

Abstract
The process of carcinogenesis is tightly regulated by antioxidant enzymes and matrix degrading enzymes, namely, matrix metalloproteinases (MMPs). Degradation of extracellular matrix (ECM) proteins like collagen, proteoglycan, laminin, elastin and fibronectin is considered to be the prerequisite for tumor invasion and metastasis. MMPs can degrade essentially all of the ECM components and, most MMPs also substantially contribute to angiogenesis, differentiation, proliferation and apoptosis. Hence, MMPs are important regulators of tumor growth both at the primary site and in distant metastases; thus the enzymes are considered as important targets for cancer therapy. The implications of MMPs in cancers are no longer mysterious; however, the mechanism of action is yet to be explained. Herein, our major interest is to clarify how MMPs are tied up with gastrointestinal cancers. Gastrointestinal cancer is a variety of cancer types, including the cancers of gastrointestinal tract and organs, i.e., esophagus, stomach, biliary system, pancreas, small intestine, large intestine, rectum and anus. The activity of MMPs is regulated by its endogenous inhibitor tissue inhibitor of metalloproteinase (TIMP) which bind MMPs with a 1:1 stoichiometry. In addition, RECK (reversion including cysteine-rich protein with kazal motifs) is a membrane bound glycoprotein that inhibits MMP-2, -9 and -14. Moreover, α2-macroglobulin mediates the uptake of several MMPs thereby inhibit their activity. Cancerous conditions increase intrinsic reactive oxygen species (ROS) through mitochondrial dysfunction leading to altered protease/anti-protease balance. ROS, an index of oxidative stress is also involved in tumorigenesis by activation of different MAP kinase pathways including MMP induction. Oxidative stress is involved in cancer by changing the activity and expression of regulatory proteins especially MMPs. Epidemiological studies have shown that high intake of fruits that rich in antioxidants is associated with a lower cancer incidence. Evidence indicates that some antioxidants inhibit the growth of malignant cells by inducing apoptosis and inhibiting the activity of MMPs. This review is discussed in six subchapters, as follows.
Keywords: Gastrointestinal cancer, Matrix metalloproteinase, Tissue inhibitor of matrix metalloproteinases, Reactive oxygen species, Antioxidants
Core tip: Matrix metalloproteinases (MMPs), a group of zinc dependent endopeptidases, substantially contribute to extra cellular remodelling, angiogenesis, cellular differentiation, proliferation and apoptosis. MMPs are also important regulators of tumor growth both at the primary site and in distant metastasis; thus the enzymes are considered as important targets for cancer therapy. This review describes the roles and regulation of different MMPs and their subsequent actions over different gastrointestinal cancers both in epigenetic and cellular level. Furthermore, this review summarizes the current state of knowledge of dietary antioxidants in preventing gastrointestinal cancer progression as well as mechanism of action.
EPIDEMIOLOGY AND GENETIC BASIS OF GASTRIC CANCERS
As far the incidence rate is concerned, gastric cancer holds the fourth position among the most common cancers in men and fifth in women, from a worldwide perspective. The death rate goes hand to hand with lung cancer, the most frequent cancer globally[1]. Approximately, one million cases of gastric cancer were reported in the year 2008, which accounted for almost 8% of all cancerous incidents throughout the world[1]. Regions of Asia, eastern Europe, South America were highlighted in the reports as the most affected continents[2]. Carcinogenesis in the gastrointestinal tract, accounts for marked geographic variations in incidence and shows morphological heterogeneity. Histologically, gastric cancers are mainly of two types, diffuse and localized intestinal types. Poorly differentiated cancer cells, scattered within the stromal cells diagnosed as diffuse-type gastric cancer (DGC), whereas tubular gland like structures formed by the cancer cells having a few stromal components give rise to intestinal-type gastric cancer (IGC). Recognized as a familial disease many years ago, hereditary diffuse gastric cancer has now been identified as an autosomal dominant cancer susceptibility syndrome. This familial disease was probably most elegantly demonstrated in the family of Napoleon Bonaparte[3,4].
Chromosomal anomalies leading to gastrointestinal cancers
Aneuploidy of chromosomes 4, 8, 17 and 20 in gastric cancer were reported in several studies. Researchers have been trying to identify the precise stages known from Correa’s pathway, where these chromosomal anomalies arise. Chromosome 4 and 20 were found to be amplified with the deletion of chromosome 17(p53) in multiple known progressive stages of carcinogenesis including normal gastric mucosa as well as metaplasia, dysplasia and cancer. A significant increase in the levels of aneuploidy was also reported with disease severity. Moreover, in some cases significant positive association was observed between chromosome 4 amplification and infection with Helicobacter pylori (H. pylori). In the same study, a similar kind of aneuploid condition was induced in vitro by exposing a human cell line to hydrogen peroxide suggesting that H. pylori induces gastric cancer with the help of reactive oxygen species (ROS) mediated chromosomal aneuploidy[5].
In 1988, Correa proposed the stages of human gastric cancer progression using several stages including gastritis, metaplasia, dysplasia, carcinoma, etc. In a study by Sugai et al[6] chromosomal allelic losses were tested of multiple cancer related chromosomal loci (1p, 3p, 4p, 5q, 8p, 9p, 13p, 17p, 18q and 22q). In addition, microsatellite instability (MSI) and overexpression of p53 protein were checked in all tumor samples. A prominent 3p allelic loss was observed in the cases of gastric phenotypes, whereas 5q allelic loss was highly associated to the intestinal phenotypes. Both loss of heterozygosity and microsatellite instability were observed in the genetic profiles of the mixed phenotypes. Allelic losses of 5q, 3p and 18q loci were consistent in intra-mucosal carcinomas and allelic losses of 17p, 1p and 9p were associated to submucosal carcinomas, all leading to loss of heterozygosity. MSI was observed only in 6 out of 31 cases of mixed phenotype gastric cancers, while p53 overexpression is observed in most of the cases of differentiated gastric carcinomas[6].
Specific gene mutations and their contributions
In most cases, the molecular expression of several biological markers show no link between the young (≤ 45 years) and the aged (≥ 45 years) patients, suggesting that early onset of gastric cancers possess different expression patterns of several important biomolecules[7]. Early onset gastric cancer patients may be more susceptible to the genetic factors but these individuals account for 10% or even less of all gastric cancer patients throughout the world[8,9]. Only10% of early onset gastric cancer cases belong to the inherited gastric cancer predisposition syndromes, but the genetic events taking place in the background of these remain largely unclear till date.
Development of tumors, often result from defects in several signaling pathways, including tyrosine phosphorylation, which occurs through the combined actions of protein tyrosine phosphatases (PTPs) and protein tyrosine kinases (PTKs). About 26% of colorectal cancers and a minute fraction of gastric cancers were reported to have mutations in the PTP genes. Mutated PTP originally are tumor suppressor genes, which regulate pathways associated with cell growth and differentiation. Wang et al[10] uncovered 83 such somatic mutations by mutational analysis of the six tyrosine phosphatase gene superfamily namely (PTPRF, PTPRG, PTPRT, PTPN3, PTPN13, and PTPN14). Production of truncated proteins lacking phosphatase activity was due to 15 mutations, which were nonsense, frame shift or splice site mutations. Reduced phosphatase activity was resulted in 5 missense mutations in PTPRT. Restoration of wild-type PTPRT expression in human cancer cells tended to cease cell growth[10].
Several studies reported the involvement of different biomolecules for the cause of gastric cancer. E-cadherin, p53, cycloxygenase-2 (COX-2), trefoil factor-1 (TFF-1), β-catenin, p16, c-myc, etc., are some of the known molecules. Significant difference in the expression of these markers and some other molecules are found, namely c-jun, HuR, C/EBP-β, etc., in early onset of gastric cancer, as well as regular gastric cancer patients. TFF-1 was overexpressed with a comparatively lower level of COX-2 in the early onset gastric cancer, whereas COX-2 overexpression and loss of TFF1 was found in regular cancers. Surprisingly, overexpression of COX-2, C/EBP-β in intestinal type gastric cancer was observed[7].
Risk factors for familial and non-familial gastrointestinal cancers
Gastrointestinal carcinomas like, esophageal adenocarcinoma, gastroesophageal junctional adenocarcinoma, etc., often originate from Barrett’s esophagus (BE), a chronic gastroesophageal reflux disease[11,12]. When BE and its associated diseases occur in families, they are collectively included within a syndrome named Familial Barrett’s Esophagus, categorized as a complex genetic disease[13,14].
The onset of adenocarcinomas is thought to be determined by a combined effect of genetic variation and distinct environmental factors. Chak et al[15] determined the relationship between risk factors and the age of onset of these cancers. Family history of BE/cancer occurrence, gastroesophageal reflux symptoms, obesity (defined as body mass index > 30) and other risk factors were assessed in a total of 356 gastroesophageal adenocarcinoma patients. This study reports that both familial and non-familial cancers arise at similar ages, but obesity is associated with a comparatively earlier age of onset.
Appropriate clinical counseling based on the genetics of gastrointestinal cancers always depends on well substantiated data reflecting the risk factors existing through a family. The estimated risk of gastric cancer within a family, however, may differ widely from one another. A group of researchers from Sweden used the updated Swedish Family-Cancer Database to investigate the familial risks of gastric cancer in 5358 patients among the offspring and 36486 patients among the parents. In this investigation, 133 families were identified having one parent and one offspring recognized as patients of gastric cancer, whereas 20 families had two affected offspring. The standardized incidence ratio (SIR) for the families was 1.63, when the parents displayed gastric cancer and the same was 2.93 in the families where the siblings had the disease. Cancer in the corpus (main body of the stomach) was related to high sibling risk (SIR = 7.28). Whenever gastric cancer was diagnosed in the parents, the SIR for cancer in the cardia (the area joining stomach and esophagus) was 1.54. In most cases, upper stomach cancer did show a particular association to esophageal adenocarcinoma. Histological analysis revealed an increase of signet ring cells in cancers. Among the factors, giving rise to high sibling risk in the case of corpus cancer, H. pylori infection may be an important one. The association of upper stomach cancer and esophageal adenocarcinoma in families may also lead to important clues on the aetiology of both diseases[16].
Chromatin remodeling and epigenetic modifications as etiological factors
Various carcinogenic pathways and environmental factors may contribute to the aetiologies of gastric cancers[17]. Several genes as well as some of their mutations were identified in a study by exome sequencing of 22 gastric cancer samples. In this way, genes participating in chromatin remodeling were most commonly found to be mutated, leading to alterations in specific pathway. Protein deficiency of AT-rich interactive domain-containing protein 1A (ARID1A) were observed in 83% of gastric cancers with MSI, 73% of those with Epstein-Barr virus (EBV) infection and 11% of those that were not infected with EBV and microsatellite stable. A small division of the disease may arise due to TP53 (gene encoding p53) mutations, as well as other genetic alterations and modified pathways. Occurrence of these mutations shows a negative correlation with mutations in TP53. The significance of chromatin remodeling is highlighted in the context of gastric cancers, which also reveal some new genomic landscapes[18].
Overexpression of claudin-4 (CLDN4), a protein involved in tight junctions is known to be associated to gastric cancers. Increased expression of CLDN4 on the membrane enhances the barrier like function of tight junctions which tends to prevent the migration and invasion of gastric cancer cells, without affecting cell growth. The epigenetic regulation of CLDN4 overexpression and its clinical significance as potential therapeutic targets was reported by Kwon et al[19]. DNA hypomethylation parallels to CLDN4 upregulation in both cancerous and non-cancerous gastric tissues. In normal gastric tissues, bivalent histone modifications often lead to repression of CLDN4 expression, whereas loss of repressive histone methylations results in upregulation of CLDN4 in gastric cancer cells[19].
Methylation level of long interspersed element-1 (LINE-1) is associated with esophagus gastric as well as colon cancer progression and prognosis[20]; this helps in assessing tumor heterogeneity and drug efficacy for the personalized treatment of patients with gastrointestinal cancers. Okada et al[21] documented that promoter methylation rate of seven genes TP73, BLU, FSD1, BCL7A, MARK1, SCRN1, and NKX3.1 are higher in EBV-associated gastric carcinomas compare to EBV-negative gastric carcinoma signify the viral-mediated epigenetic alteration in cancer. Report suggested that H. pyroli infection induced promoter methylations of THBS1 and GATA-4 gene in the early stages of chronic gastritis and gastric cancer development[22]. Jin et al[23,24] reported the enhanced rate of promoter methylation of a transmembrane glycoprotein endoglin and Ras-related associated with diabetes gene in human ESCC. Also, TIMP-3 hypermethylation contributes to the downregulation TIMP-3 protein expression in ESCC and is associated with poor patient survival[25]. Poplineau et al[26] reported that a DNA hypomethylation agent enhanced upregulation of MMP-1 gene expression and triggered tumor cell invasion. In contrast, treatment with S-adenosylmethionine, a methyl donor, resulted in activation of TIMP-2 and significant downregulation of MMP-2 and MT1-MMP gene in colorectal cancer[27]. Prognostic values of promoter hypermethylation in patients with gastric cancer documented that patients with higher stage of colorectal cancer possess a higher concentration of methylated APC, TIMP-3 and hMLH1 in the serum[28]. Wang et al[29] reported a frequent hypermethylation of RASSF1A gene promoter in gastric and colon cancer and predicted its utility as a diagnostic marker.
ELEVATED INDUCTION OF MMPS IN GASTROINTESTINAL CANCERS
The MMPs are comprised of a family of endopeptidases, which can cleave almost every component of the extra cellular matrix (ECM) proteins. It is documented that many non-ECM proteins can also be cleaved by selected MMPs. Structurally, they all have a zinc ion in the catalytic domain and their activity is dependent on divalent ions, mainly, Zn2+ and Ca2+[30,31]. There are about 27 different MMPs discovered so far and they are subdivided in groups according to substrate specificity and structural integrity (Table 1). Induction and expression of MMPs are regulated at the level of transcription and translation, respectively. Further complexity of MMPs is the activation from zymogen to active enzyme and, secondly, the mRNA stability of few MMPs play critical role. ProMMPs are converted to active MMPs by intra-molecular cleavage of cysteine bridge between thiol group at the prodomain and Zn2+ near the catalytic site. The overall activity depends on the availability of the substrate as well as inhibitor in pericellular space, though a high concentration of MMPs exists near the plasma membrane.
Table 1.
Major matrix metalloproteinases studied in cancer biology
| Collagenase | Gelatinase | Stromolysin | Matrilysin | Membrane type MMPs | Others |
| MMP-1 | MMP-2 | MMP-3 | MMP-7 | MMP-14 | MMP-12 |
| MMP-8 | MMP-9 | MMP-10 | MMP-26 | MMP-15 | MMP-20 |
| MMP-13 | MMP-11 | MMP-16 | MMP-28 | ||
| MMP-18 | MMP-19 | MMP-17 | |||
| MMP-24 | |||||
| MMP-25 |
MMP: Matrix metalloproteinases.
Cancer progression can be explained in six major steps: self-support in growth signals; resistance to growth-inhibitory signals; reduced apoptosis; uncontrolled replication; sustained angiogenesis; and tissue invasion followed by metastasis[32]. Considerable evidence has demonstrated that disease progression in experimental animal models of cancer invasion and metastasis correlate with enhanced secretion of specific MMPs by tumor cells and/or stromal cells. Gastrointestinal cancer can be subdivided into different types, e.g., cancers of upper digestive tract, esophageal cancer, gastric cancer, pancreatic cancer, liver cancer, gallbladder cancer and others like MALT lymphoma, gastrointestinal stromal tumors, cancer of the biliary tree, cancer of the lower digestive tract, colorectal and gastrointestinal carcinoid tumor.
Role of MMPs in esophageal and gastric cancer
Role of MMPs in gastrointestinal cancer has been well studied. IHC analysis of tumor biopsy samples suggest the expression of MMP-1 in 24% of oesophageal cancers, while MMP-2 and MMP-9 in 78% and 70% of samples respectively[33]. Similarly, studies revealed that MMP-13 is localized predominantly in tumor cells; and the presence of MMP-13 together with MT1-MMP is implicated in determining tumor aggressiveness of human oesophageal carcinomas. Etoh et al[34] found a significant correlation in survival period for subjects with the expression of MMP-13 and MT1-MMP in tumor tissue. Moreover, the activities of MMP-2, -3, -9, and -10 enzymes were detected in each of the 24 cancer cases. MMP overexpression was reported in tumors in comparison to normal tissue; having elevated levels of the activated form of MMP-3 and -10 in tumors. In addition, MMP-3 and -10 mRNA levels were significantly higher in tumors than paired normal tissues in both the stromal and epithelial component of tissues[35].
