

Abstract
Neuronal processes (neurites and axons) have an important role in brain cells communication and, generally, they are damaged in neurodegenerative diseases. Recent evidence has showed that the activation of PPARγ pathway promoted neuronal differentiation and axon polarity. In addition, activation of PPARγ using thiazolidinediones (TZDs) prevented neurodegeneration by reducing neuronal death, improving mitochondrial function, and decreasing neuroinflammation in neuropathic pain. In this review, we will discuss important evidence that supports a possible role of PPARγ in neuronal development, improvement of neuronal health, and pain signaling. Therefore, activation of PPARγ is a potential target with therapeutic applications against neurodegenerative disorders, brain injury, and pain regulation.
1. Introduction
1.1. Peroxisome Proliferator Activated Receptors
Peroxisome proliferator activated receptors (PPARs) are nuclear receptors that induce signaling and transcription of different pathways [1]. Generally, they participate in the regulation of lipids metabolism and glucose homeostasis, and they also are activated by specific ligands [1–3]. The family of PPARs is mostly composed of three known isoforms: PPARα, PPARβ/δ, and PPARγ. These receptors share a structural homology that consists of four functional units (A, B, C, and D) [1–3]. Unit A/B, located in N-terminal region of the receptor, controls the activation domain by AF-1 ligand, and Units C and D present a DNA binding domain that includes two zinc fingers motives and a docking domain [1–3]. The C-terminal region contains a specific binding domain and a transactivation domain for AF-2 [2]. This region is very important for nuclear localization of the PPARs and other interactions with activator proteins [1–3].
The binding of specific agonists activates the PPARs response, forming a heterodimer complex between PPARs and retinoic acid receptor (RXR), and then this complex will bind to specific PPRE regions in the DNA to activate different target genes [4]. In addition, this dimer can interact with other coactivators proteins like CBP/p300, SRC1, PBP, and PGC-1α to induce a specific gene expression (Figure 1) [3, 4].
Figure 1.
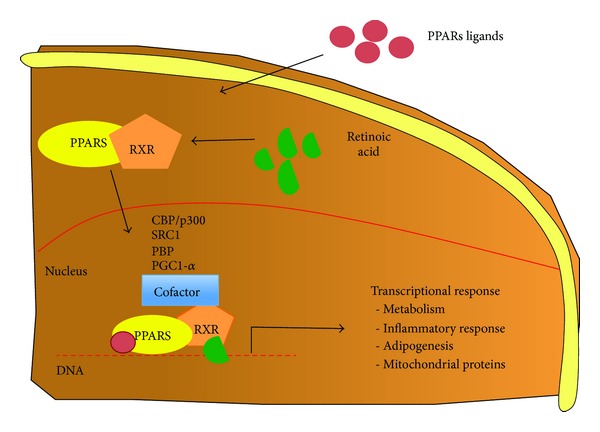
Overview of PPARs signaling and activation. The figure shows how PPARs are activated. PPARs ligand enters the cell where it binds to promote receptor dimerization with receptor of 9-cis-retinoic acid (RXR). This complex migrates to the nucleus where it binds to DNA and to different cofactors proteins (CBP/p300, SRC1, PBP, and PGC1-α), to induce the expression of several genes involved in metabolism, inflammatory response, and antioxidant defense.
PPARα expression is abundant in liver, kidney, and heart and commonly is present in tissues with high metabolic rate [1, 4]. PPARα is activated by polyunsaturated fatty acids, like docosahexaenoic acid (DHA) and icosapentaenoic acid (EPA), and by fibrate drugs like gemfibrozil and fenofibrate, which are currently used as a treatment for dyslipidemia, metabolic syndrome, and cardiovascular damage [1, 3, 4].
PPARβ expression is ubiquitous and their abundance depends on the tissue [5]. To this date, evidence suggests that PPARβ is activated like PPARα and apparently plays a role in embryo development [5].
