

Abstract
Erythropoietin, discovered for its indispensable role during erythropoiesis, has been used in the therapy for selected red blood cell disorders in erythropoietin-deficient patients. The biological activities of erythropoietin have been found to extend to non-erythroid tissues due to the expression of erythropoietin receptor. We previously demonstrated that erythropoietin promotes metabolic activity and white adipocytes browning to increase mitochondrial function and energy expenditure via peroxisome proliferator-activated receptor alpha and Sirtuin1. Here we report that AMP-activated protein kinase was activated by erythropoietin possibly via Ca2+/calmodulin-dependent protein kinase kinase in adipocytes as well as in white adipose tissue from diet induced obese mice. Erythropoietin increased cellular Nicotinamide adenine dinucleotide via increased AMP-activated protein kinase activity, possibly leading to Sirtuin1 activation. AMP-activated protein kinase knock down reduced erythropoietin mediated increase in cellular oxidative function including the increased oxygen consumption rate, fatty acid utilization and induction of key metabolic genes. Under hypoxia, adipocytes were found to generate more reactive oxygen species, and erythropoietin reduced the reactive oxygen species and increased antioxidant gene expression, suggesting that erythropoietin may provide protection from oxidative stress in adipocytes. Erythropoietin also reversed increased nicotinamide adenine dinucleotide by hypoxia via increased AMP-activated protein kinase. Additionally, AMP-activated protein kinase is found to be involved in erythropoietin stimulated increase in oxygen consumption rate, fatty acid oxidation and mitochondrial gene expression. AMP-activated protein kinase knock down impaired erythropoietin stimulated increases in antioxidant gene expression. Collectively, our findings identify the AMP-activated protein kinase involvement in erythropoietin signaling in regulating adipocyte cellular redox status and metabolic activity.
Keywords: Adipocytes, Erythropoietin, AMP-activated protein kinase, Oxidative metabolism, reactive oxygen species
1. Introduction
Erythropoietin (EPO) binds to its cell surface receptor, EpoR to promote early erythroid progenitor cell survival, proliferation and differentiation. EPO is produced in the adult kidney and regulated by hypoxia inducible factor (HIF). However, EPO signaling is not restricted to the erythroid lineage and can be found in many non-hematopoietic cells including endothelial, muscle, adipocytes, cardiovascular and renal tissue (Noguchi et al., 2008; Teng et al., 2011). EPO stimulation of mitochondrial biogenesis in part by enhancement of Peroxisome proliferator-activated receptor-gamma coactivator ((PGC)-1α) was suggested to mediate its cardioprotective activity (Carraway et al., 2010). EPO activity has also been reported for other non-hematopoietic tissues including brain protection against ischemia, enhanced neural progenitor production and anti-inflammatory effect (Sakanaka et al., 1998; Shingo et al., 2001; Tsai et al., 2006). Recently, a metabolic effect of EPO signaling in adipocytes was reported to provide protection against diet-induced obesity, increase glucose tolerance and mitochondrial function (Teng et al., 2011; Wang et al., 2013b). However, the detailed mechanism by which EPO regulates energy expenditure and metabolic activity still remains open for investigation. Although EPO was reported to promote metabolic activity of adipocytes via increasing Sirt1 and PGC-1α activity (Wang et al., 2013b), the involvement of other pathways in EPO/EpoR signaling in adipocytes remains largely unknown.
AMP-activated protein kinase (AMPK), a serine/threonine kinase, is an evolutionarily conserved energy metabolic sensor and an important regulator of energy homeostasis. AMPK can be activated to block body weight gain and increase fatty acid oxidation by inducing transcription regulators involved in energy homeostasis such as peroxisome proliferator-activated receptor alpha (PPARα) and peroxisome proliferator-activated receptor gamma (PPARγ) co-activator 1α (PGC-1α) (Canto and Auwerx, 2009; Giri et al., 2006; Hardie, 2007; Lage et al., 2008; Rodgers et al., 2005; Towler and Hardie, 2007). AMPK activator, metformin and thiazolidinediones have shown important therapeutic benefits in the treatment of type 2 diabetes and metabolic syndrome (Fryer et al., 2002; Hardie, 2007; Zhou et al., 2001). AMPK is also implicated in the appearance of brown features and increased mitochondrial activity in white adipose tissue (WAT) via regulating or interacting with factors such as PR domain containing 16 (PRDM16), a master regulator of brown fat determination and uncoupled protein 1 (UCP1), a key regulator of brown fat thermogenesis (Ahmadian et al., 2011; Bostrom et al., 2012; Kajimura et al., 2008; Seale et al., 2008; Seale et al., 2011; Sun et al., 2011). In addition, AMPK also regulates Sirt1, another metabolic sensor and a Nicotinamide adenine dinucleotide (NAD+) dependent type III deacetylase sirtuin, that activates PGC-1α and various substrates including PPARγ (Canto and Auwerx, 2009; Canto et al., 2009). We previously demonstrated that EPO enhances AMPK activity in C2C12 myoblasts, which may mediates the EPO effect to promote slow muscle fiber specification (Wang et al., 2013a). EPO is also reported to trigger AMPK activity, leading to enhanced phosphorylation of β common receptor (βCR) and endothelial nitric oxide synthase (eNOS) to stimulate NO production and, ultimately, angiogenesis (Su et al., 2012). However, how EPO triggers AMPK activity is still largely unknown so far.
In this study, we demonstrate a novel action of endogenous EPO in WAT to facilitate energy expenditure by regulating AMPK via Calcium/calmodulin-dependent protein kinase kinase (CaMKK). EPO alters cellular nicotinamide adenine dinucleotide (NADH) and NAD+ levels and modulates NAD+/NADH ratio through regulating AMPK activity, which may lead to increased Sirt1 activity. EPO mediated activation of AMPK also contributes to energy expenditure and reduction of hypoxia induced oxidative stress of adipocytes. These effects of EPO in adipocyte may account for the beneficial metabolic effects of EPO.
2. Materials and Methods
2.1 Animal studies
C57BL/6 mice (4 weeks; NCI-Frederick) were maintained under a 12-hour light/dark cycle with free access to food and drinking water except as indicated for paired-fed mice. For pair-fed study, because foods are voluntarily took in animals but not forced to be given, the intake becomes same when the animals would finish all amounts given. Therefore, amounts of foods are given according to amounts took by animals having lesser intake. The animal group having lesser intake is used as “control” and the animal group given the same amount is called as “pair-fed”. In this study, one group of EPO treated mice had free access to food while the other was pair-fed to the ad libitum intake of the EPO treated mice. Food intake was measured before the pair-fed study in the EPO treated mice and the quantity given to pair-fed mice was constantly adjusted to that of the EPO treated mice. Mice were fed normal chow (NC) NIH-07 diet (Zeigler Brothers) or high fat diet (HFD) (60 kcal% fat) D12451 (Research Diets Inc.) as indicated and separated into different groups. EPO treatment (3,000 Units/kg; 3 times/week for 2.5 to 5 weeks as indicated, Epoetin alpha, Amgen Manufacturing, Thousand Oaks, CA) is administrated via subcutaneous injection. The control and paired-fed mice were injected with PBS as vehicle control. The Animal Care and Use Committee of the National Institute of Diabetes and Digestive and Kidney Diseases approved all animal procedures and studies were conducted in accordance to National Institutes of Health guidelines.