One of the important features of the malignant phenotype in both colorectal and gastric cancer is the overexpression of MMP-2 and -9 as well as activation of proMMP-2 to active MMP-2. Expression of MMP-2, -1 and -9 was found in 94%, 73% and 70% respectively in gastric specimens when studied in 74 patients. Conversely, MMP-3 was only present in 27% of tumors while MMP-1 and -9 were present mostly in all intestinal phenotypes of gastric cancer. In addition to MMPs, TIMP-1 and TIMP-2 were detected in approximately 50% of gastric tumors. Progression of gastric cancer is associated with MMP-13 expression along with its coexpression with MT1-MMP and/or MMP-2 that may have a synergistic effect in the progression of the disease[36]. The expression of TIMP-3 was significantly higher than that of MMP-3, and MMP-3/TIMP-3 was lower in gastric cancer tissue at the early stages (n = 18) than in that of the advanced stage group (n = 26) (P < 0.05)[37]. MMP-7 expression has been found to be prognostic marker for metastasis of gastric carcinoma because MMP-7 mRNA as well as protein was pronounced in aggressiveness carcinoma tissues.
Role of specific MMPs in colorectal cancer
The critical event in the process of cancer invasion and metastasis is the degradation of the ECM surrounding the tumor tissue[38]. This ECM is degraded by the action of a set of proteases, in which several types of MMPs play major role, of which MMP-2 and -9 are most prominent. The basement membrane which prevents an invading epithelial tumor is mainly made up of type IV collagen, which is substrate of MMP-2 and -9. The event of basement membrane degradation promotes epithelial tumor invasion. Higher levels of MMP-1, -2, -3, -7, -9, -13, and MT1-MMP expression have been documented in human colorectal. Murray et al[33] demonstrated that MMP-1 in colorectal cancer specimens was linked to a poor prognosis of the disease. This study was later confirmed by performing IHC, FISH, and RT-PCR on 142 samples of colorectal carcinomas[39]. The latent form of MMP-2, i.e., proMMP-2, is expressed in significant levels almost in all normal tissues. MMP-2 acts as the ‘house-keeping’ gene due to its importance in normal cellular physiology. While active MMP-2 is found in neoplastic tissues, it is lacking in most normal tissues. Parsons et al[40] in 1992, were the first who described the role of MMP-2 in colorectal cancer and the ratio of MMP-2 to proMMP-2 was 20 fold higher in colorectal cancer specimens in comparison to non-malignant biopsies as judged by gelatin zymography. Parsons et al[40] demonstrated increased expression of proMMP-9 in colorectal cancer. The increased activity of proMMP-9 from inflammatory cells may cause an early change in progression from adenoma to carcinoma, when colonic adenoma is compared to normal mucosa. Increased co-expression of MMP-3 and MMP-9 has been found in colorectal tumors. Co-expression of uPA with MMP-9 in colorectal cancers is responsible for the activation of plasminogen to plasmin[41]. Plasmin stimulates proMMP-3 to active MMP-3 which in turn promotes proMMP-9 to active MMP-9, thus, resulting in colorectal cancer progression[41]. Excess MMP-9 expression in colorectal cancer contributes to the inflammation related to neoplasms but not to aggressive tumors[42]. Low levels of microsatellite instability and poor prognosis is observed with increased expression of MMP-3 in colorectal cancer. Moreover 90% of colonic adenocarcinomas demonstrated high levels of MMP-7 expression. Studies on surgically resected colorectal cancer specimens elucidated the clinical importance of MMP-7 expression in this cancer type. It demonstrated that overexpression of MMP-7 in colorectal cancer (measured by IHC and in situ hybridization) directly relates to nodal or distant metastasis[43,44]. On the contrary, MMP-12 overexpression is associated with increased survival in colorectal cancer because of its influence as protective factor presumably by inhibiting tumor angiogenesis[43]. In fact, inhibition of tumor growth with upregulation of MMP-12, also known as macrophage metalloprotease, is well accepted. It was reported by Dong et al[45] that macrophages capable of producing MMP-12 in tumors that are responsible for increased production of angiostatin, an inhibitor of tumor neovascularization[45,46]. High expression of MMP-13 results in poor survival of colon cancer patients. Colorectal tumor biopsy specimens were examined for the identification of MMP-13 by Leeman et al[47]. MMP-13 was found in 91% of cases and was localized to cytosol of tumor tissues. Significantly higher activity of MMP-13 was observed in malignant than the non-malignant tissues. Moreover, plasmin, MT1-MMP and MMP-2 are key molecules in the production of active MMP-13. Active MMP-13 was found to be responsible for activation of MMP-9 during cancer progression[48].
MMPs polymorphism in tumor formation
Unlike classical oncogenes, MMPs are not upregulated by gene amplification or mutations. The increased MMP expression in tumours is mainly due to transcriptional changes rather than genetic alterations. The only two reported genetic alterations in cancer cells are translocation of the MMP-23 gene in neuroblastoma[49] and amplification of the MT5-MMP gene[50]. Polymorphisms in MMP promoters also affect gene transcription and influence cancer susceptibility (Figure 1). The estimated number of single nucleotide polymorphisms (SNPs) in the human genome is 10 million, while only a small part of these polymorphisms are functionally relevant. The differences in allele transcription caused by polymorphisms in the MMP promoters are subtle compared with the overexpression that arises from the amplification of oncogenes. Most of the functional SNPs are located in the promoter region of the MMP-1,-2,-3,-9 and -7 genes that are associated with gastric cancer risk.
Figure 1.
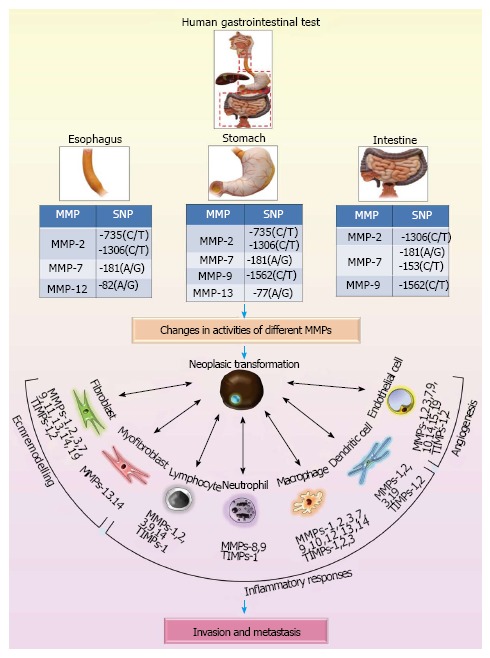
Matrix metalloproteinases polymorphism in gastrointestinal cancers. Single nucleotide polymorphism (SNP) for matrix metalloproteinases (MMP) genes in gastrointestinal organs (e.g., esophagus, stomach and intestine) of human has been reported. These SNPs are involved in changing MMPs activities in neoplastic transformation of gastric tissues in cancer patients. In addition to cancerous cells, the secretion of MMPs by fibroblasts, myofibroblasts, lymphocytes, neutrophils, macrophages, dendritic cells and endothelial cells has been documented. Both MMPs and tissue inhibitor of metalloproteinases (TIMPs) are important in regulation of extracellular matrix (ECM) remodeling, inflammatory responses and angiogenesis for cancer invasion and metastasis.
MMP1-1607 1G/2G polymorphism was found to be associated with gastric cancer risk as presence of extra guanine (2G) creates a binding site for Ets-1 transcription factor that enhances transcription of MMP-1. Bradbury et al[51] reported an elevated esophageal cancer risk in 1G/2G and 2G/2G carrier. MMP-1 protein expression is higher in tumors from gastric cancer patients who carry the 2G allele not 1G homozygotes[52]. Moreover, 2G homozygotes are more likely to develop invasive tumors. Dey et al[52] reported that MMP-1 promoter polymorphism is significantly associated with lower stomach tumor formation. MMP-1 -1607 1G/2G polymorphism is also involved with colon cancer risks.
MMP-2 polymorphism was investigated mainly in the promoter region, e.g., MMP-2 -1306 C/T, -735 C/T, -790T/G, -955A/C, and -1575G/A in the context of gastrointestinal cancer risk[53]. Studies reported association of gastrointestinal cancer risk with -1306 C/T and -735 C/T polymorphic site worldwide. Price et al[54] characterized genetic variants in the human MMP-2 -1306 C/T allele-specific transcriptional regulation. The common C>T transition at -1306 disrupts a Sp1-type promoter site (CCACC box), leading to lower promoter activity with the T allele[54]. On the other hand, G to A substitution at the MMP-2 -1575 site reduces gene expression due to a reduction of estrogen receptor-α binding to A allele[55]. Fruh et al[56] found the presence of CC allele at MMP-2 -1306 position in H. pylori infected individuals which provide protection against esophagus adenocarcinoma. Studies also reported that presence of CC allele at -1306 site increases the risk of ESCC, although Chen et al[54,57] did not observe any positive association of MMP-2 -1306 C/T polymorphism with ESCC in Mongolian population suggests differences in genetic susceptibility between Han-ethnic Chinese and the Mongolian population. However, both positive and negative influences of MMP-2 -1306 C/T polymorphism with gastric cancer in Asian and Caucasian population were reported[58]. Studies were performed to evaluate the association of MMP-2 -1306 C/T polymorphism with colon cancer risk[59]. Langer et al[53,60] reported that presence of CC or CT genotype enhances the survival rate of colon cancer patients. There is also a significant association of MMP-2 -735 C/T polymorphism with esophageal cancer risks[54].
MMP-9 over expression is associated with almost all the hallmark steps of cancer progression that make MMP-9 an ideal candidate gene for genetic association studies. Functional polymorphisms, e.g., MMP-9 promoter (-90CA(n), -1562C/T) as well as structural region (R279Q, P574R, R668Q) polymorphism were studied, assuming it might influence the MMP-9 gene expression or protein activity. MMP-9 polymorphism and gastrointestinal cancer risk is apparent in both Chinese and Caucasian populations[61,62]. In a hospital based case control study in Chinese population, MMP-9 polymorphism in individual carrying RR genotype at P574R have increased risk of ESCC while R279Q and R668Q polymorphism has no association with cancer risk. In contrast Fang et al[63] reported that individuals having RR genotype at R279Q site have enhanced risk towards colon cancer. Tang et al[62] showed R279Q and P574R polymorphism were associated with lymph node metastasis of gastric cancer. Positive association of MMP-9 -1562 C/T polymorphism and colon cancer risk has been reported by Woo et al[61] in Korean population. Other studies reported a higher incident of lymph node metastasis in gastric and colon cancer patients having CC genotype at -1562 bp[64,65].
Association of SNPs in MMP-3 promoter and gastrointestinal cancer has been investigated in MMP-3 -1171 6A/5A site, since transcription repressor bind strongly with 6A allele leading to reduced gene expression. Bradbury et al[51] suggested a positive association of EA risk with 6A/5A or 5A/5A carrier. In addition, Zhang et al[64] found a higher ESCC risk among smokers having the 5A allele and reported that an elevated risk of lymph node metastasis in patients having 5A allele instead of the 6A allele. Interestingly, Dey et al[52] reported that the frequency of homozygotes for the 6A allele is lower in gastric cancer patients than in controls of eastern Indian population[66]. On contrary, only one study performed in Japanese population that showed higher incidents of colon cancer in individuals having 6A/6A genotype.
Two common functional MMP-7 SNPs (-181A/G, -153C/T) are believed to control gene expression in several diseases, including gastrointestinal cancer[67-69]. MMP-7 up-regulation was significantly related to the promoter activity variation of the –181A/G alleles. Jormsjö et al[68] reported that the expression and promoter activity of the MMP7 -181G allele was higher in G over A and, attributed to the formation of a putative binding site (NGAAN) for a heat-shock transcription factor to G-allele. On the contrary, Richards et al[69] reported that elevated plasma MMP-7 level in AA genotype was governed by the G to A transition in -181 bp resulting in higher binding for the forkhead box A2 transcription factor to AA genotype. Studies reported a positive association of MMP-7 -181A/G polymorphism in esophagus, and gastric adenocarcinoma in Chinese population with an increased gastric cancer risk in G allele carrier[70]. However, in contrast, Kubbent et al[58] reported more AA and less AG in gastric cancer group. Moreover, MMP-7, -181 A/G and -153 C/T polymorphism is also significantly associated with colon cancer risk[71].
REGULATION OF MMPS IN GASTROINTESTINAL CANCER
An understanding of the MMP regulation in different cellular processes, e.g., apoptosis, angiogenesis, invasion, metastasis as well as immune function is important for early prognosis and better therapeutics of gastrointestinal cancers. The regulation of MMPs goes awry at any or all cellular processes during cancer development[30]. The regulatory mechanisms shared among different cellular processes might control the invasive property of cells. The presence of MMP-1, -2, -3, -9, and MT1-MMP mRNA and protein in gastric and colorectal cancer tissues are evident from IHC and FISH assay. It is also known that MMPs are produced by inflammatory and fibroblast cells, in the vicinity of cancer cells. Among various signaling pathways, mitogen-activated protein kinases (MAPKs) pathways are important in the regulation of MMP induction as most of MMP promoters contain AP-1 and NFκB-binding sites, the downstream target of MAPK pathways. NFκB and AP-1 activity are significantly enhanced during cancer progression. JNK pathway induces MMP expression through activation as well as nuclear translocation of multiple transcription factors such as Jun D, ATF and most of the MMP promoter contain putative-binding sites for these DNA-binding proteins[72]. It is now conceivable that the function of MMPs is not only confined to invasion and metastasis steps of cancer but they also facilitate initial phases of cancer development. In cancer, special emphasis has been placed on the degradation of type IV collagen, a major protein component of basement membranes that can be cleaved by MMP-2 and -9. Disease progression in experimental animal models of cancer invasion and metastasis correlate with enhanced secretion of specific MMPs by tumor cells and/or stromal cells. Specific MMPs appear to have different functions depending on the stage of cancer and tissue type.
MMPs regulate apoptosis
MMPs especially, MMP-3, -7, -9 and -11 regulate apoptosis by degrading matrix protein. MMPs have both apoptotic and anti-apoptotic actions on endothelial and epithelial cells by cleaving adhesion molecule, e.g., VE-cadherin[73], PECAM-1[74] and E-cadherin[75]. Detachment of adhesion molecules from the membrane is prerequisite for apoptosis to occur. Degradation of laminin by MMP-3 is another example of enhanced apoptosis in mammary epithelial cells possibly by degrading laminin[76]. MMP-7 releases the Fas ligand from the membrane which then induces apoptosis of neighboring cells, or decreases cancer-cell apoptosis, depending on the system[77]. MMPs might also negatively regulate cancer-cell growth, by means of activation of TGF-β or generation of pro-apoptotic molecules such as Fas ligand or TNF-α. By producing heparin-binding epidermal growth factor (HBEGF) from the latent form, i.e., pro-HBEGF, MMP-7 promotes cell survival which is opposes the apoptosis via tyrosine kinase-mediated pathway. Moreover, MMP-11 also inhibits cancer cell apoptosis, as indicated by Wu et al[78], who showed that over expression of MMP-11 decreases spontaneous apoptosis in tumor xenografts. In contrast, MMP11-null mice show a higher rate of apoptosis compared to wild-type when challenged with cancer cells. MMP-11 inhibits apoptosis by the mechanism of releasing IGFs, known to can act as survival factors[79]. Although MMP-9 and -11 decrease cancer cell apoptosis, they increase apoptosis during development[78,80].
MMPs regulate angiogenesis
Angiogenesis, the formation of new capillaries from pre-existing vessels, is associated with several physiological processes as well as pathological conditions. Vascular endothelial growth factor (VEGF) is a mitogen for endothelial cells and is required for tumor-induced angiogenesis. The activation of MMPs is governed by VEGF and then activated MMPs degrade collagen and ECM proteins of basement membrane thereby aiding in the migration of endothelial cells. Cleavage of collagen type I allow endothelial cells to invade the tumor stroma during vessel formation and MMP promote the process by degrading collagen[81]. MMP-2, -9 and -14 directly regulate angiogenesis, and MMP-19 might also be important as it is expressed in blood vessel[82]. Furthermore, reduced MMP-2 expression resulted in decreased angiogenesis in cancer cells in chicken chorioallantoic membrane model. Tumor angiogenesis is significantly inhibited in mice deficient in MMP-2 in comparison with wild type mice[83]. Cleavage of collagen type IV by MMP-2 exposes a cryptic, αvβ3 integrin binding site within the collagen that helps in migration of endothelial cells both in vitro and in vivo model[84].