PPARγ is expressed principally in fatty and vascular tissue [6, 7]; however, it has showed their presence in heart and brain tissue, where their activation reduced cardiovascular damage and neurodegeneration [6, 7]. PPARγ is activated by natural ligands like linoleic acid (9- and 13-HODE) and by prostaglandin derivative, 15-deoxi-Δ12,14-prostaglandin J2 (15d-PGJ2), which regulates inflammatory response [3]. Clinical importance of PPARγ has risen especially after the use of TZDs drugs for the treatment of diabetes mellitus type 2 [8, 9]. Treatment with TZDs activates PPARγ pathway reducing insulin resistance and blood glucose levels in patients with diabetes type 2 [9].
Interestingly, the activation of PPARγ receptors by TZDs prevented neurodegeneration and promoted neuronal development in primary neuronal cultures [10].
2. PPARγ Activation Prevents Neurodegeneration
In this review, we will discuss evidence that suggests an important role of PPARγ in brain function, neuronal development, and pain signaling. For the studies that cover neuroprotective effects of PPARγ, we will discuss evidence produced in different study models (animal, cells, and patients), which supports the use of PPARγ activation against neurodegeneration. For the effects of PPARγ promoting neuronal development, we will describe evidence obtained from neuronal stem cells (NSCs) and primary neuronal cultures. Finally, as an extension of this review, we will discuss other neurological pathways where PPARγ could be an important player.
2.1. Animal Studies
Agonists of PPARγ have been used to reduce neurodegenerative changes in mice models that mimic several neurodegenerative diseases [10]. For example, studies performed in a transgenic mouse that resembles experimental autoimmune encephalomyelitis (EAE) showed that treatment with PPARγ activators delayed neurodegeneration [10]. PPARγ agonists have also showed benefits in experimental models of stroke and ischemia [11–13]. In general, activation of these receptors reduced inflammation and apoptosis and improved memory function [11–14]. Moreover, in a mice model of amyotrophic lateral sclerosis (ALS), a disease that causes paralysis due to loss of motor neurons, treatment with PPARγ agonists like rosiglitazone extends survival and decreases motor neuron loss [15–17].
Beneficial effects of PPARγ activators also have been studied in mice models of Alzheimer's disease (AD). AD is a neurodegenerative disorder that affects a large segment of older population and clinically is characterized by a progressive memory decline of the patient and, later, the presence of brain aggregates of a protein called amyloid-β (Aβ) and the accumulation of the hyperphosphorylated form of the tau protein, which later forms intraneuronal aggregates known as neurofibrillary tangles (NFTs) [18]. Some studies explored the possibility that PPARγ activation reduced neuropathological changes in different AD mice models. For instance, PPARγ activation by some TZDs drugs reduced amyloid deposition and reversed cognitive and memory decline in some AD transgenic mice models [18–21]. Treatment with PPARγ agonist rosiglitazone improved hippocampus cognition in the Tg2576 AD mice with no effect on wild type mice [21]. Tg2576 transgenic mouse is an AD study model that presents amyloidosis (accumulation of Aβ), neuronal loss, and cognitive decline [21]. In a different study, oral treatment with PPARγ agonist pioglitazone reduced the Aβ levels within the cortex in the APPswe/PSEN1δE9 (APP/PS1) mice, another mouse model that accumulates Aβ plaques similar to neuropathological features presented in AD [22]. In addition, chronic pioglitazone treatment reduced expression of inflammatory cytokines and enhanced phagocytosis of deposited forms of Aβ [22]. More importantly, reduction in amyloid plaque levels was associated with a reduction of cognitive defects in the drug-treated APP/PS1 mice [22]. Further studies in Tg2576 AD mice extend the use of PPARγ activators against neurodegeneration [23]. Tg2576 mice showed significant impairment in memory and cognition compared with wild type mice [23]. Tg2576 AD mice treated with rosiglitazone improved neuronal function indicated by an increase in neuronal activity [23]. These effects were correlated with an increase in the expression of presynaptic proteins that are reduced in patients with AD [23].
