

Abstract
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common malignancy worldwide, and patient outcomes using current treatments remains poor. Tumor development is etiologically associated with tobacco or alcohol use and/or HPV infection. HPV positive HNSCCs, which frequently harbor wild-type p53, carry a more favorable prognosis and are a biologically distinct subgroup when compared to their HPV negative counterparts. HPV E7 induces expression of the human DEK gene, both in vitro and in vivo. In keratinocytes, DEK overexpression is sufficient for causing oncogenic phenotypes in the absence of E7. Conversely, DEK loss results in cell death in HPV positive cervical cancer cells at least in part through p53 activation, and Dek knockout mice are relatively resistant to the development of chemically induced skin papillomas. Despite the established oncogenic role of DEK in HPV associated cervical cancer cell lines and keratinocytes, a functional role of DEK has not yet been explored in HNSCC. Using an established transgenic mouse model of HPV16 E7 induced HNSCC, we demonstrate that Dek is required for optimal proliferation of E7-transgenic epidermal cells and for the growth of HNSCC tumors. Importantly, these studies also demonstrate that DEK protein is universally up-regulated in both HPV positive and negative human HNSCC tumors relative to adjacent normal tissue. Furthermore, DEK knockdown inhibited the proliferation of HPV positive and negative HNSCC cells, establishing a functional role for DEK in human disease. Mechanistic studies reveal that attenuated HNSCC cell growth in response to DEK loss was associated with reduced expression of the oncogenic p53 family member, ΔNp63. Exogenous ΔNp63 expression rescued the proliferative defect in the absence of DEK, thereby establishing a functional DEK-ΔNp63 oncogenic pathway that promotes HNSCC. Taken together, our data demonstrate that DEK stimulates HNSCC cellular growth and identify ΔNp63 as a novel DEK effector.
Keywords: DEK, p63, HPV, head and neck cancer
Introduction
Head and neck cancer is a devastating disease with approximately 60% of patients presenting with locally advanced disease.1 Over 600,000 new head and neck cancers are diagnosed each year worldwide, the vast majority representing squamous cell carcinomas (SCCs).2 HNSCC has traditionally been associated with risk factors such as tobacco and alcohol consumption; however, in recent years human papillomavirus (HPV), a well-known cause of cervical cancer, has emerged as a new etiological pathogen. It is now established that at least 25% of all HNSCCs carry HPV genomic DNA, predominantly of the high risk HPV16 subtype.3 HPV positive HNSCCs are associated with improved survival for affected patients, and such tumors are more sensitive to available therapies. This increased tumor response is due at least in part to p53 tumor suppressor activation despite the presence of E6, a viral oncogene which targets p53 through physical interactions and degradation.4, 5 Thus, HPV positive and negative head and neck tumors are biologically distinct subtypes of HNSCC. Regardless of HPV status however, achieving efficacy with radiation and chemotherapy treatments remains notoriously difficult. This is due to the inherent molecular heterogeneity of HNSCC, combined with patients presenting at late stage disease where there is a high likelihood of local recurrence and metastasis. Thus, survival rates for patients with HNSCC have not significantly improved in decades, highlighting the urgent need for novel biomarkers and therapeutic targets.6 Recent limited analysis of HNSCC tissue arrays by our laboratory had suggested that DEK expression was up-regulated in the majority of HPV negative HNSCCs.7 Herein we extend these studies by analyzing a panel of primary human tumors to determine DEK expression levels in both HPV positive and negative HNSCCs, and define the functional role of DEK in regulating HNSCC growth in vitro and in vivo.
The human DEK oncogene was first identified as a fusion with the CAN nucleoporin in acute myeloid leukemia, and the DEK protein was subsequently purified based on its ability to modulate DNA topology.8, 9 DEK is located primarily in the nucleus where it is bound to chromatin and is rarely amplified or mutated, but frequently transcriptionally up-regulated in a variety of malignancies. Transcriptional induction of DEK can occur through Rb-dependent mechanisms in several tumor types; a finding which is consistent with the original observation that DEK is induced by high risk HPV E7 expression in human keratinocytes and up-regulated in HPV positive cervical cancer cells.10 DEK knockdown in cervical cancer cells expressing the HPV E6 and E7 oncogenes, results in apoptosis that was at least partially mediated through the stabilization of the p53 tumor suppressor.11 Conversely, DEK overexpression in near-diploid immortalized keratinocytes that form skin (NIKS) stimulated proliferation and suppressed differentiation in organotypic epithelial rafts through a p53 independent mechanism.12 Effects of DEK loss beyond those dependent on p53, however, remain undefined in human SCCs.
Dek−/− mice are viable and are relatively resistant to benign papilloma formation in a chemically induced skin carcinogenesis model, thus implicating DEK in tumor initiation.13 In order to test the requirement for Dek in a malignant SCC model system in vivo, we utilized a transgenic HNSCC mouse model. Therein, HPV E7 is targeted to the stratified squamous epithelium with the keratin 14 promoter (K14E7) and mice develop rapid and highly penetrant tongue and esophageal tumors upon addition of the mutagen 4-nitroquinoline-1-oxide (4-NQO) to the drinking water.14 To determine whether Dek expression is important for HNSCC development, K14E7 mice in a Dek proficient and deficient background were subjected to 4-NQO treatment. Transgenic K14E7 Dek−/− mice exhibited decreased epidermal cell proliferation and attenuated tumor growth as compared to K14E7 Dek+/+ mice. Importantly, Dek was not required for proliferation in non-transgenic mice, indicating that cellular growth suppression by Dek is specific to the oncogenic stimulus induced by E7. Complementary studies in primary human HNSCC tumor tissue and cells demonstrate that DEK protein expression is universally up-regulated regardless of HPV status and that DEK supports tumor cell proliferation. Finally, we show for the first time that DEK promotes HNSCC growth through a ΔNp63 dependent mechanism, thus identifying ΔNp63 as a novel downstream effector of DEK function.
Results
Dek knockout mice exhibit attenuated HNSCC development in vivo
High risk E7 protein was previously shown to stimulate DEK expression in vitro and in vivo, and DEK up-regulation was sufficient to stimulate oncogenic phenotypes in cellular models of SCC.10, 13 To directly determine the functional role of Dek in HPV E7 driven HNSCC, we tested the requirement for Dek in an established K14E7-driven mouse model of HNSCC.15 K14E7 transgenic mice were crossed into a Dek knockout background to generate K14E7 Dek−/− which were compared to K14E7 Dek+/+ mice. Non-transgenic Dek wild-type and knockout mice were generated as controls. All mice were treated with 4-NQO for 16 weeks followed by normal drinking water for 8 weeks. At this 24 week time point, all mice were sacrificed and analyzed for tumor development. K14E7 Dek−/− mice trended towards increased survival compared to the K14E7 Dek+/+ mice; 82% of the K14E7 Dek+/+ mice died prior to sacrifice, compared to 43% of the K14E7 Dek−/− mice (Figure 1a). The cause of premature death for the K14E7 Dek+/+ and one of three K14E7 Dek−/− mice was likely due to tumor burden. The cause of death for the remaining two K14E7 Dek−/− mice is unknown. A representative section from a K14E7 Dek+/+ mouse confirms Dek is highly expressed in the tumor (Figure 1b). The results of the pathological analyses for these tumors are summarized in Figure 1c. As expected from the literature,15 all 11 of the K14E7 Dek+/+ mice developed SCCs of the tongue and esophagus (Figure 1c), whereas the non-transgenic mice had no macroscopic tumors at the time of sacrifice. In contrast to K14E7 Dek+/+ mice, only one of seven K14E7 Dek−/− mice developed a visible tumor. K14E7 Dek−/− mice did have microscopic tumors indicating that Dek is not required for tumor initiation in this model (Figure 1b). Importantly, Dek loss was associated with smaller tumors indicating that Dek promotes HNSCC growth.
Figure 1. Dek loss protects from HNSCC tumor promotion in vivo.