MMP-9 is a key player for pathological angiogenesis as revealed by experiments done in transgenic models of tumour progression, in the K14-HPV16 skin cancer model[85]. In contrast, the angiogenesis process is unaffected by MMP-2 in skin cancer. Both MMP-14 and MMP-9 null mice have impaired angiogenesis during development[86]. Cleavage of plasminogen by MMP-2, -3, -7, -9 and -12 generates angiostatin[87], and MMP-3, -9, -12, -13 and -20 are involved in the generation of endostatin, a C-terminal fragment of the basement membrane collagen type XVIII[45,88]. Both angiostatin and endostatin reduce endothelial cell proliferation[46]; endostatin also inhibits endothelial cell invasion by acting as an inhibitor of MMP-14 and -2[89]. By degrading fibrin matrix of blood vessels, MMP-14 promotes cell invasion and thus increase angiogenesis. In contrast, MMP-12 inhibits tumor angiogenesis by inhibiting endothelial cell invasion via a different pathway that mediated by urokinase-type plasminogen activator receptor.
MMPs regulate invasion and metastasis
One of the foremost and major steps in invasion is migration of cancer cells from the site of origin to the docking site. The cleavage of ECM is essential for detachment of cancerous cells from neighboring. Cleavage of laminin-5 by MMP-2 and -14 generates a cryptic site that facilitates cell migration[90]. This is supported by the fact of colocalization of laminin-5 and MMP-14 in human cancer specimen and abundance of degraded laminin in tumor tissues[91]. In addition, the main receptor for hyaluronan, CD44 is cleaved by MMP-14, thus, extracellular domain is released, thereby facilitating tissue invasion (Figure 2). Moreover, cell migration is hampered when the cleavage site is mutated[92]. In addition to binding to the ECM, CD44 also binds MMP-9, thereby localizing the enzyme to the cell surface that promotes tumor invasion and angiogenesis, as confirmed by overexpression of the extracellular domain of CD44 and suppression of tumor invasiveness[93]. Thus, cancer cell migration is regulated by cycles of MMP activity or localized MMP activity, not by continuously high activity. During metastasis, cancer cells first cross the epithelial basement membrane and invade the surrounding stroma, followed by invasion to blood or lymphatic vessels; then extravasate to new tissues and establish growth of new proliferating cells in new tissues. The role of MMPs in metastasis was evidenced by in vitro invasion assays and in vivo xenograft metastasis assays. Overexpression of MMP-2, -3, -13 and -14 promotes invasion as documented through either optic nerve explants or cell culture in matrigel[94-96]. In addition, metastasis is reduced in the MMP-2 and MMP-9 null mice as compared with wild-type mice. There is no linear relationship between MMP-2 expression and cell invasion, rather cells expressing intermediate levels of active MMP-2 are the most invasive. The localization of specific MMPs to specialized surfaces on the cell membrane is necessary for their ability to promote invasion. Endothelial cell-adhesion molecule E-cadherin is associated with cancer progression, as it is cleaved by MMP-3 or -7[97]. The released fragment of E-cadherin promotes tumor cell invasion in a paracrine manner in vitro[98,99]. MMP-2, -9 and -14 are known to localize to invadopodia. Moreover, MMP-2 is recruited to invadopodia by either binding to α5β3 integrin[100] or by binding to MMP-14. MMP-14 is recruited to invadopodia by means of its transmembrane and cytoplasmic domains. Overexpression of MMP-14 increases the number of cancer cells in an experimental metastasis assay. Furthermore, the docking of cancer cells at the secondary site, the late events in the metastatic process also involves MMP activity.
Figure 2.
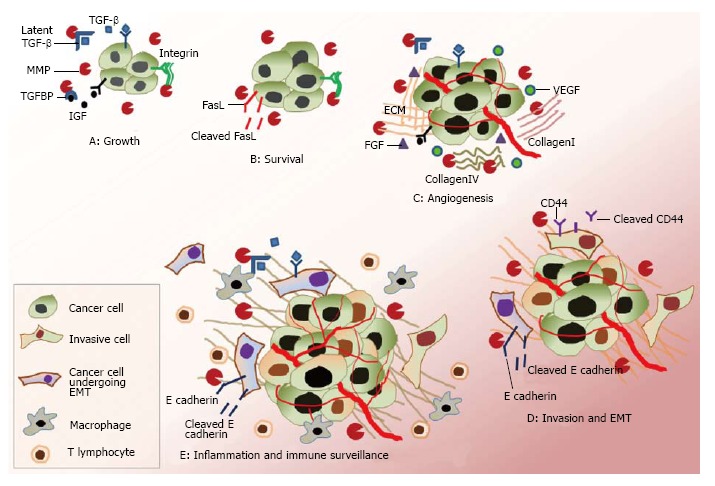
Roles of matrix metalloproteinases in cancer progression. The matrix metalloproteinases (MMPs) play complex but important roles during different stages of cancer progression. A and B: Growth and survival. MMP modulates cellular growth by cleaving different cellular components. It promotes cellular growth by releasing IGF from insulin growth factor-binding protein (IGF-BP). MMP-7 promotes cell survival by resisting apoptosis through cleaving Fas ligand (FasL). MMP modulates integrin signalling by regulating the extracellular matrix (ECM), which regulates growth. MMP activates transforming growth factor-β (TGF-β) from its latent TGF-β complex, which plays important roles in tumour development; C: Angiogenesis. MMP promotes angiogenesis through recruitment of VEGF, FGF. Angiogenesis is further promoted by degradation of extracellular component like collagen I, IV, fibrin, etc., which also act as pro-angiogenic factors; D: Invasion and epithelial to mesenchymal transition (EMT). MMP modulates invasion by degrading specific cellular components, including E-cadherin and CD-44. MMP is involved in mesenchymal transition through cleavage of E-cadherin and modulating TGF-β signaling. MMP-3 is directly involved in EMT, whereas MMP-9 has roles in differentiation; E: Inflammation and immune surveillance. MMPs also regulate immune reactions against the cancer cells. MMP-mediated TGF-β activation inhibits T lymphocyte proliferation. MMPs also modulate cancer-cell sensitivity to natural killer cells and leukocyte accumulation by cleaving different chemokines and cytokine families. VEGF: Vascular endothelial growth factor; FGF: Fibroblast growth factors.
MMPs and the immune responses to cancer
Inflammatory reactions by tumour-specific cytotoxic T lymphocytes, natural killer cells, neutrophils and macrophages are a key mechanism of cellular carcinoma[101]. The immune system is capable of recognizing and attacking cancer cells, while cancer cells somehow escape immune surveillance. MMPs are involved in the escape mechanisms. Although the immune response helps to delay tumour progression, chronic inflammation is also associated with various cancers including cancers those of gastric mucosa, large bowel, and liver[101]. In animal models, mast cells, neutrophils and macrophages are contributors to the progression of cancer. Inflammatory cells synthesize several MMPs, including -9, -12 and -14 and stimulate cancer progression. Indeed, MMP-9 null mice are less prone to skin cancer[85]. Especially, MMP-9 can cleave interleukin-2 receptor (IL-2R)-α and thereby suppress the proliferation of the T lymphocytes[102]. MMP-9 also can act on IL-8, and, thus increases the activity by several folds. MMP-2 cleaves the monocyte chemoattractant protein-3, and the cleaved fragment not only is inactivated but also becomes an antagonist to the receptors[103]. Furthermore, CXCL12 (also known as stromal-cell-derived factor 1) is cleaved and inactivated by MMP-1, -3, -9, -13 and -14[104]. CXCL12 is a ligand for the CXC chemokine receptor 4 (CXCR4) on leukocytes. Inhibition of the binding of CXCL12 to CXCR4 greatly reduces metastasis to lung and lymph nodes in breast cancer[105]. MMP-11 acts on α1-proteinase-inhibitor and the cleaved product altered sensitivity of tumor cells towards natural killer cells[106]. Moreover, few MMPs also activate TGF-β an important inhibitor of the T-lymphocyte response against tumors[107]. MMPs play indirect roles in proliferation of cancer cells by acting on growth factors that entangled into ECM (Figure 3). First, membrane-bound precursors of some growth factors, e.g., TGF-β, are released by MMPs or ADAMs[107]. Second, degradation of growth factors by MMPs makes them available in pericellular space, e.g., MMPs can cleave IGF-BP to release IGF[108]. Finally, cell proliferation by growth factors occurs through integrin signaling.
Figure 3.
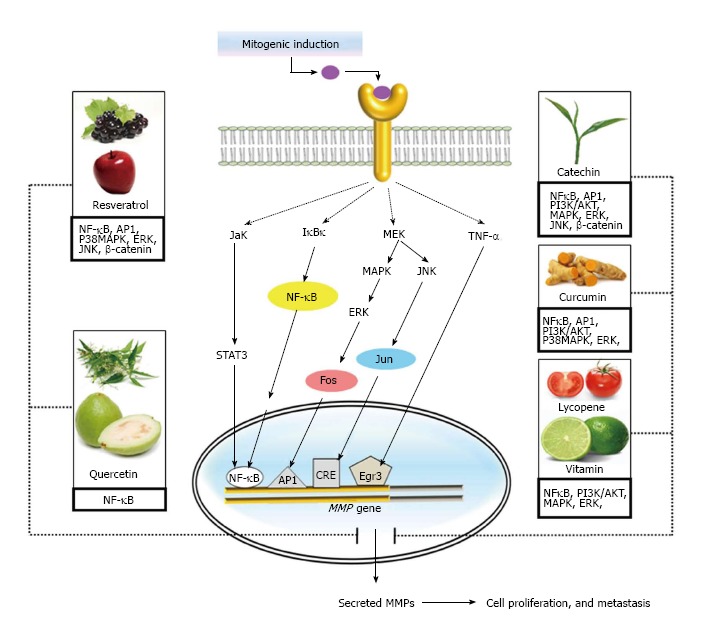
Role of dietary antioxidants in modulating matrix metalloproteinases action. Different ingredients in diet possess antioxidant activity. They act on various signalling pathways and transcription factors which modulate the synthesis and secretion of matrix metalloproteinases (MMPs). Therefore regulates MMP-dependent cellular processes, including proliferation and metastasis. TNF: Tumor necrosis factor; MAPK: Mitogen-activated protein kinases; MEK: Methyl ethyl ketone.
MAJOR IMPACT OF OXIDATIVE STRESS ON GASTROINTESTINAL DISEASES
For many years, researchers have recognized ROS only as causative factor in pathological processes, although the opinion has now changed. ROS were shown to meet the criteria for signalling molecules and they have significant roles in biological functions. This section deals with involvement of ROS in physiological and pathological processes and, subsequent responses in cancer cells under oxidative stress.
Generation of ROS in biological systems
ROS are often byproducts of mitochondrial function that are constantly generated and eliminated from physiological systems. ROS are oxygen containing reactive species, that may contain an unpaired electron, e.g., superoxide radical (O2·-), hydroxyl radical (OH·) and/or non-radical molecules, such as hydrogen peroxide (H2O2). O2·- are formed by chemical reduction of molecular oxygen, by electrons that escape from complex I and III of electron transport chain and O2·- is than dismutated to H2O2. Nearly 2% of molecular oxygen consumed during mitochondrial respiration ends up as O2·-[109,110]. Apart from mitochondria, ROS can be generated from a family of trans-membrane proteins, known as NADPH oxidases (Nox). These enzymes catalyse NADPH dependent reduction of molecular oxygen to O2·-[111].
Increased ROS generation can induce lipid peroxidation and protein oxidation, hampering normal cellular processes. In addition, ROS can target mitochondrial DNA (mDNA) more effectively due to its proximity and lack of protective histones and limited repair mechanism. mDNA does not contain any introns and encodes for 13 respiratory complexes essential for electron transport chain and oxidative phosphorylation. Mutation of mDNA results in aberrant mitochondrial proteins during ATP generation; this induces further ROS production. Moreover, ROS cause nuclear DNA damage through DNA oxidation (by formation of 8-oxo-G, 8-oxo-dG), which contributes to mutation[109].
Cancer cells are known to be metabolically active and require elevated amount of energy to support their functionality. To meet their ATP need, cancer cells exhibit an increased dependency to glycolysis which slows down mitochondrial energy production. This phenomenon of stimulation of glycolysis in cancer cells that inhibit mitochondrial respiration, is known as “Crabtree effect”[112]. However, inhibition of glycolysis in tumor cells may result impairment of mitochondrial oxidative phosphorylation and subsequently associated with hypoxia and higher amounts of ROS generation and accumulation of ROS-mediated reaction products[113].
Oxidative stress and gastrointestinal diseases
Oxidative stress plays important roles in pathogenesis of gastro-intestinal diseases, which include mucosal damage, gastro-intestinal ulcers, and cancer. Although gastric ulcer can be generated by different factors, e.g., non-steroidal anti-inflammatory drugs (NSAIDs), thermal stress, ethanol, and H. pylori infection, leading to oxidative damage through free radical generation (specially OH·) and subsequent apoptotic responses of gastric mucosa[114]. In addition, gastro-intestinal diseases are associated to increased oxidative stress and oxidants levels, such as glutathione, lipid peroxidation, myeloperoxidases, protein carbonyl, etc[115]. Furthermore, pathogens are directly involved in aggravating oxidative stress; for example, complete eradication of H. pylori is reported to attenuate oxidative stress in gastric mucosa[116].
Although certain types of gastro-intestinal inflammations, like ulcerative colitis, hepatitis, H. pylori infection, are more prone to develop cancer, the reasons are still not well elucidated. Inflammation and subsequent elevated oxidative stress might be the factors for aggravating chronic inflammation and inducing malignant transformation. Transgenic mice expressing hepatitis B protein in liver develop chronic hepatitis with elevated levels of 8-oxo-dG, leading to hepatocellular carcinoma[117]. It is well accepted that inflammation is always accompanied with elevated oxidative stress in cancer. Gastric cancer patients (with normal renal and hepatic functions) are found to have significantly increased lipid peroxidation levels[118]. Gastric carcinoma patients have significantly higher myeloperoxidase activity than controls, both before and after operation, although total antioxidant status was decreased post-operation[119]. Gastric cancers are also associated with augmented protein oxidation, although no differences are found in oxidative stress parameters and antioxidant enzyme activities between anti-H. pylori IgG positive and negative gastric cancer patients[120].
Cellular responses in cancer cells under oxidative stress
ROS, such as O2·- and H2O2, have roles in proliferation and cellular growth that contribute to the development of cancer. Cancer cells also exhibit different cellular responses under oxidative stress, which include senescence, autophagy and apoptotic responses; although it is still not well understood, how these cellular pathways assume priority to become activated under particular conditions. H2O2 can directly modulate autophagic responses during nutritional starvation through Atg4 (autophagin-1) expression and accumulation of LC3-PE on the autophagosomal membrane[121]. In addition, ROS-induced autophagy is dependent on constitutive expression of Atg, baclin, hypoxia induced factor-1 (HIF-1) and BNIP3[122]. Mutations in ATG genes (like ATG2B, ATG5, ATG9B, and ATG12) are involved in gastric and colorectal carcinomas and may contribute to cancer development by deregulating the autophagy process. Moreover, increased autophagy (through upregulation of Atg5 and Atg7) is also involved in in vitro malignant transformation by K-Ras[123]. ROS and ROS-mediated signalling pathways are also involved in senescence responses. Overexpression of p21, along with increased ROS production, directly induces the senescence phenotype; while inhibition of p21-mediated ROS accumulation can rescue cells from senescence[124]. In addition, sub-lethal doses of H2O2 can induce senescence-like growth arrest at the G1 stage via up regulation of p53 and p21. Anti-apoptotic Bcl-2 family members antagonise ROS generation during apoptosis. Moreover, ROS generation is accompanied by cellular apoptosis due to lipid peroxidation and mitochondrial depolarization.