In AD, the inflammatory response is exacerbated in glia and astrocytes and accumulative evidence indicates that neuroinflammation contributes to neuronal dysfunction [18]. Interestingly, Heneka et al. studied if PPARγ activation reduces the expression of proinflammatory cytokines, in order to improve neuronal injury observed in AD [18]. Acute treatment of 10-month-old APPV717I mice with pioglitazone significantly decreased the number of activated microglia and astrocytes in hippocampus and cortex [18]. In addition, pioglitazone treatment reduced the expression of the proinflammatory enzymes like cyclooxygenase 2 (COX2) and inducible nitric oxide synthase (iNOS). Finally, pioglitazone treatment reduced amyloid deposits in the hippocampus and cortex [18]. Complementary studies by Yamanaka et al., using the PPARγ agonist pioglitazone and a novel selective PPARγ modulator, DSP-8658, observed an increase in microglial activation and phagocytosis in the APPV717I mice [24]. PPARγ activators increased Aβ phagocytosis through the upregulation of scavenger receptor CD36 [24]. Furthermore, DSP-8658-treated mice showed improvement in spatial memory performance in APPV717I mice [24].
Further studies explored the role of PPARγ on cyclin-dependent kinase 5 (Cdk5) pathway. Cdk5 is a kinase that apparently plays an important role in neurogenesis and its deregulation is involved in the pathogenesis of AD [25]. Interestingly, effects of early lethality, astrogliosis, and increased neuroinflammation were observed in Cdk5 conditional knock-out mice [25]. More importantly, these effects were significantly reduced with the pioglitazone treatment [26].
Despite large evidence discussed above where PPARγ activation ameliorates neurodegenerative effects in AD and other neurological diseases, some studies had showed opposite results. For example, a recent study published by Dumont et al. explored the effects of peroxisome proliferator-activated receptor coactivator-α (PGC-1α) expression in the Tg19959 mice, another AD mice model that has increased Aβ levels and memory deficits [27]. Binding of PGC-1α with PPARγ induces the expression of different proteins involved in the regulation of mitochondrial biogenesis [28]. Other studies in Huntington disease (HD) showed a significant reduction in mRNA PGC-1α levels, the event that could contribute to the pathogenesis of this disease [28]. Surprisingly, these studies showed that the crossing of the Tg19959 mice with a mouse that overexpresses human PGC-1α exacerbated amyloid and tau pathology [27]. AD-like pathology was accompanied by mitochondrial dysfunction, neuronal death, and an exacerbated hyperactivity in the Tg19959/PGC1-α mice [27].
Beneficial effects of PPARγ had been investigated in other neurological conditions. The stroke is a devastating disease with limited treatment options. In this context, several groups have explored the use of PPARγ activators against neuronal injury [29]. For example, treatment with PPARγ agonists reduced injury and inflammation in a rat model of transient cerebral ischemia [29]. Complementary studies examined PPARγ expression, DNA binding, and PPARγ transcriptional activity after stroke induced in rats [30]. PPARγ expression was dramatically increased in ischemic neurons and the treatment with T0070907, a PPARγ antagonist, reversed rosiglitazone-mediated protection after stroke [29].
Beneficial effects of PPARγ activators were investigated in mice models subjected to damage for ischemia [29–31]. In response to ischemia, expression of PPARγ gene was significantly increased in neurons, suggesting that neuronal PPARγ may be a primary target for PPARγ-agonist-mediated neuroprotection [30, 31]. In other studies, Zhao et al. evaluated the contribution of PPARγ to ischemic injury, generating conditional neuron-specific PPARγ knock-out mice (PPARγ-KO) [32]. PPARγ deficiency caused increased brain damage and oxidative stress in response to cerebral artery occlusion [32]. Primary cortical neurons from PPARγ-KO mice showed increased neuronal death, reduced expression of SOD1 (superoxide dismutase 1), catalase, glutathione S-transferase, and uncoupling protein-1 (UCP-1) after ischemia [32], suggesting that PPARγ is an important factor in the regulation of the antioxidant response in the brain.