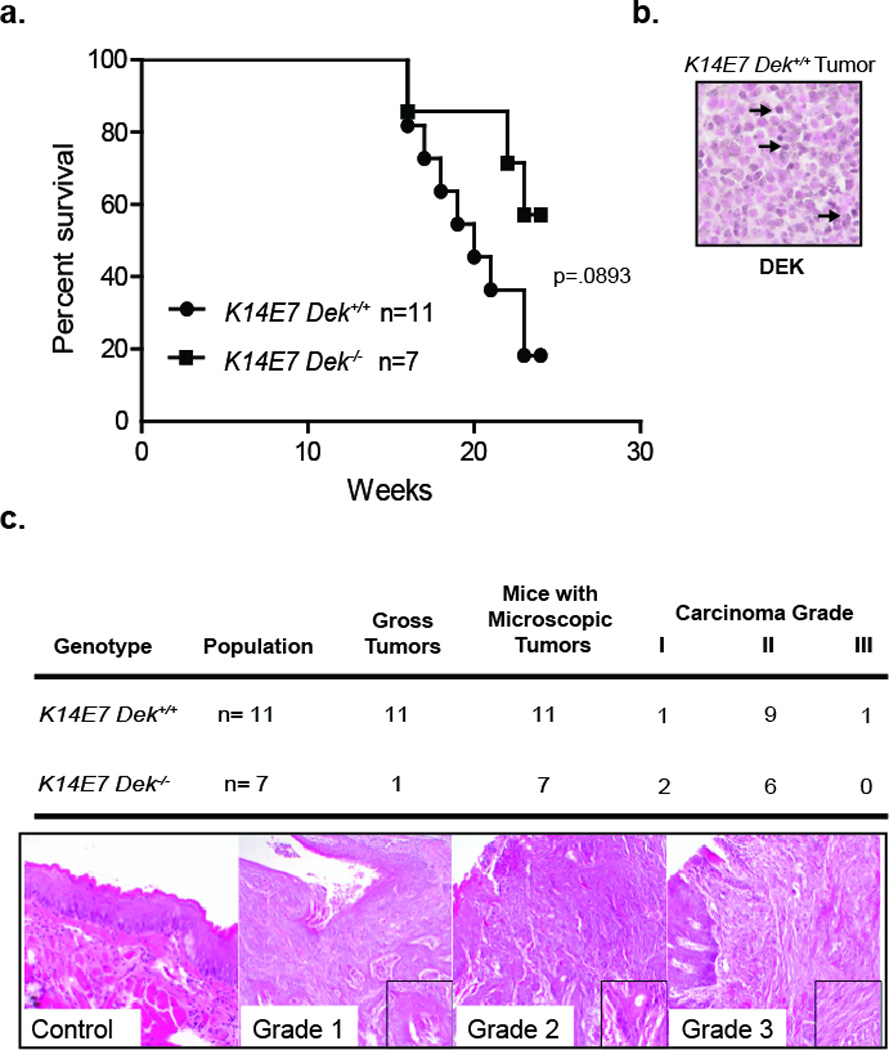
(a) Dek loss prolongs survival in a murine model of HNSCC. Nine of eleven K14E7 Dek+/+ were sacrificed prematurely due to moribund appearance, whereas only three out of seven K14E7 Dek−/− mice were sacrificed early. (b) Immunohistochemistry depicting a K14E7 Dek+/+ mouse tumor that stains positive for DEK, with arrows representing examples of positive cells. (c) Mice were sacrificed at 24 weeks for gross macroscopic and microscopic tongue and esophagus lesion analysis. Tumors grade was determined histologically as previously describe for this 4-NQO-driven HNSCC models42 and representative images are shown. One K14E7 Dek−/− mouse had microscopic tumors in both the tongue and esophagus, hence the number of microscopic tumors in this group totals 8 for the 7 mice. Original magnifications are at 200×. p=.0893 as determined by log-rank (Mantel-Cox) test.
Dek loss suppresses E7-dependent proliferation in the tongue epidermis
We next sought to determine the underlying cellular mechanism responsible for the observed HNSCC growth suppression in K14E7 Dek−/− mice. DEK was shown to inhibit apoptosis and stimulate proliferation in vitro, depending on the cellular and experimental context. Accordingly, we assessed for both biological processes in E7-transgenic Dek proficient and deficient epidermis. Apoptotic cells were not detected by cleaved caspase-3 detection (data not shown) indicating that increased cellular death did not account for the decrease in tumor growth in Dek knockout mice. As was previously published, K14E7 transgene expression was associated with hyperplasia and an increase in BrdU positive cells in both the basal and suprabasal compartments as compared to the non-transgenic controls (Figure 2a and 2b).16 Importantly, K14E7 Dek−/− mice displayed decreased proliferation when compared to K14E7 Dek+/+ epidermis, with proliferative rates repressed to baseline levels observed in non-transgenic mice (Figure 2b). Dek loss alone was not sufficient to decrease cellular proliferation in the absence of E7 expression. Taken together, these findings support the notion that Dek up-regulation is required for E7 driven epidermal proliferation, and that cellular growth suppression in response to Dek loss in vivo occurs specifically in the oncogenic environment induced by E7.
Figure 2. Dek loss attenuates E7-driven proliferation in tongue epidermis.
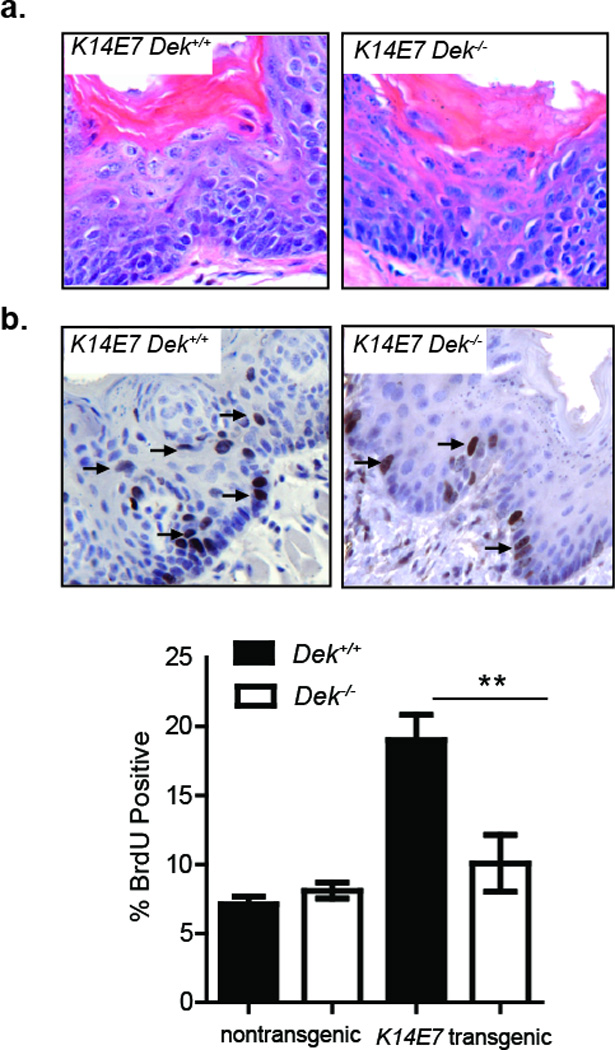
(a) H&E stained images of Dek-proficient and Dek-deficient E7-trangenic tongue epidermis showing epithelial hyperplasia are shown at a total magnification of 200×. (b) Mice were injected with BrdU prior to sacrifice, and immunohistochemistry done to detect BrdU positive cells in the pre-malignant tongue epidermis. Representative images with arrows denoting BrdU positive cells are shown. The percentage of epidermal BrdU positive cells was determined by counting >1000 cells in sections from 4 K14E7 Dek+/+, 4 K14E7 Dek−/−, 4 nontransgenic Dek+/+, and 3 nontransgenic Dek−/− mice. Tongues from K14E7 Dek+/+ mice harbored significantly more BrdU positive cells as compared to K14E7 Dek−/− mice. **=p≤0.01 by two-tailed t-test.