2.2 Cell Culture
Adipocyte differentiation of 3T3-L1 fibroblast cells (ATCC) was induced as described (Teng et al., 2011). At the beginning of differentiation induction, cells were treated with EPO, at 5 units/ml (U/ml) or at the dosage indicated, or with vehicle (PBS). 3T3-L1 adipocytes were differentiated with EPO treatment for 9 days. For hypoxia study, the 3T3-L1 adipocytes were cultured in normoxia (21% O2) or hypoxia (2% O2) condition for 24 hours or 48 hours or indicated time with or without EPO treatment. PGC-1α expression vector was purchased from Origene and transfected into 3T3-L1 adipocytes using Lipofectamine ™2000 (Invitrogen).
2.3 NAD+/NADH determination Assays
NAD+ and NADH levels were determined according to instructions provided with the NAD+/NADH assay kit purchased from Biovision. Briefly, wash cells with cold PBS. Pellet cells for each assay in a micro-centrifuge tube and extract with NADH/NAD+ Extraction Buffer and transfer the extracted NADH/NAD+ supernatant into a labeled tube. To detect total NADt (NADH and NAD+), transfer 50 μl of extracted samples into labeled 96-well plate. To detect NADH, NAD+ needs to be decomposed before the reaction. To decompose NAD+, aliquot 200 μl of extracted samples into eppendorf tubes. Heat to 60°C for 30 min. Cool samples on ice. Quick spin the samples to remove precipitates if precipitation occurs. Transfer 50 μl of NAD+ decomposed samples into labeled 96-well plate. Prepare a NAD+ Cycling Mix for each reaction and add 100 μl of the mix into each well of NADH Standard and samples. Incubate the plate at room temperature for 5 Min. Add 10 μl NADH developer into each well. Let the reaction cycling at room temperature for 1 to 4 hrs or longer. Read the plate at OD 450 nm. The reactions can be stopped by adding 10 μl of Stop Solution into each well. For unspiked samples, apply the sample readings to NADH standard curve. The amount of NADt or NADH in the sample wells can then be calculated. NAD+/NADH Ratio are calculated as: (NADt – NADH)/ NADH.
2.4 Measurement of reactive oxygen species
At the beginning of differentiation induction, 3T3-L1 adipocytes were treated with EPO (5U/ml) or with vehicle (PBS). 3T3-L1 adipocytes were differentiated with EPO treatment for 9 days. At the 9th day, 3T3-L1 adipocytes were incubated in normoxia (21% O2) or hypoxia (2% O2) for 4 hours without or with EPO treatment (5U/ml). Then 3T3-L1 adipocytes were incubated with 5uM CellRox Green (Invitrogen Corp) for 30 min. After washes, fluorescence was determined at 485/520 nm and normalized to protein concentration.
2.5 Quantitative Real-time RT- PCR
To follow MIQE guideline (Bustin et al., 2009), total RNA extracted from cells or tissues using TRIzol (Invitrogen) was treated with Turbo DNase (Ambion) and 2 μg was reverse transcribed in 40μl volume system (QuantiTect Reverse Transcription Kit (Qiagen)) for quantitative PCR assays. Displaying the integrity of RNA by checking multiple major rRNA bands and other high copy RNAs using electrophoresis method. Quantitative real-time PCR analyses were carried out using gene-specific primers (working concentration at 250nM) (the sequence of primers were listed in the Table S1) in a 7900 Sequence Detector (PE Applied Biosystems, Foster City, CA). The PCR running procedure is: 4mins denature at 95°C, followed by 40 cycles of 95°C denature for 30 seconds and 60°C annealing and extension for 1min. The fluorescent signals were collected for each cycle. Then the data were analyzed using Applied Biosystems Real-Time PCR analysis software SDS-2.2. For relative mRNA quantification of all genes (Idh3α, CytC, Cpt-1, Pgc-1α, SOD, Gpx, and Catalase), SYBR green real-time RT–PCR was used with normalization to housekeeping geneS16 as an internal control.
2.6 Western Blotting
Cells were lysed in RIPA buffer supplemented with protease and phosphatase inhibitors (Sigma-Aldrich). Lysates were sonicated for 1 min and centrifuged at 14, 000 × g for 10 min at 4 °C. Lysates were resolved by 4–20% Tris-glycine SDS/PAGE and transferred to nitrocellulose membranes. Total AMPKα and p-AMPKα antibodies, total ACC and p-ACC antibodies (Cell Signaling Technology) and pan acetylated lysine (4G12) antibody and PGC-1α antibody (Millipore), SOD, Gpx, Catalase and β-actin (Santa Cruz Biotechnology) were used for Western blotting. Horseradish peroxidase conjugated secondary antibody and the Amersham ECL Advance Western Blotting Detection System (GE Healthcare Bio-Sciences Corp) were used. Quantitative analysis was performed by measuring integrated density with the NIH image J system and normalized with β-actin.
2.7 Oxygen Consumption and fatty acid oxidation
A Seahorse Bioscience XF24-3 Extracellular Flux Analyzer was used to measure the oxygen consumption rate and fatty acid oxidation. 3T3-L1 adipocytes were seeded in XF24-well microplates at 2.5 × 104 cells per well. Respiration was measured under different conditions. To assess fatty acid oxidation, DMEM media containing 11 mM glucose and 0.5 mM carnitine was employed as an assay media, and sodium palmitate was administered at a final concentration of 200 μM. All media included 2mM L-glutamine.
2.8 RNAi
For knockdown experiments, siRNAs specific for LKB1 (“Smart Pool” of several siRNAs selected by Dharmacon) and AMPKα (“Smart Pool” of several siRNAs selected by Dharmacon) and negative control siRNA: 5′-AAUUCUCCGAACGUGUCACGU-3′ (Thermo Scientific Dharmacon) were transfected into 3T3-L1 adipocytes via DharmaFECT transfection reagent.
