

Summary
Corticobasal syndrome (CBS) is characterized by asymmetric involuntary movements including rigidity, tremor, dystonia, and myoclonus, and often associated with apraxia, cortical sensory deficits, and alien limb phenomena. Additionally, there are various nonmotor (cognitive and language) deficits. CBS is associated with several distinct histopathologies, including corticobasal degeneration, other forms of tau-related frontotemporal lobar degeneration such as progressive supranuclear palsy, and Alzheimer disease. Accurate antemortem diagnosis of underlying pathology in CBS is challenging, though certain clinical and imaging findings may be helpful. Five recent advances in the understanding of CBS are reviewed, including clinical and pathologic features, imaging and CSF biomarkers, the role of specific genes, and the concept of a spectrum of tauopathies.
In 1967 and 1968, Rebeiz et al.1,2 reported clinicopathologic findings of 3 patients with a previously undescribed syndrome they named corticodentatonigral degeneration with neuronal achromasia. Over ensuing years, several reports emerged, culminating in the present-day body of knowledge that reveals a fascinating, yet challenging, diagnostic conundrum. Indeed, it has become clear that the clinical features described by Rebeiz et al. are associated with various pathologies and conversely, that the pathology first described by Rebeiz et al. can be seen in patients with a wide array of neurologic signs and symptoms. This clinical-pathologic diversity has motivated adoption of 2 distinct terms: corticobasal degeneration (CBD), in which pathologic findings described by Rebeiz et al. are present, and corticobasal syndrome (CBS), which refers to the clinical phenotype independent of underlying histopathology. Thus, in this article, reference to CBS implies no assumptions regarding the underlying pathology, and when reference to specific pathologies such as CBD is made, evidence from the literature is drawn from studies in which pathologic confirmation of the diagnosis was available.
Therapeutics in neurodegenerative diseases are increasingly targeting the underlying pathologic process.3 Thus, it is essential to distinguish between the various types of neurodegenerative disorders, with accurate antemortem diagnosis and definition of diagnostic criteria assuming increasing importance. Given the etiologic heterogeneity of CBS, characterization of this disorder has been the focus of substantial effort, and important new data and concepts have emerged in regard to it in recent years. This review discusses 5 new aspects of CBS: clinical features, pathologic features, biomarkers, the role of specific genes, and the concept of a tauopathy spectrum.
Clinical features of CBS
Motor features
CBS is considered to be an atypical parkinsonian syndrome. Prominent features include marked asymmetry in motor abnormalities, including dystonia, rigidity, myoclonus, and tremor, as well as a “useless arm” with or without alien limb phenomena. While myoclonus was not reported in the original description, others have considered reflex, stimulus-sensitive myoclonus to be suggestive of CBS.4 Several other motor manifestations have been reported (table 1).
Table 1.