DEK protein expression is up-regulated in human HNSCC regardless of HPV status
DEK mRNA and protein expression is up-regulated in multiple cancer types including breast cancer, melanoma, and hepatocellular carcinoma17–20 and is also induced in response to E7 expression.10 We previously reported DEK up-regulation in high risk HPV positive and HPV negative HNSCC tissue microarrays; however, analyses were limited for HPV positive tumors since only 4/44 (9%) of the examined samples were HPV positive by in situ hybridization.7 Additionally, potential associations with demographic and clinical information were not examined. The current analysis extends these prior studies by performing rigorous HPV testing with maximal sensitivity for the detection of HPV16 genome sequences. HPV16 is the most prevalent HPV type in HNSCCs. We also analyzed relevant clinical characteristics including parameters characteristically associated with HPV status. Additionally, optimal tissue preservation was ensured by analyzing human HNSCC specimens that were freshly biopsied or resected with immediate processing and uniform preparation for IHC analysis to detect DEK protein expression. Table 1 depicts HPV status as determined by in situ hybridization by two independent institutions, along with highly sensitive RT-PCR for HPV16 sequences, as well as primary tumor site, tumor stage, p16 status, history of cigarette and alcohol consumption, and determination of DEK protein expression. HPV presence and DEK expression are quantified in the table, and a detailed description of the relevant values is provided in the Materials and Methods. Expression of the cyclin-dependent kinase inhibitor p16 is a clinical marker for HPV presence. Chart review indicated that 10/18 (56%) of HNSCCs tested were positive for p16 expression. HPV positive status was detected in 8/18 (44%) specimens by in situ hybridization at Site 1 (Figure 3a), which detects multiple high-risk HPV types, with examples of HPV negative and HPV positive specimens shown. Highly sensitive in situ hybridization for HPV16 at Site 2 identified two additional HPV positive specimens (10/18 or (56%)). Finally, RT-PCR identified one additional HPV16 positive specimen, (11/18) resulting in 61% of the tumors being HPV positive. All samples were subjected to IHC for DEK expression (Figure 3a). The intensity of DEK protein expression and the proportion of tumor cells positive for DEK were determined relative to adjacent normal tissue using the grading system depicted in Figure 3b and summarized in Table 1. Interestingly, strong DEK expression was detected in all tumors regardless of HPV status, tumor stage or demographic information. Neither the level of DEK expression nor the DEK-positive proportion of tumor cells correlated with HPV status (Table 1, Figure 3a, b). Taken together, these data indicate that DEK protein is universally over-expressed in HPV16 positive and negative HNSCCs.
Table 1.
Clinical characteristics of primary human tumor samples
| CCHMC ID | Primary Tumor Location |
Stage | Smoking (Y/N) |
Drinking (Y/N) |
Clinical p16 Status |
HPV in situ | RT-PCR | HPV Copies Per Cell |
DEK Levels |
||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Site 1 | Site 2 | ERV-3 | HPV-16 | ||||||||
| CCHMC-HNSCC1 | Tonsillar | IV | Y | N | + | + | + | 2.41×104 | 1.77×106 | 123 | 3S |
| CCHMC-HNSCC2 | BOTa | IV | Y | N | − | − | − | 1.77×103 | - | - | 3S |
| CCHMC-HNSCC3 | Lateral Tongue | II | Y | N | ukb | − | − | 1.74×104 | - | - | 3S |
| CCHMC-HNSCC4 | BOT | IV | Y | Y | − | − | − | 8.45×103 | - | - | 3S |
| CCHMC-HNSCC6 | BOT | III | N | N | + | + | + | 8.11 | 4.12×102 | 102 | 3S |
| CCHMC-HNSCC7 | BOT | IV | N | Y | + | − | + | 1.47×104 | 1.08×103 | 1 | 3S |
| CCHMC-HNSCC8 | Tonsillar | IV | N | N | + | + | + | 3.44×103 | 2.77×105 | 82 | 3S |
| CCHMC-HNSCC9 | Oropharyngeal | IV | N | N | + | − | − | 3.65×103 | 6.68×103 | 2 | 3S |
| CCHMC-HNSCC10 | Tonsillar | IV | Y | N | + | + | + | 5.21×103 | 4.05×104 | 10 | 3S |
| CCHMC-HNSCC11 | BOT | IV | N | N | uk | + | + | 2.32×103 | 8.19×104 | 39 | 3S |
| CCHMC-HNSCC12 | Tonsillar | IV | Y | N | + | + | + | 7.57×103 | 3.03×105 | 50 | 3V |
| CCHMC-HNSCC13 | Tonsillar | I | N | N | + | − | − | 1.41×104 | - | - | 3S |
| CCHMC-HNSCC14 | Oropharyngeal | IV | N | N | + | + | + | 6.74×103 | 3.02×104 | 6 | 2S |
| CCHMC-HNSCC15 | Tonsillar | IV | Y | Y | − | − | − | 5.25×103 | - | - | 3S |
| CCHMC-HNSCC17 | Oropharyngeal | IV | Y | Y | uk | uk | + | 1.51×104 | 5.83×103 | 1 | 2S |
| CCHMC-HNSCC18 | BOT | III | Y | Y | − | − | − | 4.70×103 | - | - | 3S |
| CCHMC-HNSCC20 | Oropharyngeal | IV | Y | Y | + | + | + | 8.19×103 | 3.13×104 | 6 | 3V |
| CCHMC-HNSCC21 | Tonsillar | IV | Y | Y | − | − | − | 1.61×104 | - | - | 3V |
Note: CCHMC-HNSCC5, 16, and 19 were removed due to a lack of embedded or malignant tissue Site 1 indicates in situ performed at CCHMC; Site 2 indicates in situ performed at OSU
BOT=base of tongue,
uk=unknown,
Please refer to the materials and methods for classifiers used to determine DEK levels
Figure 3. DEK protein expression is universally up-regulated in human HNSCC.
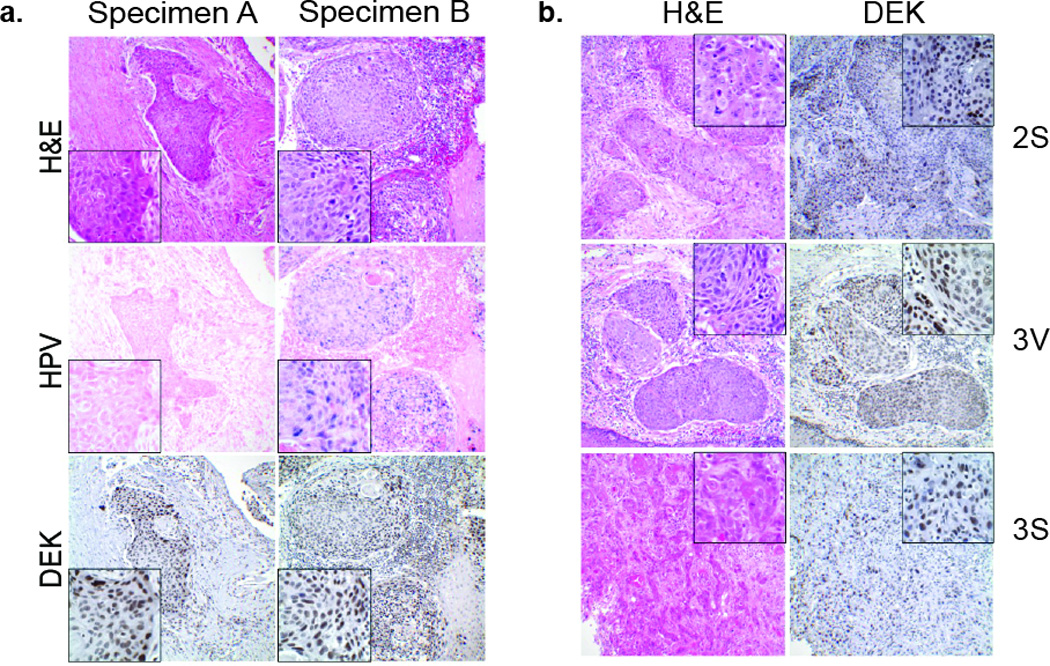
(a) Tumor specimens were sectioned, H&E stained for morphology to confirm the diagnosis of HNSCC, and subjected to in situ hybridization for dectection of high-risk HPV sequences. Representative HPV positive (Specimen B) and HPV negative (Specimen A) tumors are shown. Strong DEK expression was observed in both HPV positive and negative tumors as compared to adjacent non-neoplastic epithelium. (b) The percentage of DEK positive cells on each section was estimated and staining intensity determined as described in the Materials & Methods. Representative images and scores are shown along with the corresponding H&E image. Original magnification 200×; insets 1000×.