2.9 Statistical analyses
Values are expressed as mean ± SEM. Comparisons between two groups were made using two-tailed non-paired Student’s t-test. Statistical differences between three or more groups were evaluated by one-way analysis of variance (ANOVA) with Dunnet’s multiple comparison post hoc tests at α = 0.05. A P value of italic> 0.05 was regarded to be statistically significant.
3. Results
3.1. EPO increases AMPK activity in adipocytes
AMPK activation is able to increase fatty acid oxidation and increase the NAD+/NADH ratio (Canto et al., 2009). To determine if AMPK activity is involved in adipocyte response to EPO, 3T3-L1 preadipocytes were allowed to differentiate for 9 days and then treated with EPO. We found EPO stimulation increased AMPKα activity reflected in the two-fold increase in the ratio of phosphorylated AMPKα to total AMPKα (p-AMPKα/AMPKα), while total AMPKα protein levels remained unchanged (Figure 1A).
Figure 1. EPO increases AMPK activity in vitro and in vivo.

(A–B) Western blotting to determine total AMPKα (t-AMPK) and phosphorylated AMPKα protein (p-AMPKα) levels in 3T3-L1 adipocytes treated without (PBS) and with EPO (5U/mL) at times indicated (A) and in S-WAT and V-WAT from DIO mice with EPO treatment for 5 weeks (B) (n=4). One-way ANOVA was used in B and other statistics were performed using Student’s t-test, and bar graphs are mean±s.e.m. In vitro data are means of three independent experiments. *P bold> 0.05; **P < 0.01
EPO treatment can protect mice from diet induced obesity (DIO) (Teng et al., 2011; Wang et al., 2013b). To test if EPO can increase AMPK activity in vivo, we treated DIO mice with EPO and found EPO treatment increased AMPKα phosphorylation in inguinal subcutaneous white fat tissue (S-WAT) and visceral white fat tissue (V-WAT) (Figure 2B) compared to vehicle treatment. We previously found that EPO treatment in DIO mice reduces food intake. To demonstrate that the increase in AMPK activity is beyond that due to the loss of body weight from decreased food intake, we performed pair-fed studies in DIO mice based on the amount of food ingested with EPO treatment. Importantly, EPO treatment increased AMPKα phosphorylation in S-WAT and V-WAT (Figure 2B) but not in vehicle treated and pair-fed DIO mice, suggesting that EPO effect on increased AMPK activity is not body weight dependent.
Figure 2. EPO regulated NAD+/NADH ratio and PGC-1α deacetylation was inhibited by AMPK inhibitor.
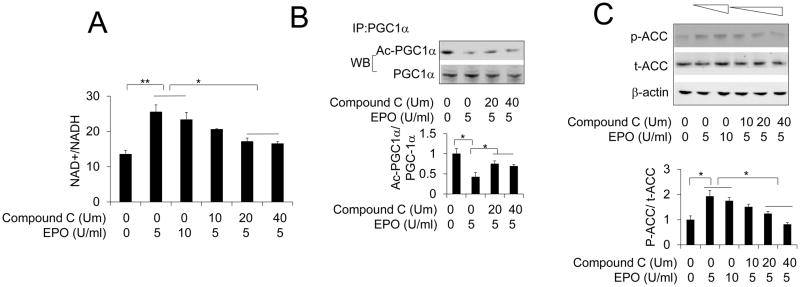
(A) NAD+/NADH ratio was determined in 3T3-L1 adipocytes exposed to EPO at indicated dosage in the absence or presence of AMPK inhibitor Compound C at indicated dosage. (B) PGC-1α deacetylation was determined in 3T3-L1 adipocytes with PGC-1α overexpression exposed to EPO (5U/ml) in the absence or presence of AMPK inhibitor Compound C at indicated dosage. (C) Total ACC (t-ACC) and phosphorylated ACC protein levels were determined in 3T3-L1 adipocytes without or with EPO treatment at indicated dosage in the absence or presence of AMPK inhibitor Compound C at indicated dosage. One-way ANOVA was used in A, B and C. And the bar graphs are mean±s.e.m. The data are means of three independent experiments. *P < 0.05; **P < 0.01
3.2. EPO regulated cellular redox status and PGC-1α acetylation is mediated by AMPK
As a master regulator of metabolic activity, AMPK activity was investigated to determine if EPO effects are mediated by AMPK. We previously demonstrated EPO increased the NAD+/NADH ratio (Wang et al., 2012; Wang et al., 2013b). Here, we observed that inhibition of AMPK activity using AMPK activity inhibitor Compound C partially attenuated the EPO stimulated increase in NAD+/NADH ratio in 3T3-L1 adipocytes (Figure 2A). As a dominant transcriptional cofactor regulating mitochondrial biogenesis and metabolic activity in WAT (Bostrom et al., 2012; Lagouge et al., 2006; Seale et al., 2011; Wu et al., 1999), PGC-1α is activated by its deacetylation (Rodgers et al., 2005). We observed that EPO treatment of 3T3-L1 adipocytes with overexpressed PGC-1α decreased acetylation of PGC-1α (Wang et al., 2013b). Sirt1, a family member of sirtuins, is a NAD+ dependent deacetylase and responsible for PGC-1α deacetylation (Rodgers et al., 2005). We found that in 3T3-L1 adipocyte with PGC-1α overexpression, Compound C treatment also partially attenuated EPO induced PGC-1α deacetylation (Figure 2B), suggesting that AMPK may be involved in the EPO activity of modulating NAD+/NADH ratio and regulating PGC-1α activity. The activity of AMPK downstream molecular acetyl-CoA carboxylase (ACC) was also increased by EPO as indicated by increased phosphorylation of ACC (Figure 2C). Compound C impaired the EPO stimulated increase in ACC phosphorylation at a dose-dependent manner (Figure 2C). These in vivo and in vitro findings link EPO activity for the first time to AMPK signaling in adipocytes and identify EPO as a novel regulator of AMPK activity independent of changes in body weight. EPO mediated activation of AMPK may also explain increased expression and activity of PGC-1α by EPO (Wang et al., 2013b) since PGC-1α is an AMPK downstream target (Canto et al., 2009; Jager et al., 2007). It is interesting to note that we used different dosage of EPO combined with Compound C to stimulate the adipocytes and found that 5U/ml EPO treatment exhibited better effect compared with increased dosage of 10U/ml, which is consistent with our previously published observations (Teng et al., 2011; Wang et al., 2013b). However, increased dosage of Compound C did not display significant impairment of EPO modulated NAD+/NADH ratio and deacetylation of PGC-1α although increased Compound C showed increased inhibition of EPO mediated AMPK activity as indicated by abolished phosphorylation of AMPK direct target, ACC at a dose-dependent manner. These data suggest that EPO modulated NAD+/NADH ratio and regulated Sirt1 activity may be partially mediated by AMPK activity in adipocytes and some unknown pathways could be involved in the EPO activity in adipocytes.