Clinical features described in corticobasal syndrome5–10
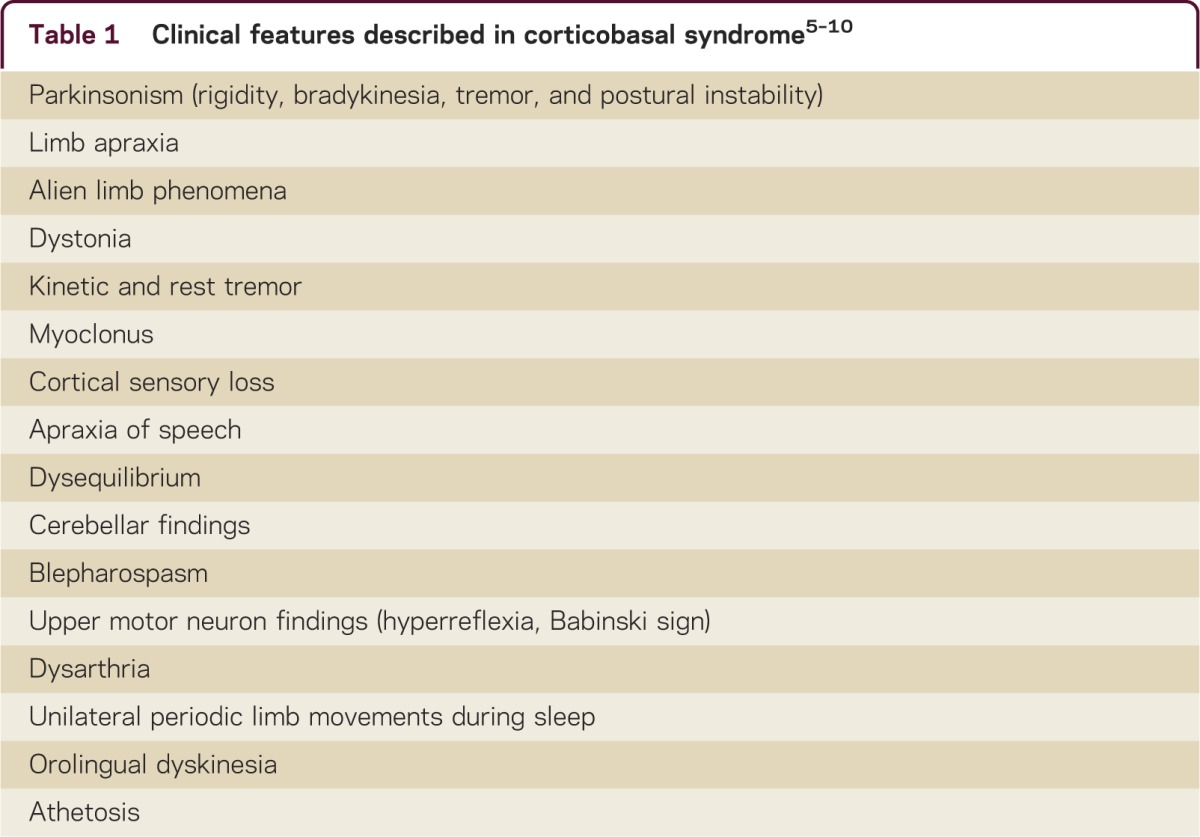
In a recently proposed diagnostic classification scheme,5 core motor features of probable CBS include (1) an asymmetric presentation including ≥2 of the following: limb rigidity/akinesia, dystonia, or myoclonus, as well as (2) ≥2 of the following features: orobuccofacial or limb apraxia, cortical sensory deficit, or alien limb phenomena.5 Further criteria based on associated motor, cognitive, and language deficits were included.5
Some motor features may help predict underlying pathology in cases of CBS. For example, while a subset of CBS patients (with various underlying pathologies) transiently respond to levodopa and may even have dyskinesias,7 a sustained response to levodopa (for over 2 years) suggests, in the right clinical context, a diagnosis of idiopathic Parkinson disease. While there are no eye movement abnormalities that reliability distinguish underlying pathology in CBS, reduced velocity of vertical saccades is most consistent with progressive supranuclear palsy (PSP).11 Overall, however, it is increasingly recognized that motor features do not reliably distinguish between CBS cases with various underlying pathologies.5,8,9 For example, cortical sensory loss and myoclonus, classically considered key features of CBD, have in fact been shown to be associated with either CBD or Alzheimer disease (AD) pathology.7,10
While the motor syndrome of CBS is distinct from other neurodegenerative parkinsonian syndromes, there are few specific motor features that reliably predict underlying pathology.