PPARγ effects were also investigated in animals submitted to spinal cord injury (SCI) [33]. Compared with the control groups, rosiglitazone treatment significantly increased locomotor recovery, reduced NF-κB expression, and increased the proliferation of endogenous neuronal precursors cells (NPCs) in animals subjected to spinal injury [33].
Finally, the protective role of a natural PPARγ ligand and 15-deoxy-delta12,14-prostaglandin J2 (15-PGJ2) in ischemia-reperfusion has been reported [34]. The treatment with 15d-PGJ2 decreased expression of autophagic proteins (LC3-II, Beclin 1, cathepsin-B, and LAMP1) in ischemic cortex of animals with artery occlusion, exerting neuroprotection through the inhibition of neuronal autophagy [34].
2.2. Neuronal Cells Studies
As we described before, PPARγ has been proposed as a therapeutic target against neurodegenerative diseases because of its anti-inflammatory action in glial cells [18, 22]. However, several reports indicate that PPARγ agonists induce neuroprotective actions through an independent pathway [35, 36]. For example, Fuenzalida et al. showed that the rosiglitazone protected hippocampal neurons against Aβ toxicity and apoptosis induced by nerve growth factor (NGF) deprivation [35]. The protective effects of rosiglitazone were associated with an increase in the expression of Bcl-2, an antiapoptotic protein [35]. Interestingly, PC12 cells expressing a dominant negative mutant of PPARγ showed an enhanced sensitivity to neurotoxic changes induced by Aβ, including apoptosis, neurites damage, oxidative stress, and mitochondrial injury [35]. In the same context, our group explored the effects of PPARγ activators on hippocampal neurons treated with Aβ [36]. These studies showed that activation of PPARγ by troglitazone and rosiglitazone protects hippocampal neurons against Aβ-induced neurodegeneration [36]. In addition, PPARγ activation results in the modulation of Wnt signaling components, including the inhibition of glycogen synthase kinase-3β (GSK-3β) and an increase of the β-catenin levels [20, 36]. GSK-3β is a kinase that has been suggested to be responsible for the anomalous tau hyperphosphorylation in AD [37], and the activation of Wnt pathway by proper Wnt ligands protected hippocampal neurons and AD mice exposed to Aβ [38].
Also, protective effects of PPARs activators on neuronal cells have been related with an increase in the antioxidant response [16, 17, 39]. For instance, Santos et al. showed that an increase in peroxisomal proliferation attenuated Aβ toxicity in hippocampal neurons [39]. Pretreatment with Wy-14.463 (Wy), a peroxisome proliferator and a PPARα activator, prevented neuronal death and neurites loss induced by the Aβ [39]. Moreover, neurons treated with this compound showed an increase in the number of peroxisomes, with a concomitant increase in catalase activity, reducing the production of intracellular reactive oxygen species (ROS), and prevented mitochondrial dysfunction in neurons exposed to both H2O2 and Aβ [39].
PPARγ agonists have also been tested in neuronal cells treated with acetaldehyde, a toxin that mimics Parkinson disease (PD) neurodegeneration [40]. Acetaldehyde is an inhibitor of mitochondrial function and induced oxidative stress and apoptosis in neuronal cells [41]. In these studies, the apoptosis induced by acetaldehyde was moderately reversed by rosiglitazone treatment in human neuroblastoma SH-SY5Y cells [41]. In addition, the treatment with rosiglitazone induced the expression of antioxidant proteins like Bcl-2 and Bax [41]. Complementary studies examined the role of PPARγ activation against PD in neuronal cells treated with 1-methyl-4-phenylpyridinium ion (MPP+). MPP is an inhibitor of mitochondrial complex I that has been widely used as a neurotoxin that mimics PD-like syndrome [42]. Human neuroblastoma SH-SY5Y cells treated with both MPP+ and rosiglitazone showed a reduction of apoptosis and an increase in the expression of superoxide dismutase (SOD) and catalase [42].