3.3. CaMKK is involved in the EPO effect on the AMPK activity
Two upstream kinases have been reported to activate AMPK activity, LKB1 and Ca2+/calmodulin-dependent kinase kinase (CaMKK) (Hawley et al., 2005; Woods et al., 2005; Woods et al., 2003). In erythroblasts and myoblasts, EPO can stimulate intracellular cytosolic calcium concentration [Ca2+]i (Miller et al., 1989; Ogilvie et al., 2000). Therefore, we investigated AMPK activity in the adipocytes treated with EPO in conjunction with CaMKK inhibitor STO-609 treatment or knock-down of LKB1 (Figure S1A). We found that activation of AMPK by EPO was impaired by CaMKK inhibition (Figure 3A). However, knock-down of LKB1 did not affect AMPK activation by EPO (Figure 3A), indicating that EPO may activate AMPK via CaMKK but not LKB1. We demonstrated that in adipocytes, EPO enhanced mitochondrial gene expression and cellular oxygen consumption rate (OCR) (Wang et al., 2013b), which can result in increased mitochondrial activity and metabolic activity. However, CaMKK activity inhibition also impaired EPO mediated increase in mitochondria metabolic genes and OCR (Figure 3B and C), but LKB1 knockdown did not (Figure S1B and C). Taken together, these data indicate that EPO may activate AMPK possibly via CaMKK but not LKB1 pathway. However, the substrates of CaMKK include CaMK I and CaMK IV in addition to AMPK (Soderling, 1999), we cannot rule out the possibility that STO-609 blocked EPO activity may be mediated by some other pathways. Further study will help to illustrate the relationships among EPO, CaMKK and its downstream signal pathways including AMPK.
Figure 3. EPO regulated AMPK activity is mediated by CaMKK.
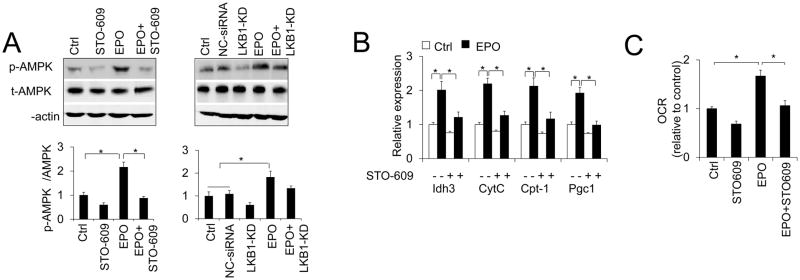
(A) Total AMPKα and p-AMPKα protein levels were determined in adipocytes without or with EPO treatment in the absence or presence of CaMKK inhibitor STO-609 or siRNA knock-down of LKB1 (LKB1-KD), Ctrl (no siRNA) and negative control siRNA (NC-siRNA) were used as control for LKB1 knock down. (B) The expression of mitochondrial genes was determined in adipocytes without or with EPO treatment in the absence or presence of CaMKK inhibitor STO-609. (C) OCR was determined in adipocytes without or with EPO treatment (5U/ml) in the absence or presence of CaMKK inhibitor STO-609. One-way ANOVA was used in A, B and C. And the bar graphs are mean±s.e.m. The data are means of three independent experiments. *P < 0.05.
3.4. EPO alleviates the oxidative stress of adipocytes under hypoxia
Obesity is associated with insulin resistance and type 2 diabetes. Adipose tissue in obese mice is in hypoxia and local hypoxia in adipose tissue increases Endoplasmic reticulum stress responsive marker genes (Hosogai et al., 2007; Jiang et al., 2011). Hypoxia has been shown to activate AMPK via the inhibition of mitochondrial respiration or oxidative stress (Gusarova et al., 2011; Liu et al., 2006; Mungai et al., 2011). It was also reported that hypoxia can reduce mitochondrial activity and number (Qatanani and Lazar, 2007). In addition, the mitochondrial respiration and biogenesis are also inhibited by hypoxia (Kim et al., 2006; Zhang et al., 2007). The number of mitochondria in adipose tissue is also reduced in obese individuals (Keijer and van Schothorst, 2008). Furthermore, CaMKK-mediated but not AMP and LKB1 dependent AMPK activation was associated with hypoxia (Gusarova et al., 2011; Mungai et al., 2011). EPO which is hypoxia inducible, can stimulate intracellular cytosolic calcium concentration [Ca2+]i in erythroblasts and myoblasts (Miller et al., 1989; Ogilvie et al., 2000). We have reported that EPO increased oxygen consumption rate under hypoxia (Wang et al., 2013b). Here, we observed that hypoxia (2% O2) increased AMPK activity and EPO further elevated AMPK activity as shown by increased AMPKα phosphorylation under hypoxia (Figure 4A). Interestingly, the intracellular NAD+/NADH ratio in differentiated 3T3-L1 adipocytes cultured under hypoxia (2%O2) for 24h was dramatically decreased due to an increase in NADH level (Figure 4B) which is consistent with reports of the hypoxia induced increase in intracellular NADH levels by inhibiting NADH oxidation and thereby decreasing the NAD+/NADH ratio (Di et al., 2010; Nyengaard et al., 2004). EPO treatment abrogated the increase in NADH level at hypoxia and increased the NAD+/NADH ratio by about 150% compared with untreated culture at hypoxia (Figure 4B). EPO also increased the NAD+ level that further contributes to the increase in the NAD+/NADH ratio compared with untreated cultures (Figure 4B). It is possible that the increased EPO response at hypoxia activates AMPK activity to counteract the obesity and resultant inhibition of mitochondrial respiration.
Figure 4. EPO regulates ROS production and expression of Antioxidant gene.

(A) Total AMPKα (t-AMPKα) and p-AMPKα protein levels were determined in the adipocyte under normoxia (21%O2) and hypoxia (2%O2) without or with EPO treatment. (B) NAD+/NADH ratio, NAD+ and NADH level were shown in 3T3-L1 adipocytes under normoxia and hypoxia without or with EPO treatment (5U/ml). (C) 3T3-L1 adipocytes were incubated with CellRox Green for 30 min after being incubated for 4 hours in normoxia or hypoxia without or with EPO treatment (5U/ml). The fluorescence was measured. Results are presented as the fold of stimulation compared with control. (D) Antioxidant gene expression level in 3T3-L1 adipocytes under normoxia and hypoxia without or with EPO treatment (5U/ml). (E) Western blotting shows SOD, Gpx and Catalase protein levels in in 3T3-L1 adipocytes under normoxia and hypoxia without or with EPO treatment (5U/ml). (F) SOD, Gpx and Catalase protein levels were determined in white fat tissue from the normal chow (NC) and high fat diet (HFD) induced obese mice with EPO treatment for 5 weeks. One-way ANOVA was used in A–D. And the bar graphs are mean±s.e.m. In vitro data are means of three independent experiments. *P < 0.05; **P < 0.01.