Cognitive features
Rebeiz et al.1 specifically noted absence of cognitive involvement in early stages of the disease, but it is now clear that cognitive and language disturbances are prominent in CBS, even in those cases that are secondary to underlying CBD pathology. Among patients with neurodegenerative disorders who manifest features of CBS in the course of their disease, motor features of CBS are the presenting signs and symptoms in some cases, with language or cognitive syndromes, or both, subsequently developing. However, in other patients, the reverse occurs9,12–14; specific focal dementia syndromes such as behavioral-variant frontotemporal dementia (FTD), primary progressive aphasia including progressive nonfluent aphasia, and posterior cortical atrophy8,9 could be the presenting and most prominent features, with motor features of CBS then emerging later, as a secondary or tertiary syndrome.13
Like motor features, there are no cognitive or language manifestations that reliably distinguish between underlying pathologies in patients with CBS. One study suggested that episodic memory complaints may predict AD pathology in patients with CBS, and evidence for frontal lobe dysfunction (such as nonfluent language deficits and utilization behavior) may predict CBD.10 However, others have not demonstrated distinct cognitive profiles in CBS cases with underlying AD vs CBD pathology.15
Clinicopathologic correlates of CBS
A multitude of etiologies have been reported in patients with CBS (table 2). As mentioned above, predicting underlying pathology in CBS is difficult,14,e12 particularly in the first 3 years of symptoms. For example, the sensitivity of clinical findings for predicting underlying CBD pathology ranges from 26.3% to 56.0%.7–9,e13 While some series have suggested that dementia syndromes (FTD or AD) are the most common misdiagnoses in cases of CBD,8,9 in others, CBD was most often misdiagnosed as PSP during life.7 Retrospective case series are frequently confounded by a referral bias that influences relative proportions of the various clinical presentations.
Table 2.
Diagnoses reported in patients with corticobasal syndrome (pathologically proven or diagnosed based on laboratory or genetic testing)