Important evidence indicates that PPARγ activators can ameliorate neurodegeneration in HD [28, 43–45]. HD is a neurodegenerative disease caused by the pathological elongation of CAG repeats in exon 1 of the huntingtin protein gene and is characterized by dysfunction and loss of striatal and cortical neurons [44]. Accumulative evidence suggests that mitochondrial impairment could be part of neuropathological mechanisms behind HD [28, 44]. In this context, previous findings of our group studied the potential neuroprotective role of PPARγ activation on preventing the loss of mitochondrial function in HD [43, 44]. PPARγ activation by rosiglitazone prevented the mitochondrial failure and reduced oxidative stress that occurred when striatal cells that express mutant huntingtin were challenged with calcium stress [43].
2.3. Clinical Studies
Positive results of PPARγ activators against neurodegeneration in cell lines and animal models have encouraged testing these compounds in patients affected by neurodegenerative disorders [18–21]. For instance, the effects of the PPARγ agonist pioglitazone on cognition, cerebral blood flow (CBF), and plasma levels of Aβ were tested in a controlled trial in patients with mild AD during 6 months [29]. Patient group treated with pioglitazone improved cognition and CBF, while untreated group showed no improvement [29]. The plasma Aβ40/Aβ42 ratio increased in the control group but showed no significant changes in the pioglitazone group [29]. In another study, authors evaluated the effects of rosiglitazone on cognition and plasma levels of Aβ in AD patients [30]. Patients with AD that received rosiglitazone exhibited an improvement in memory (at the 4th and 6th months) and selective attention (the 6th month) compared to untreated patients [30]. Plasma Aβ levels were unchanged in subjects treated with rosiglitazone but decreased for untreated subjects [30].
In addition, in a case report, Sundararajan et al. investigated the therapeutic potential of pioglitazone in a patient with secondary progressive multiple sclerosis (MS) [31]. Treatment with pioglitazone attenuated her cognitive decline and improved fine coordination, and after 3 years of treatment the patient continued being clinically stable, with no adverse events [31]. Also, in a randomized controlled trial of 5238 patients with diabetes type 2 who had evidence of macrovascular disease, treatment with pioglitazone for 34 months reduced the combined risk of heart attacks, strokes, and death by 16% in high-risk patients [32]. In a different study, 30 stroke patients received treatment with pioglitazone or rosiglitazone for 36 days after accident [33]. Treatment with PPARγ agonists showed a significant improvement in functional independence measure (FIMTM), indicating that the administration of TZDs drugs improved their functional recovery by the modulation of the neuroinflammatory response following stroke [33].
2.4. The Unsuccessful Use of PPARγ Activators against Neurodegeneration
Despite all evidence that suggests PPARs activators prevented or delayed neurodegenerative changes, several studies delivered conflict results [29, 45–47]. For instance, in phase III trial studies, memory cognition was not significantly improved in AD patients treated with PPARγ activators [47]. This evidence suggests that perhaps the mechanism of the action of PPARγ agonists in animal models of amyloid deposition may differ from those in humans [48]. Other studies showed no evidence of improvement in cognition and functional tasks, in AD patients treated with rosiglitazone [47, 48], and in AD patients positive for apolipoprotein ε4 allele, the treatment with rosiglitazone showed a significant decline in cognition [48]. Complementary studies in ALS showed that, in a phase II clinical trial, the treatment with pioglitazone had no positive effects on the survival of ALS patients treated with riluzole (a drug that extends lifespan of ALS patients) [49].
In addition, studies using different neuronal cell models showed no benefit of PPARγ activation against neurodegeneration [50]. For instance, studies that explored positive effects of troglitazone and pioglitazone against ALS showed that treatment with both drugs did not promote the survival of hippocampal neurons and rat motoneurons [50]. Also, it has reported that treatment with 15d-PGJ2 induced neurite degeneration and nuclear fragmentation, in primary neurons and SH-SY5Y neuroblastoma cells [51]. Moreover, the combined treatment with both ciglitazone (another PPARγ agonist) and 15d-PGJ2 generated neurotoxicity in cultured cerebellar granule neurons, in a dose response manner [52].