Insulin resistance has been associated with the generation of reactive oxygen species (ROS) and hypoxia can induce ROS production in 3T3-L1 adipocytes (Regazzetti et al., 2009). The induced systemic oxidative stress in accumulated fat can partially cause dysregulation of adipocytokines and development of metabolic syndrome (Furukawa et al., 2004). To investigate if EPO reduces the oxidative stress in adipocytes, we incubated 3T3-L1 adipocytes under hypoxia condition (2% O2) for 4 hours. We then monitored the formation of ROS using a fluorogenic probe in Live cells (CellRox Green oxidative stress reagent, Invitrogen). Hypoxia stimulated the generation of ROS and the production of ROS was reduced by EPO (Figure 4C). Next we investigated the expression of antioxidant enzymes including superoxide dismutase (SOD), glutathione peroxidase (GPx) and catalase in 3T3-L1 adipocytes cultured under hypoxia for 48h. The expression of these antioxidant enzymes were down-regulated by hypoxia but elevated by EPO stimulation in 3T3-L1 adipocyte compared with the 3T3-L1 adipocyte without EPO treatment (Figure 4D). At hypoxia, the protein levels of these antioxidant enzymes were also higher in EPO stimulated adipocytes (Figure 4E). The increased oxidative stress in accumulated fat in obese mice was also reported and recognized as an early instigator of metabolic syndrome (Furukawa et al., 2004). Here we found that the amounts of antioxidant factor SOD, Gpx and Catalase were decreased in WAT in DIO mice and that EPO treatment enhanced their protein levels (Figure 4F). Increased ROS production in excessively accumulated fat may promote ROS in circulating blood and harmfully affect other organs including skeletal muscle, liver and cardiovascular system. Here, the demonstrated effects of EPO imply that EPO can alleviate the oxidative stress in accumulated fat to offset the adverse effect of ROS.
3.5. EPO response requires AMPK in adipocytes
To determine whether AMPK activity is required for EPO stimulation of mitochondrial activity including both oxygen consumption and fatty acid oxidation, and the relieving effect from oxidative stress in adipocytes, we knocked down AMPKα (Figure 5A), a catalytic subunit and primary regulator of AMPK activity in adipocytes (Daval et al., 2005; Lihn et al., 2004). EPO increased basal oxygen consumption rate (OCR) in 3T3-L1 adipocytes (Figure 5B). The EPO-stimulated increase in OCR was still evident in the presence of oligomycin (OM), which uncouples phosphorylation from mitochondrial respiration by blocking mitochondrial complex V, and with treatment of carbonyl cyanide p-trifluoromethoxy phenylhydrazone (FCCP), which is used to assess maximal oxidative phosphorylation capacity. Indeed, knock down of AMPKα in 3T3-L1 adipocytes attenuated the EPO effect on the increased oxygen consumption rate and fatty acid oxidation (Figure 5B and C). The EPO-driven increased expression of genes related to mitochondrial function, including CytC and IDH3a, was prevented by reduction in AMPKα (Figure 5D). EPO induction of fatty acid utilization gene, CPT-1, and PGC-1α (PPARgc1) gene expression was also attenuated by decreasing AMPKα (Figure 5D), indicating that EPO activity on energy expenditure and mitochondrial function is, at least in part, dependent on AMPK activity in adipocytes. Under hypoxia, the increased expression of antioxidant enzymes SOD, Gpx and catalase by EPO were impaired by AMPKα knockdown (Figure 5E). These observations indicate AMPK is an essential mediator of EPO function in adipocytes.
Figure 5. AMPK mediates EPO effects in adipocytes.

(A) Total AMPKα protein level was determined in 3T3-L1 adipocyte with AMPKα knock down (KD). Ctrl (no siRNA) and negative control siRNA (NC-siRNA) were used as control for AMPKα knock down. (B–D) OCR (B) of basal and with presence of oligomycin (OM) and carbonyl cyanide p-trifluoromethoxy phenylhydrazone (FCCP), the fatty acid oxidation (C) of basal and with the presence of palmitate (PALM) and expression of mitochondrial genes (D) were determined in adipocytes with knock down of AMPKα without (open bar; control) and with EPO treatment (5U/ml; closed bar). (E) Antioxidant gene expression was determined in 3T3-L1 adipocytes with knock down of AMPKα without (open bar; control) and with EPO treatment (5U/ml; closed bar). One-way ANOVA was used in B–E. And the bar graphs are mean±s.e.m. The data are means of three independent experiments.*P < 0.05; **P < 0.01
Taken together, these data highlight a potential regulatory mechanism of EPO activity via regulating the energy sensor AMPK in adipocytes to either regulate metabolic activity or release the oxidative stress caused by increased ROS in adipocytes.
4. Discussion
In this study, we provide evidence to suggest that in white adipocytes, AMPK phosphorylation is activated in response to EPO, possibly through the CaMKK pathway, to modulate the redox status as reflected by changed NAD+/NADH ratio resulting in stimulation of downstream Sirt1 deacetylation activity and other metabolism related factors including PGC-1α (Figure 6). AMPK is required for EPO effect including increasing metabolic activity and reducing oxidative stress in vitro in adipocytes and in vivo in obese white adipose tissue.
Figure 6. The crosstalk between EPO and important energy sensors.
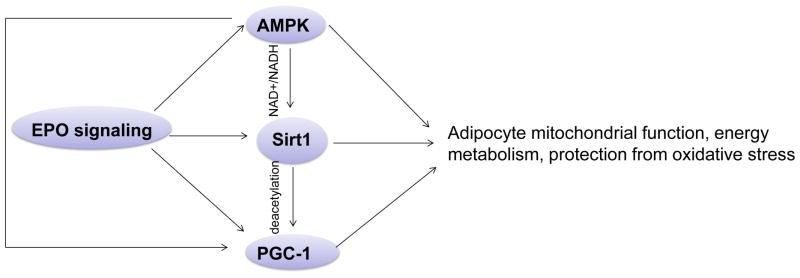
EPO regulated AMPK activity is involved in adipocyte energy metabolism, mitochondrial function regulation and protection from oxidative stress. EPO regulated AMPK activity may, at least in part, regulate Sirt1 activity via modulating NAD+/NADH ratio, which contributes to energy metabolism and mitochondrial function in adipocytes. As the downstream target of Sirt1 and AMPK, PGC-1α may also be directly regulated by EPO or via regulating Sirt1 and AMPK activity to promote adipocyte oxidative metabolism, mitochondrial biogenesis and protection from oxidative stress.