While distinguishing between etiologies of CBS is clinically challenging, neuropathologic diagnosis of the most common neurodegenerative causes of CBS is facilitated by specific findings in each case. As elaborated on further below, the majority of causes of CBS are tauopathies, and emergence of tau staining over the past decade has dramatically enhanced immunohistochemical characterization of these disorders (table 3). Tau exists in various isoforms, and each of the neurodegenerative disorders that can cause CBS have been associated with specific tau isoforms. CBD and PSP are associated with a tau isoform with 4 microtubule-binding repeats (4R tau) in neuronal and glial structures.e17 By comparison, other tauopathies such as dementia with Pick bodies are associated with 3 microtubule-binding repeats (3R). In AD, tau tangles consist of both 3R and 4R isoforms of tau. This may ultimately have therapeutic implications if isoform-specific tau therapies are developed.
Table 3.
Distinguishing neuropathologic features of the most common forms of frontotemporal lobar degeneration that may present as corticobasal syndrome
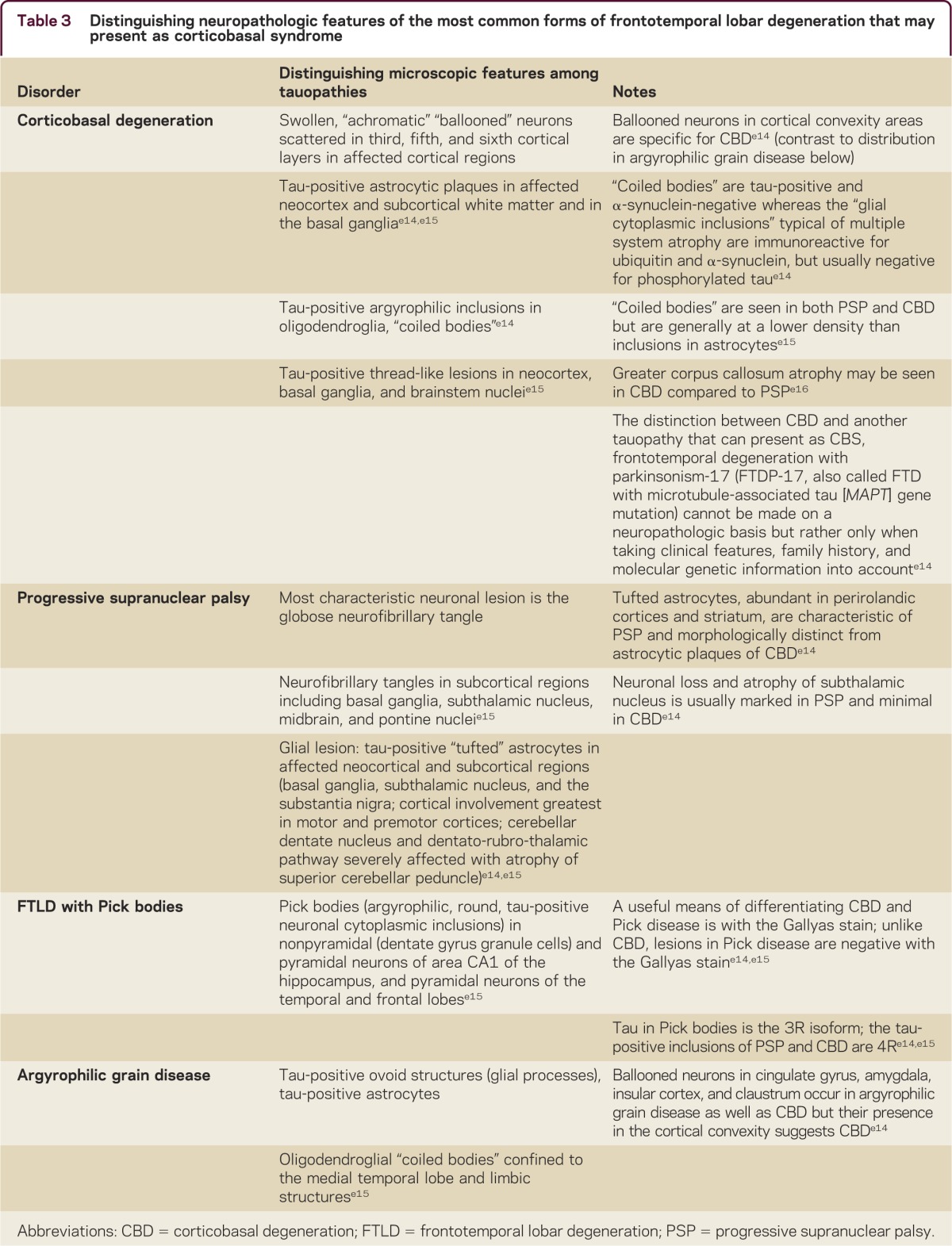
In addition to immunohistochemical features that distinguish the different pathologic causes of CBS, distinct microscopic findings also characterize the neurodegenerative causes. For example, in CBD, achromatic “ballooned” neurons are present in the cortex, cingulate gyrus, amygdala, insula, and claustrum (table 3). The presence of thread-like tau-positive neuronal and glial lesions and astrocytic plaques is also considered highly suggestive of CBD. Astrocytic plaques distinguish CBD from PSP, in which tufted astrocytes are present. “Coiled bodies,” or oligodendroglial tau-positive argyrophilic inclusions, are common in CBD, and are distinct from the tau-negative oligodendroglial lesions (glial cytoplasmic inclusions) of multiple system atrophy, another atypical parkinsonian syndrome.
Diagnosis of the underlying cause of CBS can only definitively be made postmortem, and given the degree of clinical-pathologic mismatch that exists, the term CBS should be used to describe the antemortem clinical syndrome in all cases. Given this diagnostic challenge, efforts have been directed at identifying biomarkers (e.g., CSF proteins, imaging, genetic) that can predict pathology in patients with CBS.