DEK expression stimulates growth of HPV positive and negative human HNSCC tumor cells
In order to determine the importance of DEK expression in human HNSCC cells, we next analyzed a panel of HNSCC cell lines that were transduced with non-targeting or DEK knockdown vectors. One HPV positive line, 93VU147T, and two HPV negative lines, UMSCC1 and UMSCC6, were analyzed following DEK knockdown confirmed by western blot analysis (Figure 4a–c). DEK knockdown consistently suppressed cellular growth in both HPV positive and HPV negative cell lines (Figures 4a–c). Moreover, growth suppression was associated with reduced proliferation in vitro as was observed in the E7-positive epidermis in the in vivo mouse model. BrdU incorporation was significantly reduced in all cell lines after DEK knockdown, indicating decreased progression into S phase in the absence of DEK (Figure 4a–c). Together, these data demonstrate that DEK loss in human and murine HNSCC cells attenuates tumor cell growth through suppression of cellular proliferation.
Figure 4. DEK knockdown attenuates the proliferation of HPV positive and negative HNSCC cell lines.
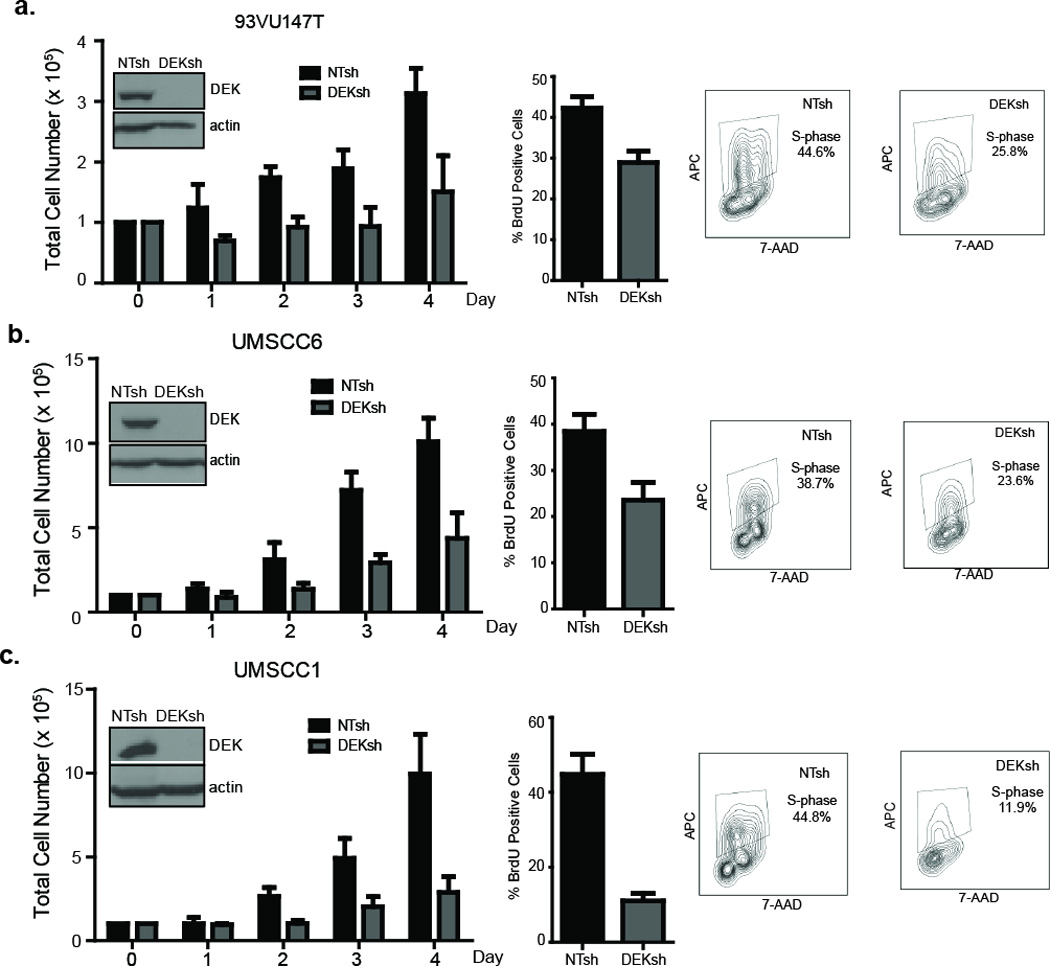
(a–c, left panels) HPV positive and HPV negative cell lines were lentivirally transduced with control or DEK-specific shRNA, selected in puromycin, and plated in equal cell numbers for cell counts taken over the course of four days. Western blot analysis indicates DEK knockdown is below the level of detection in all cell lines, with actin used as a loading control. DEK loss substantially decreased total cell numbers on day 3 and 4, regardless of HPV status. Data represents the average of two independent experiments with each sample measured in triplicate. Error bars indicate standard deviation (SD). (a–c, right panels) Control and DEK knockdown cells were incubated with BrdU for 2 hours and BrdU positive cells determined by flow cytometry. Representative contour plots show BrdU positive gating for NTsh and DEKsh cells. Data represents the average of two independent experiments with each sample measured in triplicate. Error bars indicate standard deviation (SD).
ΔNp63 protein expression is regulated by DEK and is functionally important
Our previous studies uncovered p53 dependent oncogenic function for DEK in HPV positive cervical cancer cells. p53 remains intact, yet is bound and degraded by E6 leading to suppressed p53 activity in HPV positive cells. In line with previous results in HeLa cells, DEK knockdown in HPV positive, primary CCHMC-HNSCC1 cells resulted in the up-regulation of p53 protein levels, as well as increased phosphorylation of Serine15 on p53 indicating p53 activation (Figure 5a). In contrast, p53 is mutated in the majority of HPV negative HNSCCs and is not expressed in HPV negative UMSCC1 and UMSCC6 cells (data not shown, and5, 21). Given that the growth of all three HNSCC cell lines was suppressed by DEK knockdown, p53 activity alone could not account for the proliferative defects associated with DEK depletion. The p63 family of proteins is related to p53 and expressed from two promoters, resulting in two distinct groups of p63 isoforms, TAp63 and ΔNp63, which have opposing functions. TAp63 functions as a tumor suppressor much like p53, while ΔNp63 has oncogenic functions. ΔNp63 is the predominant p63 isoform in keratinocytes and is overexpressed in the majority of HNSCCs.22 Mechanisms driving ΔNp63 up-regulation are complex and incompletely understood.22–24 Our previous data demonstrated that DEK overexpression in human keratinocytes increases ΔNp63 protein expression and the numbers of ΔNp63 positive cells in the basal layer of organotypic epithelial rafts.12 We therefore determined the effect of DEK loss in HNSCCs on ΔNp63. Lentiviral DEK knockdown in UMSCC1 and 93VU147T cells decreased ΔNp63 protein levels (Figure 5b). Furthermore, DEK loss in HPV positive or negative primary HNSCC cell populations also reduced ΔNp63 protein expression (Figure 5c) and IHC on consecutive CCHMC-HNSCC1 human tissue demonstrates DEK and ΔNp63 are strongly expressed in similar areas of the tumor (Figure 5d). Reduction of ΔNp63 was not associated with decreased mRNA levels indicating that DEK dependent control of ΔNp63 levels is not mediated through transcriptional regulation (Figure 5e); a finding which implicates DEK in a post-transcriptional mechanism controlling ΔNp63 levels. To determine whether the observed ΔNp63 repression in DEK-depleted cells was functionally relevant, HA-tagged human ΔNp63 was exogenously expressed in adenovirally DEK-depleted UMSCC1 cells to determine if the proliferation defect in DEK deficient cells could be rescued by restoring ΔNp63 levels (Figure 5f). AdDEKsh infection alone resulted in significantly reduced cell growth similar to the effects of lentiviral knockdown in Figure 4 and published data in cervical cancer cells.12 Expression of HA-ΔNp63 rescued the cellular growth defect in DEK-deficient cells, but did not increase the growth of DEK-proficient cells (Figure 5g). These data demonstrate that DEK expression in HNSCC cells is required for maintenance of ΔNp63 protein expression and indicate that DEK-dependent cellular growth in HNSCC cells is at least in part mediated by ΔNp63.