Increased ROS production in cancer cells is associated with constant activation of transcription factors including NFκB and AP-1. Moreover, a recent study found that activation of TLR4 promotes gastric cancer by increasing mitochondrial ROS generation through NFκB activation[125]. Oncogenic signals are involved in ROS generation that promote metastasis in gastric cancer[126]. The oncogene c-myc has been reported to develop genetic instability, DNA damage and mitigation of p53 function through ROS production[127]. RAS2 mutation promotes oxidative stress by restricting mitochondrial respiration into non-phosphorylating state. K-Ras is also involved in malignant transformation through ROS mediated JNK activation[123]. ROS and Rac1b are involved in MMP-3 mediated epithelial to mesenchymal transition (EMT). ROS also stimulate the expression of Snail and cause damage to DNA and subsequent genomic instability[128]. Reports have suggested a possible association of Nox and Nox-mediated ROS generation for carcinogenesis[111]. Nox1 is involved in H. pylori-induced gastric cancer and in angiogenic responses for tumour formation[129,130]. Nox2 is involved in phagocytosis; Nox4 controls cell survival in different cancers, including gastric, colon and pancreatic cancers[111,131]. Increased ROS can cause detachment of JNK associated glutathione-S-transferase (GST)-π and thus facilitates JNK activation. In addition, H2O2 mediated JNK activation also causes downregulation of JNK phosphatases[132]. Mice that have an inactive c-Jun that lacks JNK phosphorylation site or deficient in JNK displayed reduced tumor development[133,134]. Moreover, increased ROS productions in oncogenically transformed cells potentiate JNK and p38 MAP kinases activation[135].
Role of endogenous antioxidants during oxidative injury
Endogenous antioxidants are essential for maintenance and neutralization of perturbed oxidative free radical status. Superoxide dismutases (SOD) are important antioxidants, as Mn-dependent SOD (Mn-SOD) null mice cannot survive after birth[136]. Heterozygous Mn-SOD null mice can survive, however, show higher incidences of lymphomas and adenocarcinomas[137]. In addition, Zn-dependent SOD (Zn-SOD) knockdown mice develop hepatic cancer in late stages of life[138]. Mice lacking catalase also exhibit elevated cancer incidences[139]. Glutathione peroxidases (GPx) also have significant roles as antioxidants. Simultaneous knock out of GPx-1 and -2 in mice leads to gastrointestinal cancer[140].
Clinical publications have reported different levels of endogenous antioxidants in gastrointestinal cancers. GST, GPx and SOD activities are reported to be significantly elevated in colorectal cancers, than adjacent normal tissues[141]. Stomach adenocarcinoma and esophageal squamous cell carcinoma show significantly increased Mn-SOD expression as compared to noncancerous cells[142]. In clinical studies with gastric and colorectal cancer, GPx, SOD, glucose-6-phosphate dehydrogenase (G6PD), malonaldehyde and glutathione reductase were found elevated in the malignant phenotype[143]. However, because of the sustained oxidative stress conditions, these antioxidants are insufficient in cancer, eventually resulting in decreased antioxidant levels in several cancers[144]. Moreover, modulated expressions of Mn-SOD are reported due to mutations in the promoter region, abnormal methylation, loss of heterozygosity or mutation in the coding sequences[145]. These differential results of cellular antioxidant SOD cause metabolic chaos, due to different types and grades of malignancy[146].
DIETARY ANTIOXIDANTS AS A MODULATOR OF MMPs IN GASTROINTESTINAL CANCER
Human diet contains a mixture of oxidants and antioxidants substances. Dietary and endogenous antioxidants are important for cellular protection by reacting and/or eliminating oxidizing free radicals. The question of whether antioxidant supplements might protect against cancer has drawn much attention since the mid ’80s and different antioxidants were extensively studied thereafter, although the results of the investigations are mixed and contradictory. Antioxidant and endogeneous redox enzymes act as the first-line defense against ROS in all cellular compartments and also extracellularly. The most important of these enzymes include SOD, GPx, catalase and peroxiredoxins. The specific role of above enzymes in carcinogenesis is still unambiguous since their roles in ROS detoxification are well known. It is noteworthy that we do not yet fully understand the chemopreventive role of phytochemicals as antioxidants, or as modulators of other processes related to carcinogenesis. In this section, we highlight the effects of dietary antioxidants in prevention of cancers with particular emphasis on the regulation of MMPs activity.
Tea polyphenol and catechin
Tea, derived from the plant Camellia sinensis, is the most consumed beverage worldwide and it is grown in over 30 countries around the world, exclusively in the subtropical and tropical zones[147]. It is processed in different ways in different parts of the world to produce green, black, or Oolong tea. Both green and black teas have been studied for their health benefits, particularly for prevention and treatment of inflammatory diseases as well as cancer. Green tea is rich in polyphenolic substances, which include flavonoid, flavanols, and flavinidols all of which have antioxidant properties. The most common flavonal in tea is catechin. The most active and abundant catechin in green tea is epigallactocatechin-3-gallate (EGCG) that has been shown to inhibit cancer cell growth in vitro and in vivo. In black tea the major polyphenols are theaflavin and thearubigin.
During the last decade several epidemiological studies have linked tea consumption, especially green tea to a reduced risk of cancer in humans. Morse et al[148] documented the beneficial effects of the polyphenol fractions of green tea, the polyphenol fractions of black tea, i.e., EGCG and theaflavins against N-nitrosomethylbenzylamine (NMBA)-induce esophageal cancer in rat. Wang et al[149] investigated both protective and therapeutic effects of green tea and black tea extract on esophageal tumorigenesis in rats. A population-based case-control study in Shanghai indicated that tea consumption was strongly associated with reduced colorectal cancer incident. Green tea polyphenol supplementation during the initiation or postinitiation period significantly lowered azoxymethane-induced tumor incidence in rats[150]. In addition, catechin and EGCG reduced colon tumor incidence in a 1, 2-dimethylhydrazine (DMH)-induced intestinal cancer.
Green tea polyphones were shown to prevent cancer cell proliferation and invasion. EGCG has been shown to inhibit NFκB activity in human colon cancer cells[151]. Several studies indicate that chemopreventive properties of EGCG can also mediated by inhibition of MMP induction. EGCG inhibited the PMA-induced cell invasiveness and MMP-9 expression in human gastric cancer adenocarcinoma (AGS) cells[152]. Fassina et al[152] documented that EGCG (25-100 μmol/L) inhibits the MMP-2 and -9 in endothelial cells. EGCG inhibited the activity and expression of MT1MMP, a protein responsible for the activation of MMP-2 as examined by Annabi et al[153]. Onoda et al[154] found that gastric cancer cell lines, e.g., MKN-1, MKN-28, MKN-45, NUGC-3 and TMK-1 are sensitive to EGCG treatment with NUGC-3 being the most sensitive. Furthermore, EGCG suppresses Met signaling in HCT116 human colon cancer cells[155]. In another study, EGCG may exert at least part of its anticancer effect by inhibiting angiogenesis through blocking the induction of VEGF and binding to its receptors. EGCG has been shown to affect MMP-2 and -9 activities both directly and indirectly in endothelial cells thereby inhibiting or delaying cancer invasion and metastasis. Concanavalin A-induced activation of MMP-2 and activity of MT1-MMP has been reduced by EGCG[156]. Catechin, another major component of tea, prevents vascular smooth muscle cell invasion by inhibiting MT1-MMP activity and MMP-2 expression[157]. Black tea polyphenols inhibit DMH-induced colorectal cancer by inhibiting MMP-7 induction via Wnt/β-catenin pathway[158]. Hwang et al[159] showed the apoptotic effect of EGCG in HT-29 colon cancer cells that was mediated by AMPK signaling. Hence, catechin and EGCG exert strong anticancer activity by targeting transcription factors like NFκB, and AP-1 which involve in the regulation of mainly MMP-2, -9 and -7 activities. Specifically, EGCG regulates angiogenesis and apoptosis via changing expression of VEGF, uPA, IGF-1, EGFR, cell cycle regulatory proteins and in turn affects NFκB, PI3-K/Akt, ERK, JNK, Ras/Raf/MAPK and AP-1 signaling pathways, thereby acting as chemopreventive agent[160].
Curcumin
Curcuma longa (Zingiberaceae family) rhizomes have been traditionally used in the south Asian countries for the treatment of a variety of inflammatory conditions and different diseases including carcinomas[161]. The pharmacological properties of curcumin are attributed mainly to the curcumin (diferuloylmethane)-(1,7-bis (4-hydroxy-3-methoxyphenyl)-1,6-hepadiene-3,5-dione) a hydrophobic polyphenol present in the rhizome. Curcumin is a potent antioxidant that acts as a free radical scavenger[162]. Curcumin possesses a wide range of pharmacological activities including anti-inflammatory, chemo-preventive, and antimicrobial and wound healing effects[162-164]. Curcumin which is also known as turmeric, has been shown to exhibit dose dependent chemo-preventive effects in several gastrointestinal cancers including colon, duodenal, stomach, esophageal and oral carcinogenesis[165,166]. In vivo and in vitro studies have demonstrated curcumin’s ability to inhibit carcinogenesis at three stages: tumor promotion, angiogenesis and tumor growth[167]. These protective effects of curcumin are attributed mainly to its antioxidant properties and investigated for the purpose of developing novel drugs. It reduces carcinogen-induced tumorigenesis in the fore-stomach and N-ethyl-N′-nitro-nitrosoguanidine-induced duodenal tumors. Lower incidences of bowel cancer in Indians, possibly due to the use of turmeric during food preparation. The molecular basis of anti-carcinogenic and chemopreventive effects of curcumin is targeted to transcription factors, growth regulators, adhesion molecules, apoptotic genes, angiogenesis regulators and cellular signaling molecules[166].
Koo et al[168] showed that curcumin and 5-fluorouracil (5-FU) additively inhibited the growth of gastric carcinoma cells. In another study, curcumin reversed the multidrug resistance of a human gastric carcinoma cell line[169]. Curcumin exhibited both preventive and therapeutic effects on the incidence and multiplicity of fore-stomach tumors induced by benzopyrene in mice[170]. A dietary supplementation of 0.15% curcumin reduces intestinal tumor formation in Min-/- mice by 63%. Curcumin induces apoptosis and prevents adenoma development in the intestinal tract in mice[171]. Tetrahydrocurcumin (THC) significantly decreases DMH-induced colon carcinogenesis[172]. Shpitz et al[173] showed that curcumin and celecoxib synergistically inhibit colorectal cancer progression in DMH-induced rat model. Several studies documented the inhibitory effects of curcumin on AOM-induced colon cancer[174]. Curcumin inhibits the expression of MMP-9 both in vitro and in vivo and thereby inhibits tumor invasion and metastasis. Bimonte et al[175] reported that curcumin inhibits the expression of MMP-9 in orthotopically implanted pancreatic tumors. Curcumin causes a significant reduction of tumor volume, and MMP-9 activity in a xenografted model[176]. Curcumin also reduces the expression of major MMPs via reduced NFκB activity and AP-1 transcription[176]. Lin et al[177] reported that curcumin inhibits SK-Hep-1 hepatocellular carcinoma cell invasion via suppression of MMP-9 secretion. Curcumin prevents human colon cancer, colo-205 cells migration through the inhibition of NFκB/p65 and downregulation of cyclooxygenase-2 and MMP-2 expression[178]. Lin et al[179] reported that curcumin inhibits SDF-1α-induced invasion of human esophageal carcinoma cells by down regulating MMP-2 promoter activity as well as suppressing the formation of lipid raft-associated Rac1/PI3K/Akt signaling complexes. In conclusion, curcumin appears to have a significant potential in the treatment of multiple diseases that are due to oxidative stress. Thus, various inflammatory pathways ultimately act on MMP transcription or expression during prevention of various types of cancer by curcumin.
Resveratrol
Resveratrol (trans-3,4′,5-trihydroxystilbene), a non-flavonoid polyphenolic antioxidant, has attracted considerable attention due to its anti-oxidant, anti-cancer, and anti-inflammatory properties. It is in abundance in grapes and grape products such as wine, moderately abundant in blueberries, and sparsely abundant in other plants. Resveratrol is a scavenger of hydroxyl, superoxide, and other radicals and thus acts as a potent antioxidant. It protects against ROS-mediated lipid peroxidation in cell membranes and DNA damage. Resveratrol enhances the expression and/or the activity of drug metabolizing phase I/II enzymes such as Mn-SOD, GST, cytochrome P450 reductase, quinine oxidoreductase, NAD(P)H: quinone oxidoreductase (NQO1), quinone reductase (QR), heme oxygenase-1 (HO-1), glutamate cysteine ligase (GCL); thereby protecting against oxidative DNA damages[180]. Several studies demonstrated that resveratrol exhibits strong chemopreventive effects in various experimentally induced tumor models as well as inhibits proliferation and induces apoptosis of various cancerous or transformed cells. Oral or intra-peritoneal administration of resveratrol decreases expressions of COX-1, COX-2, and PGE2 reducing the number and size of esophageal tumors in N-nitrosomethylbenzylamine-induce rat tumor model[181]. Similarly, oral administration of resveratrol limits the formation of aberrant crypt foci and tumors in the colon of rats that are treated with chemical carcinogen, i.e., 1,2-dimethylhydrazine (DMH) or azoxymethane. Resveratrol prevented the formation of colon tumors and reduced the formation of small intestinal tumors by 70% in APC-/+ mice[182]. Resveratrol inhibits MMP-9 expression and invasion of human hepatocellular carcinoma cells. Resveratrol modulates all three MAP kinases namely ERK1/2, JNK, and p38 MAPK. Resveratrol impairs the expression of EMT-related genes (E-cadherin, N-cadherin, vimentin, MMP-2, and -9) in pancreatic cancer cells and inhibits proliferation, migration, and invasion[183]. Ji et al[184] demonstrated that resveratrol possesses chemopreventive effects in HCT116 CRC through inhibition of MMP-7 via Wnt/β-catenin signalling pathway. Weng et al[185] suggested that resveratrol and its related methoxy analogue MR-3 might exert anti-invasive activity against hepatoma cells through regulation of MMP-2 and -9 as well as TIMPs. Harikumar et al[186] reported that resveratrol can enhance chemopreventive activity of gemcitabine in vitro and in vivo mouse model of pancreatic cancer. In summary, anticancer activity of resveratrol is augmented by upregulation of TIMP and downregulation of MMP-9 expression.
Quercetin
Quercetin or 3,5,7,3’,4’-tetrahydroxyflavone is a flavonoid found abundantly in plant-derived foods and has been shown to possess several health beneficial activities including anti-tumor, anti-inflammation and anti-proliferation; it has recently gained attention due to its potential anticancer activity. As an antioxidant it possesses the most potent ROS scavenging activity and provides protection against the development of variety of cancers by ameliorating ROS-mediated cellular damages[187]. Moreover, it reduces the level of oxidative enzymes, such as xanthine oxidase (XOD), lipoxygenase and NADPH oxidase, thereby preventing free radical-induced cellular damage[187]. In vivo studies have been performed to depict the chemopreventive properties of quercetin on different cancers. Volate et al[188] found that food supplementation of approximately 3% quercetin exerts significantly beneficial effects by decreasing precancerous lesions through induction of cellular. Dihal et al[189] reported that quercetin inhibits azoxymethane-induced colorectal carcinogenesis in F344 rats. Quercetin was found to be protective against hepatocarcinoma that was generated by N-nitrosodiethylamine and, was accompanied by the maintenance of a correct intracellular oxidant/antioxidant status. Quercetin prevents 4-nitroquinoline 1-oxide-induced oral carcinogenesis during the initiation/post initiation phases of carcinogen treatment. A limited number of experiments were conducted to investigate the effects of quercetin in the regulation of MMP activity in gastrointestinal cancer. It has been found that quercetin supplementation did not alter MMP-2 or TIMP-2 gene transcription or plasma protein levels but TIMP-1 gene expression and plasma protein levels decreased significantly. Recently, Lai et al[190] reported that migration and invasion of SAS human oral cancer was inhibited by quercetin via downregulation of MMP-2 and -9 in a NFκB dependent pathway. They also showed significant reduction of the MMP-7, -10 protein levels by quercetin treatment.
Other promising dietary antioxidants
The roles of few more dietary antioxidants e.g., melatonin, lycopene, retinoic acid, vitamin C and vitamin E in prevention of cancer through regulation of MMPs are discussed below. Melatonin is a naturally occurring antioxidant synthesized mainly by the pineal gland of vertebrates and also found in many edible plant products[191]. Recent research documents that consumption of tropical fruit enhances the serum melatonin level as well as raises the antioxidant capacity of blood serum[191,192]. Melatonin retards the development of cancer in different animal models and possesses strong anti-proliferative and pro-apoptotic effects in various cancer cells. Sharman et al[193] reported that long term exposure to dietary melatonin reduces tumor number and size in aged male mice. In addition, melatonin inhibits the growth of murine gastric carcinoma cells by upregulation of p21, Bax and down regulation of Bcl-2. Decreased expressions of melatonin receptor in various cancers also suggest the importance of melatonin signaling in cancer development[194]. Hong et al[195] reported that melatonin treatment induces apoptosis, autophagy, and senescence in human colorectal cancer cells. A few report suggested that melatonin prevents gastrointestinal cancer development by modulation of MMP functions. Melatonin induces apoptosis and reduces invasiveness of HepG2 cells in vitro through TIMP-1 upregulation and attenuation of MMP-9 activity via NFκB signal pathway[196]. Rudra et al[192] reported that melatonin reduces MMP-9 activity in AGS cell line and binds directly to its active site.