Genetics
The overlap between CBS, PSP, AD, and FTD based on clinical, imaging, and pathologic findings has been strengthened by genetic data. While monogenic forms of these disorders are rare, familial aggregation of CBS and PSP has been reported,e18 and occurrence of PSP in the sibling of a patient with CBDe19 lends support to the occurrence of a familial tauopathy. Progranulin mutations, initially identified in patients with tau-negative FTLD with ubiquitin inclusions on pathology (now known to have TDP-43 inclusions), may be a relatively common cause of familial CBS.e20 As mentioned, the contribution of single pathogenetic mutations to the pathophysiology of these neurodegenerative disorders is likely small, but of great relevance is the predisposition that may be imposed by single nucleotide polymorphisms in specific genes. For example, the H1 haplotype in the mictrotubule-associated protein tau (MAPT) gene is strongly associated with both PSP and CBD,e21 strengthening the consideration of these diseases as lying along a pathophysiologic spectrum. A specific vascular endothelial growth factor haplotype may also confer increased risk for CBS,e22 but the relevance of this will need to be taken in context of underlying pathology, which was unknown in many of the cases for which this genetic association was found.
Biomarkers in the evaluation of CBS
Most cases of CBS appear to be sporadic. Thus, it is necessary to develop other methods for establishing the pathology associated with CBS. There is growing emphasis on disease-specific biomarkers as aids in the antemortem diagnosis of neurodegenerative disorders. While biomarkers for many of the causes of CBS, such as CBD, PSP, and AD, have yet to be validated, candidate biomarkers include those derived from CSF testing and various imaging modalities.
CSF in the evaluation of CBS
In interpreting results of biomarker studies in neurodegenerative disorders, it is key to recognize that the majority of studies are limited by the lack of pathologic diagnosis. For example, a recent studye23 found that while CSF total and phophorylated tau discriminated CBS from PD, these tests were not helpful in distinguishing CBS from PSP. However, since the underlying pathology was not known, and since it is probable that some CBS cases were due to PSP pathology, no firm conclusions can be drawn from this study. Several other studies have examined CSF tau in CBS cases,e24–e27 but only a few small studies have examined CSF tau in patients with pathologically confirmed diagnoses.e28 Of particular interest given the frequency of AD pathology in CBS cases is that a high CSF tau:amyloid-β ratio may be useful in distinguishing AD from other causes of CBS (e.g., frontotemporal lobar degeneration [FTLD])e28,e29 (note an overlap in cases reported in these 2 references). Confirmation in larger studies is needed.
Imaging in the evaluation of CBS
While there are as yet no imaging tests that can definitively identify underlying pathology in CBS, specific patterns of atrophy on MRI may be useful. For example, using MRI voxel-based morphometry, greater temporoparietal atrophy—particularly posterior temporal and inferior parietal regions, as well as the precuneus—suggests underlying AD pathology,8,e30,e31 whereas midbrain and superior cerebellar peduncle atrophy suggests PSP.e32 Focal atrophy in premotor cortex and the supplementary motor area on routine MRIe31 or frontoparietal lobe and pallidal atrophy in the absence of brainstem atrophy using MRI voxel-based morphometrye32 may suggest CBD. Although MRI, including diffusion tensor imaging,e33 has also been used to investigate white matter changes in CBS,e33–e35 we are not aware of reports involving CBS cases with known pathology.8,e32 Ultimately, multimodality imaging, assessing both gray and white matter structural changes, with incorporation of MRI quantitation (which provides information about tissue microstructure), may prove superior in predicting underlying pathology in CBS relative to either type of imaging alone.
Ligand-based nuclear imaging is another imaging modality that warrants further investigation as a tool to distinguish between different neurodegenerative disorders. Like the CSF tau:amyloid-β ratio described above, amyloid imaging may be of particular value in determining which CBS cases are due to underlying AD pathology. Tau-based ligand imaging is under active investigation.e36,e37 PET and single photon emission tomography using presynaptic and postsynaptic radioligands to components of the dopaminergic system (dopamine transporters or receptors, or both) are also of particular interest.e38–e40 Finally, studies of regional metabolic changes using fluorodeoxyglucose PET,e41 in addition to noninvasive hemodynamic assessment (through magnetic resonance or CT perfusion or newer techniques such as arterial spin labeling MRI) may be of utility in CBS as well. Application of all these imaging modalities to pathologically confirmed cases is required to further define their role.
