

Abstract
Mechanical forces influence many biological processes via activation of signaling molecules, including the family of Rho GTPases. Within the endothelium, the mechanical force of fluid shear stress regulates the spatiotemporal activation of Rho GTPases, including Rac1. Shear stress-induced Rac1 activation is required for numerous essential biological processes, including changes in permeability, alignment of the actin cytoskeleton, redox signaling, and changes in gene expression. Thus, identifying mechanisms of Rac1 activation and the spatial cues that direct proper localization of the GTPase is essential in order to gain a comprehensive understanding the role of Rac1 in shear stress responses. This commentary will highlight our current understanding of how Rac1 activity is regulated in response to shear stress, as well as the downstream consequences of Rac1 activation.
Keywords: Rac1, shear stress, mechanotransduction, polarity
Introduction
Rho GTPases are central regulators of the actin cytoskeleton and have been implicated in numerous cellular processes including proliferation, apoptosis, and changes in gene expression.1 Interestingly, all of these cellular processes are mechanically regulated. Therefore, it is not surprising that Rho GTPases, including Rac1, can be activated by mechanical forces and are essential for mediating several cellular responses to force. For instance, Rac1 is rapidly activated in response to tension on integrin receptors that couple the extraceullar matrix (ECM) to the actin cytoskeleton.2 In these studies, optical twisting cytometry was used to apply force to ECM-coated beads attached to integrin receptors on the surface of human airway smooth muscle cells. Force application on integrins induced rapid activation of Rac1.2 Other studies have highlighted the functional consequences of force-dependent Rac activation. For example, mechanical signaling via integrins promotes apoptosis by a Rac/PAK/p38 pathway.3 Rac1 has also been implicated in formation of adherens junctions at cell-cell contacts.4 Growth of adherens junctions promotes cell junction assembly by dissipating high forces that may otherwise disrupt cell-cell contacts and provides a platform for the assembly of structural and signaling complexes that are required for maintenance of cell-cell contacts. Importantly, mechanically tugging at cell-cell junctions activates Rac1 and activation of the GTPase is required for force-dependent growth of adherens junctions.5 Mechanical loading of bone influences bone growth and is essential for maintenance, and Rac1 mediates force-dependent shear stress signaling in osteoblasts.6 Rac1 is also activated in endothelial cells (ECs) as a result of fluid shear stress produced by blood flow,7-9 and flow-induced Rac1 activation in ECs is critical for numerous vascular responses. The mechanisms and consequences of flow-mediated Rac1 activation in the endothelium will be the focus of this commentary.
Mechanisms of Shear Stress-Mediated Rac1 Activation
Biochemical and immunofluorescence analyses have shown that Rac1 is activated in response to fluid shear stress. Previous studies indicated that the GTPase is activated within 5 min of exposure to shear stress and activity returns to basal levels at later time points.7,8 However, 10 y passed before the molecular mechanisms of flow-mediated Rac activation were investigated. Work over the past several years has identified a mechanosensory complex at cell-cell junctions that consists of platelet endothelial cell adhesion molecule-1 (PECAM-1), vascular endothelial cadherin (VE-cadherin), and vascular endothelial growth factor receptor 2 (VEGFR2).10 Previous studies have indicated that mechanosignaling initiated by this complex is required for two Rac-dependent processes in response to shear stress: alignment of the actin cytoskeleton in the direction of flow and activation of the transcription factor NF-κB.7,10 Thus, we recently set out to investigate the role of these junctional proteins (PECAM-1 and VE-cadherin) in shear stress-induced Rac1 activation. We revealed that PECAM-1 is required for Rac1 GTP-loading in response to shear stress, whereas, surprisingly, VE-cadherin is not.9 Our data suggest that PECAM-1 mechanosignaling results in Src activation, and Src phosphorylates and activates the Rac guanine nucleotide exchange factor (GEF) Vav2. In support of this model, ECs depleted of Vav2 do not activate Rac1 in response to shear stress. However, it is important to note that previous work indicated that new integrin-ligand binding also regulates shear-induced Rac1 activation.7 At this point, it is unknown if Vav2 activation is downstream of integrin activation or if multiple parallel mechanisms work in concert to regulate Rac1 activation in response to shear stress, but these studies are the focus of current investigations.