Multiple hormones including IL-6, leptin and adiponectin have been indicated to activate the AMPK activity in adipose tissue (Kelly et al., 2004; Orci et al., 2004; Wu et al., 2003). However, our analysis of WAT indicates that expression of these hormones is not affected by EPO signaling (Teng et al., 2011), suggesting that EPO could activate AMPK through other pathways. EPO binding in hematopoietic cells induces tyrosine phosphorylation of several proteins including EpoR itself and possibly calcium channel proteins (Miller et al., 1989). EPO has also been shown to cause a Ca2+ influx in human erythroblasts, the neuronal cell line (PC12), vascular endothelial cells and in myoblasts but not in myotubes (Masuda et al., 1993; Miller et al., 1989; Ogilvie et al., 2000; Vogel et al., 1997). Importantly, our current study uncovered how EPO activates AMPK in adipocytes as our data showed that inhibited CaMKK impaired EPO stimulated AMPK activity but knock down of LKB1 did not have influence on the EPO effect. It is likely that EPO stimulation causes a rapid calcium influx and increased calcium influx may activate CaMKK activity, leading to increased AMPK activity. Also, our finding that AMPK mediates EPO activity in modulating redox status and regulating energy metabolic activity in adipocytes expands the relationship between EPO signaling and AMPK activity to multiple tissues in addition to our previously revealed skeletal muscle (Wang et al., 2013a) and suggests a possibly widespread involvement of EPO in AMPK mediated energy sensing network.
It was demonstrated that EPO regulated Sirt1 activity is involved in PPARα activity in adipocyte response to EPO (Wang et al., 2013b). EPO regulated Sirt1 activity in adipocytes may be via changed NAD+ level and NAD+/NADH ratio. However, the mechanism by which EPO affects NAD+ level and NAD+/NADH ratio remains unknown. Our results here demonstrated that EPO activated AMPK can increase NAD+ level and change NAD+/NADH ratio. This is consistent with the previous report that AMPK modulates NAD+ metabolism to regulate energy metabolism in skeletal muscle (Canto et al., 2009). Obesity is associated with the formation of hypoxic areas within the adipose tissue. This has been demonstrated in obese mice (ob/ob and dietary induced obesity) (Furukawa et al., 2004). Hypoxia switches cell metabolism from aerobic respiration to anaerobic glycolysis and inhibits glucose transport (Simon and Keith, 2008). Hypoxia status within the adipose tissue could also contribute to the development of insulin resistance (Regazzetti et al., 2009). Of note, we previously found that under hypoxia, EPO modulated cell redox status and facilitated cellular aerobic respiration to increase oxygen utilization capacity (Wang et al., 2013b), which may promote energy metabolism and increase the utilization of extra energy, resulting in increased energy expenditure and body weight loss. These effects may explain how EPO treatment in mice decreased fat accumulation and offset the adverse effect of obesity including glucose intolerance and insulin resistance. However, it is possible that EPO also modulates the cell redox status via other pathways because inhibition of AMPK activity did not completely abrogate the increase of NAD+/NADH by EPO. It will be informative to investigate how EPO modulates the cellular redox status and NAD+ metabolism in details.
Oxidative stress, defined as a pathological state characterized by increased ROS production or decreased ability to detoxify ROS, plays a causative role in tissue injury in many disease conditions, including cardiovascular disease, neurological disorder, cancers, and aging. Obesity can induce systemic oxidative stress and increased oxidative stress in accumulated fat causes dysregulation of adipocytokines and development of metabolic syndrome (Furukawa et al., 2004). Increased ROS in excessively accumulated fat also causes increased oxidative stress in blood and has deleterious effects on other organs including the liver, skeletal muscle and cardiovascular system. It is possible that increased oxidative stress in fat is important underlying cause of obesity-associated metabolic disorder (Furukawa et al., 2004). Therefore, how to modulate the redox state and reduce ROS production in adipose tissue can be used as a potentially useful target to develop new therapies against obesity and its associated metabolism syndrome. Oxidative stress plays critical roles in the pathogenesis of various diseases. In the diabetic condition, oxidative stress impairs glucose uptake in muscle and fat and decreases insulin secretion form pancreatic β cells (Brownlee, 2001; Maddux et al., 2001; Matsuoka et al., 1997; Rudich et al., 1998). Both oxidative stress and hypoxia are known to occur in adipose tissue in response to DIO obesity as well as in genetic models of obesity (Ye et al., 2007). EPO was reported to reduce oxidative stress damage in heart disease to provide cardio protection (Kim et al., 2008; Li et al., 2006). EPO also has antioxidant effect in thalassemic blood cells (Amer et al., 2010). In this study, EPO was found to inhibit ROS production in adipocytes and the adipocyte and WAT expression of antioxidant enzymes were also increased by EPO. Insulin resistance has been associated with the generation of ROS. EPO improves insulin resistance (Teng et al., 2011; Wang et al., 2013b). It is possible that the antioxidant stress effect of EPO mediates the improvement in insulin resistance in obese mice. AMPK deletion in mice leads to the increased levels of reactive oxidative species (ROS) and oxidized proteins in erythrocytes (Wang et al., 2010), indicating that AMPK is essential in maintaining redox homeostasis in erythrocytes (Wang et al., 2010). AMPK activity can also be triggered by hypoxia through ROS mediated activation of CaMKK (Mungai et al., 2011). Here, we demonstrated that inhibition of AMPK activity partially impaired the antioxidant effect of EPO in adipocytes. These results indicate that hypoxia due to the obese status in white adipose tissue, may stimulate the ROS production. Hypoxia may also active AMPK activity that can be further enhanced by EPO. This contribution of EPO to AMPK activity may mediate the EPO stimulated protection from the metabolic syndrome caused by increased oxidative stress in accumulated fat. However, we cannot exclude the possibility that EPO may exert its antioxidant stress effect independent of AMPK activity to offset the adverse effect of obesity, hypoxia status and ROS including glucose intolerance and insulin resistance.
In conclusion, the effects of EPO on the prevention from obesity and protection from the oxidative stress are linked to AMPK activity in adipocytes. These results together with our previous observations demonstrated that EPO-AMPK-Sirt1-PGC-1α pathway may act as an energy sensing network to modulate cellular redox state and contribute to energy homeostasis in adipocytes.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases and the National Institutes of Health (ZIA/DK025102) and the Funding from University of Macau (SRG2013-00044-FHS).
