for each of these the penetration depth and signal at the detector will be different. For a planar sample inclined at an angle θ to an incident X-ray beam of intensity I0 with a detector at an angle φ to the plane of the sample, in the limit of a small solid angle β accepted by the detector, and neglecting absorption by X-ray windows, atmosphere, etc., then the fluorescence F from a homogeneous sample of thickness t will be simply given by
| 6 |
where ω is the fluorescence yield, and α incorporates the X-ray absorption coefficients at the incident X-ray energy μi and at the fluorescence energy μf:
| 7 |
The sample thickness tf required to obtain a fraction f of the maximum possible signal (that obtained with an infinitely thick sample) is given by:
| 8 |
Figure 19a and b shows a series of fluorescence curves for a selection of biologically relevant elements plotted as a function of sample thickness t, calculated using eq 6 assuming a water matrix and a 45° geometry (Figure 17a; θ = φ = 45°). Figure 19c shows curves of tf against atomic number Z for the K-edge fluorescence of midrange elements, showing that for lighter elements the surface produces most of the signal. Thus, for sulfur and selenium, in a dilute aqueous sample, 90% of the maximum possible signal will come from the first 40 μm and the first 3 mm, respectively.
Figure 19.

Fluorescence signal depth attenuation. Curves are calculated for selected biologically relevant elements, but omitting the solid angle term (β/4π) in eq 6; (a) shows the fluorescence signal without accounting for fluorescence yield ω,32 and (b) shows the same data including fluorescence yield but on a logarithmic scale. Curves of the required thickness tf for 10% increments between 90% and 10% of the maximum possible signal are shown in (c) plotted against the atomic number Z of the fluorescing element (eq 8). In all cases, the absorption was approximated by that of water, with an incident X-ray energy of 13 450 eV, and both θ and φ set to 45°.
In an elegant application of quantitative methodology, De Samber et al.53 have used the differential attenuation of the calcium Kα12 and Kβ13 fluorescence lines to correct three-dimensional data collected using confocal XFI (section 2.7) of the small freshwater crustacean Daphnia magna. These workers exploited the rapidly changing X-ray cross-section in the vicinity of the calcium fluorescence energy (the Kα12 and Kβ13 are at 3690.5 and 4012.7 eV, respectively33) plus the intensity ratio of the measured intensities F Kα12 and Kβ13 to estimate the X-ray attenuation of the fluorescence for all elements in the sample. They define the ratio R = FKα12/FKβ13, which holds a value R0 without any attenuation, and which is expected to be 11.54 for a K-edge,33 and a parameter k, which is defined as the ratio of the fluorescence energies k = EKα12/EKβ13. For an effective depth from which the signal originates d, De Samber et al. give the following approximate expression:
| 9 |
These workers use this relationship to estimate that, for their samples, the Ca Kα12 signal is attenuated to about 17% and the Zn Kα12 signal to about 87%.53
Recently, a novel approach to quantification has been reported by Kosior et al.54 These workers used magnified phase-contrast imaging to provide an indication of projected mass for lyophilized cells. This information was used in combination with XFI to provide quantitative estimates of subcellular concentration for a variety of trace elements.54
2.6. Chemically Selective Spectroscopic Imaging
Chemically selective XFI is an extension of the X-ray fluorescence microprobe method that exploits the chemically sensitive differences in X-ray absorption near-edge spectra to generate images or maps of specific chemical types.50 The method was first suggested and tested for transmittance-based imaging in a basic form by Kinney and co-workers,55,56 but more than a decade passed before the first real application using X-ray fluorescence.50 The methods rely on the chemical sensitivity of near-edge spectra. Near-edge spectra, sometimes called the X-ray absorption near-edge structure (or XANES), are often rich in structure,5 which arises from X-ray excited transitions of the core electron to unoccupied levels near to the valence levels. Intense features are usually electric dipole allowed (Δl = ±1), although weak features due to electric quadrupole transitions can also be observed,57,58 and can be used as fingerprints of overall chemical type by simple comparison with spectra of standard compounds. In the case of transmittance measurements, such as scanning transmission X-ray microscopy (STXM), the signal is sufficiently strong that very short dwell times can facilitate a data set, which comprises an image at every energy point in the near-edge spectrum, from which can be derived a near-edge spectrum at every pixel.
With X-ray fluorescence on dilute biological levels of metals, longer count times mean that usually such a complete data set cannot be collected in a tractable amount of time. Two basic methods can be used to derive similar information. The first is micro-X-ray absorption spectroscopy (μ-XAS), in which a conventional fluorescence image is taken, and then the sample is moved to locate the microfocused X-ray beam on selected points of interest and a spectrum recorded. In μ-XAS the extended X-ray absorption fine structure (EXAFS) portion of the spectrum can also be examined, provided that signal-to-noise and tolerance to radiation damage are sufficient, to give local structural information.5 Figure 20 shows an example of μ-XAS combined with an elemental image using an incident X-ray energy well above the absorption edge of the elements of interest in the sample. The spectra show that different chemical forms are present in different locations. There are two major disadvantages to this method. First, only a small number of specific locations can be interrogated by recording the μ-XAS, which gives rise to the possibility of missed detail, and second that the X-ray radiation dose to the sample is considerable because of the longer duration required to scan the μ-XAS spectra. This in turn increases the risks of radiation damage with significant chemical change to the sample (section 2.11).59
Figure 20.
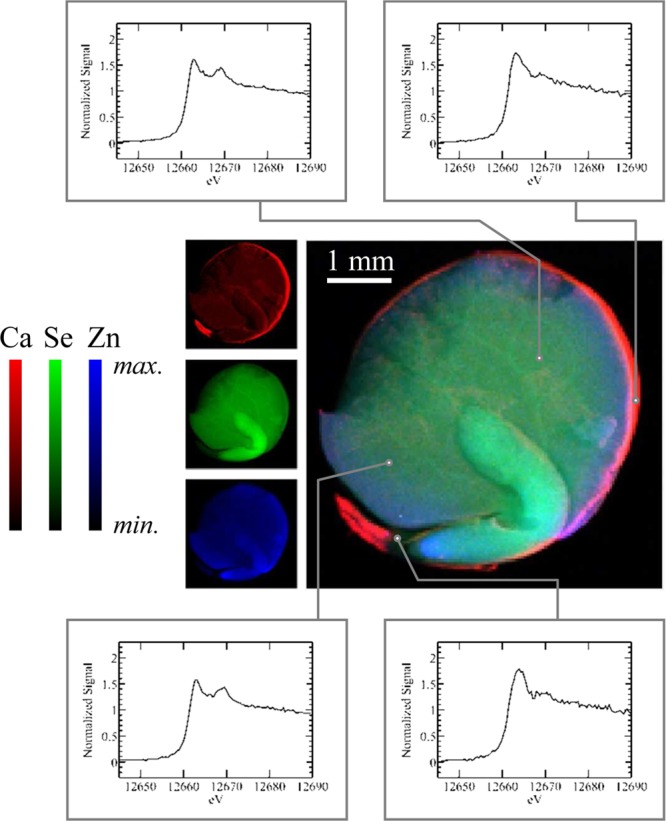
Example μ-XAS experiment on selenium in a red lentil seed (Lens culinaris). The central image shows the X-ray microprobe results with Ca, Se, and Zn, indicated as red, green, and blue, respectively. Selected locations on the image, as indicated, were then interrogated by using μ-XAS, with the different spectra indicating the presence of chemical differences between the different parts of the lentil seed. Data were collected on SSRL 9-3 using a glass monocapillary with a 6 μm exit (focal length 2 mm).
The second method of chemically selective imaging requires collection of multiple images at different energies. As we have discussed above, elemental imaging normally employs an incident X-ray energy that is well above the absorption edges of the elements to be studied. Chemically selective X-ray fluorescence imaging uses multiple incident energies across the absorption edge of the element of interest. The method exploits the aforementioned sensitivity of near-edge spectra to chemical species with incident X-ray energies carefully chosen to confer chemical selectivity. It requires prior near-edge analysis or some knowledge of the probable composition of the system so that appropriate energies can be chosen; typically X-ray absorption spectroscopy is carried out first on a bulk sample to establish candidate energies. When using an energy-dispersive detector with good background rejection, the number of necessary incident energies is minimally equal to the number of species to be mapped.50 For energies E and i components, we can write an expression for the fluorescence, F(E).
| 10 |
where ks is a quantification constant of proportionality derived from measurements of standards of known concentration, mi is the molar quantity of component i per pixel, and Ii(E) is the normalized intensity of component i at energy E, derived from measured spectra.50 This equation can be solved for the molar quantities mi. As an example we consider this method for the fern Pteris vittata.28 This plant, the Chinese brake fern, can take up arsenic from the soil in the form of the oxy-anion arsenate (a mixture of [H2AsO4]− and [HAsO4]2– at neutral pH), which is relatively nontoxic, but then transforms it into the much more toxic oxy-anion arsenite (essentially 100% [As(OH)3] at neutral pH) and hyperaccumulates it in its tissues.28,60,61 Figure 21a shows spectra of aqueous solutions of arsenite and arsenate at physiological pH, and the X-ray fluorescence imaging data at two incident X-ray energies (E1 and E2 in Figure 21) together with the transmittance data. The transmittance is essentially invariant between the two energies because the bulk of the X-ray absorbance is not specific to the relatively dilute arsenic but from the other components of the tissue. The bulk of the arsenic present is stored in the tissues as arsenite and as a result the total fluorescence at the two energies looks very similar, but solving eq 5 gives images that clearly show the arsenate to be present at very low levels in the tissues and additionally to be confined to transport vessels seen as faint lines in the center of the leaf (Figure 21).
Figure 21.
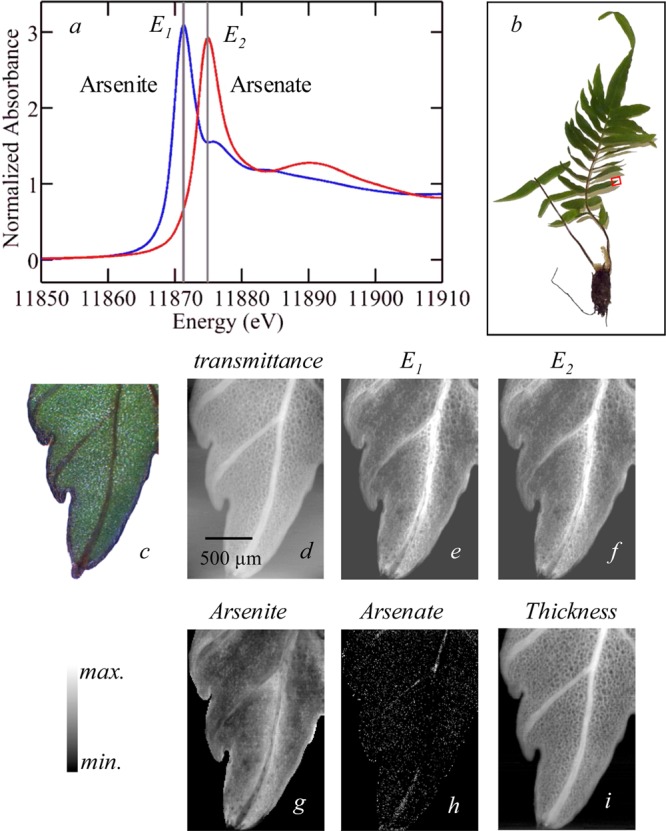
Chemically specific imaging of the fern Pteris vittata. The X-ray absorption spectra of standard solutions of arsenite and arsenate are compared in (a), and the incident X-ray energies selected for imaging are indicated as E1 and E2. The tip of one pinna (leaf) was selected for imaging (b), and the raw imaging data are shown in (d)–(f) with a micrograph (c), X-ray absorbance (d), and X-ray fluorescence at E1 (e) and at E2 (f). Using eqs 4 and 5, the analyzed data in (g)–(i) are obtained, with the concentration of arsenite (g) and arsenate (h), with maxima of 40 and 2 μM, respectively. The thickness (i) has a maximum value of 0.2 mm. Adapted with permission from ref (28). Copyright 2006 American Chemical Society.
Since the first report of the methods for quantitative analysis,50 chemically specific fluorescence imaging has been employed to follow chemical localizations of trace elements in a number of biological and environmental systems.26,50,62 Other workers have reported attempts at contrast resolution by measuring total quantities of an element with incident X-ray energies above and at a near-edge peak of one component, but in these cases quantitative analysis was lacking.63−65 In some systems, energy dispersive fluorescence detection is not as useful because of the relatively small separation between scatter and fluorescence. For example, at the lower energies of the sulfur K-edge, the separation between elastic scatter and S Kα fluorescence is almost an order of magnitude smaller than with the hard X-ray case, being only about 160 eV, which is sufficiently small that the fluorescence and scatter cannot be adequately resolved by conventional solid-state dispersive detectors.29 In this case, a slightly more complex approach can be used in which additional images at incident X-ray energies above and below the absorption edge of the element of interest are collected. We note that even though discrete fluorescence peaks may not be readily resolved, there is still important additional information from dispersive detection systems, including the fluorescence of elements of lower atomic numbers, such as phosphorus. We can assume that the total measured signal T(E) at incident X-ray energy E can be expressed as the sum of two components, F(E), the fluorescence signal of interest, and background, B(E):
| 11 |
We note that our treatment here contains no corrections for energy-dependent processes, which might affect the fluorescence intensity, such as depth of penetration of the beam into the sample, although these would be comparatively simple to include, and would be specific to the case where only one absorption edge falls within the energy range of the experiment. The background B(E) consists of contributions from both elastic and inelastic scattered X-rays, and in the case of nonenergy dispersive detectors, X-ray fluorescence arising from absorption edges other than the one of interest. B(E) is expected to vary smoothly as a function of energy and can be approximated as a polynomial function of the X-ray energy, E. A series of equations (eq 10) will be expressed, with different values of Ii(E), for each incident energy used. All forms contribute in an essentially chemically insensitive manner to images at energies that are well above the absorption edge, whereas below the edge, the intensity will equal the function at that energy B(E). However, close to or at the absorption edge itself, the near-edge structure shows considerable variation, and the intensity at a given energy will be different for each component. The equations (eq 10) can be solved by matrix inversion to yield mi, the molar amount of the element present as species i at each pixel (Figure 22), which can then easily be converted to give the fraction fi of the element that is present as each species:
| 12 |
Figure 22.
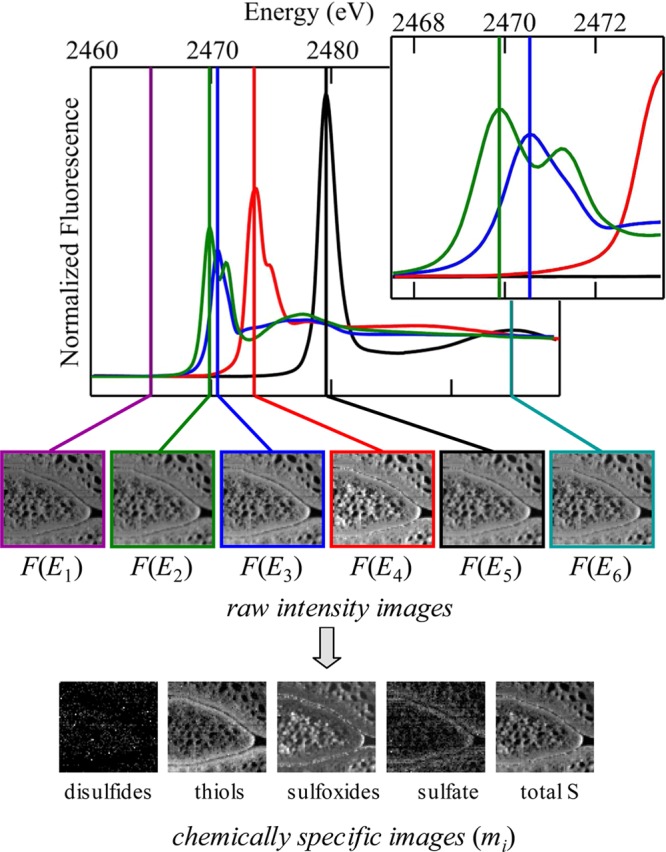
Chemically specific spectroscopic X-ray fluorescence imaging at the sulfur K-edge. The upper part shows sulfur K-edge spectra of standard solutions used to analyze the data providing criteria for selecting incident energies (see markers) E and the values for I(E). The inset shows an expanded energy scale in the region of disulfides and thiols/sulfides, illustrating the spectroscopic discrimination between these forms. The upper row of images corresponds to raw intensity data for background [F(E1)], total sulfur [F(E6)], and the spectroscopic peaks of disulfides, sulfides, sulfoxides, and sulfate [F(E2)–F(E5), respectively]. Processed images showing mole fractions mi of the different chemical species are shown in the lower part of the figure. Adapted with permission from ref (29). Copyright 2009 American Chemical Society.
The determination of the molar amount mi is dependent on measurements at all energies, rather than just on the measurement corresponding to the maximum of intensity of that component. This allows the separation of species whose spectra have quite a degree of overlap but which still show enough distinction that energies can be chosen to confer chemical sensitivity (e.g., Figure 22, inset). Because this method requires prior knowledge of the chemical forms present in a sample and their spectra, it is typically preceded by careful bulk spectroscopic measurements.29 Chemically specific imaging is one of the most important and powerful capabilities of X-ray fluorescence imaging, and while this is still a fairly specialized method, its use is increasing. There are also a number of recent reports in which quantitative analysis has been attempted using difference spectra. Although such numerically trivial approaches lack the rigor of the treatment described here, useful information can still be obtained.66
In some cases, and especially at very low X-ray energies,67 it may be possible to take images using a large number of energies so that each pixel is represented by what amounts to a complete near-edge spectrum. In this case, the number of data values considerably exceeds the number of species so that the problem is overdetermined and matrix inversion of eq 10 cannot be used to give mi values. Early applications used least-squares fitting of individual pixels to standard compound spectra, but more recently single value decomposition (SVD) has become established as the best method for efficiently providing the least-squares solution (see section 2.10 for a discussion of SVD).68 An interesting analysis variant has been reported by Lerotic et al.69 that can be used in the case where standard compounds are lacking. These workers used principal component analysis (see section 2.10) in conjunction with cluster analysis to classify the pixels in an image according to spectroscopic similarity and to extract representative cluster-averaged spectra.69
2.7. Three-Dimensional Methods
2.7.1. X-ray Fluorescence Tomography
X-ray fluorescence tomography70,71 has been employed increasingly in recent years,72−74 primarily in an elemental mapping mode to develop three-dimensional images of elements within samples. A schematic of a basic fluorescence tomography experiment is shown in Figure 23, with a micro or nanofocused X-ray beam penetrating the sample, which is raster scanned along the direction x and rotated about the angle φ. In some cases, an alternative and nearly equivalent data acquisition strategy can be to scan the detector and keep x stationary.74 The X-ray fluorescence output is monitored as a function of x and φ, and the resulting sinogram (Figure 23) is subjected to tomographic reconstruction75 to visualize a cross-sectional slice of the object. The sample can then be translated vertically along the z direction (Figure 23) and the procedure repeated to yield another slice of the object. These can be stacked and combined for three-dimensional viewing as elegantly demonstrated by de Jong et al.,76 who conducted tomographic elemental reconstruction of an air-dried freshwater diatom cell (Cyclotella meneghiniana) with 400 nm spatial resolution (Figure 24) following a total data acquisition time of 36 h. Diatoms are encapsulated by a siliceous frustule. The frustule is often exquisitely structured, and while this is best viewed in the scanning electron microscope it is also visible using light microscopy. The microstructure of the frustule was not observed by de Jong et al.,76 and while they do not comment on this, we suspect that it was likely due to the use of an air-dried sample rather than radiation damage, necessitated by the pioneering nature of the work. However, as with any X-ray exposure intensive method, the possibility of radiation damage is a significant concern, and this is discussed in section 2.11. In the interests of clarity, we note that coordinate systems alternative to that shown in Figure 23 are often employed; for example, the z-direction is often defined as the direction of the X-ray beam with x being the orthogonal horizontal axis and y being vertical.
Figure 23.

Schematic of a typical X-ray fluorescence tomography experiment. Parts (a) and (b) show schematic side and plan views, respectively. The sample is positioned on a stage capable of both rotary and Cartesian motion. It is scanned through x and rotated about φ to develop the sinogram (c), which can be subjected to tomographic reconstruction of a slice. A number of sinograms at different values of z can be collected, and their tomographic reconstructions were stacked together to make a three-dimensional rendition of the sample.
Figure 24.
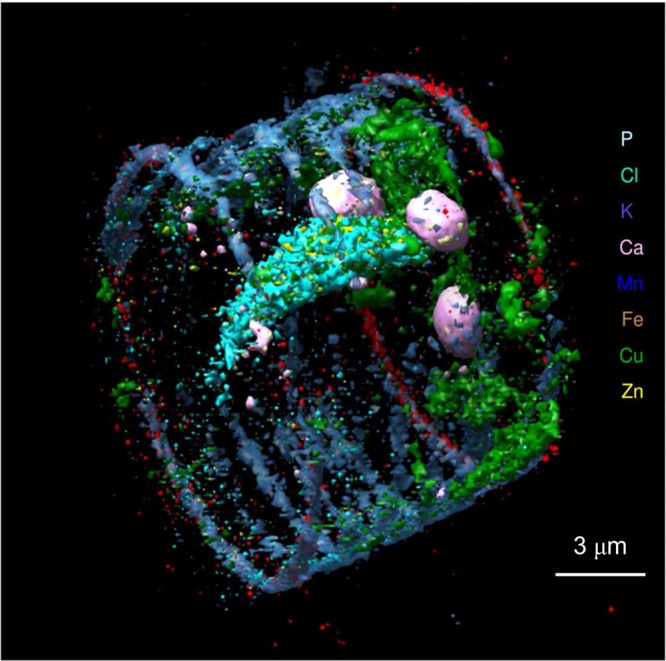
X-ray fluorescence tomography of an air-dried single-celled freshwater diatom Cyclotella meneghiniana. The silicacious frustule has been removed to show inner detail. Adapted with permission from ref (76). Copyright 2010 National Academy of Sciences.