Rac1 Function in ECs and Shear Stress-Dependent Responses
Rac1 is a multifunctional protein that has been implicated in numerous EC functions. For example, Rac1−/− ECs exhibit impaired migration, adhesion, and permeability in response to vascular endothelial growth factor (VEGF) and sphingosine-1-phosphate (S1P).11 Other studies revealed reduced endothelial nitric oxide synthase expression (eNOS) and activity in Rac1 haploinsufficient ECs.12 Our recent work showed that shear stress-dependent Rac1 activation and polarization is essential for flow-induced ROS production. Therefore, the remainder of this commentary will focus on the role of the GTPase in shear stress-dependent EC responses.
Rac1 activation has been implicated in numerous EC responses to shear stress, including regulation of endothelial barrier function. Laminar shear stress increases endothelial cell barrier function,13 and changes in junctional integrity are mediated, in part, by RhoA and Rac1. RhoA activity is tightly linked to cell-generated contractility. Therefore, increased RhoA activity is thought to disrupt junctional integrity by increasing cell-generated forces that breakdown established junctions. Conversely, increased Rac1 activity stabilizes junctions and coincides with increased transendothelial resistance.13,14 Thus, in this context shear stress-induced activation of Rac appears to have anti-inflammatory effects on the endothelium, as stabilization of endothelial cell-cell junctions corresponds with decreased permeability and leukocyte transendothelial migration. However, it is interesting to note that shear stress-mediated Rac activation has also been linked to pro-inflammatory signaling, described in further detail below.
Shear stress regulates gene expression in ECs via activation of numerous transcription factors, including NF-κB.15,16 In unstimulated cells, the NF-κB heterodimer is normally sequestered to the cytoplasm via association with members of the IκB inhibitor proteins. Upon activation, IκB is phosphorylated and degraded, allowing NF-κB to translocate to the nucleus, where it can bind promoter elements in numerous genes. The transcription factor is activated by shear stress, and regulates expression of the cell adhesion molecules ICAM-1, VCAM-1, and E-selectin, which promote leukocyte adhesion and transendothelial migration.7,17,18 To this end, NF-κB activation has been directly linked to atherosclerotic plaque formation in vivo.19 Previous studies have demonstrated that Rac1 is required for cytokine-stimulated NF-κB activation and we have shown that Rac mediates shear stress-induced NF-κB activation. Expression of a dominant negative Rac1 mutant (N17Rac) blunted flow-mediated NF-κB translocation to the nucleus and, thus, also inhibited flow-induced upregulation of the cell adhesion molecule ICAM-1.7 These data suggest that Rac1 activity may have profound effects on atherosclerotic plaque development in vivo.
Rac is a component of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase20,21 which mediates production of reactive oxygen species (ROS), and NAPDH oxidase activity is influenced by the nucleotide state of Rac1. Therefore, it is not surprising that Rac1 has been implicated in ROS production in numerous cell types and biological contexts, including ECs in response to shear stress.22,23 ROS have been implicated in numerous biological processes, including cell growth, differentiation, apoptosis, angiogenesis, and cytoskeletal remodeling. Within the endothelium, ROS are required for proliferation and migration during angiogenesis.24 In addition, ROS activate matrix metalloproteases, which are crucial during flow-mediated vascular remodeling.25 Thus, shear stress-dependent ROS production clearly has critical roles in cell biology and vascular physiology. Our recent work highlighted a novel and interesting aspect of flow-induced ROS production: bulk Rac1 activation is not sufficient to induce ROS production, but, rather, the proper spatial localization of Rac1 activity is essential for increased ROS, which we will now discuss in further detail.
Polarization of Rac1 Activity
Many ECs responses to shear stress are directional and result in polarized signaling and structural remodeling. Perhaps the best-characterized remodeling event is the breakdown of cytoskeletal filaments and re-emergence of actin stress fibers aligned in the direction of flow. Similarly, focal adhesions remodel and elongate in the direction of flow in response to shear stress26 and live-cell imaging has revealed rapid and directional rearrangement of intermediate filaments in ECs exposed to shear stress.27 Polarization of active signaling molecules has also been demonstrated and we have previously shown that Rho GTPases exhibit directional activation in response to shear stress. Cdc42 is activated in response to shear stress and active Cdc42 localizes at the downstream edge of the cell (relative to the direction of flow).28 Furthermore, localized activation of Cdc42 influences the microtubule cytoskeleton and mediates polarization of the microtubule organizing center (MTOC) to the downstream side of the nucleus.28
Similarly, Rac1 has also been implicated in directional responses to shear stress. Rac1 is required for alignment of the actin cytoskeleton in the direction of flow7 and EC polarization in response to shear.8 While ECs expressing dominant negative Rac (N17Rac) or constitutively active Rac (L61Rac) exhibit a breakdown of the actin cytoskeleton followed by the re-emergence of stress fibers in response to shear stress, the resulting stress fibers are random in orientation and do not align in the direction of flow.7,8 These results suggest that the GTPase must provide spatial cues that influence cell behavior. This result is not surprising given the polarized activation of Rac1 itself in ECs exposed to shear stress.7 In previous studies, FLAIR (Fluorescence Activation Indicator for Rho proteins) and fluorescence energy transfer (FRET) techniques were utilized in order to visualize spatial localization of activated Rac1 in response to shear stress. Interestingly, Rac1 activity is strongly polarized to the downstream edge of the cell, and localized activation of the GTPase appears to be critical for Rac1 functions in response to shear stress7 (Fig. 1).