Figure 5. DEK promotes cellular growth by up-regulating ΔNp63.
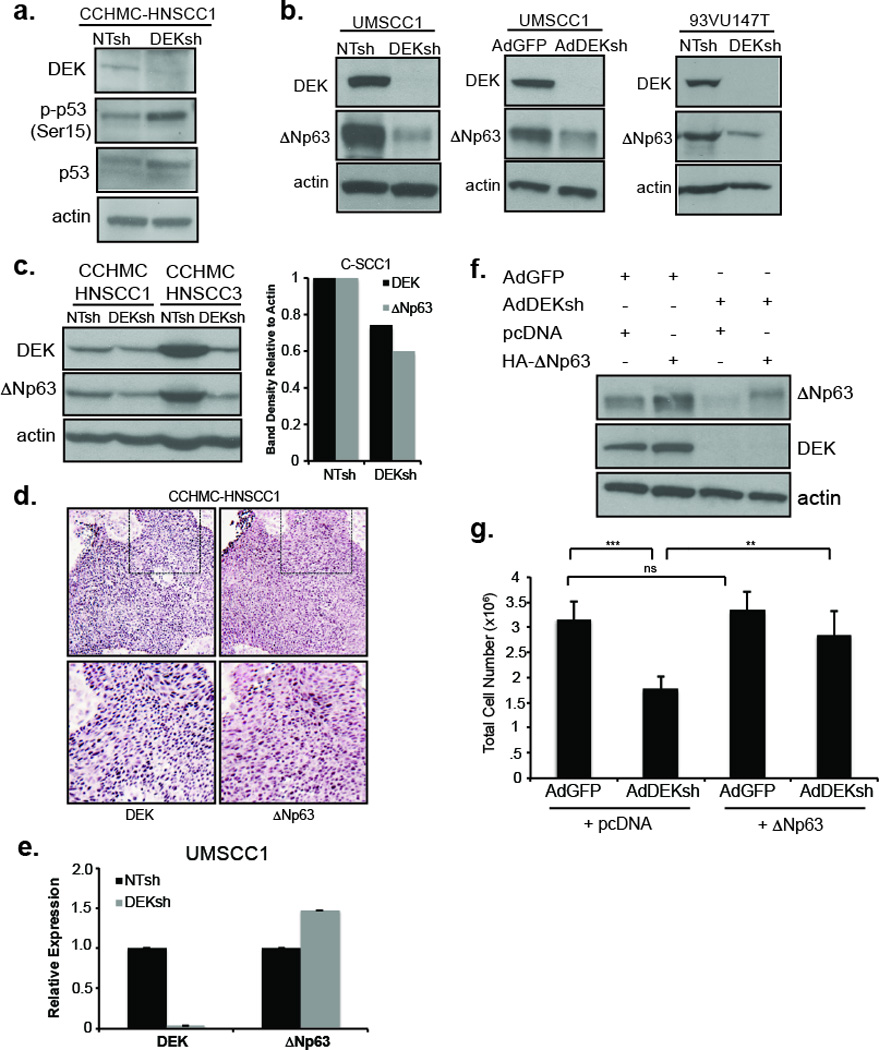
(a) Western blot analysis using CCHMC-HNSCC1 cell lysates shows phospho-ser15 p53 and total p53 up-regulation after DEK knockdown with shRNA as compared to control shRNA. (b) Western blot analysis of UMSCC1 and 93VU147T cells shows reduced ΔNp63 protein levels in DEKshRNA compared to control shRNA lentiviral infected cells and similarly with adenovirally infected UMSCC1 cells. (c) Primary human-derived cell populations CCHMC-HNSCC1 (HPV positive) and CCHMC-HNSCC3 (HPV negative) also show reduced levels of ΔNp63 with DEKshRNA as compared to NTsh controls, with densitometry depicted in the graph to the right for CCHMC-HNSCC1. (d) Immunohistochemistry for DEK and ΔNp63 have similar expression patterns in the CCHMC-HNSCC1 tumor specimen. Dashed boxes indicate expanded area with representative pictures below. Original magnifications at 200×. (e) qRT-PCR indicates ΔNp63 mRNA levels are not reduced with DEK loss in either cell line. GAPDH was used as the internal control with relative expression determined by the ΔΔCt method. (f) UMSCC1 cells were transduced with AdDEKsh or control AdGFP adenovirus and transfected with control (pcDNA) or human HA-ΔNp63 constructs on the following day. dsRed was used in all samples as a transfection efficiency control. DEK and ΔNp63 expression levels were verified by western blot analysis. (g) Viable cells from panel (f) were counted on day 3. Exogenously expressed ΔNp63 increased the total number of cells in the presence of AdDEKsh but not in control cells. Error bars indicate standard error of the mean (SEM) from 3 independent experiments. NS=not significant, **=p≤.01 and ***=p≤.001 as determined by paired two-tail t-test.
Discussion
HNSCC is a devastating disease causing approximately 350,000 deaths per year.25 Thus, development of novel therapies and identification of clinically relevant disease biomarkers and molecular targets is of paramount importance. Current chemotherapeutic strategies result in major toxicity, and surgical resection leads to debilitating physiological side effects and permanent disfigurement. Although there have been promising advancements in the field, it is unlikely that any singular treatment will drastically increase survival, and early tumor detection remains a priority and major challenge. While HPV positive tumors are associated with an improved prognosis over their HPV negative counterparts, outcomes still remain less than optimal. Biological and molecular commonalities and differences between HPV positive and HPV negative HNSCCs are only beginning to emerge.5, 26 Thus, studies of molecular pathways that mark and drive HNSCC need to be examined in parallel in these two biologically distinct subtypes. Therefore, our analyses of the expression and role of the human DEK protein were carried out in tumors that were carefully analyzed for the presence of high-risk HPV, with a focus on HPV16, together with associated patient and tumor characteristics. Expression of p16 is considered an excellent clinical marker of HPV positive tumor status. Indeed, positive staining for p16 was in agreement with the detection of HPV sequences by in situ hybridization in 70% (7/10) and 80% (8/10) of the specimens at two testing sites. However, one of ten p16 positive specimens (CCHMC-HNSCC13) was HPV16 negative by three different analyses, suggesting a need for independent confirmation of HPV status to avoid false positive results. Two specimens of unknown p16 status were additionally identified as HPV positive. All together, 61% of the HNSCC tumor specimens analyzed were determined to harbor high risk HPV16 DNA sequences. Importantly, 100% of the tumor samples showed strong DEK over-expression regardless of HPV status. However, it is important to note that our studies were primarily limited to oropharyngeal HNSCC tumors and may not represent HNSCCs as a whole. Comparisons with published DEK expression studies in breast cancer suggest some notable distinctions in HNSCCs which need to be investigated further. One report on breast cancer by our laboratory demonstrated that the proportion of DEK-expressing cells, but not per-cell staining intensity, increases with disease progression from normal breast epithelium to hyperplastic, and finally to malignant tumors.18 A second report by Liu et al. associated increasing DEK expression levels with increasing tumor stage.27 We note that for stage I–III specimens, the proportion of DEK positive tumor cells, as well as the staining intensity of the DEK oncogene, were already maximal (Table I), thus suggesting that DEK may be particularly useful as a biomarker to detect early stage HNSCCs where identification is most beneficial for the patient. Regarding the molecular mediators driving DEK protein over-expression, transcriptional up-regulation of DEK message has been reported for a multitude of human tumors and has been ascribed to distinct transcriptional mechanisms in different cell and tumor types.10, 28, 29 In HPV positive cervical cancer cells, DEK expression is driven by the high risk HPV E7 oncoprotein through Rb pocket protein dependent mechanisms. It is likely that in HPV positive HNSCCs, E7-dependent DEK regulation occurs in a similar fashion, whereas in their HPV negative counterparts, Rb mutations might be at play.