Two separate studies reported the chemo-preventive effects of lycopene in human colon cancer. Tang et al[197] evaluated the chemo-preventive effects of lycopene and fish oil in a mouse xenograft model of colon cancer. They found that inhibition of tumor growth and progression by the augmenting p21 (CIP1/WAF1) and p27 (Kip1) expression, and suppression of MMP-7, MMP-9, COX-2 and PGE2, PCNA, β-catenin, cyclin D1 and c-Myc proteins[197]. In vitro study by Lin et al[198] suggested the inhibitory effects of lycopene on tumor progression, where cell invasion was inhibited by down regulation of MMP-7 expression via blocking MAPK/ERK and PI3K/Akt signaling pathways. Adachi et al[199] reported that naturally occurring retinoid, all-trans retinoic acid (ATRA), 9-cis retinoic acid and 13-cis retinoic acid are helpful in the prevention and therapy of colon cancer. ATRA prevents tumor invasion in mice and inhibits in vitro invasion of colon cancer cells by down regulation of MMP-7 expression. Moreover, Park et al[200] reported that retinol reduces the invasive potential of retinoic acid resistance colon cancer cells by decreasing MMP-1,-2,-7,-9 expressions and activity. Retinol reduces the metastatic potential of colon cancer cells via down regulation of MMP induction in a retinoic acid receptor-independent mechanism. β-ionone, the derivative product of carotenoids, is the precursor of vitamin A also possesses anti-proliferative activity in cancer cells[201]. Liu et al[202] found that that γ-tocotrienol inhibit gastric cancer cell (SGC-7901) proliferation by reducing MMP-1 and -2 activity via modulating the expression of their inhibitor TIMP-1 and TIMP-2. Dietary supplementation of naturally occurring antioxidants, ascorbic acid reduces the size of colon xenograft cancer by downregulation of MMP-9 and VEGF in nude mice[203]. Vitamin E (γ-tocotrienol) effectively inhibits the growth of human gastric cancer in a xenograft mouse model. γ-tocotrienol inhibited the proliferation of gastric cancer cell lines, via inhibiting the NFκB mediated upregulation of MMP-9 and VEGF[204].
CONCLUSION
The immense complexity of cancer disease is not yet fully characterized despite numerous advances in modern molecular biology. Complexity in cancer cells arise from heterogeneity of tumor microenvironment, inflammatory stimuli, immune responses, diet effects as well as intestinal micobiota. All these factors determine whether the fate of cancer cells undergo apoptosis or proliferation or even develop resistance to drugs. Cancer is a multi-factorial disease, which varies from patient to patient. Even the complexity lies in a certain tumor cells that may refer to tumors with diverse genetics. In fact, every cancer types is unique. Hence, intramolecular heterogeneity poses another dimension during cancer progression. Significant intra-tumor heterogeneity is present in many patients, thus drug resistance may develop. Sequencing technology is used to monitor clonal dynamics of cancer cells. In this context, careful attention should be given to detect minor clones of clinical significance. Tumor heterogenity was documented by the Cancer Genome Atlas and the Cancer Genome Analysis projects.
In the future, personalised cancer medicine may be possible by accounting for both interpatient and intrapatient heterogeneity. Additionally, new therapeutic strategies are important for targeting cellular conditions like cellular senescence rather than targeting a particular biomolecule. Given the complexity of cancer, it is unlikely universal therapeutic strategy will be employed for different cancer types and stages. The MMP family of enzymes occupies a major importance in the field of gastric cancer research. Literature in last two decades of MMP biochemistry and cancer biology supports the possibility of particular MMP in particular types of cancer, including gastrointestinal cancer. Both basic and applied research is needed to decipher the mechanisms of cancer progression and regulatory roles of particular MMP associated with different cancer types. Drug discovery efforts have uncovered pharmacological inhibitors of different MMPs. Specific MMP inhibitors at a specific dose would be an important achievement to treat particularly gastric cancer and to halt to the progression of these diseases.
ACKNOWLEDGMENTS
Authors are thankful to Dr. Russel J Reiter, University of Texas, United States for critical reading of the manuscript.
Footnotes
Supported by Council of Scientific and Industrial Research, India; (CSIR)-INDEPTH and HUM projects
P- Reviewer: Akbulut S, Casadesus D, Sun XY S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Wadhwa R, Song S, Lee JS, Yao Y, Wei Q, Ajani JA. Gastric cancer-molecular and clinical dimensions. Nat Rev Clin Oncol. 2013;10:643–655. doi: 10.1038/nrclinonc.2013.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murakami T. Pathomorphological diagnosis. Definition and gross classification of early gastric cancer. Gann Monogr Cancer Res. 1971:11. [Google Scholar]
- 4.Sokoloff B. Predisposition to cancer in the bonaparte family. Am J Surgery. 1938;40:673–678. [Google Scholar]
- 5.Williams L, Jenkins GJ, Doak SH, Fowler P, Parry EM, Brown TH, Griffiths AP, Williams JG, Parry JM. Fluorescence in situ hybridisation analysis of chromosomal aberrations in gastric tissue: the potential involvement of Helicobacter pylori. Br J Cancer. 2005;92:1759–1766. doi: 10.1038/sj.bjc.6602533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugai T, Habano W, Uesugi N, Jao YF, Nakamura S, Abe K, Takagane A, Terashima M. Three independent genetic profiles based on mucin expression in early differentiated-type gastric cancers--a new concept of genetic carcinogenesis of early differentiated-type adenocarcinomas. Mod Pathol. 2004;17:1223–1234. doi: 10.1038/modpathol.3800170. [DOI] [PubMed] [Google Scholar]
- 7.Milne AN, Carvalho R, Morsink FM, Musler AR, de Leng WW, Ristimäki A, Offerhaus GJ. Early-onset gastric cancers have a different molecular expression profile than conventional gastric cancers. Mod Pathol. 2006;19:564–572. doi: 10.1038/modpathol.3800563. [DOI] [PubMed] [Google Scholar]
- 8.Kokkola A, Sipponen P. Gastric carcinoma in young adults. Hepatogastroenterology. 2001;48:1552–1555. [PubMed] [Google Scholar]
- 9.Kikuchi S, Nakajima T, Nishi T, Kobayashi O, Konishi T, Inaba Y, Wada O, Satou H, Ishibashi T, Ichikawa S, et al. Association between family history and gastric carcinoma among young adults. Jpn J Cancer Res. 1996;87:332–336. doi: 10.1111/j.1349-7006.1996.tb00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, Shen D, Parsons DW, Bardelli A, Sager J, Szabo S, Ptak J, Silliman N, Peters BA, van der Heijden MS, et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 2004;304:1164–1166. doi: 10.1126/science.1096096. [DOI] [PubMed] [Google Scholar]
- 11.Shaheen N, Ransohoff DF. Gastroesophageal reflux, barrett esophagus, and esophageal cancer: scientific review. JAMA. 2002;287:1972–1981. doi: 10.1001/jama.287.15.1972. [DOI] [PubMed] [Google Scholar]
- 12.Lagergren J, Bergström R, Lindgren A, Nyrén O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N Engl J Med. 1999;340:825–831. doi: 10.1056/NEJM199903183401101. [DOI] [PubMed] [Google Scholar]
- 13.Chak A, Ochs-Balcom H, Falk G, Grady WM, Kinnard M, Willis JE, Elston R, Eng C. Familiality in Barrett’s esophagus, adenocarcinoma of the esophagus, and adenocarcinoma of the gastroesophageal junction. Cancer Epidemiol Biomarkers Prev. 2006;15:1668–1673. doi: 10.1158/1055-9965.EPI-06-0293. [DOI] [PubMed] [Google Scholar]
- 14.Sappati Biyyani RS, Chessler L, McCain E, Nelson K, Fahmy N, King J. Familial trends of inheritance in gastro esophageal reflux disease, Barrett’s esophagus and Barrett’s adenocarcinoma: 20 families. Dis Esophagus. 2007;20:53–57. doi: 10.1111/j.1442-2050.2007.00651.x. [DOI] [PubMed] [Google Scholar]
- 15.Chak A, Falk G, Grady WM, Kinnard M, Elston R, Mittal S, King JF, Willis JE, Kondru A, Brock W, et al. Assessment of familiality, obesity, and other risk factors for early age of cancer diagnosis in adenocarcinomas of the esophagus and gastroesophageal junction. Am J Gastroenterol. 2009;104:1913–1921. doi: 10.1038/ajg.2009.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hemminki K, Sundquist J, Ji J. Familial risk for gastric carcinoma: an updated study from Sweden. Br J Cancer. 2007;96:1272–1277. doi: 10.1038/sj.bjc.6603722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bosman F, Carneiro F, Hruban R, Theise N. Who classification of tumours of the digestive system iarc. The International Agency for Research on Cancer; 2010. p. 3. [Google Scholar]
- 18.Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, Chan TL, Kan Z, Chan AS, Tsui WY, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43:1219–1223. doi: 10.1038/ng.982. [DOI] [PubMed] [Google Scholar]
- 19.Kwon MJ, Kim SH, Jeong HM, Jung HS, Kim SS, Lee JE, Gye MC, Erkin OC, Koh SS, Choi Y-L, et al. Claudin-4 overexpression is associated with epigenetic derepression in gastric carcinoma. Lab Invest. 2011;91:1652–1667. doi: 10.1038/labinvest.2011.117. [DOI] [PubMed] [Google Scholar]
- 20.Baba Y, Watanabe M, Murata A, Shigaki H, Miyake K, Ishimoto T, Iwatsuki M, Iwagami S, Yoshida N, Oki E, et al. LINE-1 hypomethylation, DNA copy number alterations, and CDK6 amplification in esophageal squamous cell carcinoma. Clin Cancer Res. 2014;20:1114–1124. doi: 10.1158/1078-0432.CCR-13-1645. [DOI] [PubMed] [Google Scholar]
- 21.Okada T, Nakamura M, Nishikawa J, Sakai K, Zhang Y, Saito M, Morishige A, Oga A, Sasaki K, Suehiro Y, et al. Identification of genes specifically methylated in Epstein-Barr virus-associated gastric carcinomas. Cancer Sci. 2013;104:1309–1314. doi: 10.1111/cas.12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alvarez MC, Ladeira MS, Scaletsky IC, Pedrazzoli J, Ribeiro ML. Methylation pattern of THBS1, GATA-4, and HIC1 in pediatric and adult patients infected with Helicobacter pylori. Dig Dis Sci. 2013;58:2850–2857. doi: 10.1007/s10620-013-2742-6. [DOI] [PubMed] [Google Scholar]
- 23.Jin Z, Zhao Z, Cheng Y, Dong M, Zhang X, Wang L, Fan X, Feng X, Mori Y, Meltzer SJ. Endoglin promoter hypermethylation identifies a field defect in human primary esophageal cancer. Cancer. 2013;119:3604–3609. doi: 10.1002/cncr.28276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin Z, Feng X, Jian Q, Cheng Y, Gao Y, Zhang X, Wang L, Zhang Y, Huang W, Fan X, et al. Aberrant methylation of the Ras-related associated with diabetes gene in human primary esophageal cancer. Anticancer Res. 2013;33:5199–5203. [PubMed] [Google Scholar]
- 25.Ninomiya I, Kawakami K, Fushida S, Fujimura T, Funaki H, Takamura H, Kitagawa H, Nakagawara H, Tajima H, Kayahara M, et al. Quantitative detection of TIMP-3 promoter hypermethylation and its prognostic significance in esophageal squamous cell carcinoma. Oncol Rep. 2008;20:1489–1495. [PubMed] [Google Scholar]
- 26.Poplineau M, Schnekenburger M, Dufer J, Kosciarz A, Brassart-Pasco S, Antonicelli F, Diederich M, Trussardi-Régnier A. The DNA hypomethylating agent, 5-aza-2’-deoxycytidine, enhances tumor cell invasion through a transcription-dependent modulation of MMP-1 expression in human fibrosarcoma cells. Mol Carcinog. 2013 doi: 10.1002/mc.22071. [DOI] [PubMed] [Google Scholar]
- 27.Hussain Z, Khan MI, Shahid M, Almajhdi FN. S-adenosylmethionine, a methyl donor, up regulates tissue inhibitor of metalloproteinase-2 in colorectal cancer. Genet Mol Res. 2013;12:1106–1118. doi: 10.4238/2013.April.10.6. [DOI] [PubMed] [Google Scholar]
- 28.Leung WK, To KF, Chu ES, Chan MW, Bai AH, Ng EK, Chan FK, Sung JJ. Potential diagnostic and prognostic values of detecting promoter hypermethylation in the serum of patients with gastric cancer. Br J Cancer. 2005;92:2190–2194. doi: 10.1038/sj.bjc.6602636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang YC, Yu ZH, Liu C, Xu LZ, Yu W, Lu J, Zhu RM, Li GL, Xia XY, Wei XW, et al. Detection of RASSF1A promoter hypermethylation in serum from gastric and colorectal adenocarcinoma patients. World J Gastroenterol. 2008;14:3074–3080. doi: 10.3748/wjg.14.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 31.Tezvergil-Mutluay A, Agee KA, Hoshika T, Carrilho M, Breschi L, Tjäderhane L, Nishitani Y, Carvalho RM, Looney S, Tay FR, et al. The requirement of zinc and calcium ions for functional MMP activity in demineralized dentin matrices. Dent Mater. 2010;26:1059–1067. doi: 10.1016/j.dental.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Susnow N, Zeng L, Margineantu D, Hockenbery DM. Bcl-2 family proteins as regulators of oxidative stress. Semin Cancer Biol. 2009;19:42–49. doi: 10.1016/j.semcancer.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murray GI, Duncan ME, O’Neil P, McKay JA, Melvin WT, Fothergill JE. Matrix metalloproteinase-1 is associated with poor prognosis in oesophageal cancer. J Pathol. 1998;185:256–261. doi: 10.1002/(SICI)1096-9896(199807)185:3<256::AID-PATH115>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 34.Etoh T, Inoue H, Yoshikawa Y, Barnard GF, Kitano S, Mori M. Increased expression of collagenase-3 (MMP-13) and MT1-MMP in oesophageal cancer is related to cancer aggressiveness. Gut. 2000;47:50–56. doi: 10.1136/gut.47.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mukherjee S, Roth MJ, Dawsey SM, Yan W, Rodriguez-Canales J, Erickson HS, Hu N, Goldstein AM, Taylor PR, Richardson AM, et al. Increased matrix metalloproteinase activation in esophageal squamous cell carcinoma. J Transl Med. 2010;8:91. doi: 10.1186/1479-5876-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elnemr A, Yonemura Y, Bandou E, Kinoshita K, Kawamura T, Takahashi S, Tochiori S, Endou Y, Sasaki T. Expression of collagenase-3 (matrix metalloproteinase-13) in human gastric cancer. Gastric Cancer. 2003;6:30–38. doi: 10.1007/s101200300004. [DOI] [PubMed] [Google Scholar]
- 37.Liu HQ, Song S, Wang JH, Zhang SL. Expression of MMP-3 and TIMP-3 in gastric cancer tissue and its clinical significance. Oncol Lett. 2011;2:1319–1322. doi: 10.3892/ol.2011.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liabakk N-B, Talbot I, Smith RA, Wilkinson K, Balkwill F. Matrix metalloprotease 2 (mmp-2) and matrix metalloprotease 9 (mmp-9) type iv collagenases in colorectal cancer. Cancer Res. 1996;56:190–196. [PubMed] [Google Scholar]