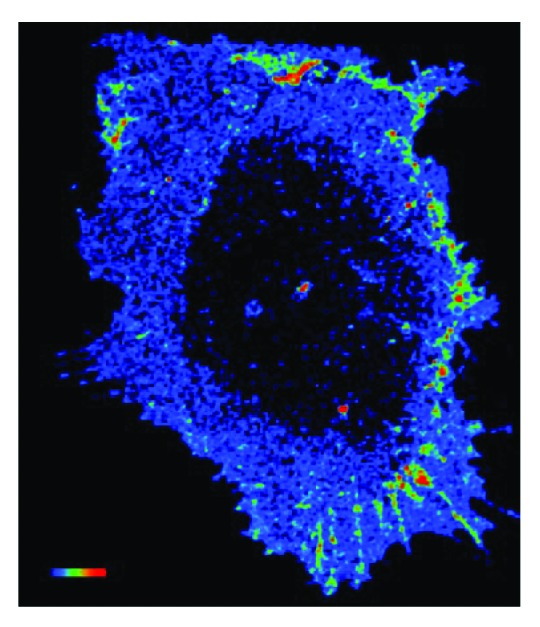
Figure 1. Shear stress induces polarized activation of Rac1 in endothelial cells. Confluent bovine aortic endothelial cells were transiently transfected an expression vector for GFP fused to wild-type Rac1 (GFP–WTRac) and were shear loaded with Alexa-PBD. Cells were sheared for 30 min. Rac activation (FRET) is shown. In the color intensity scale, red represents high and blue low (image courtesy of Bill Kiosses at The Scripps Research Institute).
Our recent work further highlights the importance of polarized Rac1 activation in redox signaling. As previously mentioned, Rac1 activity is also linked to activity of the NAPDH oxidase and ROS production. Exposure to shear stress results in an increase in ROS production in ECs and is dependent on Rac1 activity.23 Interestingly, ECs expressing a constitutively active Rac (V12Rac) exhibit increased basal ROS production, but do not increase ROS production in response to shear stress.9 These results indicate that directional activation of the GTPase (rather than bulk activation) is essential for shear stress-induced redox signaling.
How is Rac1 activity polarized in response to shear stress? As previously indicated, we recently investigated the roles of the junctional proteins PECAM-1 and VE-cadherin in flow-mediated Rac activation. This work revealed that PECAM-1 is essential for shear stress-induced Rac1 GTP loading via activation of Vav2.9 Given that PECAM-1 and VE-cadherin are part of a mechanosensory complex that transduces hemodynamic forces, we hypothesized that VE-cadherin is also essential for flow-dependent Rac1 activation. Surprisingly, loss of VE-cadherin did not affect flow-mediated changes in Rac GTP loading. However, FRET analyses revealed that VE-cadherin is essential for polarized Rac activation, as VE-cadherin−/− ECs displayed random, rather than polarized Rac1 activity in response to shear stress (Fig. 1). Further immunofluorescence and biochemical studies revealed that VE-cadherin functions as a platform that provides a docking site for the recruitment of other proteins to the downstream edge of the cell, and shear stress-induced association of this complex mediates polarized Rac1 activation. Components of this novel polarity complex include VE-cadherin, Par3/6 (which are thought to provide the polarity cue), p67phox (of the NAPDH oxidase), and perhaps most surprisingly, the Rac GEF Tiam19 (Fig. 2). Furthermore, Tiam1 appears to play a non-canonical role in shear stress-induced Rac activation, as depletion of the GEF does not inhibit GTP loading of Rac1, but only disrupts its polarization to downstream edge of the cell. In further support of this non-canonical role for the GEF, expression of a Tiam1 mutant lacking GEF-activity does not affect Rac1 activation or polarization in response to shear. These results indicate that, in certain contexts, GEFs may play a critical role in regulating GTPase localization, rather than activity. Indeed, this is not the first time that a GEF has been shown to regulate GTPase localization: Tiam1 helps restrict the activity of Rac to dendritic spines via its interaction with the PDZ protein Par3,29 and PDZRhoGEF cooperatively works with myosin II to polarize RhoA activity in neutrophil-like cells.30 Furthermore, more recent work has suggested that p190RhoGEF is required for proper FAK localization to peripheral adhesions in mouse embryonic fibroblasts.31 Thus, it may be interesting to investigate a role for other GEFs in directional EC signaling in response to shear stress in future studies.