Just as with two-dimensional chemically specific imaging, chemically specific fluorescence tomography is a practical experiment, and at least one report has been published.77 An example is shown in Figure 25, of a seedling of the two-grooved milk vetch (Astragalus bisulcatus) germinated in 5 mM selenate solution. Unlike adult plants grown in the absence of selenate,26 seedlings tolerate high concentrations of selenate without any apparent ill effects. The tomography in this example clearly shows the selenate in the transport vessels of the seedling with organic selenium forms in the tissues. We note that μ-XAS in which an XAS spectrum of a specific voxel can be acquired is not practical with fluorescence tomography, although this has been reported for transmittance tomography.72,78 Tomographic reconstruction artifacts develop if inadequate numbers of angles φ are collected, and these appear as radial lines (spokes) in tomographic reconstructions. We note that these are present to a small degree in Figure 25. Artifacts due to attenuation of the incident and fluorescent X-rays are not likely to be important for small samples such as the diatom examined by de Jong et al.,76 but for large samples correction for attenuation of both the incident and the fluorescent X-ray beams may be important. For samples that have significant internal variation in X-ray cross section, this will be a function both of X-ray energy and of the orientation of the sample within the experiment, so that different corrections may be needed for different values of x, φ, and y (Figure 23), making absorption correction a very convoluted problem. Not surprisingly, applications to date have not attempted rigorous corrections of this type.
Figure 25.

Chemically specific fluorescence tomographic reconstruction of a seedling from the selenium hyperaccumulating plant Astragalus bisulcatus. Absorbance (a) and fluorescence X-ray maps at low (b) and high energies (c) (12 661.08 and 12 667.28 eV, respectively) together with thickness (d) and chemically specific maps (amounts) for Se-methyl-selenocysteine (e), and selenate (f). The radicle (r) and cotyledons (ct) are shown in (a). Sinograms for absorbance (g) and fluorescence at low (h) and high (i) energies are also shown. The dotted lines in (a)–(c) show the z-value used for the tomography. Tomographic reconstruction of the data in (a)–(i) is shown in (k)–(n), the tomogram for Se-methyl-selenocysteine (k), selenate (l), and transmittance (m). The tricolor plot in (n) compares the localizations of Se-methyl-selenocysteine (red), selenate (green), and calcium (blue). The circle visible in the tomographic reconstruction of transmittance (m) corresponds to the sample support, a clear plastic metal-free drinking straw. The faint radial structures visible in the figure are artifacts of the tomographic reconstruction. Data were collected on SSRL 9-3.
2.7.2. Confocal X-ray Fluorescence Detection
This technique can give three-dimensional information that is very similar to that from fluorescence tomography and is illustrated schematically in Figure 26. It has two advantages, that μ-XAS spectra of specific voxels can readily be recorded, and that correction for X-ray attenuation is comparatively simple. Its main disadvantage is that the spatial resolution is often limited instrumentally in one plane. The method uses two different focusing optics, one upstream of the sample to generate the microfocused X-ray beam incident on the sample (as with all other applications addressed herein) and another placed between the sample and the detector to restrict the X-ray light accepted by the detector to a narrow volume where the two foci intersect (Figure 26). Typically this would be a polycapillary optic (section 2.1) or alternatively a microchannel array, which can give a tighter focus yielding a narrower probe volume (Figure 26), although with the disadvantage that the distance between the optic and the sample is small.79 The sample can be scanned, as indicated in Figure 26, to interrogate a plane and the detector translated toward or away from the sample to interrogate a different plane. Figure 27 shows an example of a three-dimensional confocal reconstruction of a portion of a head of a zebrafish larvae following treatment with methylmercury chloride. Confocal X-ray fluorescence has been elegantly applied to probe the elemental composition of different tissue types in the water flea Daphnia magna.53
Figure 26.

Confocal X-ray fluorescence experiment. A schematic diagram of the confocal experiment is shown in plan-view, with a blow-up of the sample area (inset), illustrating the confocal layer within the sample that is probed by the combined focal regions of the incident X-ray beam and the detector polycapillary.
Figure 27.
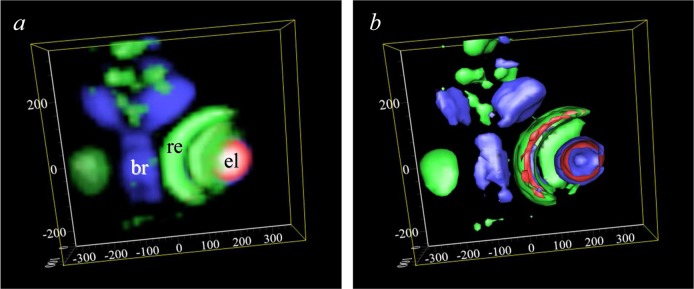
Confocal X-ray fluorescence imaging of a methylmercury-chloride treated zebrafish larvae. The sample is viewed from approximately midway between the dorsal and ventral surfaces down to the ventral surface. Both volume-rendered (a) and iso-surface-rendered (b) images are shown, with zinc depicted in green, selenium in red, and mercury in blue. All axis scales are in micrometer relative to an arbitrary zero point. The eye-lens (el), brain (br), and retina (re) are marked on (a). Data were collected on APS 20ID.
2.8. Fluorescence Imaging at High X-ray Energies
Most of the XFI of interest to us in this Review is that conducted using X-ray energies in the middle to hard X-ray regime, with incident X-ray energies between 5 and 15 keV. The upper bound of this energy range is partly governed by the normal working range of many beamlines with silicon double crystal monchromators and partly because it is comfortably above the absorption edges of the majority of biologically important elements, including all of the first transition row elements and the p-block elements through bromine. The use of higher energies, in the vicinity of 33 keV, is common in synchrotron radiation-based biomedical imaging, often using iodine-containing commercial intravenous radiological contrast agents, all of which are substituted 2,4,6-tri-iodobenzoates (e.g., diatrizoate or 3,5-bis(acetylamino)-2,4,6-triiodobenzoic acid) combined with K-edge subtraction (KES imaging) in which transmittance images above and below the iodine K-edge are subtracted to improve contrast.80 This method has been recently applied at lower X-ray energies, such as the Sr K-edge81 (16.1 keV), but as expected penetration depth is a restriction.81 The use of high X-ray energies is beneficial relative to lower energy experiments because the radiation dose is substantially decreased due to the much lower X-ray absorption cross sections at high energies, which means that measurements on live vertebrate animals become possible. The use of iodine X-ray fluorescence to improve sensitivity at low levels has recently been explored,82,83 and a combined small animal K-edge subtraction and fluorescence imaging system has been developed.83 The major problem at these higher energies and with large objects is that the X-ray inelastic (Compton) scattering tends to be very broad and extends by several keV, overlapping and adding a substantial background to the iodine Kα fluorescence. To alleviate this problem, Zhang et al.82 developed fluorescence subtraction imaging (FSI) in which incident beam energies just above and below the iodine K-edge (e.g., 33.25 and 33.10 keV) are used to obtain a Compton background image that can be subtracted.82 We note that the simplest method of removing this Compton background would be to use an incident energy that is more than 10 keV above the iodine K-edge, and a similar approach has been employed in Cd K-edge XFI, discussed in section 3.4.84,85 In cases where the main method of data acquisition is KES, the use of higher energies may add more than acceptable time to data acquisition. Alternatively, beam energies of ∼43 keV may be outside the working range of the monochromator, and application of FSI may be the best strategy. Finally, the advantages of high energies are in lower radiation dose and high penetration, which are reversed at low energies. Below 2.5 keV, nearly all of the beam will be absorbed within the first 50 μm of a sample, and the radiation dose and the potential for sample damage are correspondingly multiplied (section 2.11).
2.9. Fluorescence Imaging at Low X-ray Energies
Challenges for fluorescence imaging at low incident X-ray energies (below 5 keV) include several factors not present at higher X-ray energies. (i) Fluorescence yield (as opposed to electron yield) is much lower at low energies; (ii) the depth of penetration is very low, making sample preparation more problematic; and (iii) when using the near-edge, the energy separation between scatter and fluorescence peaks is less, leading to more overlap of these peaks. Conversely, the longer core-hole lifetimes at lower energies mean that near-edge spectra are sharper and better resolved, and so are more sensitive to chemical environment. Thus, the sensitivity of the sulfur K near-edge spectra allows ready discrimination of a number of individual chemical types, as we have discussed above (section 2.6). There are only a handful of XFI beamlines worldwide that have been specifically designed to operate in the range 2–5 keV; the ideal beamline in this range would offer all of high spatial resolution (e.g., <100 nm), high spectral brightness, and high energy resolution. In most cases, experiments are conducted in a helium filled flight path with thin polymer windows, because of the high attenuation of the X-ray beam by air and other materials, although a small air flight path has been used in some cases.29 Recent advances at scanning transmission X-ray microscopy (STXM) beamlines have added a fluorescence detector capability, effectively turning them into beamlines for XFI, although in many cases conducting experiments in the vicinity of 2 keV places STXM beamlines near their upper operational bound, and occasionally there are difficulties associated with this. These beamlines typically operate in a vacuum, and the difficulties associated with using helium are further compounded. This particularly rich spectroscopic region shows significant promise as an area of research growth in the coming years.
2.10. Other Data Analysis Methods
2.10.1. Correlation Plots
The simple expedient of plotting the levels of two different elements for each pixel in an XFI data set can often provide important chemical information. Figure 28 shows an example for the XFI of insect larvae (Chironomus dilutus) showing correlated iron and nickel localization in the gut, but not with zinc that is localized in the tissues.86 In some cases, application of a mask in which pixels within a certain correlation range are specifically selected or suppressed can be used to reveal more detail.
Figure 28.

Use of correlation plots to show colocalization of two elements, nickel and iron, in the gut of a larval Chironomus dilutus, a species of nonbiting midge.86 The linear appearance of the nickel versus iron plot indicates correlation and colocalization of the two elements. A correlation plot of zinc versus iron is also shown, illustrating the appearance when there is little or no colocalization of two elements. Data were collected on SSRL 10-2.
2.10.2. Principal Component Analysis
XFI data typically consist of several thousand individual pixels, each with an intensity corresponding to fluorescence measurements at a specific physical position on the sample. If the entire output of an energy dispersive detector is used, each of these pixels has an associated emission spectrum, itself consisting of between 1024 and 4096 energy points, and if an array detector is used (section 2.3), these data are accordingly multiplied by the number of array elements. Thus, the size of the data set can be considerable; it is not uncommon for gigabytes of data to be accumulated, and a high resolution image collected using the Maia detector can approach terabytes in size. However, this substantial volume of data contains fluorescence contributions from only a limited number of elements, probably numbering less than 20, together with the inelastic and elastic scatter, and background. Because of this, principal component analysis (PCA) can be used to carry out data reduction. The method employs single value decomposition (SVD) with data arranged as a two-dimensional matrix, A, each column containing an energy dispersive spectrum of n points at a particular pixel. Spatial correlation is not important in the SVD calculation so that these spectra are arranged by a pixel index number, up to a total m, and not as two- or three-dimensional spatially arranged data. These can be written using a single-value decomposition in which A is written as the product of an m × n matrix U, an m × m diagonal matrix V, and the transpose of an m × m orthogonal matrix W. The matrix U contains columns of orthonormal eigenvectors, called the principal components, and V is a diagonal matrix that contains the eigenvalues.
| 13 |
The principal components (eigenvectors) thus obtained are not physical component spectra, but instead are mathematical constructs. As discussed by Vogt et al.,87 the eigenvectors can be remapped as images to yield what are called the eigenimages,87 and these can be arranged in order of significance in the data set, with the data set as a whole being represented by a comparatively small number of eigenvectors or eigenimages. The photon noise in the image is not correlated between pixels and does not contribute to the important principal components. Moreover, fitting the eigenvectors can provide images of elemental components without the computational burden of peak-fitting the emission spectra of single pixels.87
2.10.3. Soft Clustering Methods
We have discussed in section 2.6 the “hard” cluster analysis of Lerotic et al.69 This approach assigns pixels to groups based on a measure of similarity between pixels. Recently, Ward et al.88 have described methods using a soft clustering approach employing multiple Gaussian fits to elemental correlation plots, and assignment of pixel-probability to groups (each represented by a Gaussian) to effectively relax group membership criteria. As a test of the method, Ward et al. compared rigid cluster analysis and soft clustering with a four Gaussian fit for Fe and Zn levels in XFI data of the malaria parasite Plasmodium falciparum, and found that soft cluster analysis could readily discriminate between the parasite, the parasite’s food vacuole, the host erythrocyte, and the background.88 In comparison, hard cluster analysis failed to discriminate these regions.88 The method was found to accurately segment images into biologically relevant subregions, and is as accurate as manual image segmentation, but considerably faster. As pointed out by Ward et al.,88 as the speed of XFI data acquisition increases, automated methods such as soft cluster analysis will become increasingly useful.
2.11. Radiation Damage and Prevention
Sample damage due to ionizing radiation such as X-rays is a recurring theme in studies of biological samples. The worst case is undoubtedly that of living samples, with vertebrate cells and tissues representing the extreme in sensitivity. The only report of XFI of living vertebrates is that of Korbas et al.,89 who reported XFI of intact living zebrafish larvae maintained under anesthesia. Invertebrates such as insects and plants are comparatively resistant and can survive extended exposure with no apparent short-term effects. In the case of aqueous samples, the radiation chemistry of water can be a major source of problems, especially with long dwell times.59 The use of microfocused X-ray beams gives a very high dose, although this is countered by the short exposure times generally used in XFI experiments, especially in rapid scan experiments. For three-dimensional imaging (either tomography or confocal, section 2.7), preventing dehydration, heating, or beam-damage during longer data acquisition times can be a particular challenge. With fixed samples, three common embedding alternatives are acrylic, wax, and epoxy. Epoxy is traditionally used for electron microscopy and is appropriately used for very thin sections of tissue, suitable for the highest XFI resolutions. For more commonly used XFI resolutions, acrylic and wax are the most appropriate choices, and of these we have found that acrylic is the most susceptible to beam damage, showing discoloration and deformation more rapidly on exposure to beam than the old-fashioned wax embedding material. Low temperatures are often employed to prevent beam damage, and a number of commercial stages designed for other spectroscopic or microscopic applications, such as the Linkam FTIR600 (Linkam Scientific Instruments, Guildford, Surrey, UK), have been adapted for XFI experiments.
2.12. Sample Preparation
Sample preparation is one of the most important factors in an XFI experiment, but has often been neglected in reviews. There are many issues to be considered, depending on the capabilities and physical setup of the beamline, the type of sample being analyzed, and the type of insight being sought. As with most sample preparations for analysis using synchrotron X-ray methods, minimal pretreatment is preferred. Chemical pretreatment of samples has the potential to alter the distribution of elements as well as their chemical speciation, while the ubiquitous use of phosphate buffered formalin for tissue fixation is also a confounding factor for phosphorus imaging and speciation. Early work by Bush et al.90 reported that trace elements were not influenced by formalin fixation; however, this has been refuted in later work by Gellein et al. where longer durations were considered.91 Using ICP-MS, Gellein et al. have demonstrated that leaching of most elements, such as Mg, Mn, Fe, Cu, Zn, As, Cd, and Hg, from biological tissue occurs,91 although the most significant changes are largely associated with long-term storage. Leaching of trace Cr and Ni from tissues stored in formalin, however, was minimal.91,92
Because of problems encountered with irregular surfaces or other issues such as edge effects, the surface to be mapped is typically flat or reasonably uniform, although corrections can be applied to account for irregularities or structured surfaces. Edge effects (discussed earlier in section 2.4) can arise in thick samples or those with irregular or rough surfaces and can result in an increased fluorescence signal from edges or decreased signal in an area that is shadowed from the detector or incident beam, both of which will alter the apparent fluorescence intensity of the sample. Similar edge effects can arise in sectioned material, such as the inadvertent introduction of small wrinkles and folds during collection of cryo-sectioned tissue onto a mounting medium. For thin sections, this may mean there are areas within a map of a 10 μm thick section that are significantly thicker, giving rise to significantly higher fluorescence and therefore greater apparent areal densities of elements within the defect region. Inspection of the X-ray scatter map can be a useful tool for identifying such edge and surface irregularities, and in some instances it may be possible to use the scatter map to normalize data channels during postprocessing. Szczerbowska-Boruchowska has reported a detailed investigation of sample thickness effects for quantitiative XFI as well as composition details for reference human tissues.93
Depending on the necessities of the experiment, samples may be windowless, with the surface of the sample exposed, or else they may be sealed, either to prevent dehydration during data acquisition or to prevent contamination. Windowless is typically preferred for imaging of light elements, for which the window material can significantly attenuate the fluorescence signal and also contribute to X-ray scatter. For soft X-ray imaging, such as that done using incident X-ray energies in the vicinity of the sulfur K-edge, enclosure of the sample and detector within a He-filled container is necessary to minimize attenuation of the sample fluorescence at these energies. A proper purge with He is absolutely required following sample changes with such a setup; otherwise, the attenuation will drift for a period at the beginning of the scan as the system equilibrates. In some cases, samples for soft X-ray experiments can be maintained in air, but in this case the bulk of the apparatus is contained in a helium chamber with a very small air gap (ca. 0.1 mm) between a thin polymer X-ray window and the sample surface to minimize air absorption.29 For similar reasons, window material containing high atomic number elements should be avoided as this can also attenuate the fluorescence signal from the sample arising from lower atomic number elements.
2.12.1. Sample Preparation/Fixation
X-ray fluorescence imaging of cells and tissues demands careful consideration of sample preparation protocol, because measurements are typically done on ex vivo samples as opposed to living systems. To avoid misinterpretation of the ex vivo elemental distributions, which may not reflect the in vivo state, care must be taken during such steps as anesthetization, the time between death and tissue fixation (sometimes called that agonal period), storage and preservation protocols (such as chemical fixation or cryo-preservation at LN2 temperatures), the method of tissue preparation, and the analysis conditions (analysis of fully hydrated tissue at cryogenic temperatures or air-dried and dehydrated samples at room temperature).
The use of aqueous environments to culture cells, or animal models that involve small organisms (Caenorhabditis elegans, zebrafish, etc.), simplifies the choices somewhat. Fixation can be achieved by passive diffusion with cell cultures or organisms in liquid fixation or anesthetic media.94−96 However, despite the simplicity with which chemical fixation can be completed for cell cultures and small organisms, elemental distributions particularly for elements such as Cl, K, and Ca that are present as readily diffusible ions are best obtained by cryo-preservation and analysis of frozen hydrated samples.97
Increasing organism complexity (e.g., mouse or rat models) brings increasing complexity to sample preparation and a greater chance for altering elemental concentrations and distributions relative to the in vivo state. In larger animals, simple diffusion of a chemical fixative is not possible, and if chemical fixation is to be performed it should be done via perfusion-fixation.98 Perfusion with saline is also often performed99,100 and has the advantage of removing blood from the tissue, which can be a major source of iron contamination. There may be significant drawbacks with this method, however. It has been established that elements such as K and Cl that are present as ions are mobilized by aqueous fixation media and are rapidly redistributed and leached from biological samples.97,101 The process of perfusion also disrupts blood supply to tissues, creating ischemic conditions (insufficient oxygen and energy supply), and although no XFI studies have systematically investigated the changes in elemental distribution as a consequence of perfusion, it is known that disrupted oxygen supply drastically alters elemental distributions in cultured cells.96 It is also well established that many cell types, particularly brain cells, release intracellular molecular stores into the extracellular space during perfusion, which could lead to confounding results. The possibility for altered elemental distribution during perfusion or perfusion-fixation of animals should always be kept in mind.
An obvious but often overlooked fact is that buffers and fixation media must be kept free of exogenous contaminants that may be inadvertently deposited in tissues, although to our knowledge few laboratories check perfusion buffers for possible contamination from trace elements such as metal ions.101 Elevated levels of metals (i.e., Fe, Cu, Zn) are often observed in formalin-fixed tissues,101,102 which have been demonstrated to result from metal contamination in the perfusion, fixation, or buffer media.101 Furthermore, contamination is highly unlikely to be uniform across biological samples, giving rise to regional differences in contamination due to different binding affinities of metals for different tissue types. For example, contamination of brain fixation media with Fe, Cu, and Zn results in the greatest increase in Fe and Zn levels in the gray matter, and the greatest increase in Cu levels in the white matter.101 Therefore, as significant molecular differences exist between different tissue types, and possibly between the same tissue type when effected by different physiological or disease processes, it is not necessarily valid to assume that consistent sample preparation across all samples will yield consistent or similar effects in all samples.
In addition to alterations in elemental distribution as a consequence of sample preparation, chemical speciation may also change. Specifically, the results of XFI employing multiple energies (section 2.6) may be significantly impacted by sample preparation. Drastic alterations in the speciation of sulfur have been observed to occur as a consequence of formalin-fixation (relative to cryo-preserved tissue), and subtle alterations also occur as a consequence of air-drying tissue sections.103 As imaging of living cells or tissues is often impractical with XFI, the possibility for some degree of chemical alteration from the in vivo state should always be considered, highlighting the importance for a thorough understanding of the likely chemical alterations, which may occur with the specific sample preparation protocol employed.
Although analysis of frozen-hydrated cryo-preserved samples yields both elemental distribution and chemical speciation closest to the in vivo state,97,103,104 this method requires specialized sampling accessories compatible with analysis at cryogenic temperatures. Implementation of such an approach is not compatible with all studies (for example, post-mortem autopsy human tissue), nor with physical or experimental limitations that may be imposed by the beamline at which the experiments will be carried out. Biological samples may also be present in some form of matrix, such as embedded in paraffin or methacrylate (acrylic). Such embedding media are highly compatible with XFI, as these matrixes contain low Z elements that do not contribute to the energy ranges generally considered for such experiments. However, as discussed above, the possibility for altered chemical speciation, elemental redistribution, leaching, or contamination from the long list of reagents or preparation steps used in paraffin and methacrylate embedding processes should always be considered.
2.12.2. Sample Thickness
Samples for biological applications are usually relatively thin and of uniform thickness. For cell cultures, for example, monolayers are the preferred choice as these typically will be on the order of 10–20 μm thick (per cell) with the resulting image not being complicated by signal from overlapping cells. Additionally for cell cultures, nonmotile cells (in the event of live cell imaging) or cells that have been fixed are necessary as the potentially long scan times provide ample opportunity for cells to move within the beam.
Larger biological specimens typically are sectioned to the desired thickness. These sections are often preferred for quantification as their thickness, and self-absorption issues can be neglected (see section 2.5).93 Further, as samples can be mounted at an angle of 45° to the incident beam, loss of spatial resolution is minimized or negligible if the sample thickness is comparable to or less than the spatial resolution of the experiment. However, for dilute levels of trace elements, the trade-off is reduced sample volume and therefore lower fluorescence signal (assuming complete penetration of the incident beam as well as negligible loss of fluorescence signal escaping from the sample), which results in low signal-to-noise and necessitates longer dwell times.