- 39.Shiozawa J, Ito M, Nakayama T, Nakashima M, Kohno S, Sekine I. Expression of matrix metalloproteinase-1 in human colorectal carcinoma. Mod Pathol. 2000;13:925–933. doi: 10.1038/modpathol.3880169. [DOI] [PubMed] [Google Scholar]
- 40.Parsons SL, Watson SA, Collins HM, Griffin NR, Clarke PA, Steele RJ. Gelatinase (MMP-2 and -9) expression in gastrointestinal malignancy. Br J Cancer. 1998;78:1495–1502. doi: 10.1038/bjc.1998.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Inuzuka K, Ogata Y, Nagase H, Shirouzu K. Significance of coexpression of urokinase-type plasminogen activator, and matrix metalloproteinase 3 (stromelysin) and 9 (gelatinase B) in colorectal carcinoma. J Surg Res. 2000;93:211–218. doi: 10.1006/jsre.2000.5952. [DOI] [PubMed] [Google Scholar]
- 42.Nielsen BS, Timshel S, Kjeldsen L, Sehested M, Pyke C, Borregaard N, Danø K. 92 kDa type IV collagenase (MMP-9) is expressed in neutrophils and macrophages but not in malignant epithelial cells in human colon cancer. Int J Cancer. 1996;65:57–62. doi: 10.1002/(SICI)1097-0215(19960103)65:1<57::AID-IJC10>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 43.Zucker S, Vacirca J. Role of matrix metalloproteinases (MMPs) in colorectal cancer. Cancer Metastasis Rev. 2004;23:101–117. doi: 10.1023/a:1025867130437. [DOI] [PubMed] [Google Scholar]
- 44.Adachi Y, Yamamoto H, Itoh F, Hinoda Y, Okada Y, Imai K. Contribution of matrilysin (MMP-7) to the metastatic pathway of human colorectal cancers. Gut. 1999;45:252–258. doi: 10.1136/gut.45.2.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dong Z, Kumar R, Yang X, Fidler IJ. Macrophage-derived metalloelastase is responsible for the generation of angiostatin in Lewis lung carcinoma. Cell. 1997;88:801–810. doi: 10.1016/s0092-8674(00)81926-1. [DOI] [PubMed] [Google Scholar]
- 46.O’Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, Lane WS, Cao Y, Sage EH, Folkman J. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79:315–328. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 47.Leeman MF, McKay JA, Murray GI. Matrix metalloproteinase 13 activity is associated with poor prognosis in colorectal cancer. J Clin Pathol. 2002;55:758–762. doi: 10.1136/jcp.55.10.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knäuper V, Will H, López-Otin C, Smith B, Atkinson SJ, Stanton H, Hembry RM, Murphy G. Cellular mechanisms for human procollagenase-3 (MMP-13) activation. Evidence that MT1-MMP (MMP-14) and gelatinase a (MMP-2) are able to generate active enzyme. J Biol Chem. 1996;271:17124–17131. doi: 10.1074/jbc.271.29.17124. [DOI] [PubMed] [Google Scholar]
- 49.Gururajan R, Lahti JM, Grenet J, Easton J, Gruber I, Ambros PF, Kidd VJ. Duplication of a genomic region containing the Cdc2L1-2 and MMP21-22 genes on human chromosome 1p36.3 and their linkage to D1Z2. Genome Res. 1998;8:929–939. doi: 10.1101/gr.8.9.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Llano E, Pendás AM, Freije JP, Nakano A, Knäuper V, Murphy G, López-Otin C. Identification and characterization of human MT5-MMP, a new membrane-bound activator of progelatinase a overexpressed in brain tumors. Cancer Res. 1999;59:2570–2576. [PubMed] [Google Scholar]
- 51.Bradbury PA, Zhai R, Hopkins J, Kulke MH, Heist RS, Singh S, Zhou W, Ma C, Xu W, Asomaning K, et al. Matrix metalloproteinase 1, 3 and 12 polymorphisms and esophageal adenocarcinoma risk and prognosis. Carcinogenesis. 2009;30:793–798. doi: 10.1093/carcin/bgp065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dey S, Ghosh N, Saha D, Kesh K, Gupta A, Swarnakar S. Matrix metalloproteinase-1 (MMP-1) Promoter polymorphisms are well linked with lower stomach tumor formation in eastern Indian population. PLoS One. 2014;9:e88040. doi: 10.1371/journal.pone.0088040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langers AM, Verspaget HW, Hommes DW, Sier CF. Single-nucleotide polymorphisms of matrix metalloproteinases and their inhibitors in gastrointestinal cancer. World J Gastrointest Oncol. 2011;3:79–98. doi: 10.4251/wjgo.v3.i6.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen XB, Chen GL, Liu JN, Yang JZ, Yu DK, Lin DX, Tan W. [Genetic polymorphisms in STK15 and MMP-2 associated susceptibility to esophageal cancer in Mongolian population] Zhonghua Yu Fang Yi Xue Za Zhi. 2009;43:559–564. [PubMed] [Google Scholar]
- 55.Harendza S, Lovett DH, Panzer U, Lukacs Z, Kuhnl P, Stahl RA. Linked common polymorphisms in the gelatinase a promoter are associated with diminished transcriptional response to estrogen and genetic fitness. J Biol Chem. 2003;278:20490–20499. doi: 10.1074/jbc.M211536200. [DOI] [PubMed] [Google Scholar]
- 56.Früh M, Zhou W, Zhai R, Su L, Heist RS, Wain JC, Nishioka NS, Lynch TJ, Shepherd FA, Christiani DC, et al. Polymorphisms of inflammatory and metalloproteinase genes, Helicobacter pylori infection and the risk of oesophageal adenocarcinoma. Br J Cancer. 2008;98:689–692. doi: 10.1038/sj.bjc.6604234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Y, Sun DL, Duan YN, Zhang XJ, Wang N, Zhou RM, Chen ZF, Wang SJ. Association of functional polymorphisms in MMPs genes with gastric cardia adenocarcinoma and esophageal squamous cell carcinoma in high incidence region of North China. Mol Biol Rep. 2010;37:197–205. doi: 10.1007/s11033-009-9593-4. [DOI] [PubMed] [Google Scholar]
- 58.Kubben FJ, Sier CF, Meijer MJ, van den Berg M, van der Reijden JJ, Griffioen G, van de Velde CJ, Lamers CB, Verspaget HW. Clinical impact of MMP and TIMP gene polymorphisms in gastric cancer. Br J Cancer. 2006;95:744–751. doi: 10.1038/sj.bjc.6603307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saeed HM, Alanazi MS, Parine NR, Shaik J, Semlali A, Alharbi O, Azzam N, Aljebreen A, Almadi M, Shalaby MA. Matrix metalloproteinase-2 (-1306 c& gt; t) promoter polymorphism and risk of colorectal cancer in the Saudi population. Asian Pac J Cancer Prev. 2013;14:6025–6030. doi: 10.7314/apjcp.2013.14.10.6025. [DOI] [PubMed] [Google Scholar]
- 60.Langers AM, Sier CF, Hawinkels LJ, Kubben FJ, van Duijn W, van der Reijden JJ, Lamers CB, Hommes DW, Verspaget HW. MMP-2 geno-phenotype is prognostic for colorectal cancer survival, whereas MMP-9 is not. Br J Cancer. 2008;98:1820–1823. doi: 10.1038/sj.bjc.6604380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woo M, Park K, Nam J, Kim JC. Clinical implications of matrix metalloproteinase-1, -3, -7, -9, -12, and plasminogen activator inhibitor-1 gene polymorphisms in colorectal cancer. J Gastroenterol Hepatol. 2007;22:1064–1070. doi: 10.1111/j.1440-1746.2006.04424.x. [DOI] [PubMed] [Google Scholar]
- 62.Tang Y, Zhu J, Chen L, Chen L, Zhang S, Lin J. Associations of matrix metalloproteinase-9 protein polymorphisms with lymph node metastasis but not invasion of gastric cancer. Clin Cancer Res. 2008;14:2870–2877. doi: 10.1158/1078-0432.CCR-07-4042. [DOI] [PubMed] [Google Scholar]
- 63.Fang WL, Liang WB, He H, Zhu Y, Li SL, Gao LB, Zhang L. Association of matrix metalloproteinases 1, 7, and 9 gene polymorphisms with genetic susceptibility to colorectal carcinoma in a Han Chinese population. DNA Cell Biol. 2010;29:657–661. doi: 10.1089/dna.2010.1017. [DOI] [PubMed] [Google Scholar]
- 64.Matsumura S, Oue N, Nakayama H, Kitadai Y, Yoshida K, Yamaguchi Y, Imai K, Nakachi K, Matsusaki K, Chayama K, et al. A single nucleotide polymorphism in the MMP-9 promoter affects tumor progression and invasive phenotype of gastric cancer. J Cancer Res Clin Oncol. 2005;131:19–25. doi: 10.1007/s00432-004-0621-4. [DOI] [PubMed] [Google Scholar]
- 65.Xing LL, Wang ZN, Jiang L, Zhang Y, Xu YY, Li J, Luo Y, Zhang X. Matrix metalloproteinase-9-1562C& gt; T polymorphism may increase the risk of lymphatic metastasis of colorectal cancer. World J Gastroenterol. 2007;13:4626–4629. doi: 10.3748/wjg.v13.i34.4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dey S, Stalin S, Gupta A, Saha D, Kesh K, Swarnakar S. Matrix metalloproteinase3 gene promoter polymorphisms and their haplotypes are associated with gastric cancer risk in eastern Indian population. Mol Carcinog. 2012;51 Suppl 1:E42–E53. doi: 10.1002/mc.21837. [DOI] [PubMed] [Google Scholar]
- 67.Ke P, Wu ZD, Wen HS, Ying MX, Long HC, Qing LG. Current evidence on associations between the MMP-7 (-181A& gt; G) polymorphism and digestive system cancer risk. Asian Pac J Cancer Prev. 2013;14:2269–2272. doi: 10.7314/apjcp.2013.14.4.2269. [DOI] [PubMed] [Google Scholar]
- 68.Jormsjö S, Whatling C, Walter DH, Zeiher AM, Hamsten A, Eriksson P. Allele-specific regulation of matrix metalloproteinase-7 promoter activity is associated with coronary artery luminal dimensions among hypercholesterolemic patients. Arterioscler Thromb Vasc Biol. 2001;21:1834–1839. doi: 10.1161/hq1101.098229. [DOI] [PubMed] [Google Scholar]
- 69.Richards TJ, Park C, Chen Y, Gibson KF, Peter Di Y, Pardo A, Watkins SC, Choi AM, Selman M, Pilewski J, et al. Allele-specific transactivation of matrix metalloproteinase 7 by FOXA2 and correlation with plasma levels in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;302:L746–L754. doi: 10.1152/ajplung.00319.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang J, Jin X, Fang S, Wang R, Li Y, Wang N, Guo W, Wang Y, Wen D, Wei L, et al. The functional polymorphism in the matrix metalloproteinase-7 promoter increases susceptibility to esophageal squamous cell carcinoma, gastric cardiac adenocarcinoma and non-small cell lung carcinoma. Carcinogenesis. 2005;26:1748–1753. doi: 10.1093/carcin/bgi144. [DOI] [PubMed] [Google Scholar]
- 71.Ghilardi G, Biondi ML, Erario M, Guagnellini E, Scorza R. Colorectal carcinoma susceptibility and metastases are associated with matrix metalloproteinase-7 promoter polymorphisms. Clin Chem. 2003;49:1940–1942. doi: 10.1373/clinchem.2003.018911. [DOI] [PubMed] [Google Scholar]
- 72.Cirillo G, Casalino L, Vallone D, Caracciolo A, De Cesare D, Verde P. Role of distinct mitogen-activated protein kinase pathways and cooperation between Ets-2, ATF-2, and Jun family members in human urokinase-type plasminogen activator gene induction by interleukin-1 and tetradecanoyl phorbol acetate. Mol Cell Biol. 1999;19:6240–6252. doi: 10.1128/mcb.19.9.6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Herren B, Levkau B, Raines EW, Ross R. Cleavage of beta-catenin and plakoglobin and shedding of VE-cadherin during endothelial apoptosis: evidence for a role for caspases and metalloproteinases. Mol Biol Cell. 1998;9:1589–1601. doi: 10.1091/mbc.9.6.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ilan N, Mohsenin A, Cheung L, Madri JA. PECAM-1 shedding during apoptosis generates a membrane-anchored truncated molecule with unique signaling characteristics. FASEB J. 2001;15:362–372. doi: 10.1096/fj.00-0372com. [DOI] [PubMed] [Google Scholar]
- 75.Steinhusen U, Weiske J, Badock V, Tauber R, Bommert K, Huber O. Cleavage and shedding of E-cadherin after induction of apoptosis. J Biol Chem. 2001;276:4972–4980. doi: 10.1074/jbc.M006102200. [DOI] [PubMed] [Google Scholar]
- 76.Sympson CJ, Talhouk RS, Alexander CM, Chin JR, Clift SM, Bissell MJ, Werb Z. Targeted expression of stromelysin-1 in mammary gland provides evidence for a role of proteinases in branching morphogenesis and the requirement for an intact basement membrane for tissue-specific gene expression. J Cell Biol. 1994;125:681–693. doi: 10.1083/jcb.125.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Powell WC, Fingleton B, Wilson CL, Boothby M, Matrisian LM. The metalloproteinase matrilysin proteolytically generates active soluble Fas ligand and potentiates epithelial cell apoptosis. Curr Biol. 1999;9:1441–1447. doi: 10.1016/s0960-9822(00)80113-x. [DOI] [PubMed] [Google Scholar]
- 78.Wu E, Mari BP, Wang F, Anderson IC, Sunday ME, Shipp MA. Stromelysin-3 suppresses tumor cell apoptosis in a murine model. J Cell Biochem. 2001;82:549–555. doi: 10.1002/jcb.1181. [DOI] [PubMed] [Google Scholar]
- 79.Baserga R. The contradictions of the insulin-like growth factor 1 receptor. Oncogene. 2000;19:5574–5581. doi: 10.1038/sj.onc.1203854. [DOI] [PubMed] [Google Scholar]
- 80.Swarnakar S, Paul S, Singh LP, Reiter RJ. Matrix metalloproteinases in health and disease: regulation by melatonin. J Pineal Res. 2011;50:8–20. doi: 10.1111/j.1600-079X.2010.00812.x. [DOI] [PubMed] [Google Scholar]
- 81.Seandel M, Noack-Kunnmann K, Zhu D, Aimes RT, Quigley JP. Growth factor-induced angiogenesis in vivo requires specific cleavage of fibrillar type I collagen. Blood. 2001;97:2323–2332. doi: 10.1182/blood.v97.8.2323. [DOI] [PubMed] [Google Scholar]
- 82.Kolb C, Mauch S, Peter HH, Krawinkel U, Sedlacek R. The matrix metalloproteinase RASI-1 is expressed in synovial blood vessels of a rheumatoid arthritis patient. Immunol Lett. 1997;57:83–88. doi: 10.1016/s0165-2478(97)00057-6. [DOI] [PubMed] [Google Scholar]
- 83.Itoh T, Tanioka M, Yoshida H, Yoshioka T, Nishimoto H, Itohara S. Reduced angiogenesis and tumor progression in gelatinase A-deficient mice. Cancer Res. 1998;58:1048–1051. [PubMed] [Google Scholar]
- 84.Xu J, Rodriguez D, Petitclerc E, Kim JJ, Hangai M, Moon YS, Davis GE, Brooks PC. Proteolytic exposure of a cryptic site within collagen type IV is required for angiogenesis and tumor growth in vivo. J Cell Biol. 2001;154:1069–1079. doi: 10.1083/jcb.200103111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell. 2000;103:481–490. doi: 10.1016/s0092-8674(00)00139-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhou Z, Apte SS, Soininen R, Cao R, Baaklini GY, Rauser RW, Wang J, Cao Y, Tryggvason K. Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc Natl Acad Sci USA. 2000;97:4052–4057. doi: 10.1073/pnas.060037197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cornelius LA, Nehring LC, Harding E, Bolanowski M, Welgus HG, Kobayashi DK, Pierce RA, Shapiro SD. Matrix metalloproteinases generate angiostatin: effects on neovascularization. J Immunol. 1998;161:6845–6852. [PubMed] [Google Scholar]
- 88.Gorrin-Rivas MJ, Arii S, Furutani M, Mizumoto M, Mori A, Hanaki K, Maeda M, Furuyama H, Kondo Y, Imamura M. Mouse macrophage metalloelastase gene transfer into a murine melanoma suppresses primary tumor growth by halting angiogenesis. Clin Cancer Res. 2000;6:1647–1654. [PubMed] [Google Scholar]
- 89.Kim YM, Jang JW, Lee OH, Yeon J, Choi EY, Kim KW, Lee ST, Kwon YG. Endostatin inhibits endothelial and tumor cellular invasion by blocking the activation and catalytic activity of matrix metalloproteinase. Cancer Res. 2000;60:5410–5413. [PubMed] [Google Scholar]