Figure 2. Current understanding of flow-induced Rac1 polarization in endothelial cells. Mechanotransduction of shear stress results in PECAM-1-dependent Src activation. Src activates the GEF Vav2, which promotes Rac1 GTP-loading. Rac associates with a polarity complex consisting of VE-cadherin, Tiam1, p67phox, and Nox and the downstream edge of the cell and results in ROS production.
It is also important to note that other pathways have also been shown to influence Rac1 localization in response to shear stress. Previous work has demonstrated that flow-induced PKA-dependent phosphorylation of α4 integrin at the downstream edge of the cells promotes polarized Rac activation and is required for alignment of the actin cytoskeleton.32 In addition, in subconfluent ECs, Rac1 activity is decreased at the upstream edge of ECs exposed to flow via a paxillin-p130Cas pathway.33 Thus, multiple pathways may influence flow-induced Rac polarization in different biological contexts.
Mechanical activation of Rac1 has been implicated in numerous cell types and contexts and flow-mediated Rac1 activation is clearly crucial for numerous EC responses to shear stress. Activation of the GTPase is required for alignment of the actin cytoskeleton, NF-κB activation, changes in gene expression, permeability and ROS production. Interestingly, all of these processes can also influence onset and progression of disease. In this vein, endothelial-specific haploinsufficient mice have been shown to have mild hypertension due to decreased expression and activity of eNOS.12 In concordance, recovery from hindlimb ischemia was impaired in endothelial-specific haploinsufficient Rac1 mice, suggesting that therapeutic strategies to enhance Rac1 function might be important for preventing diseases associated with EC dysfunction.12 On the other hand, Rac1 might also have roles in disease via its role in ROS production; for example, EC ROS production (which is influenced by Rac1 activity and localization) influences cell growth, hypertrophy, apoptosis, and EC adhesiveness which can contribute to the development of numerous diseases, including diabetes, heart failure, ischemia reperfusion, and atherosclerosis.34 While these are all cardiovascular-related diseases, it is important to note that Rac1 has also been implicated in numerous other diseases and many types of cancers. Nonetheless, tightly controlled Rac1 activity and localization is clearly critical for EC and cardiovascular function. Our recent work highlights the notion that spatial activation of Rac1 is also essential for some responses to shear stress, including ROS production. This underscores the importance of studies that are geared toward understanding the molecular mechanisms that govern both spatial and temporal activation of Rho GTPases in mechanotransduction.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by NIH grant HL088632 (to E.T.). E.T. is an Ellison Medical Foundation New Scholar, and C.C. is American Heart Predoctoral Fellow (12PRE11930000).