2.12.3. Sample Substrate
Samples, especially sections, must be mounted on some type of support such as a coverslip, slide, or other reasonably X-ray transparent material. Careful choice of metal-free, sulfur-free, or halogen-free mounting material is required, although many commercial vendors will not be able to ensure their products meet these requirements. Therefore, screening of mounting materials prior to experiments is always recommended. As a general rule, samples should be mounted on a material that is thinner than the sample to be mapped, as this will present minimal background scatter (e.g., 10 μm thick tissue on 6 μm polypropylene). In practice, however, good success has been achieved with thicker mounting materials, such as 20 μm thick tissue mounted on 200 μm plastic coverslips, provided the coverslip is comprised of low atomic number material (i.e., carbon, oxygen, hydrogen). Plastic materials often have the advantage of being relatively inexpensive, durable, and able to accommodate a wide range of sample types and sizes. Plastic supports supplied by vendors can have variable levels of trace impurities, such as one instance in the authors’ experience of commercial “metal-free” microscope mounting materials that contained substantial levels of cobalt which varied from batch to batch. Conventional glass materials used for optical microscopy are incompatible choices for XFI measurements because of their high content of zinc and other elements; additionally, substantial silicon fluorescence from glass slides may saturate detectors. Electron microscopy grids can also be employed; however, careful consideration must be given to the proximity of the area to be mapped with the grid, as the gridbars may also contain elements of interest to the experiment. Silicon nitride (Si3N4) windows are a close to ideal mounting material from an imaging perspective, are compatible with cell cultures, and allow a range of spectroscopic measurements to be performed on the same sample.105 However, the use of silicon nitride windows is often limited by their small size (generally 2 × 2 mm2 or 5 × 5 mm2), their fragile nature, and higher cost.
2.13. Pitfalls and Problems
2.13.1. Overlapping Fluorescence Lines and Contamination
One of the advantages of XFI is that in most cases elements cannot be confused, as the technique depends on basic atomic physics. A common question among newcomers to the technique is whether chemically similar metal ions, such as Cd2+ and Hg2+, can be confused. In this, and in almost all other cases, there is no possibility of confusion, as the fluorescence lines of cadmium and mercury are very well separated in energy. However, the fluorescence lines of a few pairs of elements overlap sufficiently that additional measurements are needed to distinguish them. A very well-known and sometimes encountered example is that of arsenic and lead; the centroid of the As Kα12 fluorescence is 10 532 eV, which is insufficiently separated from the Pb Lα12 at 10 518 eV to be distinguished [note that solid-state detectors cannot typically resolve α1 and α2 fluorescence lines (section 2.3) and so we use the intensity weighted centroid of these lines here]. Both arsenic and lead may be present in a sample, and in addition lead X-ray fluorescence contamination is sometimes seen due to lead’s widespread use in beamline radiation shielding. In the case of discriminating As from Pb, simply changing the X-ray monochromator to move the incident X-ray energy below the lead LIII edge at 13 035 eV but above the As K-edge at 11 867 eV would eliminate Pb Lα12 fluorescence without removing As Kα12 fluorescence, effectively allowing determination of As in the presence of Pb. Determination of Pb in the presence of As can also be accomplished by moving the monochromator energy to above the Pb LII edge at 15 202 eV, which would excite the intense primary LII fluorescence, the Lβ1 at 12 614 eV; unfortunately confusion of the Lβ1 with the Se Kβ at 12 496 eV is also possible, although the Se Kα12 at 11 209 eV would clearly indicate the presence of Se. Studies of samples containing As, Se, and Pb require some care to unambiguously determine the different elements present. In many cases, simply changing the monochromator energy will not be sufficient to eliminate overlaps in fluorescence because of the energy ranges of the beamline. Thus, hard X-ray beamlines that typically operate at 10–15 keV may not be able to operate at energies much below 5 keV. For example, XFI investigation of organo-tin toxicology might be somewhat problematic on a hard X-ray beamline because the monochromator would excite all of the L-edges, with the tin LII primary fluorescence, Sn Lβ1 (3663 eV), substantially overlapping with the Ca Kα12 (3691 eV). Another noteworthy case is the proximity of the Zn Kβ (9572 eV) to the Hg Lα12 (9980 eV); while these are easily resolvable, Zn can be so abundant in biological samples that the tail of the Zn Kβ can add a background to the Hg Lα12 and confuse inattentive researchers, especially if simple binning is used.
Components of the beamline can give rise to contaminating signals that may confuse the analysis. Examples include iron, lead, copper, bromine, and antimony. As discussed above, lead is found in shielding and is second only to iron, which is present in a very large number of beamline components, including most vacuum-compatible X-ray windows fabricated from beryllium, the element of choice due to its low X-ray cross section. The authors have also encountered copper contamination arising from exposed wiring internal to a new array detector, which was subsequently returned to the manufacturer and the problem eliminated. Bromine at first seems a somewhat unlikely contaminant, but it is often present in plastics such as acrylics, because ethyl 2-bromoisobutyrate and ethyl 2-bromopropionate are used as polymerization initiators.106 Similarly, antimony compounds such as antimony trioxide (Sb2O3) or antimony triacetate are used as condensation catalysts in the production of poly ethylene terepthallate (PET).107 PET is most commonly found in plastic bottles used to contain drinks such as bottled water or carbonated beverages, and we note in passing that antimony leaching from PET is a potential source of exposure to compounds of this highly toxic element.108 PET is often used in plastic coverslips as sample supports in XFI, and if used for this purpose care must be taken to obtain antimony-free PET as the Sb Lα12 (3604 eV) and Ca Kα12 (3691 eV) overlap enough to make determination of biological calcium difficult.
2.13.2. X-ray Fluorescence Self-Absorption
The phenomenon known as fluorescence self-absorption is an attenuation of the X-ray fluorescence due to high X-ray absorption coefficient, and is only a problem for concentrated samples that are also thick. The common description of this phenomenon as “self-absorption” is somewhat inaccurate, and it could also be referred to as fluorescence thickness effects. Although thickness effects, including self-absorption, are discussed at length in section 2.5, we mention self-absorption briefly here because it represents a potential pitfall. With adequately dilute or thin samples, the X-ray fluorescence from the sample is proportional to the absorption coefficient μ(E), eq 3, which is the condition desired. In most cases, X-ray fluorescence self-absorption can be avoided with good experimental practice, especially with biological samples that are usually in the dilute limit. In some cases, however, self-absorption may be unavoidable, such as calcium from bone in sections of whole animals, and comparison with optically thin standards must be done with care.
3. Applications in the Study of Plant Physiology
Plants were used in many of the early XFI studies of living systems. While plants are interesting in their own right, early XFI studies were enabled by the fact that plants are highly resistant to radiation damage from the X-ray beam so that they often can be observed in a living state. Moreover, no protocols are required for their study, minimizing paperwork and eliminating ethical review, which is mandatory for animal and human tissue work. Applications of XFI to metal ion plant physiology have recently been reviewed at length by Sarret et al.,109 and for this reason we will only discuss selected topics to provide examples. For a more comprehensive review, the reader is referred to Sarret et al.109
3.1. Hyperaccumulating Plants
Of particular note in the field of plant metal ion physiology are those plants known as hyperaccumulators, which can assimilate very high levels of metals or metalloids from their environment. They are of interest with respect to the bioremediation method called phytoremediation,110,111 in which plants are used to sequester or modify an environmental contaminant, thereby alleviating an environmental problem without the need for excavating large amounts of soil or rock. The levels of accumulation can be quite extreme; for example, the New Caledonian tree Sebertia acuminata can have vivid blue-green latex due to its startlingly high nickel concentration of about 2 M.112,113 Other examples include the nickel hyperaccumulating penny-cress Thlaspi goesingense,114,115 the arsenic hyperaccumulating brake fern Pteris vitata,28,60,61,116 and the selenium hyperaccumulating two-grooved milk vetch Astragalus bisulcatus.117 Some tropical trees accumulate selenium in their tissues to a surprising extent, including the brazil nut tree (Bertholletia excelsa), which concentrates l-selenomethionine in the nuts,118 and the Coco de Mono or Monkey Pot tree (Lecythis ollaria).119−121 The nuts of the Coco do Mono tree, also known as Paradise Nuts, have a very high l-selenocystathionine content,122,123 and are a noteworthy source of human toxic exposure to selenium, possibly due to their agreeable taste. Kerdel-Vegas has described a number of case histories of people who consumed Coco de Mono nuts, nearly all of whom were warned by locals of dire consequences but nonetheless ate the nuts, with the unpleasant symptoms of selenium intoxication as their reward.120 Because the first and one of the best-studied hyperaccumulator plants is A. bisulcatus, we will initially discuss XFI of selenium hyperaccumuators.
3.2. Selenium Hyperaccumulators and Accumulators
Selenium hyperaccumulation was first noted by Moxon in the 1930s,124 while investigating the cause of the livestock disease called the “blind staggers” or “locoism”. The blind staggers are now thought of as several unrelated diseases in which an unsteady gait and seeming blindness are apparent. In the North American plains, a common cause may be selenium intoxication from ingestion of selenium hyperaccumulating range plants, including certain Astragali (in particular A. bisulcatus), Aster, Stanleya, and Oonopsis.124 While selenium intoxication by feeding animals A. bisulcatus does produce the symptoms of the blind staggers,125 O’Toole et al. have criticized the hypothesis that the bind staggers are due to selenium, but they also comment that it is not yet discredited.126 The two-grooved milk vetch A. bisulcatus can take up selenium from the soil;127 it is accumulated as a mixture of Se-methyl-seleno-l-cysteine and γ-glutamyl-Se-methyl-seleno-l-cysteine.128 These compounds are generated from seleno-l-cysteine by a specific selenocysteine methyltransferase.129A. bisulcatus is also known as the poison-vetch and by cattle ranchers as “locoweed” due to its association with the blind staggers. The first XFI study of any hyperaccumulating plant was of A. bisulcatus, by Pickering et al.,50 who showed that the plant stored different forms of selenium in leaves of different ages. Newly grown or young leaves were found to contain high levels of Se-methyl-seleno-l-cysteine, while older mature leaves contained predominantly selenate. Leaves of intermediate age were found to contain a mixture of these species. Pickering et al. also reported that the overall selenium content of the leaves decreased with their age.50 Some Astragali are well-known for their malodorous nature, which is due to loss of volatile selenium compounds, among which dimethyldiselenide is prominent.130 Pickering et al. have also reported that there is correspondence between sulfur and selenium chemical forms in old and young A. bisulcatus leaves.131 It has long been thought that selenium hyperaccumulation might be a defense mechanism aimed at insect herbivores, and it seems likely that the production of volatile selenium compounds such as dimethyldiselenide130 plays a role in this. The mechanism by which dimethyldiselenide is generated from Se-methyl-seleno-l-cysteine is still unknown, but Pickering et al. noted that both are relatively nontoxic as compared to selenate, the other major selenium species present. These workers also commented that the young leaves in which these less toxic forms predominate are those that the plant can least afford to lose, but that the foul smell of the volatile selenium species might serve to redirect herbivorous insects to the older, less pungent, but more toxic leaves. Freeman et al. have expanded this early work, and also studied a different selenium hyperaccumulator, a mustard known as prince’s plume (Stanleya pinnata).132 They used XFI with improved spatial resolution to show that organic selenium was concentrated in the trichomes (leaf hairs) of plants thought to be A. bisulcatus. These workers also harvested large numbers of trichomes, and employed conventional analysis to show that this was Se-methyl-seleno-l-cysteine and γ-glutamyl-Se-methyl-seleno-l-cysteine.132 Se-methyl-seleno-l-cysteine was found to be present in the vascular tissues of a S. pinnata young leaf petiole as well as in guttation fluid.132,133 These results contrast with the results of Pickering and co-workers26 (also including unpublished data), who did not find organic selenium in the trichomes but did observe selenium within cells, and found high levels of calcium in the trichomes (Figure 10). The two groups used different culture methods; Freeman et al. used a commercial horticultural growing mix,132 while Pickering et al. used hydroponic growth media.50,131 The plants were also harvested from different locations; Freeman et al. obtained theirs from Fort Collins, CO, and Pickering et al. from Big Hollow, WY, and from Saskatoon, Saskatchewan, Canada. Moreover, Freeman et al. did not observe calcium in the trichomes of plants identified as A. bisulcatus,132 but did for the trichomes of the nonhyperaccumulator Indian mustard, Brassica juncia. Given that large numbers of related vetches of similar appearance exist, it seems plausible that one group or other misidentified A. bisulcatus, and that two discrete species were in fact studied. At present, more work is required to resolve this discrepancy. More recently, XFI has been used to examine Astragalus roots134,135 and two Se-resistant herbivorous moths, one of which was parasitized by a wasp.134,136 XFI of the adult moths, larvae, and wasps showed that each accumulated organo selenium compounds, with no apparent ill effects. The studies of roots showed that elemental selenium producing endosymbionts may be able to affect selenium speciation in hyperaccumulators.134,135
In contrast to the selenium hyperaccumulators, which have special selenium biochemistry, selenium uptake by nonhyperaccumulating species appears to be solely as a consequence of the similarities between sulfur and selenium chemistry.137 Thus, selenate uptake is thought to occur through sulfate transporters, and selenate reduction is thought to occur via the normal route for sulfate, involving the ATP sulfurylase/APS reductase pathway.137 In nonhyperaccumulating plants, a predominant form in tissues is often l-selenomethionine.138 While a number of studies have been directed at phytoremediation,138 XFI has been used to investigate the physiology of selenium uptake and enrichment in putative biofortified food plants,139,140 a particularly interesting application may be ordinary crop plants141 grown on naturally selenium-rich areas as a potential selenium supplement for parts of the world that are selenium deficient.142
3.3. Arsenic Hyperaccumulators and Accumulators
Arsenic accumulation and hyperaccumulation is rare among plants, and is only known for a handful of ferns, of which Pteris vittata is the most noteworthy.28,60,61 Pickering et al.28 used chemically selective XFI to show that the chemical form present in the transport vessels was arsenate (a mixture of [HAsO4]2– and [H2AsO4]− at physiological pH), which was transformed to arsenite (As(OH)3) in the tissues (Figure 29). They also clearly detected a small amount of thiolate coordinated As(III),28 which had been previously observed in bulk measurements.60 Pickering et al. hypothesized that this was an intermediate in the reduction of the As(V) arsenate to arsenite.28 By enhancing the intensity threshold, Pickering et al. observed a line of arsenite following the periphery of the arsenate-containing transport vessels,28 which suggested transport of arsenite from the photosynthetic tissues to the rest of the plant. The same species of fern was subsequently studied using XFI by Hokura et al.,143 who used large area imaging to view substantial parts of the plant. Recently, a different hyperaccumuating fern, the gold dust fern Pityrogramma calomelanos var. austroamericana, has been studied using XFI by Kachenko et al.144 These workers also observed arsenate in the transport vessels, and detected thiolate coordinated species in the tissues. The clear physiological picture that is apparent from these studies is that arsenate is taken up from the soil by the roots and transported through the vascular tissue from the roots to the fronds (leaves), where it is reduced to arsenite and stored at high concentrations. The arsenic-thiolate species surrounding veins may be intermediates in this reduction. The life cycle of the fern145 can be described in terms of two discrete generations; the familiar leafy fern is the diploid sporophyte generation that generates haploid spores through meiosis. These spores are contained in capsules known as sporangia, which in turn are localized within spore dispersal regions called sori on the underside of mature fronds.145 Following dispersal and germination, the spores develop into small free-living photosynthetic haploid gametophyte plants,145 only a few millimeters across and one cell thick throughout most of their structure, which develop sperm within antheridia and egg cells within archegonia. Fertilization results in a new sporophyte plant, which grows directly through the gametophyte.145 In addition to examining the sporophytes, Pickering et al. also examined the sporophyte sporangia and gametophytes of Pteris vittata.28 In both cases, the reproductive tissues, or the areas in which reproductive tissues would develop, were found to be free of arsenite. Figure 30 shows the XFI data of the P. vittata sporangia and a typical gametophyte. Thus, in the sporophyte sori and vicinity, arsenite is excluded from the spores and the sporangia, but it is preferentially concentrated within the paraphyses (Figure 30), sterile hair-like structures adjacent to but not directly associated with the sporangia. Arsenite also accumulates within the pseudoindusium, the reflexed margin of the lamina that partially covers and protects the sori. Exclusion of toxic arsenite from the plant reproductive tissues may be needed to protect these from genetic damage.28 The images of the gametophyte also give clues to the subcellular localization of arsenite. Plant cells typically have a large centralized vacuole with the majority of the cytoplasm contained in the periphery;145 the images clearly show arsenite localized within the central cellular vacuole. Arsenate levels are very low in the gametophyte and, as suggested by Pickering et al., may be confined to the numerous plant cell Golgi bodies, but further high-resolution imaging is needed to determine this.28
Figure 29.
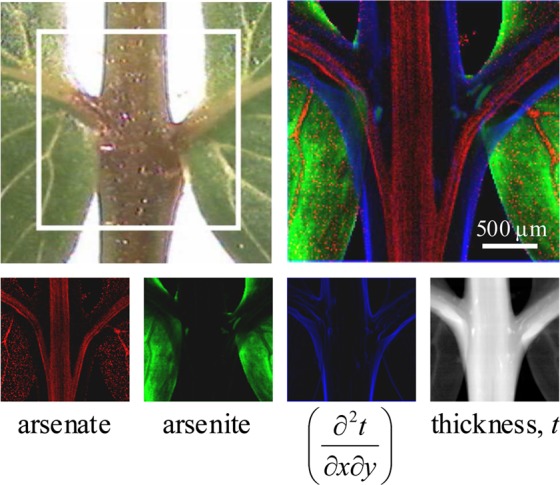
Chemically specific X-ray fluorescence imaging of Pteris vittata, showing the presence of arsenate in the transport vessels, and biotransformation to arsenite in the tissues.28 The figure shows XFI of the highlighted area in the optical micrograph, showing a central rachis (stem) with two branching rachilla supporting the pinnae (leaves). Arsenate is depicted in red, arsenite in green, and the derivative of the transmittance-derived thickness t in blue. The derivative of t is shown rather than t itself so as to better indicate the tissue periphery. Adapted with permission from ref (28). Copyright 2006 American Chemical Society.
Figure 30.
X-ray fluorescence imaging of Pteris vittata reproductive tissues.28 In the figure, (a), (c), and (e) show optical micrographs, and (b), (d), and (f) show XFI images with arsenate in red, arsenite in green, and thickness (transmittance) in blue. The sporangia of the sporophyte generation are shown at different resolutions in (a)–(d) and the gametophyte in (e) and (f). The future site of archegonial development is shown in (f) (ar), and several rhizoids (rh) are visible in (e). Adapted with permission from ref (28). Copyright 2006 American Chemical Society.
3.4. Cadmium Hyperaccumulators and Accumulators
Cadmium is generally thought of as a toxic element and has a demonstrated functional role in only a single case, in a carbonic anhydrase of the marine diatom Thalassiosira weissflogii.146 Cadmium hyperaccumulators are included here as a further example, primarily because they provide an example of an element where relatively inconvenient X-ray energies are involved. The cadmium K-edge at 26 714.0 eV is high for a conventional XFI experiment (section 2.8), although still within the reach of many beamlines, and the LIII and LII-edges at 3537.6 and 3728.0 eV, respectively, are close to our lower bound in X-ray energy (section 2.9). The high X-ray energies of the K-edge mean that short core-hole lifetimes give broad near-edge features,147 which in turn means that any chemical differences give changes in the spectra that are quite subtle and difficult to exploit. Conversely, the LIII and LII-edges are at low energies,147 and so the problems we have previously discussed associated with beam attenuation will be prominent. Moreover, the Cd Lα12 and Lβ1 fluorescence lines at 3133 and 3317 eV, respectively (the primary lines of the LIII and LII edges, respectively; Figure 8), are inconveniently close to the Kα12 of biologically common potassium at 3313 eV. Cadmium is therefore an element that poses some challenges for XFI, but despite this the method has been used to provide important information on cadmium in both hyperaccumulating and nonhyperaccumulating plants.
Early work on the nonhyperaccumulator Brassica juncea identified that Cd was bound by sulfur donors in the root and by hard ligands such as oxygen or nitrogen in the xylem sap with transpiration-driven mobilization into the leaves.148 This work also conclusively showed that Cd was localized in trichomes, but rather than XFI these investigators employed a combination of109Cd autoradiography to probe localization and Cd K-edge XAS to probe the chemical form.148 Subsequent XFI combined with μ-diffraction of the nonhyperaccumulating tobacco plant (Nicotiana tabacum L.) showed that when Cd2+ was given in the presence of excess Ca2+, Cd-containing crystalline deposits were exuded from the tips of trichomes.149 The accumulator willow (Salix sp.) has been explored as a potential cadmium phytoremediator; XFI has been used to show accumulation of cadmium at the tips of the serrations in leaves, and in the bark.150,151 Tian et al.152 have used both XFI and LA-ICP-MS (section 5.2) to compare hyperaccumulator and nonaccumulator ecotypes of Sedum alfredii. This study showed that the hyperaccumulator S. alfredii ecotype accumulates cadmium in parenchymal cells, especially in the stems, but that cadmium was restricted to the vascular bundles in the nonhyperaccumulating ecotype.152 Tian et al. also showed a distinctive correlation between the levels of cadmium and calcium, but not cadmium and zinc, in the hyperaccumulator,152 and used EXAFS spectroscopy to show that cadmium was bound by oxygen donors.152 These workers suggested that cadmium metabolism in the hyperaccumulating S. alfredii is associated with calcium pathways, and that the metal is bound with oxygen donors from malate.152 Elegant studies using Cd K-edge XFI of the cadmium hyperacculator Arabidopsis halleri spp. gemmifera with an incident X-ray energy of 37 keV also clearly showed cadmium in the trichomes.84,85 XFI indicated that Cd was localized in highly specific regions within the trichome, bilaterally disposed, straddling the stem of the Y-shaped trichomes, just below the branch.85 Moreover, the Cd was colocalized with manganese and zinc, but not with calcium, and μ-XAS showed that, as in other species, Cd was bound to hard ligands such as oxygen or nitrogen.85
Pickering et al. have suggested what may be a general rule,28 which is that the chemistry underlying hyperaccumulating plants, or plants otherwise adapted to high levels of metal or metalloids, lacks sulfur coordination but instead shows binding by oxygen or nitrogen.153,154 This appears to be true even for chemical species that have an inherently high affinity for thiolates, and chalcogenides in general, such as the arsenite stored in P. vittata.28 Conversely, sulfur is often involved when plants that are poorly adapted to toxic metals are exposed.155−157
4. Applications in the Health Sciences
XFI is finding increasing application in the field of health science. Examples include characterization of disease and dyshomeostasis, as well as molecular therapy and correlation with other imaging modalities, with XFI providing fresh insights into such critical topics as heavy metal toxicology, Alzheimer’s disease, amyotrophic lateral sclerosis, stroke, and multiple sclerosis. We will review pertinent examples taken from each of these fields.