- 90.Giannelli G, Falk-Marzillier J, Schiraldi O, Stetler-Stevenson WG, Quaranta V. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science. 1997;277:225–228. doi: 10.1126/science.277.5323.225. [DOI] [PubMed] [Google Scholar]
- 91.Koshikawa N, Giannelli G, Cirulli V, Miyazaki K, Quaranta V. Role of cell surface metalloprotease MT1-MMP in epithelial cell migration over laminin-5. J Cell Biol. 2000;148:615–624. doi: 10.1083/jcb.148.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kajita M, Itoh Y, Chiba T, Mori H, Okada A, Kinoh H, Seiki M. Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J Cell Biol. 2001;153:893–904. doi: 10.1083/jcb.153.5.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yu Q, Stamenkovic I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1999;13:35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Beliën AT, Paganetti PA, Schwab ME. Membrane-type 1 matrix metalloprotease (MT1-MMP) enables invasive migration of glioma cells in central nervous system white matter. J Cell Biol. 1999;144:373–384. doi: 10.1083/jcb.144.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Deryugina EI, Luo GX, Reisfeld RA, Bourdon MA, Strongin A. Tumor cell invasion through matrigel is regulated by activated matrix metalloproteinase-2. Anticancer Res. 1997;17:3201–3210. [PubMed] [Google Scholar]
- 96.Ala-Aho R, Johansson N, Baker AH, Kähäri VM. Expression of collagenase-3 (MMP-13) enhances invasion of human fibrosarcoma HT-1080 cells. Int J Cancer. 2002;97:283–289. doi: 10.1002/ijc.1619. [DOI] [PubMed] [Google Scholar]
- 97.Noë V, Fingleton B, Jacobs K, Crawford HC, Vermeulen S, Steelant W, Bruyneel E, Matrisian LM, Mareel M. Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. J Cell Sci. 2001;114:111–118. doi: 10.1242/jcs.114.1.111. [DOI] [PubMed] [Google Scholar]
- 98.Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, Gray JW, Pinkel D, Bissell MJ, Werb Z. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 1999;98:137–146. doi: 10.1016/s0092-8674(00)81009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lochter A, Galosy S, Muschler J, Freedman N, Werb Z, Bissell MJ. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J Cell Biol. 1997;139:1861–1872. doi: 10.1083/jcb.139.7.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brooks PC, Strömblad S, Sanders LC, von Schalscha TL, Aimes RT, Stetler-Stevenson WG, Quigley JP, Cheresh DA. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell. 1996;85:683–693. doi: 10.1016/s0092-8674(00)81235-0. [DOI] [PubMed] [Google Scholar]
- 101.Coussens LM, Werb Z. Inflammatory cells and cancer: think different! J Exp Med. 2001;193:F23–F26. doi: 10.1084/jem.193.6.f23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sheu BC, Hsu SM, Ho HN, Lien HC, Huang SC, Lin RH. A novel role of metalloproteinase in cancer-mediated immunosuppression. Cancer Res. 2001;61:237–242. [PubMed] [Google Scholar]
- 103.McQuibban GA, Gong JH, Wong JP, Wallace JL, Clark-Lewis I, Overall CM. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood. 2002;100:1160–1167. [PubMed] [Google Scholar]
- 104.McQuibban GA, Gong JH, Tam EM, McCulloch CA, Clark-Lewis I, Overall CM. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science. 2000;289:1202–1206. doi: 10.1126/science.289.5482.1202. [DOI] [PubMed] [Google Scholar]
- 105.Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 106.Kataoka H, Uchino H, Iwamura T, Seiki M, Nabeshima K, Koono M. Enhanced tumor growth and invasiveness in vivo by a carboxyl-terminal fragment of alpha1-proteinase inhibitor generated by matrix metalloproteinases: a possible modulatory role in natural killer cytotoxicity. Am J Pathol. 1999;154:457–468. doi: 10.1016/s0002-9440(10)65292-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 108.Mañes S, Mira E, Barbacid MM, Ciprés A, Fernández-Resa P, Buesa JM, Mérida I, Aracil M, Márquez G, Martínez-A C. Identification of insulin-like growth factor-binding protein-1 as a potential physiological substrate for human stromelysin-3. J Biol Chem. 1997;272:25706–25712. doi: 10.1074/jbc.272.41.25706. [DOI] [PubMed] [Google Scholar]
- 109.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria in cancer cells: what is so special about them? Trends Cell Biol. 2008;18:165–173. doi: 10.1016/j.tcb.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 111.Meitzler JL, Antony S, Wu Y, Juhasz A, Liu H, Jiang G, Lu J, Roy K, Doroshow JH. NADPH oxidases: a perspective on reactive oxygen species production in tumor biology. Antioxid Redox Signal. 2014;20:2873–2889. doi: 10.1089/ars.2013.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ibsen KH. The Crabtree effect: a review. Cancer Res. 1961;21:829–841. [PubMed] [Google Scholar]
- 113.Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:C125–C136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- 114.Ding SZ, Minohara Y, Fan XJ, Wang J, Reyes VE, Patel J, Dirden-Kramer B, Boldogh I, Ernst PB, Crowe SE. Helicobacter pylori infection induces oxidative stress and programmed cell death in human gastric epithelial cells. Infect Immun. 2007;75:4030–4039. doi: 10.1128/IAI.00172-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ganguly K, Swarnakar S. Chronic gastric ulceration causes matrix metalloproteinases-9 and -3 augmentation: alleviation by melatonin. Biochimie. 2012;94:2687–2698. doi: 10.1016/j.biochi.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 116.Banerjee A, Mukhopadhyay AK, Paul S, Bhattacharyya A, Swarnakar S. Unveiling the intricacies of helicobacter pylori-induced gastric inflammation: T helper cells and matrix metalloproteinases at a crossroad. Current topics in Gastritis-2012 Mozsik G, editor. InTech. 2013 [Google Scholar]
- 117.Hagen TM, Huang S, Curnutte J, Fowler P, Martinez V, Wehr CM, Ames BN, Chisari FV. Extensive oxidative DNA damage in hepatocytes of transgenic mice with chronic active hepatitis destined to develop hepatocellular carcinoma. Proc Natl Acad Sci USA. 1994;91:12808–12812. doi: 10.1073/pnas.91.26.12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Reddy EP, Reddy VS, Chandra Mouli K, Rao P. Physiological antioxidant system and oxidative stress in stomach cancer patients with normal renal and hepatic function. Online J Health Allied Sci. 2010:8. [Google Scholar]
- 119.Czygier M, Kamocki Z, Ławicki S, Szmitkowski M. [The plasma level of myeloperoxidase (MPO) and total antioxidant status (TAS) in gastric cancer patients after surgery] Przegl Lek. 2010;67:443–445. [PubMed] [Google Scholar]
- 120.Noyan T, Guducuoglu H, Ilhan M. A study of oxidative stress parameters in anti-helicobacter pylorus immunoglobulin g positive and negative gastric cancer patients. Yonsei Med J. 2009;50:677–682. doi: 10.3349/ymj.2009.50.5.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Huang J, Lam GY, Brumell JH. Autophagy signaling through reactive oxygen species. Antioxid Redox Signal. 2011;14:2215–2231. doi: 10.1089/ars.2010.3554. [DOI] [PubMed] [Google Scholar]
- 122.Gibson SB. A matter of balance between life and death: targeting reactive oxygen species (ROS)-induced autophagy for cancer therapy. Autophagy. 2010;6:835–837. doi: 10.4161/auto.6.7.13335. [DOI] [PubMed] [Google Scholar]
- 123.Kim MJ, Woo SJ, Yoon CH, Lee JS, An S, Choi YH, Hwang SG, Yoon G, Lee SJ. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J Biol Chem. 2011;286:12924–12932. doi: 10.1074/jbc.M110.138958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Macip S, Igarashi M, Fang L, Chen A, Pan ZQ, Lee SW, Aaronson SA. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002;21:2180–2188. doi: 10.1093/emboj/21.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yuan X, Zhou Y, Wang W, Li J, Xie G, Zhao Y, Xu D, Shen L. Activation of TLR4 signaling promotes gastric cancer progression by inducing mitochondrial ROS production. Cell Death Dis. 2013;4:e794. doi: 10.1038/cddis.2013.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu L, Sun L, Zhao P, Yao L, Jin H, Liang S, Wang Y, Zhang D, Pang Y, Shi Y, et al. Hypoxia promotes metastasis in human gastric cancer by up-regulating the 67-kDa laminin receptor. Cancer Sci. 2010;101:1653–1660. doi: 10.1111/j.1349-7006.2010.01592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, Wahl GM. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9:1031–1044. doi: 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- 128.Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tominaga K, Kawahara T, Sano T, Toida K, Kuwano Y, Sasaki H, Kawai T, Teshima-Kondo S, Rokutan K. Evidence for cancer-associated expression of NADPH oxidase 1 (Nox1)-based oxidase system in the human stomach. Free Radic Biol Med. 2007;43:1627–1638. doi: 10.1016/j.freeradbiomed.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 130.Arbiser JL, Petros J, Klafter R, Govindajaran B, McLaughlin ER, Brown LF, Cohen C, Moses M, Kilroy S, Arnold RS, et al. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc Natl Acad Sci USA. 2002;99:715–720. doi: 10.1073/pnas.022630199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Block K, Gorin Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat Rev Cancer. 2012;12:627–637. doi: 10.1038/nrc3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chen YR, Shrivastava A, Tan TH. Down-regulation of the c-Jun N-terminal kinase (JNK) phosphatase M3/6 and activation of JNK by hydrogen peroxide and pyrrolidine dithiocarbamate. Oncogene. 2001;20:367–374. doi: 10.1038/sj.onc.1204105. [DOI] [PubMed] [Google Scholar]
- 133.Chen N, Nomura M, She QB, Ma WY, Bode AM, Wang L, Flavell RA, Dong Z. Suppression of skin tumorigenesis in c-Jun NH(2)-terminal kinase-2-deficient mice. Cancer Res. 2001;61:3908–3912. [PubMed] [Google Scholar]
- 134.Behrens A, Jochum W, Sibilia M, Wagner EF. Oncogenic transformation by ras and fos is mediated by c-Jun N-terminal phosphorylation. Oncogene. 2000;19:2657–2663. doi: 10.1038/sj.onc.1203603. [DOI] [PubMed] [Google Scholar]
- 135.Benhar M, Dalyot I, Engelberg D, Levitzki A. Enhanced ROS production in oncogenically transformed cells potentiates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase activation and sensitization to genotoxic stress. Mol Cell Biol. 2001;21:6913–6926. doi: 10.1128/MCB.21.20.6913-6926.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 137.Van Remmen H, Ikeno Y, Hamilton M, Pahlavani M, Wolf N, Thorpe SR, Alderson NL, Baynes JW, Epstein CJ, Huang TT, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics. 2003;16:29–37. doi: 10.1152/physiolgenomics.00122.2003. [DOI] [PubMed] [Google Scholar]
- 138.Elchuri S, Oberley TD, Qi W, Eisenstein RS, Jackson Roberts L, Van Remmen H, Epstein CJ, Huang T-T. Cuznsod deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2004;24:367–380. doi: 10.1038/sj.onc.1208207. [DOI] [PubMed] [Google Scholar]
- 139.Ishii K, Zhen LX, Wang DH, Funamori Y, Ogawa K, Taketa K. Prevention of mammary tumorigenesis in acatalasemic mice by vitamin E supplementation. Jpn J Cancer Res. 1996;87:680–684. doi: 10.1111/j.1349-7006.1996.tb00277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chu FF, Esworthy RS, Chu PG, Longmate JA, Huycke MM, Wilczynski S, Doroshow JH. Bacteria-induced intestinal cancer in mice with disrupted Gpx1 and Gpx2 genes. Cancer Res. 2004;64:962–968. doi: 10.1158/0008-5472.can-03-2272. [DOI] [PubMed] [Google Scholar]
- 141.Kanbagli O, Ozdemirler G, Bulut T, Yamaner S, Aykaç-Toker G, Uysal M. Mitochondrial lipid peroxides and antioxidant enzymes in colorectal adenocarcinoma tissues. Jpn J Cancer Res. 2000;91:1258–1263. doi: 10.1111/j.1349-7006.2000.tb00912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Janssen AM, Bosman CB, van Duijn W, Oostendorp-van de Ruit MM, Kubben FJ, Griffioen G, Lamers CB, van Krieken JH, van de Velde CJ, Verspaget HW. Superoxide dismutases in gastric and esophageal cancer and the prognostic impact in gastric cancer. Clin Cancer Res. 2000;6:3183–3192. [PubMed] [Google Scholar]
- 143.Scibior D, Skrzycki M, Podsiad M, Czeczot H. Glutathione level and glutathione-dependent enzyme activities in blood serum of patients with gastrointestinal tract tumors. Clin Biochem. 2008;41:852–858. doi: 10.1016/j.clinbiochem.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 144.Oberley LW, Bize IB, Sahu SK, Leuthauser SW, Gruber HE. Superoxide dismutase activity of normal murine liver, regenerating liver, and H6 hepatoma. J Natl Cancer Inst. 1978;61:375–379. [PubMed] [Google Scholar]
- 145.Hernandez-Saavedra D, McCord JM. Paradoxical effects of thiol reagents on Jurkat cells and a new thiol-sensitive mutant form of human mitochondrial superoxide dismutase. Cancer Res. 2003;63:159–163. [PubMed] [Google Scholar]
- 146.Hwang TS, Choi HK, Han HS. Differential expression of manganese superoxide dismutase, copper/zinc superoxide dismutase, and catalase in gastric adenocarcinoma and normal gastric mucosa. Eur J Surg Oncol. 2007;33:474–479. doi: 10.1016/j.ejso.2006.10.024. [DOI] [PubMed] [Google Scholar]
- 147.Yang CS, Landau JM. Effects of tea consumption on nutrition and health. J Nutr. 2000;130:2409–2412. doi: 10.1093/jn/130.10.2409. [DOI] [PubMed] [Google Scholar]
- 148.Morse MA, Kresty LA, Steele VE, Kelloff GJ, Boone CW, Balentine DA, Harbowy ME, Stoner GD. Effects of theaflavins on N-nitrosomethylbenzylamine-induced esophageal tumorigenesis. Nutr Cancer. 1997;29:7–12. doi: 10.1080/01635589709514595. [DOI] [PubMed] [Google Scholar]
- 149.Yang G, Zheng W, Xiang YB, Gao J, Li HL, Zhang X, Gao YT, Shu XO. Green tea consumption and colorectal cancer risk: a report from the Shanghai Men’s Health Study. Carcinogenesis. 2011;32:1684–1688. doi: 10.1093/carcin/bgr186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Caderni G, De Filippo C, Luceri C, Salvadori M, Giannini A, Biggeri A, Remy S, Cheynier V, Dolara P. Effects of black tea, green tea and wine extracts on intestinal carcinogenesis induced by azoxymethane in F344 rats. Carcinogenesis. 2000;21:1965–1969. doi: 10.1093/carcin/21.11.1965. [DOI] [PubMed] [Google Scholar]
- 151.Peng G, Dixon DA, Muga SJ, Smith TJ, Wargovich MJ. Green tea polyphenol (-)-epigallocatechin-3-gallate inhibits cyclooxygenase-2 expression in colon carcinogenesis. Mol Carcinog. 2006;45:309–319. doi: 10.1002/mc.20166. [DOI] [PubMed] [Google Scholar]