The tauopathy spectrum
The designation of tauopathy, defined as a neurodegenerative disorder characterized pathologically by the presence of neuronal and glial deposits of tau protein, encompasses CBD, PSP, Pick disease, and other forms of tau-related FTLD including FTLD associated with MAPT mutations.e42,e43 The clinical and pathologic overlap between CBD and other tauopathies has led CBD to be grouped with these other conditions under the pathologic rubric of FTLD-tau.e42,e44
While predicting the specific underlying pathology in CBS has proven difficult, data suggest that predicting an underlying tauopathy on clinical grounds may be achieved with relatively high sensitivity,14,e45 although longitudinal data are lacking. It has been estimated that 83% of cases presenting with CBS can be accurately classified as tauopathies.e45 Tau-negative CBS may have a younger age at onset,14 although others have demonstrated shorter disease duration in tauopathies.e46
The phenotype of neurodegenerative disorders relates in large part to the anatomic distribution of the underlying pathology rather than the underlying pathology per se.e45 Several lines of evidence suggest that CBD, PSP, and FTLD lie along a spectrum, with shared clinical, genetic, and pathologic findings. Moreover, AD can present in protean fashions, including CBS. While clinical characterization continues to be an important part of neurology, incorporation of imaging, biochemical, and genetic markers into disease classification will be essential for accurate diagnosis and appropriate patient selection for clinical trials, particularly those targeting specific underlying proteinopathies such as a tauopathy.
DISCUSSION
Understanding of CBS has evolved dramatically since its description over 40 years ago. While certain features have traditionally been associated with this condition, a wide spectrum of clinical presentations can occur, ranging from purely motor to predominantly cognitive and language-related syndromes. CSF, imaging, and genetic markers are likely to be of great utility in predicting risk for and diagnosis of the underlying neurodegenerative disorders implicated in CBS. The key to establishing these as reliable diagnostic tests will be further studied in clinically well-characterized patients with known pathology. The underlying proteinopathy in many causes of CBS is tau-related, and in the future, tau-targeted therapeutics will benefit many patients with CBS. Correctly identifying those patients who may benefit from such interventions will be essential for therapeutic success.
Corticobasal syndrome: Five new things.
The term CBS should be used to describe the antemortem clinical syndrome
There are no specific clinical features that reliably predict underlying pathology in CBS
Recent efforts have been directed at identifying biomarkers (e.g., CSF proteins, imaging) that can predict pathology in patients with CBS, and definitive diagnosis of the underlying cause of CBS can only be made postmortem
Specific single nucleotide polymorphisms may contribute to the risk of several causes of CBS, including PSP and CBD
While predicting the specific underlying pathology in CBS has proven difficult, the vast majority of cases presenting with CBS can be accurately classified as tauopathies, and as such may benefit from tau-targeted therapies in the near future
Supplementary Material
Footnotes
Supplemental data at Neurology.org/cp
STUDY FUNDING
No targeted funding reported.
DISCLOSURES
L. Chahine receives publishing royalties for Comprehensive Review in Clinical Neurology: A Multiple Choice Question Book for the Wards and Boards (LWW/Wolters Kluwer Health, 2011) and has received research support from the Philadelphia VA Parkinson's Disease Research, Education, and Clinical Center, and the University of Pennsylvania Institute for Translational Medicine and Therapeutics (CTRC Junior Investigator Preliminary/Feasibility Grant). T. Rebeiz and J. Rebeiz report no disclosures. M. Grossman serves on scientific advisory boards for TauRx and Bristol Myers Squibb; serves on the editorial boards of Neurology® and Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration; and receives research support from the NIH and the Wyncote Foundation. R.G. Gross receives research support from the NIH/National Institute of Neurological Disorders and Stroke, American Academy of Neurology/American Academy of Neurology Foundation (clinical research training fellowship), and Parkinson Council. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.