4.1. Mercury Toxicology
The compounds of mercury in general are more toxic than those of any other nonradioactive heavy element (Figure 31). Despite this extreme toxicity, human and environmental exposure is surprisingly widespread, with significant sources ranging from dental amalgam to predatory marine fish. Moreover, despite widespread human exposure and health concerns for many populations worldwide, there remain many unanswered questions about the mechanism of action of mercury and its compounds. As a further consequence of its high toxicity, mercury levels in biological tissues are usually very low, making this a difficult element to study. Added to this, the mercury LIII near-edge shows only subtle spectroscopic variability with chemical form. These issues combine to effectively make studies of mercury toxicology particularly challenging. Two broad chemical forms are of interest, the inorganic form, exposure to which might arise from industrial activities or from dental amalgam, and the organometallic form, found in marine fish, which is known to be predominantly monomethylmercury covalently bound to a single aliphatic thiolate (probably l-cysteineate).158 Methylmercury compounds are some of the most dangerous neurotoxicants because they can cross both the blood–brain barrier and the blood–placental barrier, accumulating in the brain and resulting in severe damage. Indeed, while under some circumstances the placenta can provide protection against other toxic metal species, such as thallium,159 the consequences of in utero exposure are particularly disastrous for the developing fetus. The toxic potential of mercury is demonstrated most graphically by large-scale catastrophic acute mercury poisonings that have occurred in Japan and in Iraq.160 These tragic incidents revealed the insidious and debilitating nature of mercury poisoning with its creeping but deadly neurotoxicity, particularly in fetuses and young children.160−163 While adults can be severely impacted by methylmercury exposure, effects on the developing fetus are devastating, and can result in microcephaly, cerebropalsy, seizures, mental retardation, blindness, paralysis, and other very severe consequences.164 The extreme toxicity of methylmercury to vertebrate fetuses was unknown until the Minamata mass-poisoning, where many severely handicapped children were born to relatively healthy mothers.163 Numerous additional consequences of mercury poisoning include central nervous system defects, arrhythmias and cardiomyopathies, and kidney damage. Mercury inhalation can result in necrotizing bronchitis and pneumonitis that in turn can result in respiratory failure.160,161 Mercury can also act as either an immunostimulant or an immunosuppressant, depending on the nature of the exposure, leading to a number of pathologic consequences.160,161 One of the most perplexing phenomena in the toxicology of methylmercury species is that there can be significant latency between administration and onset of toxic symptoms,165 which in humans can be as long as 150 days.166 The cause of this latency remains one of the major unanswered questions in this field, and while different possible mechanisms have been suggested,165 at present the cause remains unknown. The toxicology of mercury and methylmercury has been studied by XFI of larval stage zebrafish as an animal model and of human brain samples.
Figure 31.
A comparison of the relative toxicities of some heavy elements that pose human health problems. Toxicities are expressed as the reciprocal of values of the LD50 (rat, oral, mg/kg) taken from MSDS forms (Sigma-Aldrich, Acros Organics) for a common toxic inorganic form of each metal or metalloid. The compounds used were as follows: Na2CrO4, NaAsO2, Na2SeO3, CdCl2, SnCl2, HgCl2, and PbCl2.
Larval zebrafish (Danio rerio) are increasingly used as a model organism to study embryonic development. Their many advantages include high fecundity, ease of maintenance, and well-characterized developmental stages. They are easily treated with exogenous agents by simple addition to their habitat water. Zebrafish are used increasingly in vertebrate toxicology167 and by the pharmaceutical industry for de facto first-pass drug screening.168 Korbas et al. used XFI of both anaesthetized larvae and prepared sections to show that fish treated with methylmercury-l-cysteinate accumulate Hg preferentially in the outer layers of eye lens (Figure 32).89 This compound was selected in initial studies because of its stability in aqueous solution, so that the larvae were presented with essentially a single molecular entity. The comparison of the XFI of anaesthetized and fixed sectioned specimens indicated that no large-scale redistribution of the metals had been caused by the sample preparation.89 These workers also observed differential mercury accumulation in the brain and various other organs. Subsequent work studied the dynamics of methylmercury accumulation and redistribution in the lens following embryonic and larval exposure.169 The accumulation of mercury in the lens continued well after removal of the fish from treatment solutions, thus significantly increasing the postexposure loading of mercury in the lens. This indicated that mercury is redistributed from the original target tissue to the eye lens, identifying the developing lens as a major sink for methylmercury in early embryonic and larval stages. Subsequent comparative XFI work revealed substantial differences in the tissue-specific accumulation patterns of mercury in zebrafish larvae exposed to four different mercury formulations in water.51 For nonthiolate coordinated mercury compounds, multiple species are expected to be present in solution, and Korbas et al. calculated the expected species distributions for the four reagents added to the fish culture water (HgCl2, CH3HgCl, Hg(l-cysteineate)2, and CH3Hg-l-cysteineate). Thus, for 1 μM HgCl2 solutions, Korbas et al. calculated that solutions would consist of 90.0% Hg(OH)2, 9.5% Hg(OH)Cl, 0.3% HgCO3, and 0.2% HgCl2. XFI showed that methylmercury species exhibited higher mercury uptake and targeted different cells and tissues than inorganic mercury, revealing a significant role of speciation in cellular and molecular targeting and mercury sequestration. For methylmercury species, the highest concentrations of mercury were observed in the surface of the eye lens, independent of the formulation ligand. For inorganic mercury species, the olfactory epithelium and kidney accumulated the greatest amounts of mercury.51 However, with Hg(II)-bis-l-cysteineate, uptake was significantly decreased. This work showed that the common differentiation between organic and inorganic mercury is insufficient to determine the toxicity of various mercury species. To determine whether physical chemical factors alone could account for the chemical sensitivity observed with XFI, Korbas et al. estimated diffusion coefficients and membrane permeabilities for all of the significant solution species present for each treatment group.51 They concluded that a simple passive diffusion model of the compounds across the lipid part of the cell membranes cannot explain the differential accumulation of inorganic and organic mercury observed by XFI, so that other processes such as active transport must play a role. Most recently, high-resolution XFI has been used to study the uptake and localization of methylmercury species in larval zebrafish photoreceptors.15 This work addressed a long-standing assumption, which is that the visual disturbances that are a known symptom of organo-mercury exposure were due to damage to the visual cortex of the brain. The XFI study showed that photoreceptors take up methylmercury compounds directly, indicating an additional possible mechanism by which methymercury species might cause visual disturbances.15
Figure 32.

XFI of mercury in methylmercury-l-cysteinate treated zebrafish larvae. Part (a) shows an intact tricaine-anesthetized larva showing localization of Ca, Zn, and Hg plus an optical micrograph (opt); the developing fish otoliths are prominent in the Ca image. Part (b) shows a typical head section displaying the same chemical elements, plus a histologically stained optical micrograph together with a line rendition of the stained micrograph showing the cornea (co), eye-lens (el), retina (re), optic nerve (on), and the fore-brain (br) (diencaphalon). Intensity scales are shown with linear scaling (lin), and with logarithmic scaling (log) to allow better visualization of the lower intensities. Part (c) shows a high-resolution image of the surface regions of the eye lens indicating that the mercury is localized in a layer beneath the lens epithelium (ep). Data were collected on SSRL 9-3 (a),89 APS 20ID (b),51 and APS 2ID (c).15
XFI has also been used in conjunction with XAS to study samples of human brain following poisoning or environmental exposure.170 Samples from individuals poisoned with high levels of methylmercury species showed high levels of both mercury and selenium colocalized in the gray matter, with a substantial fraction of nano-particulate mercuric selenide (HgSe) together with inorganic mercury and methylmercury bound to organic sulfur. Mercury exposure did not perturb observed organic selenium levels. Individuals with a lifetime of high fish consumption showed much lower HgSe levels and dominant methylmercury-l-cysteineate. These results provide the first evidence that HgSe formation may act as a detoxification mechanism in humans, at least under conditions of acute exposure.
XFI has provided evidence that may challenge the perceived benign nature of mercury amalgam dental restoratives. Loss of Hg and biotransformation of mercury from amalgam fillings has been observed,171 and Harris et al.172 have used XFI to show that Hg can be detected several millimeters away from the site of the filling, associated with dental tubules, and that the chemical form of Hg had also been altered.172 Their work also identified potential routes of exposure to Hg from fillings, as Hg was localized in the calculus (suggesting it may at one time have been in a mobile form within the mouth) and highly innervated areas of the tooth with circulating blood flow, potentially affording a route to enter the bloodstream.
To date, almost all of the studies on mercury toxicology to date have employed acute mercury exposure, in which organisms experience relatively high doses for a short time. However, most human and environmental exposure is chronic in nature, involving much lower doses over considerably longer exposure times. Whether the lessons learned from acute exposure can be translated to the chronic case remains unclear. Irrespective of this, the high sensitivity and specificity of XFI will ensure that it will continue to play an essential role in understanding the risks posed by this most problematic and enigmatic of the toxic elements.
4.2. Arsenic Toxicology
As compared to mercury, arsenic is an element that is very well suited to study by XFI, with a K-edge energy convenient for most beamlines and a rich and highly structured spectroscopy. Despite its reputation as a deadly poison, it is very much less toxic than mercury (Figure 31), which means that rather higher levels can be tolerated, with a consequence that it is much easier to study. Also, in contrast to mercury, at least insofar as acute toxicity is concerned, the mechanism of arsenic toxicity is relatively well understood, so that there are fewer fundamental questions that need to be answered. Arsenic, as the trivalent arsenite, is the most toxic environmentally common form of the element. It is often written as [AsO2]−, although no such molecular entity actually exists,173 and more correctly as As(OH)3 (at physiological pH). Similarly to mercury, As(III) has a high affinity for thiols; in acute toxicity, arsenite acts as a respiratory poison by chelation of essential dithiols and in particular reduced α-lipoic acid in the pyruvate dehydrogenase complex to form a stable six-membered chelate ring. Arsenic-based chemical warfare agents owe their lethality to the same mechanism.174 Chronic arsenic exposure, for which the mechanisms of action are comparatively poorly understood, poses enormous human health problems for communities in developing countries that consume contaminated drinking water. Such exposure results in arsenicosis, a syndrome featuring characteristic patterns of hypermelanosis and hyperkeratosis, vascular and endocrine pathologies, several types of cancer, and increased mortality.175 While the biochemical mechanisms underlying arsenicosis are still substantially unknown, arsenic is a known carcinogen; there is extensive literature that links human exposure to inorganic arsenic with a variety of cancers, including lung cancers,176 skin cancers,177 and urinary bladder carcinoma.178 Moreover, arsenic may act as a cocarcinogen in urinary and bladder cancer with cigarette smoking178,179 and in skin cancer with UV light exposure, even at low levels.180,181 Several possible modes of action may be involved in arsenic carcinogenesis,182 but the underlying mechanisms remain poorly defined. Probably in excess of 100 million individuals worldwide have severe health impacts from arsenicosis, with the crisis in Bangladesh representing an extreme example. There are thus many areas of arsenic toxicology in which XFI could make a significant impact, although to date only a few XFI studies of arsenic in animal systems have been reported, including some studies of insects,62 with most vertebrate data limited to such samples as fur and feathers.183 Insofar as studies relevant to human health are concerned, Munro et al.184 used XFI and μ-XAS to investigate the fate of arsenite and arsenate in cultures of human liver carcinoma cells (HepG2) and observed accumulation in the euchromatin region of the cell nucleus following arsenite exposure, which suggested arsenic targeting of either DNA or proteins involved in DNA transcription. μ-XAS and XAS of arsenite-treated cells showed predominance of arsenic sulfur-coordinated species, supporting the hypothesis that the mechanism of action may concern nuclear proteins, that this occurs through arsenic’s affinity for chalconide donors, and that this nuclear interaction may be a key factor in arsenic-induced toxicity.
Arsenic and selenium have long been known to have intertwined toxicities,185,186 wherein a lethal dose of arsenic can be counteracted by an equal and otherwise lethal dose of selenium.186 The mechanism of this surprising antagonism was found to be the formation of a novel arsenic selenium species, the seleno-bis(S-glutathionyl) arsinium ion,187 which is formed in the liver187 and possibly in erythrocytes188 and excreted in the bile,189 probably through the MRP2 transporter.190 The organic selenium compound 1,4-phenylene bis(methylene)selenocyanate and vitamin E have both been shown to block the cocarcinogen activity of arsenic in mice with ultraviolet irradiation, although only vitamin E had beneficial effects with ultraviolet irradiation in the absence of arsenic.191 XFI has been used to probe this protective effect of selenium; Burns et al. found elevated levels of arsenic were present in hair follicles and epidermis of mice treated with both arsenite and ultraviolet light, yet when 1,4-phenylene bis(methylene)selenocyanate was given, arsenic was entirely absent in these tissues, with only diffuse low levels in the liver.192 Arsenic K near-edge μ-XAS of the liver was consistent with the presence of the seleno-bis(S-glutathionyl) arsinium ion, suggesting that the mechanism underlying the protective effect of selenium in arsenic/ultraviolet cocarcinogenesis was through removal and biliary excretion of arsenic.192
An intriguing example in which XFI provided critical information is that of Phar Lap. Phar Lap was a successful and famous racehorse that ran between 1929 and 1932, gaining acclaim for a spectacular series of wins. He became a cultural icon of the Australian sporting community, but while in California he died suddenly and painfully following a spectacular win in Mexico. While poisoning was suspected, necropsies were inconclusive, and Phar Lap’s death remained a mystery for nearly eight decades. Phar Lap’s hide is preserved on display at Melbourne Museum, and a recent XFI and μ-XAS study used hair taken from the preserved hide to partly resolve the mystery.193 Because of Phar Lap’s symptoms, arsenic was suggested early as a possible poison. However, providing conclusive evidence of this proved difficult because arsenic can be used in taxidermy and in the 1930s routinely was administered as a tonic to race horses, in the form of Fowler’s solution. Kempson and Dermot used XFI to study Phar Lap’s hair, employing four samples that included intact bulb and root tissue.193 Longitudinal scans indicated low arsenic levels within the hair but with an abrupt rise in the subcutaneous region followed by decrease in level toward the root tip, consistent with a large dose followed by a slow decay due to metabolism and excretion.193 Kempson and Dermot also detected external arsenic that had likely been applied during taxidermy, but at much lower levels than that contained within the hair. The subcutaneous arsenic-rich regions of the hair were shown by μ-XAS to contain As(III) coordinated by three thiolates,193 which would be expected if the poison was administered as arsenite or arsenic trioxide. The study showed that Phar Lap had received a sudden and substantial dose of trivalent arsenic that was likely responsible for his death, making the case for poisoning stronger, although exactly who administered the poison will likely remain a mystery.193
4.3. Iatrogenic Toxic Metals
In rare cases, harmful effects from heavy metal ion intoxication have resulted from medical treatment, which is known as iatrogenic, or physician induced, exposure. We note in passing that mercury exposure from dental amalgam, considered in section 4.1, constitutes a potential iatrogenic exposure to a toxic metal, although the negative consequences of this are still a matter of debate, so this may not be the best example. One example in which XFI has provided definitive information is that of the gadolinium contrast agents. Trivalent gadolinium magnetic resonance imaging contrast agents have many excellent properties, including enhancing water T1 relaxation through interaction with the strongly paramagnetic Gd(III).194 These Gd(III) agents are clinically presented in the form of chelation complexes, and are in general very well tolerated with only a very low incidence of anaphylaxis.195 Indeed, they are considered better tolerated than the very commonly used iodinated X-ray CT contrast agents.195 The Gd drugs are typically eliminated in the urine with a half-life of only 80 min in healthy patients.196 However, in patients with kidney insufficiency, elimination is prolonged and with chronic renal failure can be between 30 and 130 h.196 In such patients, repeated administration of Gd contrast agents may trigger nephrogenic systemic fibrosis (NSF).197 NSF is a very serious syndrome characterized by red skin areas or plaques that successively develop to painful thickened skin with a wood-like texture developing over several weeks. The fibrosis may occur many months after the use of the contrast agent and typically starts at the extremities, may spread to the trunk, and may also progressively affect skin, joints, eyes, and internal organs.198,199 XFI has been used to definitively identify Gd deposition within biopsied tissue samples from NSF patients.200,201 Moreover, George et al. have determined that the deposited Gd was no longer bound to the original contrast agent chelator, and was instead within a Na-, P-, and Ca-rich region,201 which was spectroscopically consistent with a GdPO4-type chemical form.200,201 NSF is thus thought to be associated with release of Gd(III) from the chelator, and recommendations as to choice of contrast agent are now in place based on perceived in vivo stability of the various chelators. Optimization of a high-affinity Gd(III)-specific chelator using criteria similar to those developed for other metals202,203 might prove useful in this area.
4.4. Neurodegenerative Diseases
Recent advances in understanding the role of trace metals in neurodegerative diseases have been gained through the application of XFI. A good general overview of XFI in neurodegenerative diseases has recently been provided by Bourassa and Miller,204 and the potential of XFI has been eloquently discussed by Popescu, Nichol, and co-workers.205,206
There remains much groundwork to be done in this area, including determination of elemental distributions, concentrations, and chemical species within cultured neurological cells and from ex vivo tissues. Examples of the type of groundwork being carried out come from Ortega and Carmona et al.,207,208 who have mapped the elemental distribution in dopaminergic cells and neuritic processes at high spatial resolution (90 nm).207,208 Their results have identified iron-rich structures collocated with neurovesicles,207 as well as elevated Cu, Zn, and Pb within neuritic processes, which, excluding the latter, may be intrinsic components related to the protein machinery involved in these outgrowths.
The infectious miss-folded form of the prion protein is well-known as the cause of fatal neurodegenerative prion diseases,209 including Creutzfeldt–Jakob disease in humans and bovine spongiform encephalopathy, commonly known as “mad cow disease” in cattle. The normal function of the properly folded nonpathogenic form of the prion protein has been the focus of debate. Prion protein is highly expressed in the brain and central nervous system and has been proposed to have a role, at least in part, in regulating trace metals.210 Pushie and co-workers have used XFI to map the trace metal distribution in the brains of transgenic mice expressing different levels of the prion protein, comparing a prion knockout strain, wild-type, and a prion overexpressing strain.211 The levels of iron, copper, and zinc were all found to be altered upon changing prion expression level, although their overall localization within the brain was not altered. Pushie et al. also used immunohistochemical staining in combination with XFI to reveal that regions of the brain showing pronounced differences in prion expression mirrored relative changes in metal levels, although the changes in metal ion levels were more pronounced and widespread than the differences in detectable prion expression alone.211 In particular, XFI indicated dramatically elevated copper levels in periventricular regions.211 The extent to which metal ions play a role in the development and pathology of neurodegenerative prion diseases is at present unknown; however, the XFI results clearly indicate a role for prion protein in the regulation of specific metals within the central nervous system. This in turn raises the possibility that prion levels might modulate the progression of diseases in which altered metal homeostasis is thought to play a pathogenic role such as Alzheimer’s, Parkinson’s, and Wilson’s diseases, and disorders such as hemochromatosis.
Parkinson’s disease (PD) is characterized by the loss of dopaminergic neurons from the substantia nigra. Popescu et al.13 have used XFI to characterize the iron accumulation associated with PD in post mortem autopsy sections from human brain, confirming earlier investigations by alternate methods such as magnetic resonance imaging. Indeed, the major point of this work was to present rapid scan XFI as a new method for investigation of neurodegenerative disease, as the findings were essentially confirmatory in nature, although they were at rather higher spatial resolution than previous work. High-resolution XFI has recently been reported for small regions of PD brain, targeting specific brain regions that are known to be vulnerable.212 This work compared the surviving neurons in normal and PD brains from the substantia nigra and locus coeruleus, and employed both XFI and conventional methods to show that both copper and copper transporter protein 1 were substantially reduced in PD, but that copper-containing superoxide dismutase was not.212
Alzheimer’s disease (AD) is associated with the formation of plaques that are known to accumulate trace metals; however, some animal models of disease have shown significant differences in the levels of detectable metals accumulated. The ability of XFI to directly measure the total trace metal content associated with plaques can clarify ambiguities that may arise through alternative metal detection methods, such as the use of metal-specific fluorescent probes. Miller and colleagues have pioneered synchrotron-based imaging investigations of AD tissue, using a combination of Fourier transform infrared (FTIR) and XFI.213 Their XFI results have confirmed the colocalization of elevated levels of Fe, Cu, and Zn within human plaques associated with AD214 and have also identified significant differences in the extent of metal accumulation between several mouse lines used in AD research.215 Other examples of the use of XFI include investigation of Zn distribution in the hippocampus in relation to memory loss and delayed neurodegeneration associated with stroke,216 epilepsy,217−220 and mechanical brain injury.221
4.5. Pharmacological Applications
The ability to characterize elemental distributions and chemical forms under conditions of dyshomeostasis as well as to track the uptake of drug and probe molecules is particularly attractive in cancer research. Typical approaches use metal-labeled drugs or other tagged molecules that are anticipated to be preferentially taken up by the cancerous cells, often by dint of some facet related to the cancerous cell’s growth or nutrient requirements; the targeted location within the tumor or tumor cells can be identified through tracking the tagging element.222 XFI has been used to identify the localization of Gd and Pt within treated tumor cells,223 where incorporation of these heavy elements is ultimately anticipated to enhance neutron capture therapy and synchrotron stereotactic radiotherapy treatments. XFI tomography has been used to map the platinum distribution in DLD-1 colorectal cancer cell spheroids that had been exposed to platinum-containing anticancer drugs.224,225 This work showed for the first time that the anticancer agent can accumulate in the central hypoxic and necrotic region of the tumor spheroids, and that the charge on the complex can affect cellular uptake and modulate tumor penetration.224 Two Ru(III)-based drugs, NAMI-A and KP1019, are structurally similar but possess vastly different anticancer properties. NAMI-A has been shown to inhibit metastasis formation,226 whereas KP1019 shows efficacy in inducing apoptosis in primary tumors.227 Aitken et al.228 have investigated the cellular fate of both Ru-based drugs via intact single cell XFI and found that uptake of KP1019 appears to disrupt Fe metabolism. The KP1019-treated cells showed ruthenium apparently localized to the nucleus, which was consistent with its wide range of applicability against primary tumors, whereas NAMI-A did not show any significant Ru uptake into cells, consistent with its proposed extracellular level of action.228 Ruthenium anticancer drugs usually contain N-heterocyclic ligands to Ru, such as the bis-indazole coordination in the case of the drug KP1019, in which the two heterocycles bind axially with the pseudo-octahedral coordination being completed by four equatorial chloride ligands. One important question has been whether the Ru remains coordinated to the N-heterocycle in the cell. Recent XAS data229 clearly indicate that substantial chemical modification rapidly occurs in biological media, but these measurements are most sensitive to the loss of coordinating chloride and sulfur donors, so whether the heterocycle remains coordinated to the metal was not clear. This problem has recently been addressed in novel experiments using iodinated analogues of ruthenium anticancer drugs.230 By quantitative monitoring of both the ruthenium and the iodine X-ray fluorescence, colocalization of the two elements was clearly demonstrated,230 indicating that the Ru–N bonds likely remain intact. These results are expected to be important in future refinements of these promising anticancer drugs.