A requirement for Dek expression in genetic mouse models of cancer, and specifically during SCC development in vivo, has remained unexplored. We utilized an established E7-driven, 4-NQO dependent mouse model of HNSCC and demonstrate that Dek expression is important for the proliferation of epidermal keratinocytes and for sustained tumor cell growth. The presence of microscopic tumors in the Dek knockout tongues shows that Dek is not required for tumor initiation in this 4-NQO-dependent system. The fact that Dek loss attenuated cellular proliferation specifically in the E7 oncogene expressing, but not in control epidermis, indicates the existence of a therapeutic window for targeting DEK. DEK depletion may represent a reasonably tumor specific approach that can be exploited for tumor prevention and cancer therapy in HNSCC patient populations and beyond. Along these lines, a degree of specificity for targeting DEK functions has been supported by earlier in vitro studies, where DEK expression was highest in HPV positive cervical cell lines, lower in normal, primary keratinocytes, and lowest in differentiated keratinocytes.10, 12 Broad HNSCC cancer cell addiction to DEK over-expression was observed in both HPV positive and negative HNSCC lines. Previously published data demonstrate that in HeLa cells, DEK loss was associated with apoptosis and senescence by a mechanism that at least partially involved up-regulation and activation of p53.11 Similarly, DEK depletion in HPV positive HNSCCs led to p53 up-regulation. However, in light of the fact that HPV negative HNSCCs respond to DEK knockdown, we sought to identify p53-independent DEK effectors.
Expression of the ΔNp63 member of the p53 protein family has been implicated in the maintenance of stem and progenitor cells in various epithelia. In fact, ΔNp63 was recently shown to be essential for reprogramming mouse embryonic fibroblasts as measured by pluripotent stem cell phenotypes such as colony formation by alkaline phosphatase stain and teratoma formation.30 Such activities in cellular stemness are supported by the restricted expression of ΔNp63 in the basal cell layer of the epidermis where keratinocyte stem and progenitor cells reside. On the other hand, ΔNp63 has been implicated in triggering terminal differentiation.31, 32 The role of ΔNp63 in stem cell functions is therefore complex, and the extent to which ΔNp63 regulates stem cell proliferation, differentiation or a combination of both remains controversial. Apart from pluripotent stem cells, ΔNp63 is now recognized as a somatic stem cell marker in other types of epithelium, highlighting the importance of this protein in multiple developmental contexts.33 p63−/− mice are well characterized for their severe developmental defects at birth including the lack of appendages, squamous epithelium, and hair follicles, and not surprisingly, recent work implicates the loss of the ΔNp63 isoform as the largest contributor to these phenotypes.34–36 Not surprisingly, maintaining appropriate levels of ΔNp63 is critical for preserving epithelial integrity and suppressing transformation. Indeed, overexpression of ΔNp63 is considered oncogenic in HNSCC, and p63 amplification and mutation have been reported in a subset of HNSCCs.22, 23, 37 Regulation of ΔNp63 expression is complex and can occur at both transcriptional and post-transcriptional levels. Here, we report that DEK controls ΔNp63 expression via a post-transcriptional mechanism. Importantly, exogenous ΔNp63 expression in DEK-depleted cells rescued cancer cell growth demonstrating that DEK-dependent ΔNp63 expression is functionally important for promoting HNSCC growth. DEK activities can thus be placed upstream of ΔNp63, where targeting DEK results in decreased levels of this recognized oncogene that contributes to SCC pathogenesis.22 Future work will examine the mechanism(s) whereby DEK regulates ΔNp63. Given that ΔNp63 expression in DEK deficient tumor cells did not completely restore proliferation to the level of control cells, we cannot rule out the possibility that other DEK effectors exist and contribute to tumor cell growth in vitro and in vivo. Taken together, our results suggest the existence of a new ΔNp63-DEK signaling axis, which promotes proliferation in HNSCC.
Materials and Methods
Cell culture
HPV negative UMSCC1 and UMSCC6 head and neck cancer cells were maintained in Dulbecco’s modified Eagle medium (DMEM, Gibco, New York, USA) supplemented with 1% hydrocortisone, 10% fetal bovine serum (FBS), antibiotics and antifungals. HPV positive 93VU147T head and neck cancer cell lines were maintained in the same media but without hydrocortisone.
Lentiviral transduction
Cells were transduced at 30% confluency with lentiviral pLKO.1 non-targeting control (NTsh) or pLKO.1 DEK832 (DEKsh2) (Sigma-Aldrich Mission shRNA library) as previously described.18, 38 A final concentration of 8 µg/mL polybrene was added for each transduction. Cells were selected and carried in 1ug/mL of puromycin for primary cultures or in 1.25 µg/mL for established cell lines, and were independently transduced for each experiment.
Adenoviral transduction
Cells were transduced at 40% confluency with adenoviral control vector (AdGFP) or adenoviral DEK knockdown (AdDEKsh) at 10 infectious units per cell as previously published.11 Protein lysates were collected 72 hours post-infection.
Primary human head and neck cancer specimens
Human HNSCC tissues were freshly obtained with IRB approval at the time of surgical resection or biopsy. Chart review was performed to determine patient demographics, risk factors and histopathological characteristics. Tissues were immediately placed in F-media.12 A portion of the tissues was fixed in 4% paraformaldehyde and embedded in paraffin for immunohistochemical analysis. A second portion of each specimen was also cultured in F-media on irradiated J2–3T3 mouse fibroblasts for primary cell culture as described previously.39 Resulting cell populations were considered primary if passaged fewer than 15 times. Tissue sections were subjected to hemotoxylin and eosin (H&E) staining to confirm the presence of squamous cell carcinoma.
HPV detection
For in-house HPV analysis (Site 1), tissue sections were deparaffinized and subjected to in situ hybridization using standard DNA probes specific for 12 high risk HPV types including 16 and 18 types provided by Ventana Medical Systems (INFORM HPV III Family 16 Probe (B)). Sections were then incubated with a fluorescein-tagged DNA probe and counterstained using the automated Ventana BenchMark instrument (Ventana Medical System, Tucson, AZ, USA). Further HPV detection by in situ hybridization and real-time TaqMAN PCR assays (Site 2) was previously described by the Gillison laboratory.40 Briefly, all 21 CCHMC-HNSCC specimens were evaluated for HPV16 using a biotinlyated probe (Genpoint, Dako, Copenhagen, Denmark) and were considered positive based on nuclear staining in tumor cells. Real-time PCR was performed on formalin-fixed, paraffin-embedded specimen (FFPE) extracted DNA, and interrogated for HPV16 sequences. Human endogenous retrovirus-3 (ERV3) was used as the internal control. Threshold for positivity is any value above 1 HPV copy as determined by calculating the ratio of HPV16/ERV3 and accounting for total percentage of tumor present.
Evaluation of patient clinical characteristics
Chart review was performed to extract relevant clinical information for each patient. For Table 1, any patient who currently smokes or who smoked previously was designated as a smoker. Alcohol consumption was assigned to patients who self-reported moderate to heavy drinking and not social or occasional drinkers. Patients who answered no to either smoking or drinking were designated as such. Expression of p16, a marker of HPV positivity, was also noted if clinically determined. Quantitation of DEK positive staining by immunohistochemistry was carried out as follows: The number of cells with positive staining for DEK was quantified as: 3= >90% positive tumor cells, 2= 10–50% positive tumor, 1= <10% positive tumor cells. The overall intensity of DEK staining was determined as W (weak), V (variable) or S (strong) as compared to surrounding non-neoplastic squamous epithelium. The overall DEK status is then labeled by a combination of these values. For instance, ‘3S’ in Table 1 indicates this tumor specimen is maximally positive for DEK staining intensity and for the proportion of DEK-expressing cells.