REFERENCES
- 1.Rebeiz JJ, Kolodny EH, Richardson EP., Jr Corticodentatonigral degeneration with neuronal achromasia. Arch Neurol 1968;18:20–33 [DOI] [PubMed] [Google Scholar]
- 2.Rebeiz JJ, Kolodny EH, Richardson EP., Jr Corticodentatonigral degeneration with neuronal achromasia: a progressive disorder of late adult life. Trans Am Neurol Assoc 1967;92:23–26 [PubMed] [Google Scholar]
- 3.Gozes I. Microtubules (tau) as an emerging therapeutic target: NAP (davunetide). Curr Pharm Des 2011;17:3413–3417 [DOI] [PubMed] [Google Scholar]
- 4.Litvan I, Agid Y, Goetz C, et al. Accuracy of the clinical diagnosis of corticobasal degeneration: a clinicopathologic study. Neurology 1997;48:119–125 [DOI] [PubMed] [Google Scholar]
- 5.Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013;80:496–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stamelou M, Alonso-Canovas A, Bhatia KP. Dystonia in corticobasal degeneration: a review of the literature on 404 pathologically proven cases. Mov Disord 2012;27:696–702 [DOI] [PubMed] [Google Scholar]
- 7.Ling H, O'Sullivan SS, Holton JL, et al. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain 2010;133:2045–2057 [DOI] [PubMed] [Google Scholar]
- 8.Lee SE, Rabinovici GD, Mayo MC, et al. Clinicopathological correlations in corticobasal degeneration. Ann Neurol 2011;70:327–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murray R, Neumann M, Forman MS, et al. Cognitive and motor assessment in autopsy-proven corticobasal degeneration. Neurology 2007;68:1274–1283 [DOI] [PubMed] [Google Scholar]
- 10.Shelley BP, Hodges JR, Kipps CM, Xuereb JH, Bak TH. Is the pathology of corticobasal syndrome predictable in life? Mov Disord 2009;24:1593–1599 [DOI] [PubMed] [Google Scholar]
- 11.Boxer AL, Garbutt S, Seeley WW, et al. Saccade abnormalities in autopsy-confirmed frontotemporal lobar degeneration and Alzheimer disease. Arch Neurol 2012;69:509–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kertesz A, Martinez-Lage P, Davidson W, Munoz DG. The corticobasal degeneration syndrome overlaps progressive aphasia and frontotemporal dementia. Neurology 2000;55:1368–1375 [DOI] [PubMed] [Google Scholar]
- 13.Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain 2005;128:1996–2005 [DOI] [PubMed] [Google Scholar]
- 14.Josephs KA, Petersen RC, Knopman DS, et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology 2006;66:41–48 [DOI] [PubMed] [Google Scholar]
- 15.Hu WT, Rippon GW, Boeve BF, et al. Alzheimer's disease and corticobasal degeneration presenting as corticobasal syndrome. Mov Disord 2009;24:1375–1379 [DOI] [PubMed] [Google Scholar]
- 16.Rinne JO, Lee MS, Thompson PD, Marsden CD. Corticobasal degeneration: a clinical study of 36 cases. Brain 1994;117:1183–1196 [DOI] [PubMed] [Google Scholar]
- 17.Boeve BF, Maraganore DM, Parisi JE, et al. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology 1999;53:795–800 [DOI] [PubMed] [Google Scholar]
- 18.Josephs KA, Holton JL, Rossor MN, et al. Neurofilament inclusion body disease: a new proteinopathy? Brain 2003;126:2291–2303 [DOI] [PubMed] [Google Scholar]
- 19.Masellis M, Momeni P, Meschino W, et al. Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain 2006;129:3115–3123 [DOI] [PubMed] [Google Scholar]
- 20.Yu CE, Bird TD, Bekris LM, et al. The spectrum of mutations in progranulin: a collaborative study screening 545 cases of neurodegeneration. Arch Neurol 2010;67:161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.