The selenium-containing drug ebselen [2-phenyl-1,2-benzoselenazol-3-one] is an antioxidant drug, which shows considerable promise in the treatment of stroke, with demonstrated reduced infarct volumes in focal ischemia.231 Ebselen has been postulated as an in situ mimic of glutathione peroxidase.232 XFI has been applied to ebselen-treated rodent neuroblastoma cells to develop an understanding of the mechanism of action of this novel drug.233 Selenium was localized in a specific region of the cell, which, through comparison with phosphorus and potassium localizations, was postulated to correspond to the cellular endoplasmic reticulum.233 Most notably, an ebselen-induced efflux of potassium, chloride, and calcium was observed in this study, changes that are characteristic of the induction of oxidative stress. Thus, almost paradoxically, the antioxidant effects of ebselen may not be a direct consequence of any antioxidant abilities of the drug itself, but rather may be due to induction of cellular antioxidant defense mechanisms through induction of an oxidative stress response.233
XFI has also been used to develop an understanding of the mechanism of action of some novel antibiotics to combat pathogenic Escherichia coli. The majority of urinary tract infections (UTI) are caused by what are known as uropathogenic E. coli.234 Orally administered gallium maltolate has been used to successfully treat UTI mice infected with a uropathogenic strain of E. coli. XFI showed that Ga was localized in the transitional epithelium of the bladder, which is a potential site of E. coli.235 These results indicate that Ga compounds may prove to be effective in antimicrobial therapy for UTI caused by uropathogenic E. coli.
As a final topic in this section, we turn to arsenical-based chemotherapy. Arsenic is best known as a poison, as we have discussed in section 4.2. Legitimate use of arsenic compounds as drugs has been extant since the advent of salvarsan in 1909.236,237 In more recent years, arsenic trioxide has become established as a highly effective treatment for acute promyelocytic leukemia, and at the time of writing there are over 100 clinical trials of arsenic compounds registered with the United States Food and Drug Administration.238 As discussed above, Munro et al. have used XFI to show that arsenite-treated human hepatoma cells present arsenic localization in the nucleus,184 and these workers have also used a combination of XAS and XFI to show that [CH3As(GS)2] and [(CH3)2As(GS)] (where GS is glutathione bound through its cysteine thiolate) were produced in the cells.239 There remains much uncertainty concerning the mechanism of action of arsenic drugs in chemotherapy,238 and, given the sensitivity of arsenic XAS as a spectroscopic probe,173 it seems very likely that applications of XFI will play an important role in understanding the mechanism of action of existing drugs, and in the development of new and more effective agents.
4.6. Selenium Biochemistry
Synchrotron XFI and spectroscopy has also been used to redefine our understanding of cellular selenium biochemistry, in part through characterization of subcellular selenium localization and speciation.240,241 A long-held belief has been that subcellular HSe– comprises the intracellular Se store; however, no evidence for this chemical form of Se has been found in human lung cancer A549 cells.242 A novel finding using the same cell system was that selenite treatment induced localized selenium deposits within cells, and that these were associated with an approximately 2-fold increase in colocalized copper.243 Despite the colocalization, no evidence was found for chemical bonding between the two elements.243 Bulk selenium EXAFS indicated the presence of Se–S and Se–Se backscattering, and μ-XAS of the highly localized selenium regions was mostly elemental selenium. Weekley et al. point out that the selenium levels were some 13-fold higher than the observed Cu levels, so that Se–Cu might not be observable in the presence of significant other types of bonding. Weekley et al.243 were not able to examine the Cu μ-XAS, so that while there is no evidence for it, a chemical association between the two elements cannot yet be ruled out. Weekley et al. also suggest that the copper accumulation might be a response to the toxicity of the selenium,243 although at the time of writing more research is required to elucidate the molecular basis of this colocalization. Weekley et al. have recently extended these cell culture studies with similar experiments on kidney tissues from rats given selenite supplements.244 In these tissues, a selenium and copper colocalization was observed, and the bulk of the selenium was found to be in species containing Se–Se bonding and no evidence for Cu–Se species was found, just as with the previous cell culture experiments,243 where Cu was predominantly bound to sulfur and nitrogen. Moreover, the expression levels of superoxide dismutase 1 did not change, so upregulation of this enzyme has no role in the observed copper accumulation.244 Selenium accumulation has been reported in neuromelanin deposits in the substantia nigra of human brain.245 In this study, XFI indicated iron-rich microdeposits associated with pigmented neuromelanin, with colocalized sulfur, calcium, manganese, copper, zinc, and selenium.245 Bohic et al. suggest that the increased selenium is likely to be associated with glutathione peroxidase.245 The results are interpreted in the context of neurological development, and, interestingly, these workers observe changes in elemental composition and in the sulfur XAS spectra of neuromelanin deposits with the age of the individual from which the brain sample was taken.245 However, post-mortem chemical changes associated with sample preparation (formalin and paraffin embedding) might distort the sulfur biochemistry (section 2.12).
XFI has been used to investigate the roles of selenium in spermatogenesis with a dramatic increase in selenium in late spermatids, with selenium concentrating at the luminal side of elongating spermatids.246 XFI of knockout mice lacking mitochondrial glutathione peroxidase 4 and lacking selenoprotein P showed no elevated selenium levels.246 Thus, selenium transport was found to be associated with selenoprotein P, and accumulation with mitochondrial glutathione peroxidase 4.246 In the spermatozoa, selenium was found to be colocalized with copper and iron, and primarily located in the mitochondria-rich sperm midpiece. These workers also estimated concentrations of selenium in the sperm cell head and the sperm cell midpiece to be 29 and 377 μM, respectively.246 These levels are considerably above what are normally considered as toxic levels of selenium, illustrating the ability of the sperm cells to handle these high levels.246
4.7. Pathological Organisms
New insight into the nutrient requirements of the erythrocyte pathogen, Plasmodium falciparum, associated with malaria, has been gained through elemental mapping of red blood cells during P. falciparum infection and may point to new biochemical avenues to combat malaria. Using a combination of XFI and ICP-MS, Marvin et al.247 have identified that the P. falciparum parasite induces massive Zn accumulation within the host erythrocyte.247 Zinc accumulation occurs late in the pathogen’s lifecycle, associated with replication of mitochondria. The accumulated Zn was integral for propagation of the P. falciparum pathogen, as depletion of free Zn through administration of a strong Zn2+ chelator halted parasite growth.
Significant areas of growth are in cellular and subcellular imaging. These areas will be particularly useful for identifying target organs and cellular fates of trace element containing drugs or probe molecules (i.e., cancer or tagged metabolites). For the foreseeable future, these experiments will be of benefit to informing better drug or target molecule design as well as improving treatments, while the high radiation doses, on the other hand, generally preclude large-scale experiments on live subjects. Using XFI imaging to identify changes in localization, concentration, and chemical speciation is readily obtainable, but a critical challenge for this field is extending the wealth of insight and information to the underlying biochemical processes and metabolic pathways from which they arise. As the field matures, additional methods will need to be integrated to provide the kinds of insight necessary for understanding the significance of XFI imaging data. One such example is the application of peripheral techniques to tissue or biological matrix XFI imaging, such as imaging of electrophoresis gels, where components can be separated and their trace element composition determined.248−250 These data can then be related back to elemental or chemical speciation maps to aid in identification and characterization.
5. Synergy with Other Methods
A number of other methods can provide information that is complementary to that available from XFI and vice versa. In this section, we will discuss these techniques with an emphasis on the information provided by XFI, and upon overlap and complementary information.
5.1. Magnetic Resonance Imaging
Magnetic resonance imaging (MRI) is a widely used and very successful clinical and research imaging technique. It has the significant advantage over XFI of being noninvasive and available for large samples, such as intact humans, with moderate spatial resolution. Metal ions can be detected by MRI but are sensed only indirectly and only when in paramagnetic oxidation states through their influence on water proton relaxation times. Hopp et al.251 and Zheng et al.252 have used XFI to calibrate the iron levels in MRI using susceptibility weighted imaging at 1.5 T and susceptibility mapping at 3 T, respectively. Susceptibility mapping estimates of iron correlate well with those determined by XFI251,252 in both cadaver brain and ferritin phantoms. This calibration provides an excellent example of translational research with direct impacts relevant to methods for diagnosis of human diseases.
5.2. Mass Spectrometry Imaging
Mass spectrometry imaging (MSI) is an increasingly used family of techniques that are capable of mapping both metals and biomolecules. Laser-ablation inductively coupled-plasma mass spectrometry (LA-ICP-MS) uses a laser beam focused to micrometer size on a sample to ablate small particles from the surface of the sample. These are then fed to an inductively coupled plasma (ICP) source, which vaporizes, atomizes, and ionizes the ablated particle. The ICP source in turn feeds a mass spectrometer, and the ions are separated and detected according to their mass to charge ratio m/z. LA-ICP-MS can provide elemental maps with resolution and sensitivity similar to that of XFI,253 but with better access to the lighter elements such as carbon.254 The ideal sample thickness for LA-ICP-MS is 20–30 μm,253 and well-defined methods for quantification have been established.254,255 In general, data collection times for LA-ICP-MS are rather longer than for XFI; for example, data acquisition of a whole transverse section of rat brain with LA-ICP-MS at a resolution of 120 μm will take 3–4 h, whereas the same resolution XFI scan might take less than 1 h. However, the LA-ICP-MS experiment has the significant advantage that commercial instruments are available and access to large specialized XFI synchrotron infrastructure is not needed. Whereas LA-ICP-MS can provide elemental mapping information essentially similar to that of XFI, other mass spectrometry methods can provide quite different and highly complementary information. Matrix-assisted laser desorption ionization (MALDI) mass spectrometry can spatially resolve molecular weights in the range of m/z ∼1000 to ∼200 000, and, in some cases, higher.256 Tissue section thicknesses of ∼10 μm are compatible with XFI, but electrically conductive sample plates are required, which may not be directly compatible with XFI. MALDI MSI also requires deposition of a compatible matrix material to aid desorption of molecules from the surface of the sample. Matrix deposition is critical for reproducibility and spatial resolution, although smaller matrix droplets reduce the detectable signal as there are fewer ions generated. Prior to matrix deposition, MSI sample preparation often includes several rinsing steps, which may include ethanol and acetic acid to improve the quality of the MS data. In our experience, rinsing tissue sections with deionized water can remove labile pools of some elements, although their originally associated proteins may persist. While these changes in elemental content may be detectable with XFI, they are unlikely to be consequential to MSI as they likely represent the loss of low molecular weight species or the loss of labile ions from sites within their parent protein, which may still persist after rinsing.
Spatial resolution in MALDI MSI can approach 10 μm, although careful consideration must be given to the method of matrix deposition, as well as the acquisition method, to ensure resulting images contain spatially relevant information at these length scales. There are also relatively fewer molecules available for desorption below 10 μm, which significantly limits detectability. Oversampling methods, where the step size of the sample position motor is smaller than the laser spot size, have been successfully applied257 in a manner analogous to that of XFI. However, unlike XFI, there is less material available for ablation and for subsequent detection from the surface in subsequent steps after the initial spot. A microscope mode method of MALDI MSI has also been developed, whereby snapshots of a large area of sample are collected and mapped to a position sensitive detector, affording significantly improved spatial resolution equivalent to ∼4 μm.258
MSI requires generating ionizable species that can be readily ablated from the sample surface; however, some biomolecular species may not be amenable to such methods. Further challenges are posed by the ion-suppression effect, the possible presence of isobaric species, as well as detection of low molecular weight species. Separation of isobaric species is possible with ion mobility mass spectrometry,259 whereby ions are separated according to their collision cross section as well as their mass-to-charge ratio, making analysis of complex mixtures readily accessible, as well as separation of low molecular weight species. Alternate methods of detection for low molecular weight species, such as drugs or probe molecules, can potentially be overcome by subsequent collision-induced dissociation of the molecular ion peak belonging to the suspected parent species, followed by confirmation of the parent ion from its characteristic molecular fragments.
Combining molecular imaging techniques, such as MSI, with the additional wealth of data afforded by XFI as well as XAS characterization of chemical species within particular regions of interest is particularly attractive and holds promise for more complete characterization of molecular species within biological tissues at the cellular (and in some cases subcellular) level. Mass spectrometric analysis consumes sample material, requiring that MSI be performed on the same sample following imaging and analysis with nondestructive methods such as XFI, or be performed on adjacent sample sections.
5.3. Fluorescent Molecular Probes
Metal ion fluorescent probes are small molecules that are specifically synthesized to bind metal ions with a degree of selectivity, and, upon binding, to change their optical fluorescent properties.260 There are significant advantages to this approach, including the convenience of optical microscopy, and the short time required to acquire an image. To illustrate the relative strengths and limitations, we will discuss three different examples: mercury, copper, and zinc.
5.3.1. Mercury
As we have already discussed XFI applications to mercury toxicology in some detail (section 4.1), as an example we will examine the comparative strengths and limitations of mercury fluorescent probes. This area has recently been reviewed by Nolan and Lippard;261 the reader is referred to this work for a comprehensive discussion. Mercury sensors can be categorized into molecules that either turn on or turn off fluorescence upon mercury exposure,262,263 including novel approaches in which mercury causes chemical modification of the sensor.264−266 The primary concerns with these approaches are the chemical modifications of the mercury, the specificity of the probe, and sensitivity. The turn-on probes developed by Chang and co-workers have excellent reported specificity, but because the in vivo levels of Hg2+ can be several orders of magnitude lower than other species such as Zn2+, despite the response of the probes for Hg2+ being many times that of other cations, some confusion may be possible. Chemical modification of the endogenous mercury that is inherent in these methods may also be a cause for concern. Thus, XFI of both mercury and methylmercury compounds has clearly shown that uptake, transport, and localization all depended heavily upon the chemical form in which mercury is presented,51 and in vivo association with a sensor molecule might change the metal distribution. Insofar as sensitivity is concerned, both XFI and fluorescent probes give sufficient response to estimate mercury in tissue samples, although the sensitivity of XFI depends strongly upon the source of X-rays and detector technology being employed.
5.3.2. Copper
Studies using copper fluorescent probes side-by-side with XFI have been reported. Price et al.267 have studied the Cu(I)-specific fluorescent probe CS1.268 Price et al. found that CS1 competed poorly with endogenous copper handling biomolecules, and that increased fluorescence from CS1 was difficult to directly attribute to coordination of the probe’s target, Cu(I).267 Their results also highlight important caveats in the use of fluorescent probes, which might not be evident without complementary XFI characterization; in their case, trafficking of CS1 to lysosomes, and its localization therein, could have ultimately altered some of the subcellular distribution of Cu(I), which was significantly different in controls as compared to the same compartment in treatments where the fluorescent chelator was employed. Thus, when compared to the use of fluorescent probes, XFI has the significant advantage that chemical treatments or chemical modification of the target metal is not required. Fluorescent probes may also specifically target a labile pool of the metal ion of interest, or those occurrences of the metal ion in which it may be competitively exchanged. In such cases, while the use of fluorescent probes alone might be misleading, the combined use of XFI and fluorescent probes can in principle provide highly complementary information, with XFI probing total metal and the sensor molecules only labile metals. The benefits of using both methods have been discussed by Yang et al.,269 who used a combination of XFI in parallel with a different Cu(I)-specific fluorescent probe CTAP-1, in addition to μ-XAS to probe chemical form. In this case, the XFI and CTAP-1 images showed similarities, and Yang et al. reported evidence for a kinetically labile copper pool, predominantly localized in mitochondria and Golgi.269
5.3.3. Zinc
As the second most abundant transition metal in the mammalian body, zinc plays many crucial roles in biological chemistry. Although the metal itself is incapable of redox chemistry under physiological conditions, depleted or elevated zinc levels are known to indirectly affect cell redox status, with carefully balanced zinc homeostasis being crucial for cellular viability. In addition, redox stability of Zn2+ is a likely reason underlying many of the biological functions of the metal, particularly redox independent cell signaling pathways. For these reasons, imaging the distribution and alterations in local concentration of zinc is of great interest. However, studies in this field may be problematic, particularly in the field of neuroscience, due to the occurrence of several Zn2+ pools having significant chemical differences. Specifically, although the majority of zinc is tightly bound within proteins, a sizable percentage of brain zinc exists as a loosely bound or “chelatable” pool of zinc, particularly in the hippocampus. This “chelatable” zinc has long been thought to play crucial roles in neuronal signaling pathways, although the complete role of this zinc pool remains to be elucidated.270,271 “Chelatable” zinc has previously been imaged using cytochemical methods (Timm’s staining),272 and, more recently, a wide range of zinc chelating fluorophores have been designed to study the chelatable zinc pool.273,274 As reviewed recently, however, nonspecific binding of Zn2+ chelators to other divalent ions (i.e., Ca2+), or to protein bound Zn2+, has the potential to confound studies of the chelatable Zn pool.273,275−277 Therefore, a combined approach with XFI to determine the distribution of total zinc, in combination with fluorescence probes and/or cyto-chemistry to study chelatable zinc, has been valuable. In an elegant example of this approach, traditional biochemical methods were used to demonstrate the location of the ZnT3 protein and chelatable zinc pool in wild-type and in ZnT3 knockout mice.278,279 XFI was then used to demonstrate that the decrease in chelatable zinc was not due to altered speciation or coordination of zinc, but due to a net decrease in total zinc in ZnT3 knockout mice.280 This important contribution conclusively demonstrated a direct role of ZnT3 as a Zn transport protein.280
Overall, it is clear that a combination of techniques provides strong mutual benefits for understanding and characterizing trace metals in biological systems. Thus, XFI and μ-XAS can probe total elemental distributions and provide chemical speciation information, while prudently chosen fluorescent probes can be used to target and identify labile or localized pools of the target metal.
5.4. Fourier Transform Infrared
Fourier transform infrared (FTIR) spectroscopy provides a wealth of molecular information for biological samples, is nondestructive, and can be performed on the same sample as XFI analyses if a suitable substrate is used.
The FTIR spectrum results from the absorption of characteristic wavelengths of light that correspond to the frequency of an oscillating molecular dipole. FTIR spectroscopic maps or images are best viewed as “functional group” maps or images, in which a variety of organic functional groups present in lipids, proteins, carbohydrates, and various metabolites can be distinguished. Detailed reviews of FTIR data acquisition methods have recently been reported,281−283 and we present only a brief discussion here. Collection of FTIR spectroscopic data may be performed in two different modes, each with specific advantages. FTIR mapping mode works in a way similar to many XFI experiments (e.g., see section 2.4); a small beam, typically defined with an aperture, is employed, and the sample is moved in a step-by-step manner to spectroscopically map an area. The second approach, known as FTIR imaging, does not use an aperture, but incorporates wide-field sample illumination and detection using a focal plane array detector to quickly acquire FTIR images of substantial areas of the sample. In general, the point-by-point mapping approach provides superior quality spectra over a larger range than can be achieved with the imaging approach, but the focal plane array detector allows substantial oversampling (e.g., pixel sizes <1 μm) relatively rapidly, so that images can be acquired in several minutes instead of hours or days for point-by-point mapping.284 With both methods, synchrotron light is needed to provide sufficient spectral brightness for diffraction-limited spatial resolution, although globar sources and benchtop instruments are well suited to imaging larger samples at spatial resolutions of approximately double the diffraction limit, making them a competitive alternative to synchrotron-based FTIR imaging for some samples.
The diffraction-limited spatial resolution of FTIR images is 2–10 μm, which is comparable to most XFI experiments. Moreover, the sample substrates, sample thicknesses, and methods of sample preparation can be similar for both methods. For example, cells grown on infrared-transparent Si3N4 membranes, or on thin carbon-polymer-based films (e.g., mylar), can be investigated with FTIR imaging as air-dried, freeze-dried, or chemically fixed samples, and the same cells can then be analyzed with XFI. Likewise, sections from hard tissues such as bone or teeth, or from soft tissues such as brain or tumors, can be mounted on Si3N4 or polymer films and analyzed in transmission geometry by FTIR, or mounted on aluminum or gold-coated slides and analyzed in transflection geometry with FTIR, prior to XFI. Transflection measurements use a reflective substrate and measure transmission spectra from the two passes through the sample, the first through sample to the reflective layer, and the second on the return out of the sample, with a very small reflection contribution from the surface of the sample itself. We note that, as discussed by Bassan et al.,285 artifacts can occur when biological samples are analyzed in transflection geometry so that such analysis should done with care. In many cases, examination of exactly the same sample by FTIR and XFI is not needed as serial sections of a sample are often available, so that alternate sections can be mounted on CaF2 or BaF2 for FTIR and on plastic or Si3N4 films for XFI.
One of the major applications of complementary FTIR and XFI analyses has been the field of neuroscience. This approach is well suited to study spatially resolved cause and effect relationships between molecular alterations such as protein misfolding and the formation of β-sheet aggregates and metal (Fe, Cu, Zn) and ionic (Cl–, K+, Ca2+) homeostasis. Miller and co-workers have successfully applied a combined approach to study both metal content and protein secondary structure within amyloid plaques in Alzheimer’s disease,286 metal content and protein secondary structure within individual neuons in Parkinson’s Disease and Amyotrophic lateral sclerosis,287 and protein aggregation and elemental content in the hippocampus after “epileptic”-like seizures.288,289 Additional examples include the use of FTIR spectroscopic imaging to identify crystalline creatine deposits in diseased brain tissue, and corresponding elemental analysis of these deposits using XFI.290
In addition to analysis of brain tissue, a combined FTIR and XFI approach has been used to monitor the molecular and elemental changes induced in tumor spheroids by chemotherapeutic drugs.291 Likewise, the use of FTIR in combination with XFI and XAS to study molecular alterations induced by metal drug uptake in cancer cells has been reviewed recently.292 A significant advantage of this combined multimodal approach is that greater insight is obtained regarding chemical alterations induced in cells at the site of drug uptake/localization, and molecular alterations that result as secondary consequences, which are not colocalized with administered drugs. Other samples that have been successfully investigated at the subcellular level using a combined FTIR and XFI approach have previously been reviewed,293 and include cultured adipocytes and cardiomyoctes.105
5.5. Novel X-ray Imaging Methods
A number of new and novel imaging modalities of a complementary nature have recently been reported. For the stable iron isotope 57Fe (2% natural abundance), nuclear resonance is readily excited by synchrotron light at 14 412 eV, and imaging with both coherent and incoherent nuclear resonant scattering has recently been explored at a spatial resolution of 5 μm.294 The coherent nuclear resonant scattering is more commonly known as synchrotron Mössbauer spectroscopy (SMS);295 it is measured in the forward direction (i.e., in the direction of the beam), and yields information on the magnetic properties of the iron in the sample. The incoherent nuclear resonant scattering is measured at 90° to the incident beam, is probably less useful than SMS imaging in a biological context, and yields information on lattice dynamics. While pilot SMS imaging studies have explored only very high concentrations, biological systems are likely to become possible with new chopper technology. The major disadvantage of SMS imaging is that, for all practical purposes, it cannot be used for elements other than iron. However, given the widespread importance of iron in biology, the technique is still likely to be important in future studies.