Transgenic and knockout mouse models
The K14E7 head and neck cancer mouse model and Dek−/− mouse were previously described.16,13 Dek−/− mice were backcrossed into an F/VBN background and E7 was maintained in a heterozygous state. For generation of K14E7 Dek−/− mice, female Dek−/− mice were crossed with K14E7 males. The following primer sets were used for genotyping: Dek wild-type allele (5’-CGA ACT CGT GAA GAG GAT CTT GA-3′, 5’-ATG TGT CAG GCT GCA TCT CCA ATG-3’), Dek knockout allele (5’-ATC CAT CAT GGC TGA TGC AAT GCG-3′, 5’-TGG AAG GTA AAG AGT GGC CCT TA-3’), E7 transgene (5’-ACT CTA CGC TTC GGT TGT GCG TA-3’, 3’-GCA CAC AAT TCC TAG TGT GCC CAT-5’). Mouse use and handling was carried out in accordance with the American Association for Accreditation of Laboratory Animal Care and the Cincinnati Children's Hospital Veterinary Care Facility according to a protocol approved by the Institutional Animal Care and Use Committee to S.I. Wells.
Head and neck cancer induction
Eight week old transgenic and non-transgenic mice were administered the chemical carcinogen and tumor initiator 4-NQO in the drinking water at 10 µg/ml as previously published14 for a period of 16 weeks and then returned to normal water for 8 weeks. One hour prior to sacrifice, mice were injected intraperitoneally with 6µg/ gram of mouse weight of bromodeoxyuridine (BrdU) (BD Biosciences, San Jose, CA, USA). Mice were analyzed either upon premature death, or at 24 weeks after timed sacrifice. Sacrifice occurred upon moribund appearance due to the tumor burden which prevented the mice from eating and drinking. In the case of the K14E7 Dek−/− mice, two of three mice died prematurely from unknown causes. Gross and microscopic tumors were recorded. Tongue and esophagus were collected for histological analyses.
Histological analysis and immunohistochemistry
Mouse tissues were fixed in 4% paraformaldehyde, embedded in paraffin, sectioned at 5 µm thickness, and fixed onto slides. Routine H&E stained sections were analyzed for histopathology.13 Paraffin sections were deparaffinized in xylene and rehydrated for antigen retrieval in sodium citrate. Sections were then treated with the Mouse on Mouse peroxidase immunostaining kit (Vector Labs, Burlingame, CA, USA). Sections were stained with diaminobenzidine (DAB) and counterstained with Vector Laboratories hematoxylin QS or Nuclear Fast Red (Poly Scientific, Bay Shore, NY, USA) and mounted with Permount (Fisher Scientific, Pittsburgh, PA, USA). Staining was detected with a Leica DM2500 microscope and LAS software (Leica Microsystems Inc., Buffalo Grove, IL, USA), and images were captured at the indicated magnifications. Antibody dilutions were used as follows: BrdU (1:100, Invitrogen), p63α (1:100, Santa Cruz (H-129), Dallas, TX, USA) and DEK (1:50 and 1:100, BD Transduction).
Western blotting
Whole cell lysates were harvested with RIPA buffer containing protease and phosphatase inhibitors or Laemmli buffer containing phosphatase inhibitors. 20 µg total protein was analyzed by western blotting as previously described.11 Membranes were probed with DEK (1:1000, BD Biosciences), p53 (1:1000, Cell Signal (1C12), Boston, MA, USA), phospho-serine15 p53 (1:1000, Cell Signal), ΔNp63 (1:200, Santa Cruz (4A4)) or actin (1:10,000; a gift from James Lessard, Cincinnati Children’s Hospital Medical Center).
Growth curves
Following selection, lentivirally transduced cells were plated in triplicate at an equal density of 100,000 cells/well. The cells were then trypsinized, counted on a hemacytometer and total cell number was calculated for each time point. Data represent two independent experiments.
Flow cytometry
BrdU incorporation was assessed as described previously.28 Briefly, HNSCC cells were plated in equal numbers and labeled with 10 µM BrdU for 2 hours. Collected cells were prepared following the BD Pharmingen APC BrdU Flow kit instructions, analyzed on a BD FacsCalibur, and data further analyzed with FlowJo software (Tree Star, Ashland, OR). Samples were analyzed in triplicate and data represents two independent experiments.
Quantitative RT-PCR
mRNA was harvested using Trizol reagent (Invitrogen, Carlsbad, CA, USA) from transduced cells immediately following puromycin selection. 1 µg of RNA was reverse transcribed to cDNA using the QuantiTect Reverse Transription kit (Qiagen, Valencia, CA, USA). cDNA expression was detected using SYBR green master mix (Applied Biosystems, Carlsbad, CA, USA) on an ABI7300 Real Time PCR machine (Applied Biosystems). Data was analyzed using the ΔΔCt method. Primers were as follows: (1) ΔNp63 Forward: 5’-GGAAAACAATGCCCAGACTC-3’ Reverse: 5’-GTGGAATACGTCCAGGTGGC-3’ (2) DEK Forward: 5’-TGT TAAGAAAGCAGATAGCAGCACC-3’ Reverse: 5’-ATTAAAGGTTCATCATCTGAACTATCCTC-3’ (3) GAPDH Forward: 5’-GGTCTCCTCTGACTTCAACA-3’ Reverse: 5’-ATACCAGGAAATGAGCTTGA-3’. ΔNp63 primers were previously published.41
Cellular proliferation rescue experiments
400,000 cells were plated and transduced with adenovirus on the following day, as described above. Twenty-four hours post-infection, cells were transfected in duplicate with 4 ug of the control (pcDNA) or ΔNp63 construct (human HA-ΔNp63), along with 1 ug of dsRed as a transfection efficiency control. DNA was added to Opti-MEM (Gibco), followed by DharmaFECT 1 Transfection Reagent (Fisher) and added drop-wise to the cells. Total cell counts were determined by hemacytometer counts 72 hours post-infection. This represents three independent experiments.
Statistics
Statistical significance was calculated using GraphPad Prism 6 software (La Jolla, CA, USA). Student’s two-tailed t-test was used for experiments where noted, with **=p≤.01 and ***=p≤.001.
Acknowledgements
This work was supported by the Clinical Scientist Training Program at the University of Cincinnati (TWD), Public Health Service Grant R01 CA116316 from the NIH (SIW), NIH Training Grant T32ES007250 (AKA) and R00 CA160639 from the NIH (RJK). All flow cytometric data were acquired using equipment in the Research Flow Cytometry Core in the Division of Rheumatology at Cincinnati Children’s Hospital Medical Center, supported in part by NIH AR-47363, NIH DK78392, and NIH DK90971.
We would like to acknowledge the CCHMC pathology core with excellent technical support by Meredith Taylor for Ventana HPV in situ hybridization, Dr. Madhavi Kadakia for providing the HA-ΔNp63 construct, Marie Matrka for providing protein lysates, and expert technical support by Dr. Ronald Waclaw for use of his Leica microscope.
Footnotes
Conflict of interest.
The authors declare no conflict of interest.