Glossary
Abbreviations:
- ECM
extracellular matrix
- EC
endothelial cell
- PECAM-1
platelet endothelial cell adhesion molecule-1
- VE-cadherin
vascular endothelial-cadherin
- VEGFR2
vascular endothelial growth factor receptor 2
- GEF
guanine nucleoide exchange factor
- NADPH
nicotinamide adenine dinucleotide phosphate oxidase
- ROS
reactive oxygen species
- MTOC
microtubule organizing center
- FLAIR
fluorescence activation indicator for Rho protein
- FRET
fluorescence resonance energy transfer
- VEGF
vascular endothelial growth factor
- S1P
sphingosine-1-phosphate
- eNOS
endothelial nitric oxide synthase
References
- 1.Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans. 2005;33:891–5. doi: 10.1042/BST20050891. [DOI] [PubMed] [Google Scholar]
- 2.Poh YC, Na S, Chowdhury F, Ouyang M, Wang Y, Wang N. Rapid activation of Rac GTPase in living cells by force is independent of Src. PLoS One. 2009;4:e7886. doi: 10.1371/journal.pone.0007886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shifrin Y, Pinto VI, Hassanali A, Arora PD, McCulloch CA. Force-induced apoptosis mediated by the Rac/Pak/p38 signalling pathway is regulated by filamin A. Biochem J. 2012;445:57–67. doi: 10.1042/BJ20112119. [DOI] [PubMed] [Google Scholar]
- 4.Yamada S, Nelson WJ. Localized zones of Rho and Rac activities drive initiation and expansion of epithelial cell-cell adhesion. J Cell Biol. 2007;178:517–27. doi: 10.1083/jcb.200701058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Z, Tan JL, Cohen DM, Yang MT, Sniadecki NJ, Ruiz SA, Nelson CM, Chen CS. Mechanical tugging force regulates the size of cell-cell junctions. Proc Natl Acad Sci U S A. 2010;107:9944–9. doi: 10.1073/pnas.0914547107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wan Q, Cho E, Yokota H, Na S. Rac1 and Cdc42 GTPases regulate shear stress-driven β-catenin signaling in osteoblasts. Biochem Biophys Res Commun. 2013;433:502–7. doi: 10.1016/j.bbrc.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tzima E, Del Pozo MA, Kiosses WB, Mohamed SA, Li S, Chien S, Schwartz MA. Activation of Rac1 by shear stress in endothelial cells mediates both cytoskeletal reorganization and effects on gene expression. EMBO J. 2002;21:6791–800. doi: 10.1093/emboj/cdf688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wojciak-Stothard B, Ridley AJ. Shear stress-induced endothelial cell polarization is mediated by Rho and Rac but not Cdc42 or PI 3-kinases. J Cell Biol. 2003;161:429–39. doi: 10.1083/jcb.200210135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y, Collins C, Kiosses WB, Murray AM, Joshi M, Shepherd TR, Fuentes EJ, Tzima E. A novel pathway spatiotemporally activates Rac1 and redox signaling in response to fluid shear stress. J Cell Biol. 2013;201:863–73. doi: 10.1083/jcb.201207115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437:426–31. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- 11.Tan W, Palmby TR, Gavard J, Amornphimoltham P, Zheng Y, Gutkind JS. An essential role for Rac1 in endothelial cell function and vascular development. FASEB J. 2008;22:1829–38. doi: 10.1096/fj.07-096438. [DOI] [PubMed] [Google Scholar]
- 12.Sawada N, Salomone S, Kim HH, Kwiatkowski DJ, Liao JK. Regulation of endothelial nitric oxide synthase and postnatal angiogenesis by Rac1. Circ Res. 2008;103:360–8. doi: 10.1161/CIRCRESAHA.108.178897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seebach J, Dieterich P, Luo F, Schillers H, Vestweber D, Oberleithner H, Galla HJ, Schnittler HJ. Endothelial barrier function under laminar fluid shear stress. Lab Invest. 2000;80:1819–31. doi: 10.1038/labinvest.3780193. [DOI] [PubMed] [Google Scholar]
- 14.DePaola N, Phelps JE, Florez L, Keese CR, Minnear FL, Giaever I, Vincent P. Electrical impedance of cultured endothelium under fluid flow. Ann Biomed Eng. 2001;29:648–56. doi: 10.1114/1.1385811. [DOI] [PubMed] [Google Scholar]
- 15.Resnick N, Gimbrone MA., Jr. Hemodynamic forces are complex regulators of endothelial gene expression. FASEB J. 1995;9:874–82. doi: 10.1096/fasebj.9.10.7615157. [DOI] [PubMed] [Google Scholar]