Another method of potential importance that to date has remained unexplored in an imaging modality is high-resolution fluorescence spectroscopy.296,297 Typically, these data are collected using a Johann geometry bent-crystal analyzer apparatus, often constructed as an array to increase the solid angle. Here, minor fluorescence lines, such as the Kβ″, which shift depending upon chemical environment, can be monitored. The major disadvantage with this method is low sensitivity as the aforementioned minor fluorescence lines are at best hundreds of times less intense than Kα1 and Kα2 lines, and it is possible that this may never become practical on the most challenging biological samples. Although they are not strictly speaking X-ray fluorescence spectroscopy, resonant inelastic X-ray scattering298 and X-ray Raman299 experiments are conducted with essentially the same bent-crystal analyzers as high-resolution fluorescence, although a different geometrical arrangement of crystals may be used for the different experiments. All of these are inherently low sensitivity methods, and very high spectral brightness X-ray sources are required for them to be viable. Despite caution about the practicability of these methods for many biological systems, the authors are aware of at least three such bent-crystal analyzers that are, or will be, available on XFI compatible beamlines, and it seems likely that this area will be explored in future applications.
6. Concluding Remarks: Refinements and Future Directions
Future applications of XFI will require an increase in the capabilities of beamlines, scanning systems, and detectors. The capabilities of synchrotron radiation sources to provide improved spectral brightness continue to advance. Modern synchrotron light sources are also considerably more stable than in previous years, and are also able to operate essentially continuously, in what is called top-up mode, without routine interruptions in beam delivery to refill the electron storage ring. As the sources improve, the technique of XFI will undoubtedly improve to keep pace. Faster and more efficient detector systems with good energy resolution, together with faster readout electronics, will be increasingly important. Also important will be improved speed of data acquisition and sample scanning systems. As we have discussed, biological samples are inherently delicate, and improved cryogenic protection for larger samples is likely to be important. The multienergy methods discussed in section 2.6 may also improve to the ideal of collecting a complete μ-XAS spectrum for every pixel within a tractable amount of time, and as throughput and data acquisition improve then so will analytical methods and automation. The ability to change spatial resolution on a sample will also likely be an important future capability. At present, if an experimenter wishes to use XFI to interrogate a region of interest within a sample at substantially higher resolution, it may require a different beamline and sometimes even necessitate relocating to a different synchrotron light source. The advantages of being able to dynamically change spatial resolutions on one experimental station would be significant. At the time of writing, the authors know of only one beamline for which this is planned (the BioXAS Imaging beamline at the Canadian Light Source). Finally, improved overlap of XFI with other methods and alternate imaging modalities are likely to continue to see growth and increasing scrutiny as these methods are employed for their mutual benefit (section 5).
Acknowledgments
Research at the University of Saskatchewan is funded by the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canadian Institutes of Health Research (CIHR), the Saskatchewan Health Research Foundation (SHRF), the University of Saskatchewan, and the Canada Foundation for Innovation (CFI). G.N.G. and I.J.P. are supported by Canada Research Chair awards. M.J.P. and M.J.H. acknowledge SHRF Postdoctoral Fellowships, CIHR Postdoctoral Fellowships, and are also CIHR-Training in Health Research Using Synchrotron Techniques (THRUST) Associate (M.J.P.) and Fellow (M.J.H.). Our research has benefited from access to and interactions with staff from the Stanford Synchrotron Radiation Lightsource (SSRL), the Advanced Photon Source (APS), and the Canadian Light Source (CLS). We are indebted to the staff of these facilities for their continued support, without which many of the results described herein would not have been possible. SSRL is a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program (P41RR001209). Use of the Advanced Photon Source, an Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract no. DE-AC02-06CH11357. The CLS is funded by the CFI, NSERC, the National Research Council Canada, CIHR, the Government of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan. We thank Ms. Sanjukta Choudhury and Ms. Tracy MacDonald, both of the University of Saskatchewan, for the data of Figure 27. We also thank Helen Nichol (University of Saskatchewan) and Roger C. Prince (ExxonMobil Biomedical Sciences Inc.) for helpful comments on the manuscript.
Glossary
Abbreviations
- AD
Alzheimer’s disease
- ADC
analog to digital converter
- APS
Advanced Photon Source
- CLS
Canadian Light Source
- EXAFS
extended X-ray absorption fine structure
- FET
field-effect transistor
- FTIR
Fourier transform infrared
- fwhm
full width at half-maximum
- FSI
fluorescence subtraction imaging
- HWHM
half-width at half-maximum
- K–B
Kirkpatrick–Baez
- KES
K-edge subtraction
- LA-ICP-MS
laser-ablation inductively coupled-plasma mass spectrometry
- MALDI
matrix-assisted laser desorption ionization
- MCA
multichannel analyzer
- MRI
magnetic resonance imaging
- MSI
mass spectrometry imaging
- NSF
nephrogenic systemic fibrosis
- PET
positron emission tomography
- PCA
principal component analysis
- PD
Parkinson’s disease
- RSXRF
rapid scan X-ray fluorescence
- SCA
single channel analyzer
- SMS
synchrotron Mössbauer spectroscopy
- SRIXE
synchrotron radiation-induced X-ray emission
- SSRL
Stanford Synchrotron Radiation Lightsource
- STXM
scanning transmission X-ray microscopy
- SVD
single value decomposition
- SXRF
synchrotron X-ray fluorescence
- SXFI
synchrotron X-ray fluorescence imaging
- UTI
urinary tract infection
- XANES
X-ray absorption near-edge fine structure
- XAS
X-ray absorption spectroscopy
- μ-XAS
micro X-ray absorption spectroscopy
- XFI
X-ray fluorescence imaging
- XFM
X-ray fluorescence mapping
- XFM
X-ray fluorescence microscopy
- XRF
X-ray fluorescence
Biographies

Jake Pushie completed his M.Sc. in Theoretical and Computational Chemistry in 2003 at the University of Calgary and his Ph.D. in Physical Bioinorganic Chemistry in 2007 at the same institution. He began a postdoctoral fellowship with Prof. George in 2007, and in 2010 Dr. Pushie was the recipient of the Top Researcher Award at the postdoctoral level from the Saskatchewan Health Research Foundation. He employs a range of spectroscopic and computational chemistry methods, as well as X-ray fluorescence imaging, to study the structural and functional aspects of metals in biological systems, particularly in regards to topics related to human health and disease. Dr. Pushie is also a CIHR-THRUST (training in health research using synchrotron techniques) Associate, and is currently a Research Officer in the Department of Anatomy and Cell Biology at the University of Saskatchewan.

Ingrid Pickering received her B.A. in Natural Sciences from the University of Cambridge (UK) in 1986 and a Ph.D. in Physical Chemistry from Imperial College-University of London (UK) in 1990, in the area of heterogeneous catalysis. After two years as a postdoctoral fellow with Exxon Research and Engineering Co. (NJ), she moved to the Stanford Synchrotron Radiation Laboratory (CA). She came to Canada and her present position at the University of Saskatchewan in 2003. Ingrid is currently Professor and Canada Research Chair (Tier 1) in Molecular Environment Science at the University of Saskatchewan. Her research investigates metals and other elements in biological systems from the environment to human health.

Malgorzata Korbas is a staff scientist at the Canadian Light Source, Canada’s national centre for synchrotron research in Saskatoon, Saskatchewan. She received her M.Sc. and Ph.D. in physics in the Department of Medical Physics at the Jagiellonian University in Krakow, Poland. After completing her fellowship at the European Molecular Biology Laboratory in Hamburg, Germany, she joined the laboratory of Profs. Graham N. George and Ingrid J. Pickering as a postdoctoral fellow at the University of Saskatchewan. In 2012 she moved to the Canadian Light Source, where she is a staff scientist for the Biological X-ray Absorption Spectroscopy (BioXAS) Facility. Her scientific interest is directed at understanding how metals and metalloids interact with biological systems. She is particularly interested in elements that pose environmental and human health risks such as mercury and selenium. Her research takes advantage of various synchrotron-based imaging and chemical speciation techniques to study the molecular basis for mercury toxicity in human brain and in a zebrafish model system.

Mark Hackett completed his B.Sc. (Hons) in 2006 within the department of chemistry at Curtin University (Perth, Australia) and completed his Ph.D. in 2011 within the department of chemistry at The University of Sydney (Sydney, Australia). Since 2011 Dr. Hackett has worked as a postdoctoral fellow with the synchrotron medical imaging of stroke team at the University of Saskatchewan, as both a Saskatchewan Health Research Foundation (SHRF) and a Canadian Institute of Health Research (CIHR) postdoctoral fellow. He is also a CIHR-THRUST fellow. Dr. Hackett’s research interests focus on the development of synchrotron spectroscopic imaging modalities for direct biochemical imaging, and integration of these techniques into the field of neuroscience. Specifically, Dr. Hackett utilizes Fourier transform infrared spectroscopy (FTIR), X-ray fluorescence imaging (XFI), and X-ray absorption spectroscopy (XAS) to image direct biochemical markers of oxidative stress and altered metabolism in the brain.

Graham George was educated at King’s College London (B.Sc., 1979) and the University of Sussex (D. Phil., 1983). After postdoctoral fellowships at the University of Sussex and at Exxon Research & Engineering Co. in New Jersey, he continued at Exxon as a Principal Investigator until moving to the Stanford Synchrotron Radiation Laboratory in 1992. In 2003 he became full professor and Tier 1 Canada Research Chair of X-ray Absorption Spectroscopy at the University of Saskatchewan. His research interests include a career-long interest in spectroscopy and bioinorganic chemistry and the development of new methods for understanding the roles and mechanisms of metals in biology.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Swart C.; Fisicaro P.; Goenaga-Infante H.; Zakel S. Metallomics 2012, 4, 1137. [DOI] [PubMed] [Google Scholar]
- Waldron K. J.; Robinson N. J. Nat. Rev. Microbiol. 2009, 6, 25. [DOI] [PubMed] [Google Scholar]
- Thomson A. J.; Gray H. B. Curr. Opin. Chem. Biol. 1998, 2, 155. [DOI] [PubMed] [Google Scholar]
- Cvetkovic A.; Lal Menon A.; Thorgersen M. P.; Scott J. W.; Poole F. L. II; Jenney F. E. Jr.; Lancaster W. A.; Praissman J. L.; Shanmukh S.; Vaccaro B. J.; Trauger S. A.; Kalisiak E.; Apon J. V.; Siuzdak G.; Yannone S. M.; Tainer J. A.; Adams M. W. W. Nature 2010, 466, 779. [DOI] [PubMed] [Google Scholar]
- Cotelesage J. J. H.; Pushie M. J.; Grochulski P.; Pickering I. J.; George G. N. J. Inorg. Biochem. 2012, 115, 127. [DOI] [PubMed] [Google Scholar]
- Andrews N. C. Curr. Opin. Chem. Biol. 2002, 6, 181. [DOI] [PubMed] [Google Scholar]
- Tuemer Z. Hum. Mutat. 2013, 34, 417. [DOI] [PubMed] [Google Scholar]
- Kodama H.; Fujisawa C.; Bhadhprasit W. Curr. Drug Metab. 2012, 13, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blindauer C. Biochemist 2012, 34, 4. [Google Scholar]
- Wang D.; Lippard S. J. Nat. Rev. Drug Discovery 2005, 4, 307. [DOI] [PubMed] [Google Scholar]
- Levina A.; Mitra A.; Lay P. A. Metallomics 2009, 1, 458. [DOI] [PubMed] [Google Scholar]
- Lengfelder E.; Hofmann W.-K.; Nowak D. Leukemia 2012, 26, 433. [DOI] [PubMed] [Google Scholar]
- Popescu B. F. G.; George M. J.; Bergmann U.; Garachtchenko A. V.; Kelly M. E.; McCrea R. P. E.; Lüning K.; Devon R. M.; George G. N.; Hanson A. D.; Harder S. M.; Chapman L. D.; Pickering I. J.; Nichol H. Phys. Med. Biol. 2009, 54, 651. [DOI] [PubMed] [Google Scholar]
- Habib C. A.; Zheng W.; Haacke M. E.; Webb S.; Nichol H. AIP Conf. Proc. 2010, 1266, 78. [Google Scholar]
- Korbas M.; Lai B.; Vogt S.; Gleber S.-C.; Karunakaran C.; Pickering I. J.; Krone P. H.; George G. N. ACS Chem. Biol. 2013, 8, 2256. [DOI] [PubMed] [Google Scholar]
- Synchrotron Radiation Sources: A Primer; Winick H., Ed.; World Scientific Publishing Co. Pte. Ltd.: Singapore, New Jersey, London, 1994. [Google Scholar]
- David C.; Weitkamp T.; Nfhammer B.; van der Veen J. F. Spectrochim. Acta, Part B 2004, 59, 1505. [Google Scholar]
- Snigireva I.; Snigirev A. J. Environ. Monit. 2006, 8, 33. [DOI] [PubMed] [Google Scholar]
- Sakdinawat A.; Attwood D. Nat. Photonics 2010, 4, 840. [Google Scholar]
- Ice G. E.; Budai J. D.; Pang J. W. L. Science 2011, 334, 1234. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick P.; Baez A. J. Opt. Soc. Am. 1948, 38, 766. [DOI] [PubMed] [Google Scholar]
- Morawe Ch.; Osterhoff M. Nucl. Instrum. Methods Phys. Res., Sect. A 2010, 616, 98. [Google Scholar]
- Liu W.; Ice G. E.; Assoufid L.; Liu C.; Shi B.; Khachatryan R.; Qian J.; Zschack P.; Tischler J. Z.; Choi J. Y. J. Synchrotron Radiat. 2011, 18, 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Ice G. E.; Assoufid L.; Liu C.; Shi B.; Zschack P.; Tischler J.; Qian J.; Khachartryan R.; Shu D. Nucl. Instrum. Methods Phys. Res., Sect. A 2011, 649, 169. [Google Scholar]
- Gao N.; Janssens K. In X-ray Spectrometry: Recent Technological Advances; Tsuji K., Injuk J., Van Grieken R., Eds.; John Wiley & Sons Ltd.: New York, 2004; pp 89–110. [Google Scholar]
- Hirsch G. X-ray Spectrom. 2003, 32, 229. [Google Scholar]
- Pickering I. J.; Hirsch G.; Prince R. C.; Sneeden E. Y.; Salt D. E.; George G. N. J. Synchrotron Radiat. 2003, 10, 289. [DOI] [PubMed] [Google Scholar]
- Pickering I. J.; Gumaelius L.; Harris H. H.; Prince R. C.; Hirsch G.; Banks J. A.; Salt D. E.; George G. N. Environ. Sci. Technol. 2006, 40, 5010. [DOI] [PubMed] [Google Scholar]
- Pickering I. J.; Sneeden E. Y.; Prince R. C.; Harris H. H.; Hirsch G.; George G. N. Biochemistry 2009, 48, 6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan G. B.; Wright J. P.; Bytchkov A.; Rossat M.; Gleyzolle H.; Snigireva I.; Snigirev A. J. Synchrotron Radiat. 2011, 18, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vladimirsky Y.; Kern D. P.; Chang T. H. P.; Attwood D. T.; Iskander N.; Rothman S.; McQuaide K.; Kirz J.; Ade H.; McNulty I.; Rarback H.; Shu D. Nucl. Instrum. Methods Phys. Res., Sect. A 1988, 266, 324. [Google Scholar]
- Krause M. O. J. Phys. Chem. Ref. Data 1979, 8, 307. [Google Scholar]
- Thompson A.; Attwood D.; Gullikson E.; Howells M.; Kim K.-J.; Kirtz J.; Kortright J.; Lindau I.; Liu Y.; Pianetta P.; Robinson A.; Scofield J.; Underwood J.; Williams G.; Winick H.. X-ray Data Booklet; Center for X-ray Optics and Advanced Light Source, Lawrence Berkeley Laboratory: Berkeley, 2009. [Google Scholar]
- Jenkins R.; Gould R. W.; Gedcke D.. Quantitative X-ray Spectrometry, 2nd ed.; Marcel Dekker: New York, 1995. [Google Scholar]
- Knoll G. F.Radiation Detection and Measurement, 4th ed.; John Wiley & Sons: NJ, 2010. [Google Scholar]
- Schlosser D. M.; Lechner P.; Lutz G.; Niculae A.; Soltau H.; Strüder L.; Eckhardt R.; Hermenau K.; Schaller G.; Schopper F.; Jaritschin O.; Liebel A.; Simsek A.; Fiorini C.; Longoni A. Nucl. Instrum. Methods Phts. Res., Sect. A 2010, 624, 270. [Google Scholar]
- Funk T.; Deb A.; George S. J.; Wang H.; Cramer S. P. Coord. Chem. Rev. 2005, 249, 3. [Google Scholar]
- Ryan C. G.; Kirkham R.; Hough R. M.; Moorhead G.; Siddons D. P.; de Jonge M. D.; Paterson D. J.; De Geronimo G.; Howard D. L.; Cleverley J. S. Nucl. Instrum. Methods Phys. Res., Sect. A 2010, 619, 37. [Google Scholar]
- Kirkham R.; Dunn P. A.; Kuczewski A. J.; Siddons D. P.; Dodanwela R.; Moorhead G. F.; Ryan C. G.; De Geronimo G.; Beuttenmuller R.; Pinelli D.; Pfeffer M.; Davey P.; Jensen M.; Paterson D. J.; de Jonge M. D.; Howard D. L.; Küsel M.; McKinlay J. AIP Conf. Proc. 2010, 1234, 240. [Google Scholar]
- Siddons D. P.; Kirkham R.; Ryan C. G.; De Geronimo G.; Dragone; Kuczewski A. J.; Li Z. Y.; Carini G. A.; Pinelli D.; Beuttenmuller R.; Elliott D.; Pfeffer M.; Tyson T. A.; Moorhead G. F.; Dunn P. A. J. Phys.: Conf. Ser. 2014, 499, 01200. [Google Scholar]
- Twomey T. R.; Keyser R. M.; Simpson M. L.; Wagner S. E. Radioact. Radiochem. 1991, 2, 28. [Google Scholar]
- Cramer S. P.; Tench O.; Yocum M.; George G. N. Nucl. Instrum. Methods Phys. Res., Sect. A 1988, 266, 586. [Google Scholar]
- Nakhostin M.; Podolyak Zs.; Regan P. H.; Walker P. M. Rev. Sci. Instrum. 2010, 81, 103507/1. [DOI] [PubMed] [Google Scholar]
- Kropf A. J.; Finch R. J.; Fortner J. A.; Aase S.; Karanfil C.; Segre C. U.; Terry J.; Bunker G.; Chapman L. D. Rev. Sci. Instrum. 2003, 74, 4696. [Google Scholar]
- Howard D. L.; de Jonge M. D.; Lau D.; Hay D.; Varcoe-Cocks M.; Ryan C. G.; Kirkham R.; Moorhead G.; Paterson D.; Thurrowgood D. Anal. Chem. 2012, 84, 3278. [DOI] [PubMed] [Google Scholar]
- Bergmann U. Phys. World 2007, 20, 39. [Google Scholar]
- Manning P. L.; Edwards N. P.; Wogelius R. A.; Bergmann U.; Barden H. E.; Larson P. L.; Schwarz-Wings D.; Egerton V. M.; Sokaras D.; Mori R. A.; Sellers W. I. J. Anal. At. Spectrom. 2013, 28, 1024. [Google Scholar]
- Edwards N. P.; Wogelius R. A.; Bergmann U.; Larson P.; Sellers W. I.; Manning P. L. Appl. Phys. A: Mater. Sci. Process. 2013, 111, 147. [Google Scholar]
- Ryan C. G.; Cousens D. R.; Sie S. H.; Griffin W. L. Nucl. Instrum. Methods Phys. Res., Sect. B 1990, 49, 271. [Google Scholar]
- Pickering I. J.; Prince R. C.; Salt D. E.; George G. N. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbas M.; MacDonald T. C.; Pickering I. J.; George G. N.; Krone P. H. ACS Chem. Biol. 2012, 7, 411. [DOI] [PubMed] [Google Scholar]
- McMaster W. H.; Kerr Del Grande N.; Mallett J. H.; Hubbell J. H.. Lawrence Livermore National Laboratory Report, 1969; UCRL-50174 Section II Revision I.