Finally, secondary effects of PPARγ activators have been reported in several neurodegenerative diseases [53]. For example, in Friedreich's ataxia the use of TZDs drugs caused a decrease in the number of fast fibers and an increase in mitochondrial biogenesis in cardiac muscle, enhancing the incidence of heart failure and thrombosis in these patients [53]. In addition, the use of pioglitazone in nondiabetic patients with AD showed a 28.6% of increase in peripheral edema compared to patients treated with placebo [54].
3. The Role of PPARγ in Neuronal Development
3.1. PPARγ and Neuronal Stem Cells
Recent evidence suggests that PPARγ could have a potential role in neuronal development [55]. In physiological conditions PPARγ expression was found in embryo mouse brain and in neuronal stem cells (NSCs) [55]. In contrast, extremely low levels of PPARγ were observed in adult mouse brain [55]. More important, PPARγ agonists promoted oligodendrocyte differentiation of mouse NSCs, by modulating expression of differentiation genes [56]. Moreover, activation of PPARγ induced expression of neurogenic differentiation factor (Neurod1), a member of the basic helix-loop-helix (bHLH) transcriptional factor that plays a role in the development of nervous and endocrine systems [56]. These studies in Neurod1-null mice exhibited behavioral abnormalities due to a reduction in the number of sensory neurons [56]. Thus, the upregulation of selective differentiation factors could be a mechanism by which PPARγ agonists promote differentiation of NSCs.
It has been suggested that activation of PPARγ could be an interesting therapeutic target against AD [17, 19]. In this context, Cannabidiol (CBD), a Cannabis derivative, has attracted much attention because of its promising neuroprotective properties [57]. New studies suggest that neuroprotective effects of CBD could be mediated through PPARγ pathway [57]. Interestingly, due to its interaction with PPARγ, CBD was able to stimulate hippocampal neurogenesis [57], indicating that CBD may exert protective functions through an increase of neuronal population by the activation of PPARγ [57].
3.2. PPARγ Activation Induces Neuronal Differentiation
An important role of PPARγ in the differentiation of neuronal cells has been demonstrated in different studies [58, 59]. PPARγ is expressed in the central nervous system [30, 36], and 15d-PGJ2, a natural PPARγ ligand, stimulated neurite outgrowth in pheochromocytoma 12 (PC12) cells stimulated with NGF [60]. In addition, we reported that PPARγ is present in rat hippocampal neurons and that its activation by TZDs prevented axon degeneration, neurite loss, and mitochondrial impairment induced by Aβ [35, 36]. Importantly, the treatment with troglitazone induced an increase in axon length and neurite outgrowth compared with untreated neurons [36, 61].
In addition, the role of PPARγ in neuronal development has been studied in neuronal cells treated with retinoic acid (RA) [62]. RA regulates gene expression by activating the nuclear retinoic acid receptor (RXR) inducing neuronal outgrowth in neuroblastoma cells [63]. Activation of RA induced differentiation of stem cells to neuronal progenitors through activation of FABP5/PPARγ pathway [61].
Complementary to studies in which PPARγ activation induces neuronal differentiation, recent evidence suggests an important role of PPARγ in neuronal polarity [61]. Studies made on hippocampal neurons indicated that activation of PPARγ by TZDs drugs enhanced axonal growth [61]. This effect on axonal growth was accompanied by an increase in PPARγ expression and was completely prevented by the use of GW9662, a specific PPARγ antagonist [61]. The enhanced axonal growth induced by PPARγ activators was prevented by SP 600125, an inhibitor of c-Jun N-terminal kinase (JNK), indicating that the effect of PPARγ on neuronal polarity was through the activation of JNK pathway [61].