Footnotes
Disclosure: The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmadian M, Abbott MJ, Tang T, Hudak CS, Kim Y, Bruss M, Hellerstein MK, Lee HY, Samuel VT, Shulman GI, et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 2011;13:739–748. doi: 10.1016/j.cmet.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amer J, Dana M, Fibach E. The antioxidant effect of erythropoietin on thalassemic blood cells. Anemia. 2010;2010:978710. doi: 10.1155/2010/978710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, Rasbach KA, Bostrom EA, Choi JH, Long JZ, et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. 2012;481:463–468. doi: 10.1038/nature10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical chemistry. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- Canto C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Current opinion in lipidology. 2009;20:98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraway MS, Suliman HB, Jones WS, Chen CW, Babiker A, Piantadosi CA. Erythropoietin activates mitochondrial biogenesis and couples red cell mass to mitochondrial mass in the heart. Circulation research. 2010;106:1722–1730. doi: 10.1161/CIRCRESAHA.109.214353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daval M, Diot-Dupuy F, Bazin R, Hainault I, Viollet B, Vaulont S, Hajduch E, Ferre P, Foufelle F. Anti-lipolytic action of AMP-activated protein kinase in rodent adipocytes. J Biol Chem. 2005;280:25250–25257. doi: 10.1074/jbc.M414222200. [DOI] [PubMed] [Google Scholar]
- Di LJ, Fernandez AG, De Siervi A, Longo DL, Gardner K. Transcriptional regulation of BRCA1 expression by a metabolic switch. Nat Struct Mol Biol. 2010;17:1406–1413. doi: 10.1038/nsmb.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer LG, Parbu-Patel A, Carling D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277:25226–25232. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri S, Rattan R, Haq E, Khan M, Yasmin R, Won JS, Key L, Singh AK, Singh I. AICAR inhibits adipocyte differentiation in 3T3L1 and restores metabolic alterations in diet-induced obesity mice model. Nutr Metab (Lond) 2006;3:31. doi: 10.1186/1743-7075-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusarova GA, Trejo HE, Dada LA, Briva A, Welch LC, Hamanaka RB, Mutlu GM, Chandel NS, Prakriya M, Sznajder JI. Hypoxia Leads to Na,K-ATPase Downregulation via Ca2+ Release-Activated Ca2+ Channels and AMPK Activation. Mol Cell Biol. 2011;31:3546–3556. doi: 10.1128/MCB.05114-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, Furukawa S, Tochino Y, Komuro R, Matsuda M, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56:901–911. doi: 10.2337/db06-0911. [DOI] [PubMed] [Google Scholar]
- Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Qu A, Matsubara T, Chanturiya T, Jou W, Gavrilova O, Shah YM, Gonzalez FJ. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes. 2011;60:2484–2495. doi: 10.2337/db11-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Seale P, Tomaru T, Erdjument-Bromage H, Cooper MP, Ruas JL, Chin S, Tempst P, Lazar MA, Spiegelman BM. Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes Dev. 2008;22:1397–1409. doi: 10.1101/gad.1666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keijer J, van Schothorst EM. Adipose tissue failure and mitochondria as a possible target for improvement by bioactive food components. Current opinion in lipidology. 2008;19:4–10. doi: 10.1097/MOL.0b013e3282f39f95. [DOI] [PubMed] [Google Scholar]
- Kelly M, Keller C, Avilucea PR, Keller P, Luo Z, Xiang X, Giralt M, Hidalgo J, Saha AK, Pedersen BK, et al. AMPK activity is diminished in tissues of IL-6 knockout mice: the effect of exercise. Biochem Biophys Res Commun. 2004;320:449–454. doi: 10.1016/j.bbrc.2004.05.188. [DOI] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Kim KH, Oudit GY, Backx PH. Erythropoietin protects against doxorubicin-induced cardiomyopathy via a phosphatidylinositol 3-kinase-dependent pathway. The Journal of pharmacology and experimental therapeutics. 2008;324:160–169. doi: 10.1124/jpet.107.125773. [DOI] [PubMed] [Google Scholar]
- Lage R, Dieguez C, Vidal-Puig A, Lopez M. AMPK: a metabolic gauge regulating whole-body energy homeostasis. Trends Mol Med. 2008;14:539–549. doi: 10.1016/j.molmed.2008.09.007. [DOI] [PubMed] [Google Scholar]
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Li Y, Takemura G, Okada H, Miyata S, Maruyama R, Li L, Higuchi M, Minatoguchi S, Fujiwara T, Fujiwara H. Reduction of inflammatory cytokine expression and oxidative damage by erythropoietin in chronic heart failure. Cardiovasc Res. 2006;71:684–694. doi: 10.1016/j.cardiores.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Lihn AS, Jessen N, Pedersen SB, Lund S, Richelsen B. AICAR stimulates adiponectin and inhibits cytokines in adipose tissue. Biochem Biophys Res Commun. 2004;316:853–858. doi: 10.1016/j.bbrc.2004.02.139. [DOI] [PubMed] [Google Scholar]
- Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006;21:521–531. doi: 10.1016/j.molcel.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddux BA, See W, Lawrence JC, Jr, Goldfine AL, Goldfine ID, Evans JL. Protection against oxidative stress-induced insulin resistance in rat L6 muscle cells by mircomolar concentrations of alpha-lipoic acid. Diabetes. 2001;50:404–410. doi: 10.2337/diabetes.50.2.404. [DOI] [PubMed] [Google Scholar]
- Masuda S, Nagao M, Takahata K, Konishi Y, Gallyas F, Jr, Tabira T, Sasaki R. Functional erythropoietin receptor of the cells with neural characteristics. Comparison with receptor properties of erythroid cells. J Biol Chem. 1993;268:11208–11216. [PubMed] [Google Scholar]
- Matsuoka T, Kajimoto Y, Watada H, Kaneto H, Kishimoto M, Umayahara Y, Fujitani Y, Kamada T, Kawamori R, Yamasaki Y. Glycation-dependent, reactive oxygen species-mediated suppression of the insulin gene promoter activity in HIT cells. J Clin Invest. 1997;99:144–150. doi: 10.1172/JCI119126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BA, Cheung JY, Tillotson DL, Hope SM, Scaduto RC., Jr Erythropoietin stimulates a rise in intracellular-free calcium concentration in single BFU-E derived erythroblasts at specific stages of differentiation. Blood. 1989;73:1188–1194. [PubMed] [Google Scholar]