- 16.Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev. 1995;9:2723–35. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 17.Marui N, Offermann MK, Swerlick R, Kunsch C, Rosen CA, Ahmad M, Alexander RW, Medford RM. Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J Clin Invest. 1993;92:1866–74. doi: 10.1172/JCI116778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schindler U, Baichwal VR. Three NF-kappa B binding sites in the human E-selectin gene required for maximal tumor necrosis factor alpha-induced expression. Mol Cell Biol. 1994;14:5820–31. doi: 10.1128/MCB.14.9.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gareus R, Kotsaki E, Xanthoulea S, van der Made I, Gijbels MJ, Kardakaris R, Polykratis A, Kollias G, de Winther MP, Pasparakis M. Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab. 2008;8:372–83. doi: 10.1016/j.cmet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 20.Abo A, Pick E, Hall A, Totty N, Teahan CG, Segal AW. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature. 1991;353:668–70. doi: 10.1038/353668a0. [DOI] [PubMed] [Google Scholar]
- 21.Knaus UG, Heyworth PG, Evans T, Curnutte JT, Bokoch GM. Regulation of phagocyte oxygen radical production by the GTP-binding protein Rac 2. Science. 1991;254:1512–5. doi: 10.1126/science.1660188. [DOI] [PubMed] [Google Scholar]
- 22.Chiu JJ, Wung BS, Shyy JY, Hsieh HJ, Wang DL. Reactive oxygen species are involved in shear stress-induced intercellular adhesion molecule-1 expression in endothelial cells. Arterioscler Thromb Vasc Biol. 1997;17:3570–7. doi: 10.1161/01.ATV.17.12.3570. [DOI] [PubMed] [Google Scholar]
- 23.Yeh LH, Park YJ, Hansalia RJ, Ahmed IS, Deshpande SS, Goldschmidt-Clermont PJ, Irani K, Alevriadou BR. Shear-induced tyrosine phosphorylation in endothelial cells requires Rac1-dependent production of ROS. Am J Physiol. 1999;276:C838–47. doi: 10.1152/ajpcell.1999.276.4.C838. [DOI] [PubMed] [Google Scholar]
- 24.Ushio-Fukai M. VEGF signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal. 2007;9:731–9. doi: 10.1089/ars.2007.1556. [DOI] [PubMed] [Google Scholar]
- 25.Castier Y, Brandes RP, Leseche G, Tedgui A, Lehoux S. p47phox-dependent NADPH oxidase regulates flow-induced vascular remodeling. Circ Res. 2005;97:533–40. doi: 10.1161/01.RES.0000181759.63239.21. [DOI] [PubMed] [Google Scholar]
- 26.Davies PF, Robotewskyj A, Griem ML. Quantitative studies of endothelial cell adhesion. Directional remodeling of focal adhesion sites in response to flow forces. J Clin Invest. 1994;93:2031–8. doi: 10.1172/JCI117197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helmke BP, Thakker DB, Goldman RD, Davies PF. Spatiotemporal analysis of flow-induced intermediate filament displacement in living endothelial cells. Biophys J. 2001;80:184–94. doi: 10.1016/S0006-3495(01)76006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tzima E, Kiosses WB, del Pozo MA, Schwartz MA. Localized cdc42 activation, detected using a novel assay, mediates microtubule organizing center positioning in endothelial cells in response to fluid shear stress. J Biol Chem. 2003;278:31020–3. doi: 10.1074/jbc.M301179200. [DOI] [PubMed] [Google Scholar]
- 29.Nishimura T, Yamaguchi T, Kato K, Yoshizawa M, Nabeshima Y, Ohno S, Hoshino M, Kaibuchi K. PAR-6-PAR-3 mediates Cdc42-induced Rac activation through the Rac GEFs STEF/Tiam1. Nat Cell Biol. 2005;7:270–7. doi: 10.1038/ncb1227. [DOI] [PubMed] [Google Scholar]
- 30.Wong K, Van Keymeulen A, Bourne HR. PDZRhoGEF and myosin II localize RhoA activity to the back of polarizing neutrophil-like cells. J Cell Biol. 2007;179:1141–8. doi: 10.1083/jcb.200706167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller NL, Lawson C, Kleinschmidt EG, Tancioni I, Uryu S, Schlaepfer DD. A non-canonical role for Rgnef (p190RhoGEF) in promoting integrin-stimulated focal adhesion kinase activation. J Cell Sci. 2013 doi: 10.1242/jcs.135509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldfinger LE, Tzima E, Stockton R, Kiosses WB, Kinbara K, Tkachenko E, Gutierrez E, Groisman A, Nguyen P, Chien S, et al. Localized alpha4 integrin phosphorylation directs shear stress-induced endothelial cell alignment. Circ Res. 2008;103:177–85. doi: 10.1161/CIRCRESAHA.108.176354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zaidel-Bar R, Kam Z, Geiger B. Polarized downregulation of the paxillin-p130CAS-Rac1 pathway induced by shear flow. J Cell Sci. 2005;118:3997–4007. doi: 10.1242/jcs.02523. [DOI] [PubMed] [Google Scholar]
- 34.Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1014–30. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]