- De Samber B.; Silversmit G.; De Shcamphelaere K.; Evens R.; Schoonjans T.; Vekemers B.; Janssen C.; Masschaele B.; Van Hoorebeke L.; Szaloki I.; Vanhaecke F.; Rickers K.; Falkenberg G.; Vincze L. J. Anal. At. Spectrom. 2010, 25, 544. [Google Scholar]
- Kosior E.; Bohic S.; Suhonen H.; Ortega R.; Devès G.; Carmona A.; Marchi F.; Guillet J. F.; Cloetens P. J. Struct. Biol. 2012, 177, 239. [DOI] [PubMed] [Google Scholar]
- Kinney J.; Johnson Q.; Nichols M. C.; Bonse U.; Nusshardt R. Appl. Opt. 1986, 25, 4583. [DOI] [PubMed] [Google Scholar]
- Kinney J. H.; Johnson Q. C.; Saroyan R. A.; Nichols M. C.; Bonse U.; Nusshardt R.; Pahl R. Rev. Sci. Instrum. 1988, 59, 196. [Google Scholar]
- Hahn J. E.; Scott R. A.; Hodgson K. O.; Doniach S.; Desjardins S. R.; Solomon E. I. Chem. Phys. Lett. 1982, 33, 595. [Google Scholar]
- Pickering I. J.; George G. N. Inorg. Chem. 1995, 34, 3142. [Google Scholar]
- George G. N.; Pickering I. J.; Pushie M. J.; Nienaber K.; Hackett M. J.; Ascone I.; Hedman B.; Hodgson K. O.; Aitken J. B.; Levina A.; Glover C.; Lay P. A. J. Synchrotron Radiat. 2012, 19, 875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb S. M.; Gaillard J.-F.; Ma L. Q.; Tu C. Environ. Sci. Technol. 2003, 37, 754. [DOI] [PubMed] [Google Scholar]
- Tu C.; Ma L. Q.; Bondada B. J. Environ. Qual. 2002, 31, 1671. [DOI] [PubMed] [Google Scholar]
- Andrahennadi R.; Pickering I. J. Environ. Chem. 2008, 5, 413. [Google Scholar]
- Blute N. K.; Brabander D. J.; Hemond H. F.; Sutton S. R.; Newville M. G.; Rivers M. L. Environ. Sci. Technol. 2004, 38, 6074. [DOI] [PubMed] [Google Scholar]
- Bulska E.; Wysocka I. A.; Wierzbicka M. H.; Proost K.; Janssens K.; Falkenberg G. Anal. Chem. 2006, 78, 7616. [DOI] [PubMed] [Google Scholar]
- Lenz M.; van Hullebusch E. D.; Farges F.; Nikitenko S.; Borca C. N.; Grolimund D.; Lens P. N. L. Environ. Sci. Technol. 2008, 42, 7587. [DOI] [PubMed] [Google Scholar]
- Szczerbowska-Boruchowska M.; Stegowski Z.; Lankosz M.; Szpaka M.; Adamek D. J. Anal. At. Spectrom. 2012, 27, 239. [Google Scholar]
- Hitchcock A. P.; Obst M.; Wang J.; Lu Y. S.; Tyliszczak T. Environ. Sci. Technol. 2012, 46, 2821. [DOI] [PubMed] [Google Scholar]
- Koprinarov I. N.; Hitchcock A. P.; McCrory C. T.; Childs R. F. J. Phys. Chem. B 2002, 106, 5358. [Google Scholar]
- Lerotic M.; Jacobsen C.; Schäfer T.; Vogt S. Ultramicroscopy 2004, 100, 35. [DOI] [PubMed] [Google Scholar]
- Goloso B.; Simionovici A.; Somogyi A.; Lemelle L.; Chukalina M.; Brunetti A. J. Appl. Phys. 2003, 94, 145. [Google Scholar]
- Lombi E.; de Jonge M. D.; Donner E.; Kopittke P. M.; Howard D. L.; Kirkham R.; Ryan C. G.; Paterson D. PLoS One 2011, 6, e20626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNear D. H.; Peltier E.; Everhart J.; Chaney R. L.; Sutton S.; Newville M.; Rivers M.; Sparks D. L. Environ. Sci. Technol. 2005, 39, 2210. [DOI] [PubMed] [Google Scholar]
- Kim S. A.; Punshon T.; Lanzirotti A.; Li L.; Alonso J. M.; Ecker J. R.; Kaplan J.; Guerinot M. L. Science 2006, 314, 1295. [DOI] [PubMed] [Google Scholar]
- de Jonge M. D.; Vogt S. Curr. Opin. Struct. Biol. 2010, 20, 606. [DOI] [PubMed] [Google Scholar]
- La Rivière P. J.; Rivers M.; Sutton S. R.; Billmire D.; Vargas P. Opt. Eng. 2005, 45, 077005. [Google Scholar]
- de Jonge M. D.; Holzner C.; Baines S. B.; Twining B. S.; Ignatyeve K.; Diaz J.; Howard D. L.; Legnini D.; Micelie A.; McNultye I.; Jacobsen C. J.; Vogt S. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 15676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amos W.; Webb S.; Liu Y.; Andrews J. C.; LeDue D. L. Anal. Bioanal. Chem. 2012, 404, 1277. [DOI] [PubMed] [Google Scholar]
- Hama K.; Butler L. G. Nucl. Instrum. Methods Phys. Res., Sect. B 2007, 262, 117. [Google Scholar]
- Woll A. R.; Agyeman-Budu D.; Bilderback D. H.; Dale D.; Kazimirov A. Y.; Pfeifer M.; Plautz T.; Szebenyi T.; Untracht G. Proc. SPIE 2012, 8502, 85020K/1.Advances in X-Ray/EUV Optics and Components VII. [Google Scholar]
- Thomlinson W.; Suortti P.; Chapman D. Nucl. Instrum. Methods Phys. Res., Sect. A 2005, 543, 288. [Google Scholar]
- Cooper D. M. L.; Chapman L. D.; Carter Y.; Wu Y.; Panahifar A.; Britz H. M.; Bewer B.; Zhouping W.; Duke M. J. M.; Doschak M. Phys. Med. Biol. 2012, 57, 5777. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Zhu Y.; Bewer B.; Zhang L.; Korbas M.; Pickering I. J.; George G. N.; Gupta M.; Chapman D. Nucl. Instrum. Methods Phys. Res., Sect. A 2008, 594, 283. [Google Scholar]
- Bewer B.; Zhang H.; Zhu Y.; Zhang L.; George G. N.; Pickering I. J.; Chapman D. Rev. Sci. Instrum. 2008, 79, 085102/1. [DOI] [PubMed] [Google Scholar]
- Hokura A.; Onuma R.; Kitajima N.; Terada Y.; Saito H.; Abe T.; Yoshida S.; Nakai I. Chem. Lett. 2006, 35, 1246. [Google Scholar]
- Fukuda N.; Hokura A.; Terada Y.; Saito H.; Abe T.; Nakai I. J. Anal. At. Spectrom. 2008, 23, 1068. [Google Scholar]
- Tse J. J.; Gallego-Gallegos M.; Franz E. D.; Liber K.; Pickering I. J. Chemosphere 2012, 89, 274. [DOI] [PubMed] [Google Scholar]
- Vogt S.; Maser J.; Jacobsen C. J. Phys. IV 2003, 104, 617. [Google Scholar]
- Ward J.; Marvin R.; O’Halloran T.; Jacobsen C.; Vogt S. Microsc. Microanal. 2013, 19, 1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbas M.; Blechinger S.; Krone P. H.; Pickering I. J.; George G. N. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush V. J.; Moyer T. P.; Batts K. P.; Parisi J. E. Clin. Chem. 1995, 41, 284. [PubMed] [Google Scholar]
- Gellein K.; Flaten T. P.; Erikson K. M.; Aschner M.; Syversen T. Biol. Trace Elem. Res. 2008, 121, 221. [DOI] [PubMed] [Google Scholar]
- Seemann J.; Wittig P.; Kollmeier H.; Müller K.-M.; Schejbal V. Pathol. Res. Pract. 1990, 186, 197. [DOI] [PubMed] [Google Scholar]
- Szczerbowska-Boruchowska M. X-ray Spectrom. 2012, 41, 328. [Google Scholar]
- McColl G.; James S. A.; Mayo S.; Howard D. L.; Ryan C. G.; Kirkham R.; Moorhead G. F.; Paterson D.; de Jonge M. D.; Bush A. I. PloS One 2012, 7, e32685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James S. A.; de Jonge M. D.; Howard D. L.; Bush A. I.; Paterson D.; McColl G. Metallomics 2013, 5, 627. [DOI] [PubMed] [Google Scholar]
- Duong T. T. H.; Witting P. K.; Antao S. T.; Parry S. N.; Kennerson M.; Lai B.; Vogt S.; Lay P. A.; Harris H. H. J. Neurochem. 2009, 108, 1143. [DOI] [PubMed] [Google Scholar]
- Matsuyama S.; Shimura M.; Fujii M.; Maeshima K.; Yumoto H.; Mimura H.; Sano Y.; Yabashi M.; Nishino Y.; Tamasaku K.; Ishizaka Y.; Ishikawa T.; Yamauchi K. X-ray Spectrom. 2010, 39, 260. [Google Scholar]
- James S. A.; Myers D. E.; de Jonge M. D.; Vogt S.; Ryan C. G.; Sexton B. A.; Hoobin P.; Paterson D.; Howard D. L.; Mayo S. C. Anal. Bioanal. Chem. 2011, 401, 853. [DOI] [PubMed] [Google Scholar]
- Leskovjan A. C.; Kretlow A.; Lanzirotti A.; Barrea R.; Vogt S.; Miller L. M. NeuroImage 2011, 55, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leskovjan A. C.; Lanzirotti A.; Miller L. M. NeuroImage 2009, 47, 1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett M. J.; McQuillan J. A.; El-Assaad F.; Aitken J. B.; Levina A.; Cohen D. D.; Siegele R.; Carter E. A.; Grau G. E.; Hunt N. H.; Lay P. A. Analyst 2011, 136, 2941. [DOI] [PubMed] [Google Scholar]
- Chwiej J.; Szczerbowska-Boruchowska M.; Lankosz M.; Wojcik S.; Falkenberg G.; Stegowski Z.; Setkowicz Z. Spectrochim. Acta, Part B 2005, 60, 1531. [Google Scholar]
- Hackett M. J.; Smith S. E.; Paterson P. G.; Nichol H.; Pickering I. J.; George G. N. ACS Chem. Neurosci. 2012, 3, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacquart T.; Deves G.; Carmona A.; Tucoulou R.; Bohic S.; Ortega R. Anal. Chem. 2007, 79, 7353. [DOI] [PubMed] [Google Scholar]
- Carter E. A.; Rayner B. S.; McLeod A. I.; Wu L. E.; Marshall C. P.; Levina A.; Aitken J. B.; Witting P. K.; Lai B.; Cai Z.; Vogt S.; Lee Y. C.; Chen C. I.; Tobin M. J.; Harris H. H.; Lay P. A. Mol. BioSyst. 2010, 6, 1316. [DOI] [PubMed] [Google Scholar]
- Matyjaszewski M.; Wang J.-L.; Grimaud T.; Shipp D. A. Macromolecules 1998, 31, 1527. [Google Scholar]
- Thiele U. K. Chem. Fibres Int. 2004, 54, 162. [Google Scholar]
- Shotyk W.; Krachler M. Environ. Sci. Technol. 2007, 41, 1560. [DOI] [PubMed] [Google Scholar]
- Sarret G.; Pilon Smits E. A. H.; Castillo Michel H.; Isaure M. P.; Zhao F. J.; Tappero R. Adv. Agronomy 2013, 119, 1. [Google Scholar]
- Salt D. E.; Blaylock M.; Kumar N. P. B. A.; Dushenkov V.; Ensley B. D.; Chet I.; Raskin I. Nat. Biotechnol. 1995, 13, 468. [DOI] [PubMed] [Google Scholar]
- Gupta A. K.; Verma S. K.; Khan K.; Verma R. K. Environ. Sci. Technol. 2013, 47, 10115. [DOI] [PubMed] [Google Scholar]
- Jaffré T.; Brooks R. R.; Lee J.; Reeves R. D. Science 1976, 193, 579. [DOI] [PubMed] [Google Scholar]
- Jaffré T.; Pillon Y.; Thomine S.; Merlot S. Front. Plant Sci. 2013, 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer U.; Pickering I. J.; Prince R. C.; Raskin I.; Salt D. E. Plant Physiol. 2000, 122, 1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na G.; Salt D. E. J. Biol. Chem. 2011, 286, 40423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu C.; Ma L. Q. J. Environ. Qual. 2002, 31, 641. [PubMed] [Google Scholar]
- Trelease S. F.; Di Somma A. A.; Jacobs A. L. Science 1960, 132, 618. [DOI] [PubMed] [Google Scholar]
- Vonderheide A. P.; Wrobel K.; Kannamkumarath S. S.; B’Hymer C.; Montes-Bayon M.; Ponce de Leon C.; Caruso J. A. J. Agric. Food Chem. 2002, 50, 5722. [DOI] [PubMed] [Google Scholar]
- Kerdel-Vegas F. J. Invest. Dermatol. 1964, 42, 91. [DOI] [PubMed] [Google Scholar]
- Kerdel-Vegas F. Econ. Bot. 1966, 20, 187. [Google Scholar]
- Mueller D.; Desel H. Hum. Exp. Toxicol. 2010, 29, 431. [DOI] [PubMed] [Google Scholar]
- Aronow L.; Kerdel-Vegas F. Nature 1965, 205, 1186. [Google Scholar]
- Kerdel-Vegas F.; Wagner F.; Russell P. B.; Grant N. H.; Alburn H. E.; Clark D. E.; Miller J. A. Nature 1965, 205, 1186. [Google Scholar]
- Moxon A. L. S. Dak. Agr. Expt. Sta. Bull. 1937, 311, 3. [Google Scholar]
- Rosenfeld I.; Beath O. A.. Selenium. Geobotany, Biochemistry, Toxicity, and Nutrition; Academic Press: New York, 1964. [Google Scholar]
- O’Toole D.; Raisbeck M.; Case J. C.; Whitson T. D. Vet. Pathol. 1996, 33, 104. [DOI] [PubMed] [Google Scholar]
- Trelease S. F.; Di Somma A. A.; Jacobs A. L. Science 1960, 132, 618. [DOI] [PubMed] [Google Scholar]
- Nigam S. N.; McConnell W. B. Biochim. Biophys. Acta 1969, 192, 185. [DOI] [PubMed] [Google Scholar]
- Neuhier B.; Thanbichler M.; Lottspeich F.; Bock A. J. Biol. Chem. 1999, 274, 5407. [DOI] [PubMed] [Google Scholar]
- Evans C. S.; Asher C. J.; Johnson C. M. Aust. J. Biol. Sci. 1968, 21, 13. [Google Scholar]
- Pickering I. J.; Wright C.; Bubner B.; Ellis D.; Persans M. W.; Yu E. Y.; George G. N.; Prince R. C.; Salt D. E. Plant Physiol. 2003, 131, 1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman J. L.; Zhang L. H.; Marcus M. A.; Fakra S.; McGrath S. P.; Pilon-Smits E. A. H. Plant Physiol. 2006, 142, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman J. L.; Tamaoki M.; Stushnoff C.; Quinn C. F.; Cappa J. J.; Devonshire J.; Fakra S. C.; Marcus M. A.; McGrath S. P.; Van Hoewyk D.; Pilon-Smits E. A. Plant Physiol. 2010, 153, 1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez Barillas J. R.; Quinn C. F.; Freeman J. L.; Lindblom S. D.; Fakra S. C.; Marcus M. A.; Gilligan T. M.; Alford E. R.; Wangeline A. L.; Pilon-Smits E. A. H. Plant Physiol. 2012, 159, 1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblom S. D.; Valdez-Barillas J. R.; Fakra S. C.; Marcus M. A.; Wangeline A. L.; Pilon-Smits E. A. H. Environ. Exp. Bot. 2013, 88, 33. [Google Scholar]
- Freeman J. L.; Marcus M. A.; Fakra S. C.; Devonshire J.; McGrath S. P.; Quinn C. F.; Pilon-Smits E. A. H. PLoS One 2012, 7, e50516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sors T. G.; Ellis D. R.; Salt D. E. Photosynth. Res. 2005, 86, 373. [DOI] [PubMed] [Google Scholar]
- de Souza M. P.; Lytle M. C.; Mulholland M. M.; Otte M. L.; Terry N. Plant Physiol. 2000, 122, 1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bañuelos G. S.; Fakra S. C.; Walse S. S.; Marcus M. A.; Yang S. I.; Pickering I. J.; Pilon-Smits E. A. H.; Freeman J. L. Plant Physiol. 2011, 155, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J.; Hu Y.; Li Y.; Li B.; Dong Y.; Chai Z. Metallomics 2013, 5, 869. [DOI] [PubMed] [Google Scholar]
- Carey A.-M.; Scheckel K. G.; Lombi E.; Newville M.; Choi Y.; Norton G. J.; Price A. H.; Meharg A. A. Environ. Sci. Technol. 2012, 46, 5557. [DOI] [PubMed] [Google Scholar]
- Thavarajah D.; Vandenberg A.; George G. N.; Pickering I. J. J. Agric. Food Chem. 2007, 55, 7337. [DOI] [PubMed] [Google Scholar]
- Hokura A.; Omuma R.; Terada Y.; Kitajima N.; Abe T.; Saito H.; Yoshida S.; Nakai I. J. Anal. At. Spectrom. 2006, 21, 321. [Google Scholar]
- Kachenko A. G.; Grafe M.; Singh B.; Heald S. M. Environ. Sci. Technol. 2010, 44, 4735. [DOI] [PubMed] [Google Scholar]
- Raven P. H.; Evert R. F.; Eichhorn S. E.. Biology of Plants, 5th ed.; Worth: New York, 1992. [Google Scholar]
- Lane T.; Saito M. A.; George G. N.; Pickering I. J.; Prince R. C.; Morel F. F. M. Nature 2005, 435, 42. [DOI] [PubMed] [Google Scholar]
- Pickering I. J.; Prince R. C.; George G. N.; Rauser W. E.; Wickramasinge W. A.; Watson A. A.; Dameron C. T.; Dance I. G.; Fairlie D. P.; Salt D. E. Biochim. Biophys. Acta 1999, 1429, 351. [DOI] [PubMed] [Google Scholar]
- Salt D. E.; Prince R. C.; Pickering I. J.; Raskin I. Plant. Physiol. 1995, 109, 1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokura A. J. Cryst. Soc. Jpn. 2011, 53, 416. [Google Scholar]
- Harada E.; Hokura A.; Takada S.; Baba K.; Terada Y.; Nakai I.; Yazaki K. Plant Cell Physiol. 2010, 51, 848. [DOI] [PubMed] [Google Scholar]
- Harada E.; Hokura A.; Nakai I.; Terada Y.; Baba K.; Yazaki K.; Shiono M.; Mizuno N.; Mizuno T. Metallomics 2011, 3, 1340. [DOI] [PubMed] [Google Scholar]
- Tian S.; Lu L.; Labavitch J.; Yang X.; He Z.; Hu H.; Sarangi R.; Newville M.; Commisso J.; Brown P. Plant Physiol. 2011, 157, 1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer U.; Pickering I. J.; Prince R. C.; Raskin I.; Salt D. E. Plant Physiol. 2000, 122, 1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salt D. E.; Prince R. C.; Baker A. J. M.; Raskin I.; Pickering I. J. Environ. Sci. Technol. 1999, 33, 713. [Google Scholar]
- Ebbs S.; Lau I.; Ahner B.; Kochian L. Planta 2002, 214, 635. [DOI] [PubMed] [Google Scholar]
- Salt D. E.; Prince R. C.; Pickering I. J.; Raskin I. Plant Physiol. 1995, 109, 1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering I. J.; Prince R. C.; George M. J.; Smith R. D.; George G. N.; Salt D. E. Plant Physiol. 2000, 122, 1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris H. H.; Pickering I. J.; George G. N. Science 2003, 381, 1203. [DOI] [PubMed] [Google Scholar]
- Hoffman R. S. J. Toxicol., Clin. Toxicol. 2000, 38, 767. [DOI] [PubMed] [Google Scholar]
- Clarkson T. W. J. Trace Elem. Exp. Med. 1998, 11, 303. [Google Scholar]
- Clarkson T. W. Environ. Health Perspect. 2002, 110, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchounwou P. B.; Ayensu W. K.; Ninashvili N.; Sutton D. Environ. Toxicol. 2003, 18, 149. [DOI] [PubMed] [Google Scholar]
- Harada M. Crit. Rev. Toxicol. 1995, 25, 1. [DOI] [PubMed] [Google Scholar]
- Clarkson T. W.; Magos L.; Myers G. J. N. Engl. J. Med. 2003, 349, 1731. [DOI] [PubMed] [Google Scholar]
- Weiss B.; Clarkson T. W.; Simon W. Environ. Health Perspect. 2002, 110, 851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nierenberg D. W.; Nordgren R. E.; Chang M. B.; Siegler R. W.; Blayney M. B.; Hochberg F.; Toribara T. Y.; Crenichiari E.; Clarkson T. N. Engl. J. Med. 1998, 338, 1672. [DOI] [PubMed] [Google Scholar]
- Hill A. J.; Teraoka H.; Heideman W.; Peterson R. E. Toxicol. Sci. 2005, 86, 6. [DOI] [PubMed] [Google Scholar]
- Zon L. I.; Peterson R. T. Nat. Rev. Drug Discovery 2005, 4, 35. [DOI] [PubMed] [Google Scholar]
- Korbas M.; Krone P. H.; Pickering I. J.; George G. N. J. Biol. Inorg. Chem. 2010, 15, 1137. [DOI] [PubMed] [Google Scholar]
- Korbas M.; O’Donoghue J. L.; Watson G. E.; Pickering I. J.; Singh S. P.; Myers G. J.; Clarkson T. W.; George G. N. ACS Chem. Neurosci. 2010, 1, 810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George G. N.; Singh S. P.; Hoover J.; Pickering I. J. Chem. Res. Toxicol. 2009, 22, 1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris H. H.; Vogt S.; Eastgate H.; Legnini D. G.; Hornberger B.; Cai Z.; Lai B.; Lay P. A. J. Synchrotron Radiat. 2008, 15, 123. [DOI] [PubMed] [Google Scholar]
- George G. N.; Pickering I. J.; Doonan C. J.; Korbas M.; Singh S. P.; Hoffmeyer R. Adv. Mol. Toxicol. 2008, 2, 125. [Google Scholar]
- Ord M. G.; Stocken L. A. Trends Biochem. Sci. 2000, 25, 253. [DOI] [PubMed] [Google Scholar]
- Saha K. C. J. Environ. Sci. Health, Part A 2003, 38, 255. [DOI] [PubMed] [Google Scholar]
- Guo H. R.; Wang N. S.; Hu H.; Monson R. R. Cancer Epidemiol., Biomarkers Prev. 2004, 13, 638. [PubMed] [Google Scholar]
- Guo H. R.; Yu H. S.; Hu H.; Monson R. R. Cancer, Causes Control 2001, 12, 909. [DOI] [PubMed] [Google Scholar]