- 152.Kim HS, Kim MH, Jeong M, Hwang YS, Lim SH, Shin BA, Ahn BW, Jung YD. EGCG blocks tumor promoter-induced MMP-9 expression via suppression of MAPK and AP-1 activation in human gastric AGS cells. Anticancer Res. 2004;24:747–753. [PubMed] [Google Scholar]
- 153.Annabi B, Lachambre MP, Bousquet-Gagnon N, Page M, Gingras D, Beliveau R. Green tea polyphenol (-)-epigallocatechin 3-gallate inhibits MMP-2 secretion and MT1-MMP-driven migration in glioblastoma cells. Biochim Biophys Acta. 2002;1542:209–220. doi: 10.1016/s0167-4889(01)00187-2. [DOI] [PubMed] [Google Scholar]
- 154.Onoda C, Kuribayashi K, Nirasawa S, Tsuji N, Tanaka M, Kobayashi D, Watanabe N. (-)-Epigallocatechin-3-gallate induces apoptosis in gastric cancer cell lines by down-regulating survivin expression. Int J Oncol. 2011;38:1403–1408. doi: 10.3892/ijo.2011.951. [DOI] [PubMed] [Google Scholar]
- 155.Larsen CA, Dashwood RH. (-)-Epigallocatechin-3-gallate inhibits Met signaling, proliferation, and invasiveness in human colon cancer cells. Arch Biochem Biophys. 2010;501:52–57. doi: 10.1016/j.abb.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cheng XW, Kuzuya M, Nakamura K, Liu Z, Di Q, Hasegawa J, Iwata M, Murohara T, Yokota M, Iguchi A. Mechanisms of the inhibitory effect of epigallocatechin-3-gallate on cultured human vascular smooth muscle cell invasion. Arterioscler Thromb Vasc Biol. 2005;25:1864–1870. doi: 10.1161/01.ATV.0000179675.49619.9b. [DOI] [PubMed] [Google Scholar]
- 157.El Bedoui J, Oak MH, Anglard P, Schini-Kerth VB. Catechins prevent vascular smooth muscle cell invasion by inhibiting MT1-MMP activity and MMP-2 expression. Cardiovasc Res. 2005;67:317–325. doi: 10.1016/j.cardiores.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 158.Patel R, Ingle A, Maru GB. Polymeric black tea polyphenols inhibit 1,2-dimethylhydrazine induced colorectal carcinogenesis by inhibiting cell proliferation via Wnt/beta-catenin pathway. Toxicol Appl Pharmacol. 2008;227:136–146. doi: 10.1016/j.taap.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 159.Hwang JT, Ha J, Park IJ, Lee SK, Baik HW, Kim YM, Park OJ. Apoptotic effect of EGCG in HT-29 colon cancer cells via AMPK signal pathway. Cancer Lett. 2007;247:115–121. doi: 10.1016/j.canlet.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 160.Shankar S, Ganapathy S, Srivastava RK. Green tea polyphenols: biology and therapeutic implications in cancer. Front Biosci. 2007;12:4881–4899. doi: 10.2741/2435. [DOI] [PubMed] [Google Scholar]
- 161.Ammon HP, Wahl MA. Pharmacology of Curcuma longa. Planta Med. 1991;57:1–7. doi: 10.1055/s-2006-960004. [DOI] [PubMed] [Google Scholar]
- 162.Barzegar A, Moosavi-Movahedi AA. Intracellular ROS protection efficiency and free radical-scavenging activity of curcumin. PLoS One. 2011;6:e26012. doi: 10.1371/journal.pone.0026012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Swarnakar S, Ganguly K, Kundu P, Banerjee A, Maity P, Sharma AV. Curcumin regulates expression and activity of matrix metalloproteinases 9 and 2 during prevention and healing of indomethacin-induced gastric ulcer. J Biol Chem. 2005;280:9409–9415. doi: 10.1074/jbc.M413398200. [DOI] [PubMed] [Google Scholar]
- 164.Jana S, Paul S, Swarnakar S. Curcumin as anti-endometriotic agent: implication of MMP-3 and intrinsic apoptotic pathway. Biochem Pharmacol. 2012;83:797–804. doi: 10.1016/j.bcp.2011.12.030. [DOI] [PubMed] [Google Scholar]
- 165.Rajasekaran SA. Therapeutic potential of curcumin in gastrointestinal diseases. World J Gastrointest Pathophysiol. 2011;2:1–14. doi: 10.4291/wjgp.v2.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Aggarwal BB, Kumar A, Bharti AC. Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res. 2003;23:363–398. [PubMed] [Google Scholar]
- 167.Nagpal M, Sood S. Role of curcumin in systemic and oral health: An overview. J Nat Sci Biol Med. 2013;4:3–7. doi: 10.4103/0976-9668.107253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Koo JY, Kim HJ, Jung KO, Park KY. Curcumin inhibits the growth of AGS human gastric carcinoma cells in vitro and shows synergism with 5-fluorouracil. J Med Food. 2004;7:117–121. doi: 10.1089/1096620041224229. [DOI] [PubMed] [Google Scholar]
- 169.Tang XQ, Bi H, Feng JQ, Cao JG. Effect of curcumin on multidrug resistance in resistant human gastric carcinoma cell line SGC7901/VCR. Acta Pharmacol Sin. 2005;26:1009–1016. doi: 10.1111/j.1745-7254.2005.00149.x. [DOI] [PubMed] [Google Scholar]
- 170.Anand P, Sundaram C, Jhurani S, Kunnumakkara AB, Aggarwal BB. Curcumin and cancer: an “old-age” disease with an “age-old” solution. Cancer Lett. 2008;267:133–164. doi: 10.1016/j.canlet.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 171.Perkins S, Verschoyle RD, Hill K, Parveen I, Threadgill MD, Sharma RA, Williams ML, Steward WP, Gescher AJ. Chemopreventive efficacy and pharmacokinetics of curcumin in the min/+ mouse, a model of familial adenomatous polyposis. Cancer Epidemiol Biomarkers Prev. 2002;11:535–540. [PubMed] [Google Scholar]
- 172.Kim JM, Araki S, Kim DJ, Park CB, Takasuka N, Baba-Toriyama H, Ota T, Nir Z, Khachik F, Shimidzu N, et al. Chemopreventive effects of carotenoids and curcumins on mouse colon carcinogenesis after 1,2-dimethylhydrazine initiation. Carcinogenesis. 1998;19:81–85. doi: 10.1093/carcin/19.1.81. [DOI] [PubMed] [Google Scholar]
- 173.Shpitz B, Giladi N, Sagiv E, Lev-Ari S, Liberman E, Kazanov D, Arber N. Celecoxib and curcumin additively inhibit the growth of colorectal cancer in a rat model. Digestion. 2006;74:140–144. doi: 10.1159/000098655. [DOI] [PubMed] [Google Scholar]
- 174.Huang MT, Lou YR, Ma W, Newmark HL, Reuhl KR, Conney AH. Inhibitory effects of dietary curcumin on forestomach, duodenal, and colon carcinogenesis in mice. Cancer Res. 1994;54:5841–5847. [PubMed] [Google Scholar]
- 175.Bimonte S, Barbieri A, Palma G, Luciano A, Rea D, Arra C. Curcumin inhibits tumor growth and angiogenesis in an orthotopic mouse model of human pancreatic cancer. Biomed Res Int. 2013;2013:810423. doi: 10.1155/2013/810423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kunnumakkara AB, Diagaradjane P, Guha S, Deorukhkar A, Shentu S, Aggarwal BB, Krishnan S. Curcumin sensitizes human colorectal cancer xenografts in nude mice to gamma-radiation by targeting nuclear factor-kappaB-regulated gene products. Clin Cancer Res. 2008;14:2128–2136. doi: 10.1158/1078-0432.CCR-07-4722. [DOI] [PubMed] [Google Scholar]
- 177.Lin LI, Ke YF, Ko YC, Lin JK. Curcumin inhibits SK-Hep-1 hepatocellular carcinoma cell invasion in vitro and suppresses matrix metalloproteinase-9 secretion. Oncology. 1998;55:349–353. doi: 10.1159/000011876. [DOI] [PubMed] [Google Scholar]
- 178.Su CC, Chen GW, Lin JG, Wu LT, Chung JG. Curcumin inhibits cell migration of human colon cancer colo 205 cells through the inhibition of nuclear factor kappa B /p65 and down-regulates cyclooxygenase-2 and matrix metalloproteinase-2 expressions. Anticancer Res. 2006;26:1281–1288. [PubMed] [Google Scholar]
- 179.Lin ML, Lu YC, Chen HY, Lee CC, Chung JG, Chen SS. Suppressing the formation of lipid raft-associated Rac1/PI3K/Akt signaling complexes by curcumin inhibits SDF-1α-induced invasion of human esophageal carcinoma cells. Mol Carcinog. 2014;53:360–379. doi: 10.1002/mc.21984. [DOI] [PubMed] [Google Scholar]
- 180.Goswami SK, Das DK. Resveratrol and chemoprevention. Cancer Lett. 2009;284:1–6. doi: 10.1016/j.canlet.2009.01.041. [DOI] [PubMed] [Google Scholar]
- 181.Li ZG, Hong T, Shimada Y, Komoto I, Kawabe A, Ding Y, Kaganoi J, Hashimoto Y, Imamura M. Suppression of N-nitrosomethylbenzylamine (NMBA)-induced esophageal tumorigenesis in F344 rats by resveratrol. Carcinogenesis. 2002;23:1531–1536. doi: 10.1093/carcin/23.9.1531. [DOI] [PubMed] [Google Scholar]
- 182.Schneider Y, Duranton B, Gossé F, Schleiffer R, Seiler N, Raul F. Resveratrol inhibits intestinal tumorigenesis and modulates host-defense-related gene expression in an animal model of human familial adenomatous polyposis. Nutr Cancer. 2001;39:102–107. doi: 10.1207/S15327914nc391_14. [DOI] [PubMed] [Google Scholar]
- 183.Li W, Ma J, Ma Q, Li B, Han L, Liu J, Xu Q, Duan W, Yu S, Wang F, et al. Resveratrol inhibits the epithelial-mesenchymal transition of pancreatic cancer cells via suppression of the PI-3K/Akt/NF-κB pathway. Curr Med Chem. 2013;20:4185–4194. doi: 10.2174/09298673113209990251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ji Q, Liu X, Fu X, Zhang L, Sui H, Zhou L, Sun J, Cai J, Qin J, Ren J, et al. Resveratrol inhibits invasion and metastasis of colorectal cancer cells via MALAT1 mediated Wnt/β-catenin signal pathway. PLoS One. 2013;8:e78700. doi: 10.1371/journal.pone.0078700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Weng CJ, Wu CF, Huang HW, Wu CH, Ho CT, Yen GC. Evaluation of anti-invasion effect of resveratrol and related methoxy analogues on human hepatocarcinoma cells. J Agric Food Chem. 2010;58:2886–2894. doi: 10.1021/jf904182y. [DOI] [PubMed] [Google Scholar]
- 186.Harikumar KB, Kunnumakkara AB, Sethi G, Diagaradjane P, Anand P, Pandey MK, Gelovani J, Krishnan S, Guha S, Aggarwal BB. Resveratrol, a multitargeted agent, can enhance antitumor activity of gemcitabine in vitro and in orthotopic mouse model of human pancreatic cancer. Int J Cancer. 2010;127:257–268. doi: 10.1002/ijc.25041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Gibellini L, Pinti M, Nasi M, Montagna JP, De Biasi S, Roat E, Bertoncelli L, Cooper EL, Cossarizza A. Quercetin and cancer chemoprevention. Evid Based Complement Alternat Med. 2011;2011:591356. doi: 10.1093/ecam/neq053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Volate SR, Davenport DM, Muga SJ, Wargovich MJ. Modulation of aberrant crypt foci and apoptosis by dietary herbal supplements (quercetin, curcumin, silymarin, ginseng and rutin) Carcinogenesis. 2005;26:1450–1456. doi: 10.1093/carcin/bgi089. [DOI] [PubMed] [Google Scholar]
- 189.Dihal AA, de Boer VC, van der Woude H, Tilburgs C, Bruijntjes JP, Alink GM, Rietjens IM, Woutersen RA, Stierum RH. Quercetin, but not its glycosidated conjugate rutin, inhibits azoxymethane-induced colorectal carcinogenesis in F344 rats. J Nutr. 2006;136:2862–2867. doi: 10.1093/jn/136.11.2862. [DOI] [PubMed] [Google Scholar]
- 190.Lai WW, Hsu SC, Chueh FS, Chen YY, Yang JS, Lin JP, Lien JC, Tsai CH, Chung JG. Quercetin inhibits migration and invasion of SAS human oral cancer cells through inhibition of NF-κB and matrix metalloproteinase-2/-9 signaling pathways. Anticancer Res. 2013;33:1941–1950. [PubMed] [Google Scholar]
- 191.Reiter RJ, Tan DX, Manchester LC, Simopoulos AP, Maldonado MD, Flores LJ, Terron MP. Melatonin in edible plants (phytomelatonin): Identification, concentrations, bioavailability and proposed functions. World Rev Nutr Diet. 2007;97:211–230. doi: 10.1159/000097917. [DOI] [PubMed] [Google Scholar]
- 192.Rudra DS, Pal U, Maiti NC, Reiter RJ, Swarnakar S. Melatonin inhibits matrix metalloproteinase-9 activity by binding to its active site. J Pineal Res. 2013;54:398–405. doi: 10.1111/jpi.12034. [DOI] [PubMed] [Google Scholar]
- 193.Xu L, Liu H, Zhang H, Wang RX, Song J, Zhou RX. Growth-inhibitory activity of melatonin on murine foregastric carcinoma cells in vitro and the underlying molecular mechanism. Anat Rec (Hoboken) 2013;296:914–920. doi: 10.1002/ar.22689. [DOI] [PubMed] [Google Scholar]
- 194.Nemeth C, Humpeler S, Kallay E, Mesteri I, Svoboda M, Rögelsperger O, Klammer N, Thalhammer T, Ekmekcioglu C. Decreased expression of the melatonin receptor 1 in human colorectal adenocarcinomas. J Biol Regul Homeost Agents. 2011;25:531–542. [PubMed] [Google Scholar]
- 195.Hong Y, Won J, Lee Y, Lee S, Park K, Chang KT, Hong Y. Melatonin treatment induces interplay of apoptosis, autophagy, and senescence in human colorectal cancer cells. J Pineal Res. 2014;56:264–274. doi: 10.1111/jpi.12119. [DOI] [PubMed] [Google Scholar]
- 196.Ordoñez R, Carbajo-Pescador S, Prieto-Dominguez N, García-Palomo A, González-Gallego J, Mauriz JL. Inhibition of matrix metalloproteinase-9 and nuclear factor kappa B contribute to melatonin prevention of motility and invasiveness in HepG2 liver cancer cells. J Pineal Res. 2014;56:20–30. doi: 10.1111/jpi.12092. [DOI] [PubMed] [Google Scholar]
- 197.Tang FY, Pai MH, Kuo YH, Wang XD. Concomitant consumption of lycopene and fish oil inhibits tumor growth and progression in a mouse xenograft model of colon cancer. Mol Nutr Food Res. 2012;56:1520–1531. doi: 10.1002/mnfr.201200098. [DOI] [PubMed] [Google Scholar]
- 198.Lin MC, Wang FY, Kuo YH, Tang FY. Cancer chemopreventive effects of lycopene: suppression of MMP-7 expression and cell invasion in human colon cancer cells. J Agric Food Chem. 2011;59:11304–11318. doi: 10.1021/jf202433f. [DOI] [PubMed] [Google Scholar]
- 199.Adachi Y, Itoh F, Yamamoto H, Iku S, Matsuno K, Arimura Y, Imai K. Retinoic acids reduce matrilysin (matrix metalloproteinase 7) and inhibit tumor cell invasion in human colon cancer. Tumour Biol. 2001;22:247–253. doi: 10.1159/000050623. [DOI] [PubMed] [Google Scholar]
- 200.Park EY, Wilder ET, Lane MA. Retinol inhibits the invasion of retinoic acid-resistant colon cancer cells in vitro and decreases matrix metalloproteinase mRNA, protein, and activity levels. Nutr Cancer. 2007;57:66–77. doi: 10.1080/01635580701268238. [DOI] [PubMed] [Google Scholar]
- 201.Tanaka T, Shnimizu M, Moriwaki H. Cancer chemoprevention by carotenoids. Molecules. 2012;17:3202–3242. doi: 10.3390/molecules17033202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Liu HK, Wang Q, Li Y, Sun WG, Liu JR, Yang YM, Xu WL, Sun XR, Chen BQ. Inhibitory effects of gamma-tocotrienol on invasion and metastasis of human gastric adenocarcinoma SGC-7901 cells. J Nutr Biochem. 2010;21:206–213. doi: 10.1016/j.jnutbio.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 203.Roomi MW, Ivanov V, Kalinovsky T, Niedzwiecki A, Rath M. In vivo antitumor effect of ascorbic acid, lysine, proline and green tea extract on human colon cancer cell HCT 116 xenografts in nude mice: evaluation of tumor growth and immunohistochemistry. Oncol Rep. 2005;13:421–425. [PubMed] [Google Scholar]
- 204.Manu KA, Shanmugam MK, Ramachandran L, Li F, Fong CW, Kumar AP, Tan P, Sethi G. First evidence that γ-tocotrienol inhibits the growth of human gastric cancer and chemosensitizes it to capecitabine in a xenograft mouse model through the modulation of NF-κB pathway. Clin Cancer Res. 2012;18:2220–2229. doi: 10.1158/1078-0432.CCR-11-2470. [DOI] [PubMed] [Google Scholar]