References
- 1.Forastiere AA, Ang KK, Brizel D, Brockstein BE, Burtness BA, Cmelak AJ, et al. Head and neck cancers. J Natl Compr Canc Netw. 2008;6:646–695. doi: 10.6004/jnccn.2008.0051. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 3.Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. 2000;92:709–720. doi: 10.1093/jnci/92.9.709. [DOI] [PubMed] [Google Scholar]
- 4.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 363:24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kimple RJ, Smith MA, Blitzer GC, Torres AD, Martin JA, Yang RZ, et al. Enhanced radiation sensitivity in HPV-positive head and neck cancer. Cancer Res. 2013;73:4791–4800. doi: 10.1158/0008-5472.CAN-13-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta S, Kong W, Peng Y, Miao Q, Mackillop WJ. Temporal trends in the incidence and survival of cancers of the upper aerodigestive tract in Ontario and the United States. Int J Cancer. 2009;125:2159–2165. doi: 10.1002/ijc.24533. [DOI] [PubMed] [Google Scholar]
- 7.Wise-Draper TM, Draper DJ, Gutkind JS, Molinolo AA, Wikenheiser-Brokamp KA, Wells SI. Future directions and treatment strategies for head and neck squamous cell carcinomas. Transl Res. 160:167–177. doi: 10.1016/j.trsl.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Lindern M, Fornerod M, van Baal S, Jaegle M, de Wit T, Buijs A, et al. The translocation (6;9), associated with a specific subtype of acute myeloid leukemia, results in the fusion of two genes, dek and can, and the expression of a chimeric, leukemia specific dek-can mRNA. Mol Cell Biol. 1992;12:1687–1697. doi: 10.1128/mcb.12.4.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alexiadis V, Waldmann T, Andersen J, Mann M, Knippers R, Gruss C. The protein encoded by the proto-oncogene DEK changes the topology of chromatin and reduces the efficiency of DNA replication in a chromatin-specific manner. Genes Dev. 2000;14:1308–1312. [PMC free article] [PubMed] [Google Scholar]
- 10.Wise-Draper TM, Allen HV, Thobe MN, Jones EE, Habash KB, Munger K, et al. The human DEK proto-oncogene is a senescence inhibitor and an upregulated target of high-risk human papillomavirus E7. J Virol. 2005;79:14309–14317. doi: 10.1128/JVI.79.22.14309-14317.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wise-Draper TM, Allen HV, Jones EE, Habash KB, Matsuo H, Wells SI. Apoptosis inhibition by the human DEK oncoprotein involves interference with p53 functions. Mol Cell Biol. 2006;26:7506–7519. doi: 10.1128/MCB.00430-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wise-Draper TM, Morreale RJ, Morris TA, Mintz-Cole RA, Hoskins EE, Balsitis SJ, et al. DEK proto-oncogene expression interferes with the normal epithelial differentiation program. Am J Pathol. 2009;174:71–81. doi: 10.2353/ajpath.2009.080330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wise-Draper TM, Mintz-Cole RA, Morris TA, Simpson DS, Wikenheiser-Brokamp KA, Currier MA, et al. Overexpression of the cellular DEK protein promotes epithelial transformation in vitro and in vivo. Cancer Res. 2009;69:1792–1799. doi: 10.1158/0008-5472.CAN-08-2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jabbar S, Strati K, Shin MK, Pitot HC, Lambert PF. Human papillomavirus type 16 E6 and E7 oncoproteins act synergistically to cause head and neck cancer in mice. Virology. 407:60–67. doi: 10.1016/j.virol.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strati K, Lambert PF. Role of Rb-dependent and Rb-independent functions of papillomavirus E7 oncogene in head and neck cancer. Cancer Res. 2007;67:11585–11593. doi: 10.1158/0008-5472.CAN-07-3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herber R, Liem A, Pitot H, Lambert PF. Squamous epithelial hyperplasia and carcinoma in mice transgenic for the human papillomavirus type 16 E7 oncogene. J Virol. 1996;70:1873–1881. doi: 10.1128/jvi.70.3.1873-1881.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khodadoust MS, Verhaegen M, Kappes F, Riveiro-Falkenbach E, Cigudosa JC, Kim DS, et al. Melanoma proliferation and chemoresistance controlled by the DEK oncogene. Cancer Res. 2009;69:6405–6413. doi: 10.1158/0008-5472.CAN-09-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Privette Vinnedge LM, McClaine R, Wagh PK, Wikenheiser-Brokamp KA, Waltz SE, Wells SI. The human DEK oncogene stimulates beta-catenin signaling, invasion and mammosphere formation in breast cancer. Oncogene. 30:2741–2752. doi: 10.1038/onc.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carro MS, Spiga FM, Quarto M, Di Ninni V, Volorio S, Alcalay M, et al. DEK Expression is controlled by E2F and deregulated in diverse tumor types. Cell Cycle. 2006;5:1202–1207. doi: 10.4161/cc.5.11.2801. [DOI] [PubMed] [Google Scholar]
- 20.Lu ZL, Luo DZ, Wen JM. Expression and significance of tumor-related genes in HCC. World J Gastroenterol. 2005;11:3850–3854. doi: 10.3748/wjg.v11.i25.3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bradford CR, Zhu S, Ogawa H, Ogawa T, Ubell M, Narayan A, et al. P53 mutation correlates with cisplatin sensitivity in head and neck squamous cell carcinoma lines. Head Neck. 2003;25:654–661. doi: 10.1002/hed.10274. [DOI] [PubMed] [Google Scholar]
- 22.Deyoung MP, Ellisen LW. p63 and p73 in human cancer: defining the network. Oncogene. 2007;26:5169–5183. doi: 10.1038/sj.onc.1210337. [DOI] [PubMed] [Google Scholar]
- 23.Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Molecular cancer research : MCR. 2004;2:371–386. [PubMed] [Google Scholar]
- 24.Su X, Chakravarti D, Flores ER. p63 steps into the limelight: crucial roles in the suppression of tumorigenesis and metastasis. Nat Rev Cancer. 2013;13:136–143. doi: 10.1038/nrc3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA: a cancer journal for clinicians. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 26.Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 11:9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 27.Liu S, Wang X, Sun F, Kong J, Li Z, Lin Z. DEK overexpression is correlated with the clinical features of breast cancer. Pathology international. 2012;62:176–181. doi: 10.1111/j.1440-1827.2011.02775.x. [DOI] [PubMed] [Google Scholar]
- 28.Privette Vinnedge LM, Ho SM, Wikenheiser-Brokamp KA, Wells SI. The DEK oncogene is a target of steroid hormone receptor signaling in breast cancer. PloS one. 2012;7:e46985. doi: 10.1371/journal.pone.0046985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sitwala KV, Adams K, Markovitz DM. YY1 and NF-Y binding sites regulate the transcriptional activity of the dek and dek-can promoter. Oncogene. 2002;21:8862–8870. doi: 10.1038/sj.onc.1206041. [DOI] [PubMed] [Google Scholar]
- 30.Alexandrova EM, Petrenko O, Nemajerova A, Romano RA, Sinha S, Moll UM. DeltaNp63 regulates select routes of reprogramming via multiple mechanisms. Cell Death Differ. 2013;20:1698–1708. doi: 10.1038/cdd.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Truong AB, Kretz M, Ridky TW, Kimmel R, Khavari PA. p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev. 2006;20:3185–3197. doi: 10.1101/gad.1463206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koster MI, Dai D, Marinari B, Sano Y, Costanzo A, Karin M, et al. p63 induces key target genes required for epidermal morphogenesis. Proc Natl Acad Sci U S A. 2007;104:3255–3260. doi: 10.1073/pnas.0611376104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pignon JC, Grisanzio C, Geng Y, Song J, Shivdasani RA, Signoretti S. p63-expressing cells are the stem cells of developing prostate, bladder, and colorectal epithelia. Proc Natl Acad Sci U S A. 2013;110:8105–8110. doi: 10.1073/pnas.1221216110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398:708–713. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- 35.Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398:714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 36.Romano RA, Smalley K, Magraw C, Serna VA, Kurita T, Raghavan S, et al. DeltaNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development. 2012;139:772–782. doi: 10.1242/dev.071191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kappes F, Fahrer J, Khodadoust MS, Tabbert A, Strasser C, Mor-Vaknin N, et al. DEK is a poly(ADP-ribose) acceptor in apoptosis and mediates resistance to genotoxic stress. Mol Cell Biol. 2008;28:3245–3257. doi: 10.1128/MCB.01921-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoskins EE, Morris TA, Higginbotham JM, Spardy N, Cha E, Kelly P, et al. Fanconi anemia deficiency stimulates HPV-associated hyperplastic growth in organotypic epithelial raft culture. Oncogene. 2009;28:674–685. doi: 10.1038/onc.2008.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chaturvedi AK, Engels EA, Pfeiffer RM, Hernandez BY, Xiao W, Kim E, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 29:4294–4301. doi: 10.1200/JCO.2011.36.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell. 2006;9:45–56. doi: 10.1016/j.ccr.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 42.Strati K, Pitot HC, Lambert PF. Identification of biomarkers that distinguish human papillomavirus (HPV)-positive versus HPV-negative head and neck cancers in a mouse model. Proc Natl Acad Sci U S A. 2006;103:14152–14157. doi: 10.1073/pnas.0606698103. [DOI] [PMC free article] [PubMed] [Google Scholar]