- Chiou H. Y.; Chiou S. T.; Hsu Y. H.; Chou Y. L.; Tseng C. H.; Wei M. L.; Chen C. J. Am. J. Epidemiol. 2001, 153, 411. [DOI] [PubMed] [Google Scholar]
- Chung C.-J.; Huang C.-Y.; Pu Y.-S.; Shiue H.-S.; Su C.-T.; Hsueh Y.-M. Toxicol. Appl. Pharmacol. 2012, 266, 254. [DOI] [PubMed] [Google Scholar]
- Rossman T. G.; Uddin A. N.; Burns F. J.; Bosland M. C. Toxicol. Appl. Pharmacol. 2001, 176, 64. [DOI] [PubMed] [Google Scholar]
- Rossman T. G.; Uddin A. N.; Burns F. J. Toxicol. Appl. Pharmacol. 2004, 198, 394. [DOI] [PubMed] [Google Scholar]
- Kitchin K. T. Toxicol. Appl. Pharmacol. 2001, 172, 249. [DOI] [PubMed] [Google Scholar]
- Smith P. G.; Koch I.; Reimer K. J. Sci. Total Environ. 2008, 390, 198. [DOI] [PubMed] [Google Scholar]
- Munro K. L.; Mariana A.; Klavins A. I.; Foster A. J.; Lai B.; Vogt S.; Cai Z.; Harris H. H.; Dillon C. T. Chem. Res. Toxicol. 2008, 21, 1760. [DOI] [PubMed] [Google Scholar]
- Moxon A. L. Science 1938, 88, 81. [DOI] [PubMed] [Google Scholar]
- Gailer J. Coord. Chem. Rev. 2007, 251, 234. [Google Scholar]
- Gailer J.; George G. N.; Pickering I. J.; Prince R. C.; Ringwald S. C.; Pemberton J.; Glass R. S.; Younis H. S.; DeYoung D. W.; Aposhian H. V. J. Am. Chem. Soc. 2000, 122, 4637. [Google Scholar]
- Manley S.; George G. N.; Pickering I. J.; Prince R. C.; Glass R. S.; Prenner E. J.; Yamdagni R.; Wu Q.; Gailer J. Chem. Res. Toxicol. 2006, 19, 601. [DOI] [PubMed] [Google Scholar]
- Gailer J.; George G. N.; Pickering I. J.; Prince R. C.; Younis H. S.; Winzerling J. J. Chem. Res. Toxicol. 2002, 15, 1466. [DOI] [PubMed] [Google Scholar]
- Carew M. W.; Leslie E. M. Carcinogenesis 2010, 31, 1450. [DOI] [PubMed] [Google Scholar]
- Uddin A. N.; Burns F. J.; Rossman T. G. Carcinogenesis 2005, 26, 2179. [DOI] [PubMed] [Google Scholar]
- Burns F. J.; Rossman T.; Vega K.; Uddin A.; Vogt S.; Lai B.; Reeder R. J. Environ. Health Perspect. 2008, 116, 703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempson I. M.; Dermot H. A. Angew. Chem., Int. Ed. 2010, 49, 4237. [DOI] [PubMed] [Google Scholar]
- Yang C.-T.; Chuang K.-H. Med. Chem. Commun. 2012, 3, 552. [Google Scholar]
- Li A.; Wong C. S.; Wong M. K.; Lee C. M.; Au Yeung M. C. Br. J. Radiol. 2006, 76, 368. [DOI] [PubMed] [Google Scholar]
- Joffe P.; Thomsen H. S.; Meusel M. Acad. Radiol. 1998, 5, 491. [DOI] [PubMed] [Google Scholar]
- Grobner T.; Prisch F. C. Kidney Int. 2007, 72, 260. [DOI] [PubMed] [Google Scholar]
- Marckmann P.; Skov L.; Rossen K.; Dupont A.; Damholt M. B.; Heaf J. G.; Thomsen H. S. J. Am. Soc. Nephrol. 2006, 17, 2359. [DOI] [PubMed] [Google Scholar]
- Grobner T. Nephrol., Dial., Transplant. 2006, 21, 1104. [DOI] [PubMed] [Google Scholar]
- High W. A.; Ranville J. F.; Brown M.; Punshon T.; Lanzirotti A.; Jackson B. P. J. Am. Acad. Dermatol. 2010, 62, 38. [DOI] [PubMed] [Google Scholar]
- George S. J.; Webb S. M.; Abraham J. L.; Cramer S. P. Br. J. Dermatol. 2010, 163, 1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George G. N.; Prince R. C.; Gailer J.; Buttigieg G. A.; Denton M. B.; Harris H. H.; Pickering I. J. Chem. Res. Toxicol. 2004, 17, 999. [DOI] [PubMed] [Google Scholar]
- Fu J.; Hoffmeyer R. E.; Pushie M. J.; Singh S. P.; Pickering I. J.; George G. N. J. Biol. Inorg. Chem. 2011, 16, 15. [DOI] [PubMed] [Google Scholar]
- Bourassa M. W.; Miller L. M. Metallomics 2012, 4, 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu B. F.; Nichol H. CNS Neurosci. Ther. 2011, 17, 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu B. F.; Robinson C. A.; Rajput A.; Rajput A. H.; Harder S. L.; Nichol H. Cerebellum 2009, 8, 74. [DOI] [PubMed] [Google Scholar]
- Ortega R.; Cloetens P.; Devès G.; Carmona A.; Bohic S. PLoS One 2007, 9, e925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona A.; Cloetens P.; Devès G.; Bohic S.; Ortega R. J. Anal. At. Spectrom. 2008, 23, 1083. [Google Scholar]
- Norrby E. J. Int. Med. 2011, 270, 1. [DOI] [PubMed] [Google Scholar]
- Brown D. R.; Qin K.; Herms J. W.; Madlung A.; Manson J.; Strome R.; Fraser P. E.; Kruck T.; von Bohlen A.; Schulz-Schaeffer W.; Giese A.; Westaway D.; Kretzschmar H. Nature 1997, 390, 684. [DOI] [PubMed] [Google Scholar]
- Pushie M. J.; Pickering I. J.; Tsutsui S.; Martin G. R.; Jirik F. R.; George G. N. Metallomics 2011, 3, 206. [DOI] [PubMed] [Google Scholar]
- Davies K. M.; Bohic S.; Carmona A.; Ortega R.; Cottam V.; Hare D. J.; Finberg J. P. M.; Reyes S.; Halliday G. M.; Mercer J. F. B.; Double K. L. Neurobiol. Aging 2014, 35, 858. [DOI] [PubMed] [Google Scholar]
- Bourassa M. W.; Miller L. M. Metallomics 2012, 4, 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller L. M.; Wang Q.; Telivala T. P.; Smith R. J.; Lanzirotti A.; Miklossy J. J. Struct. Biol. 2006, 155, 30. [DOI] [PubMed] [Google Scholar]
- Bourassa M. W.; Leskovjan A. C.; Tappero R. V.; Farquhar E. R.; Colton C. A.; Van Nostrand W. E.; Miller L. M. Biomed. Spectrosc. Imaging 2013, 2, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silasi G.; Klahr A. C.; Hackett M. J.; Auriat A. M.; Nichol H.; Colbourne F. J. Cereb. Blood Flow Metab. 2012, 32, 1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chwiej J.; Dulinska J.; Janeczko K.; Appel K.; Setkowicz Z. J. Biol. Inorg. Chem. 2012, 17, 731. [DOI] [PubMed] [Google Scholar]
- Chwiej J.; Kutorasinska J.; Janeczko K.; Gzielo-Jurek K.; Uram L.; Appel K.; Simon R.; Setkowicz Z. Anal. Bioanal. Chem. 2012, 404, 3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chwiej J.; Kutorasinska J.; Janeczko K.; Gzielo-Jurek K.; Uram L.; Appel K.; Simon R.; Setkowicz Z. Anal. Bioanal. Chem. 2012, 404, 3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chwiej J.; Winiarski W.; Ciarach M.; Janeczko K.; Lankosz M.; Rickers K.; Setkowicz Z. J. Biol. Inorg. Chem. 2008, 13, 1267. [DOI] [PubMed] [Google Scholar]
- Chwiej J.; Sarapata A.; Janeczko K.; Stegowski Z.; Appel K.; Setkowicz Z. J. Biol. Inorg. Chem. 2011, 16, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan E. J.; Austin C. J. D.; Aitken J. B.; Vogt S.; Jolliffe K. A.; Harris H. H.; Rendina L. M. J. Synchrotron Radiat. 2013, 20, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley E. L.; Aitken J. B.; Vogt S.; Harris H. H.; Rendina L. M. Angew. Chem., Int. Ed. 2010, 49, 1231. [DOI] [PubMed] [Google Scholar]
- Alderden R. A.; Mellor H. R.; Modok S.; Hall M. D.; Sutton S. R.; Newville M. G.; Callaghan R.; Hambley T. W. J. Am. Chem. Soc. 2007, 129, 13400. [DOI] [PubMed] [Google Scholar]
- Zhang J. Z.; Bryce N. S.; Lanzirotti A.; Chen C. K. J.; Paterson D.; de Jonge M. D.; Howard D. L.; Hambley T. W. Metallomics 2012, 4, 1209. [DOI] [PubMed] [Google Scholar]
- Cocchietto M.; Zorzet S.; Sorc A.; Sava G. Invest. New Drugs 2003, 21, 55–62. [DOI] [PubMed] [Google Scholar]
- Hartinger C. G.; Jakupec M. A.; Zorbas-Seifried S.; Groessl M.; Egger A.; Berger W.; Zorbas H.; Dyson P. J.; Keppler B. K. Chem. Biodiversity 2008, 5, 2140. [DOI] [PubMed] [Google Scholar]
- Aitken J. B.; Antony S.; Weekley C. M.; Lai B.; Spiccia L.; Harris H. H. Metallomics 2012, 4, 1051. [DOI] [PubMed] [Google Scholar]
- Levina A.; Aitken J. B.; Gwee Y. Y.; Lim Z. J.; Liu M.; Singharay A. M.; Wong P. F.; Lay P. A. Chem.—Eur. J. 2013, 19, 3609. [DOI] [PubMed] [Google Scholar]
- Antony S.; Aitken J. B.; Vogt S.; Lai B.; Brown T.; Spiccia L.; Harris H. H. J. Biol. Inorg. Chem. 2013, 18, 845. [DOI] [PubMed] [Google Scholar]
- O’Collins V. E.; Macleod M. R.; Donnan G. A.; Horky L. L.; van der Worp B. H.; Howells D. W. Ann. Neurol. 2006, 59, 467. [DOI] [PubMed] [Google Scholar]
- Parnham M. J.; Sies H. Biochem. Pharmacol. 2013, 86, 1248. [DOI] [PubMed] [Google Scholar]
- Aitken J. B.; Lay P. A.; Hong Duong T. T.; Aran R.; Witting P. K.; Harris H. H.; Lai B.; Vogt S.; Giles G. I. J. Biol. Inorg. Chem. 2012, 17, 589. [DOI] [PubMed] [Google Scholar]
- Rana R. R.; Zhang M.; Spear A. M.; Atkins H. S.; Byrne B. Med. Microbiol. Immunol. 2013, 202, 1. [DOI] [PubMed] [Google Scholar]
- Ball K. R.; Sampieri F.; Chirino M.; Hamilton D. L.; Blyth R. I.; Sham T. K.; Dowling P. M.; Thompson J. Antimicrob. Agents Chemother. 2013, 57, 5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich P. Wien. Med. Wochenschr. 1911, 61, 14. [Google Scholar]
- Lloyd N. C.; Morgan H. W.; Nicholson B. K.; Ronimus R. S. Angew. Chem., Int. Ed. 2005, 44, 941. [DOI] [PubMed] [Google Scholar]
- Swindell E. P.; Hankins P. L.; Chen H.; Miodragovic D. U.; O’Halloran T. V. Inorg. Chem. 2013, 52, 12292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro K. L.; Dillon C. T.; Harris H. H.; Cai Z.; Lai B.; Vogt S.; Cheah M. In Understanding the Geological and Medical Interface of Arsenic - As 2012: Proceedings of the 4th International Congress on Arsenic in the Environment, Cairns, Australia; Ng J. C., Noller B. N., Naidu R., Bundschuh J., Bhattacharya P., Eds.; CRC Press: Boca Raton, FL, 2012; pp 193–195. [Google Scholar]
- Weekley C. M.; Aitken J. B.; Finney L.; Witting P. K.; Harris H. H. Nutrients 2013, 5, 1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weekley C. M.; Harris H. H. Chem. Soc. Rev. 2013, 42, 8801. [DOI] [PubMed] [Google Scholar]
- Weekley C. M.; Aitken J. B.; Vogt S.; Finney L. A.; Paterson D. J.; de Jonge M. D.; Howard D. L.; Musgrave I. F.; Harris H. H. Biochemistry 2011, 50, 1641. [DOI] [PubMed] [Google Scholar]
- Weekley C. M.; Aitken J. B.; Vogt S.; Finney L. A.; Paterson D. J.; de Jonge M. D.; Howard D. L.; Witting P. K.; Musgrave I. F.; Harris H. H. J. Am. Chem. Soc. 2011, 133, 18272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weekley C. M.; Shanu A.; Aitken J. B.; Vogt S.; Witting P. K.; Harris H. H. Metallomics 2014, 10.1039/c4mt00088a. [DOI] [PubMed] [Google Scholar]
- Bohic S.; Murphy K.; Paulus W.; Cloetens P.; Salome M.; Susini J.; Double K. Anal. Chem. 2008, 80, 9557. [DOI] [PubMed] [Google Scholar]
- Kehr S.; Malinouski M.; Finney L.; Vogt S.; Labunskyy V. M.; Kasaikina M. V.; Carlson B. A.; Zhou Y.; Hatfield D. L.; Gladyshev V. N. J. Mol. Biol. 2009, 389, 808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin R. G.; Wolford J. L.; Kidd M. J.; Murphy S.; Ward J.; Que E. L.; Mayer M. L.; Penner-Hahn J. E.; Haldar K.; O’Halloran T. V. Chem. Biol. 2012, 19, 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega R. Metallomics 2009, 1, 137. [DOI] [PubMed] [Google Scholar]
- Finney L.; Chishti Y.; Khare T.; Giometti C.; Levina A.; Lay P. A.; Vogt S. ACS Chem. Biol. 2010, 5, 577. [DOI] [PubMed] [Google Scholar]
- Raimunda D.; Khare T.; Giometti C.; Vogt S.; Argüello J. M.; Finney L. Metallomics 2012, 4, 921. [DOI] [PubMed] [Google Scholar]
- Hopp K.; Popescu B. F.; McCrea R. P.; Harder S. L.; Robinson C. A.; Haacke M. E.; Rajput A. H.; Rajput A.; Nichol H. J. Mag. Res. Imaging 2010, 31, 1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W.; Nichol H.; Liu S.; Cheng Y.-C. N.; Haacke E. M. NeuroImage 2013, 78, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z.; Caruso J. A.; Lai B.; Matusch A.; Becker J. S. Metallomics 2011, 3, 28. [DOI] [PubMed] [Google Scholar]
- Becker J. S.; Matusch A.; Palm C.; Salber D.; Morton K. A.; Becker J. S. Metallomics 2010, 2, 104. [DOI] [PubMed] [Google Scholar]
- Hare D.; Austin C.; Doble P. Analyst 2012, 137, 1527. [DOI] [PubMed] [Google Scholar]
- Chaurand P.; Caprioli R. M. Electrophoresis 2002, 23, 3125. [DOI] [PubMed] [Google Scholar]
- Jurchen J. C.; Rubakhin S. S.; Sweedler J. V. J. Am. Soc. Mass Spectrom. 2005, 16, 1654. [DOI] [PubMed] [Google Scholar]
- Luxembourg S. L.; Mize T. H.; McDonnell L. A.; Heeren R. M. Anal. Chem. 2004, 76, 5339. [DOI] [PubMed] [Google Scholar]
- Clemmer D. E.; Jarrold M. F. J. Mass Spectrom. 1997, 32, 577. [Google Scholar]
- Terai T.; Nagano T. Pfluegers Arch. 2013, 465, 347. [DOI] [PubMed] [Google Scholar]
- Nolan E. M.; Lippard S. J. Chem. Rev. 2008, 108, 3443. [DOI] [PubMed] [Google Scholar]
- Yoon S.; Albers A. E.; Wong A. P.; Chang C. J. J. Am. Chem. Soc. 2005, 127, 16030. [DOI] [PubMed] [Google Scholar]
- Yoon S.; Miller E. W.; He Q.; Do P. H.; Chang C. J. Angew. Chem., Int. Ed. 2007, 46, 6658. [DOI] [PubMed] [Google Scholar]
- Ko S.-K.; Yang Y.-K.; Tae J.; Shin I. J. Am. Chem. Soc. 2006, 128, 14150. [DOI] [PubMed] [Google Scholar]
- Song F.; Watanabe S.; Floreancig P. E.; Koide K. J. Am. Chem. Soc. 2008, 130, 16460. [DOI] [PubMed] [Google Scholar]
- Ando S.; Koide K. J. Am. Chem. Soc. 2011, 133, 2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price K. A.; Hickey J. L.; Xiao Z.; Wedd A. G.; James S. A.; Liddell J. R.; Crouch P. J.; White A. R.; Donnelly P. S. Chem. Sci. 2012, 3, 2748. [Google Scholar]
- Miller E. W.; Zeng L.; Domaille D. W.; Chang C. J. Nat. Protoc. 2006, 1, 824. [DOI] [PubMed] [Google Scholar]
- Yang L.; McRae R.; Henary M. M.; Patel R.; Lai B.; Vogt S.; Fahrni C. J. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederickson C. J.; Koh J.-Y.; Bush A. I. Nat. Rev. Neurosci. 2005, 6, 449. [DOI] [PubMed] [Google Scholar]
- Pérez-Clausell J.; Danscher G. Brain Res. 1985, 337, 91. [DOI] [PubMed] [Google Scholar]
- Danscher G. Histochemistry 1981, 71, 1. [DOI] [PubMed] [Google Scholar]
- Meeusen J. W.; Tomasiewicz H.; Nowakowski A.; Petering D. H. Inorg. Chem. 2011, 50, 7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J. H.; Sensi S. L.; Koh J. Y. Trends Pharmacol. Sci. 2000, 21, 395. [DOI] [PubMed] [Google Scholar]
- Meeusen J. W.; Nowakowski A.; Petering D. H. Inorg. Chem. 2012, 51, 3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowski A. B.; Petering D. H. Inorg. Chem. 2011, 50, 10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman L. M.; Franz K. J. Coord. Chem. Rev. 2012, 256, 2333–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole T. B.; Wenzel H. J.; Kafer K. E.; Schwartzkroin P. A.; Palmiter R. D. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmiter R. D.; Cole T. B.; Quaife C. J.; Findley S. D. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 14934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkous D. H.; Flinn J. M.; Koh J. Y.; Lanzirotti A.; Bertsch P. M.; Jones B. F.; Giblin L. J.; Frederickson C. J. J. Histochem. Cytochem. 2008, 56, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasse M. J.; Reininger R.; Kubala T.; Janowski S.; Hirschmugl C. Nucl. Instrum. Methods Phys. Res., Sect. A 2007, 582, 107. [Google Scholar]
- Miller L. M.; Dumas P. Biochim. Biophys. Acta 2006, 1758, 846. [DOI] [PubMed] [Google Scholar]
- Lasch P.; Naumann D. Biochim. Biophys. Acta 2006, 1758, 814–829. [DOI] [PubMed] [Google Scholar]
- Nasse M. J.; Walsh M. J.; Mattson E. C.; Reininger R.; Kajdacsy-Balla A.; Macias V.; Bhargava R.; Hirschmugl C. J. Nat. Methods 2011, 8, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassan P.; Lee J.; Sachdeva A.; Pissardini J.; Dorling K. M.; Fletcher J. S.; Henderson A.; Gardner P. Analyst 2012, 138, 144. [DOI] [PubMed] [Google Scholar]
- Miller L. M.; Wang Q.; Telivala T. P.; Smith R. J.; Lanzirotti A.; Miklossy J. J. Struct. Biol. 2006, 155, 30. [DOI] [PubMed] [Google Scholar]
- Szczerbowska-Boruchowska M.; Chwiej J.; Lankosz M.; Adamek D.; Wojcik S.; Krygowaska-Wajs A.; Tomik B.; Bohic S.; Susini J.; Simionovici A.; Dumas P. X-ray Spectrom. 2005, 34, 514. [Google Scholar]
- Chwiej J.; Dulinska J.; Janeczko K.; Dumas P.; Eichert D.; Dudala J.; Setkowicz Z. J. Chem. Neuroanat. 2010, 40, 140. [DOI] [PubMed] [Google Scholar]
- Chwiej J.; Winiarski W.; Ciarach M.; Janeczko K.; Lankosz M.; Rickers K.; Setkowicz Z. J. Biol. Inorg. Chem. 2008, 13, 1267. [DOI] [PubMed] [Google Scholar]
- Kastyak M. Z.; Szczerbowska-Boruchowska M.; Adamek D.; Tomik B.; Lankosz M.; Gough K. M. Neuroscience 2010, 166, 1119. [DOI] [PubMed] [Google Scholar]
- Zhang J. Z.; Bryce N. S.; Siegele R.; Carter E. A.; Paterson D.; de Jonge M. D.; Howard D. L.; Ryan C. G.; Hambley T. W. Integr. Biol. 2010, 4, 1072. [DOI] [PubMed] [Google Scholar]
- Dillon C. T. Aust. J. Chem. 2012, 65, 204. [Google Scholar]
- Aitken J. B.; Carter E. A.; Eastgate H.; Hackett M. J.; Harris H. H.; Levina A.; Lee Y. C.; Chen C. I.; Lai B.; Vogt S. Radiat. Phys. Chem. 2010, 79, 176. [Google Scholar]
- Yan L.; Zhao J.; Toellner T. S.; Divan R.; Xu S.; Cai Z.; Boesenberg J. S.; Friedrich J. M.; Cramer S. P.; Alp E. E. J. Synchrotron Radiat. 2012, 19, 814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui T.; Kobayashi Y.; Seto M. Jpn. J. Appl. Phys. 2004, 43, 389. [Google Scholar]
- Bergmann U.; Glatzel P.; Robblee J. H.; Messinger J.; Fernandez C.; Cinco R.; Visser H.; McFarlane K.; Bellacchio E.; Pizarro S.; Sauer K.; Yachandra V. K.; Klein M. P.; Cox B. L.; Nealson K. H.; Cramer S. P. J. Synchrotron Radiat. 2001, 8, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doonan C. J.; Zhang L.; Young C. G.; George S. J.; Deb A.; Bergmann U.; George G. N.; Cramer S. P. Inorg. Chem. 2005, 44, 2579. [DOI] [PubMed] [Google Scholar]
- Glatzel P.; Yano J.; Bergmann U.; Visser H.; Robblee J. H.; Gu W. W.; de Groot F. M. F.; Cramer S. P.; Yachandra V. J. Phys. Chem. Solids 2005, 66, 2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann U.; Glatzel P.; Cramer S. P. Microchem. J. 2002, 71, 221. [Google Scholar]