- Mungai PT, Waypa GB, Jairaman A, Prakriya M, Dokic D, Ball MK, Schumacker PT. Hypoxia Triggers AMPK Activation through Reactive Oxygen Species-Mediated Activation of Calcium Release-Activated Calcium Channels. Mol Cell Biol. 2011;31:3531–3545. doi: 10.1128/MCB.05124-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi CT, Wang L, Rogers HM, Teng R, Jia Y. Survival and proliferative roles of erythropoietin beyond the erythroid lineage. Expert reviews in molecular medicine. 2008;10:e36. doi: 10.1017/S1462399408000860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyengaard JR, Ido Y, Kilo C, Williamson JR. Interactions between hyperglycemia and hypoxia: implications for diabetic retinopathy. Diabetes. 2004;53:2931–2938. doi: 10.2337/diabetes.53.11.2931. [DOI] [PubMed] [Google Scholar]
- Ogilvie M, Yu X, Nicolas-Metral V, Pulido SM, Liu C, Ruegg UT, Noguchi CT. Erythropoietin stimulates proliferation and interferes with differentiation of myoblasts. The Journal of biological chemistry. 2000;275:39754–39761. doi: 10.1074/jbc.M004999200. [DOI] [PubMed] [Google Scholar]
- Orci L, Cook WS, Ravazzola M, Wang MY, Park BH, Montesano R, Unger RH. Rapid transformation of white adipocytes into fat-oxidizing machines. Proc Natl Acad Sci U S A. 2004;101:2058–2063. doi: 10.1073/pnas.0308258100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qatanani M, Lazar MA. Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes Dev. 2007;21:1443–1455. doi: 10.1101/gad.1550907. [DOI] [PubMed] [Google Scholar]
- Regazzetti C, Peraldi P, Gremeaux T, Najem-Lendom R, Ben-Sahra I, Cormont M, Bost F, Le Marchand-Brustel Y, Tanti JF, Giorgetti-Peraldi S. Hypoxia decreases insulin signaling pathways in adipocytes. Diabetes. 2009;58:95–103. doi: 10.2337/db08-0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Rudich A, Tirosh A, Potashnik R, Hemi R, Kanety H, Bashan N. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes. 1998;47:1562–1569. doi: 10.2337/diabetes.47.10.1562. [DOI] [PubMed] [Google Scholar]
- Sakanaka M, Wen TC, Matsuda S, Masuda S, Morishita E, Nagao M, Sasaki R. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci U S A. 1998;95:4635–4640. doi: 10.1073/pnas.95.8.4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scime A, Devarakonda S, Conroe HM, Erdjument-Bromage H, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, Cohen P, Cinti S, Spiegelman BM. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest. 2011;121:96–105. doi: 10.1172/JCI44271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shingo T, Sorokan ST, Shimazaki T, Weiss S. Erythropoietin regulates the in vitro and in vivo production of neuronal progenitors by mammalian forebrain neural stem cells. J Neurosci. 2001;21:9733–9743. doi: 10.1523/JNEUROSCI.21-24-09733.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nat Rev Mol Cell Biol. 2008;9:285–296. doi: 10.1038/nrm2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderling TR. The Ca-calmodulin-dependent protein kinase cascade. Trends in biochemical sciences. 1999;24:232–236. doi: 10.1016/s0968-0004(99)01383-3. [DOI] [PubMed] [Google Scholar]
- Su KH, Yu YB, Hou HH, Zhao JF, Kou YR, Cheng LC, Shyue SK, Lee TS. AMP-activated protein kinase mediates erythropoietin-induced activation of endothelial nitric oxide synthase. Journal of cellular physiology. 2012;227:3053–3062. doi: 10.1002/jcp.23052. [DOI] [PubMed] [Google Scholar]
- Sun L, Xie H, Mori MA, Alexander R, Yuan B, Hattangadi SM, Liu Q, Kahn CR, Lodish HF. Mir193b-365 is essential for brown fat differentiation. Nat Cell Biol. 2011;13:958–965. doi: 10.1038/ncb2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng R, Gavrilova O, Suzuki N, Chanturiya T, Schimel D, Hugendubler L, Mammen S, Yver DR, Cushman SW, Mueller E, et al. Disrupted erythropoietin signalling promotes obesity and alters hypothalamus proopiomelanocortin production. Nature communications. 2011;2:520. doi: 10.1038/ncomms1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007;100:328–341. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- Tsai PT, Ohab JJ, Kertesz N, Groszer M, Matter C, Gao J, Liu X, Wu H, Carmichael ST. A critical role of erythropoietin receptor in neurogenesis and post-stroke recovery. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:1269–1274. doi: 10.1523/JNEUROSCI.4480-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel V, Kramer HJ, Backer A, Meyer-Lehnert H, Jelkmann W, Fandrey J. Effects of erythropoietin on endothelin-1 synthesis and the cellular calcium messenger system in vascular endothelial cells. American journal of hypertension. 1997;10:289–296. doi: 10.1016/s0895-7061(96)00410-4. [DOI] [PubMed] [Google Scholar]
- Wang L, Jia Y, Rogers H, Suzuki N, Gassmann M, Wang Q, McPherron AC, Kopp JB, Yamamoto M, Noguchi CT. Erythropoietin contributes to slow oxidative muscle fiber specification via PGC-1alpha and AMPK activation. The international journal of biochemistry & cell biology. 2013a;45:1155–1164. doi: 10.1016/j.biocel.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Jia Y, Rogers H, Wu YP, Huang S, Noguchi CT. GATA-binding protein 4 (GATA-4) and T-cell acute leukemia 1 (TAL1) regulate myogenic differentiation and erythropoietin response via cross-talk with Sirtuin1 (Sirt1) The Journal of biological chemistry. 2012;287:30157–30169. doi: 10.1074/jbc.M112.376640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Teng R, Di L, Rogers H, Wu H, Kopp JB, Noguchi CT. PPARalpha and Sirt1 mediate erythropoietin action in increasing metabolic activity and browning of white adipocytes to protect against obesity and metabolic disorders. Diabetes. 2013b;62:4122–4131. doi: 10.2337/db13-0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Dale GL, Song P, Viollet B, Zou MH. AMPKalpha1 deletion shortens erythrocyte life span in mice: role of oxidative stress. J Biol Chem. 2010;285:19976–19985. doi: 10.1074/jbc.M110.102467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- Wu X, Motoshima H, Mahadev K, Stalker TJ, Scalia R, Goldstein BJ. Involvement of AMP-activated protein kinase in glucose uptake stimulated by the globular domain of adiponectin in primary rat adipocytes. Diabetes. 2003;52:1355–1363. doi: 10.2337/diabetes.52.6.1355. [DOI] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab. 2007;293:E1118–1128. doi: 10.1152/ajpendo.00435.2007. [DOI] [PubMed] [Google Scholar]
- Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV, Semenza GL. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11:407–420. doi: 10.1016/j.ccr.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.