Finally, the effects of PPARγ activation have been also studied in AD mice model that expressed apolipoprotein (Apo-E4) [64]. Apo-E4 is a major genetic risk factor for AD and exerts neuropathological effects through multiple pathways, including reduction of dendritic spine density and mitochondrial dysfunction [65]. Apo-E4 fragments are neurotoxic and cause neurodegeneration and behavioral deficits in transgenic mice [66]. In this context, Brodbeck et al. studied the effects of rosiglitazone on dendritic spine density in AD mice that expressed Apo-E4 [64]. Treatment with rosiglitazone significantly increased dendritic spine density in a dose-dependent manner in cultured cortical neurons from wild type mice [64]. This effect was prevented by GW9662, suggesting that rosiglitazone exerts this effect by activating the PPARγ [64]. Furthermore, dendritic spine density was significantly decreased in cortical cultures obtained from AD Apo-E4 mice, and treatment with rosiglitazone rescued this detrimental effect [64].
4. The Role of PPARγ in Pain Signaling
Several studies using experimental models have showed that administration of PPAR ligands reduces inflammation, suggesting their possible use for treating human inflammatory and neuropathic pain [67]. Earlier in 1995, a prominent expression of PPARs in the thalamus was reported, particularly in the posterior part of the ventral medial nucleus, a site responsive to pain and cold stress, suggesting the possibility that PPARs might play a role in modulating response to thermal and pain sensations [68].
Further studies reported that the treatment with TZDs drugs, such as rosiglitazone and pioglitazone, prevented myelin loss, reduced neuropathic pain, and improved motor function recovery after spinal cord injury [69]. In addition, Ajulemic acid, which has a potent analgesic and anti-inflammatory activity, directly interacts with PPARγ suggesting that this may be a pharmacologically relevant receptor for this compound and a potential target for drug development in the treatment of pain [70]. In addition, new studies have suggested PPARγ as a new target for treating chronic pain [71, 72]. Thus, the expression and function of PPARγ in spinal cord were reported [71]. Moreover, intrathecal administration of rosiglitazone reduced allodynia (increased sensitivity to pain from a stimulus that normally does not provoke pain) and hyperalgesia (increased sensitivity to pain from a stimulus that normally provokes pain) in the spared nerve injury (SNI) mouse model of neuropathic pain [71]. These studies suggest that new or current drugs that targeted spinal PPARγ may yield important therapeutic effects for neuropathic pain [71]. Also it was reported that pioglitazone administration reduced tactile allodynia and thermal hyperalgesia in partial sciatic nerve ligation (PSL), a study model for neuropathic pain [73]. PSL-induced upregulation of TNFα and IL-6 was suppressed by pioglitazone treatment, indicating that pioglitazone alleviates neuropathic pain through attenuation of proinflammatory cytokine upregulation by PPARγ [73]. Also, systemic administration of TZDs reduces peripheral inflammation in vivo suggesting that pharmacological activation of PPARγ in the brain rapidly inhibits local edema and the spinal transmission of noxious inflammatory signals [74]. Interestingly, it was showed that PPARγ is crucial for coupling ibuprofen to RhoA inhibition and subsequently induces neurite growth in neurons, providing additional therapeutic targets to the disorders characterized by RhoA activation, including spinal cord injury and AD [75]. It was also reported that rosiglitazone attenuates postincisional pain by regulating macrophage polarization [76] and alleviated the development of inflammatory pain, possibly through regulating macrophage infiltration [77]. These observations suggest that PPARγ signaling in macrophages may be a potential therapeutic target for the treatment of acute pain development [77]. Finally, oral or intraperitoneal administration of pioglitazone prevents multiple behavior sings of somatosensory hypersensitivity [72]. Thus, pioglitazone reduces spinal glial and stimulus-evoked p-ERK activation and reduced neuropathic pain [72].
5. Conclusions
Evidence discussed here clearly shows the importance of PPARγ promoting the development and health of neurons. Accumulative evidence suggests that PPARγ induces neuronal differentiation by a mechanism that implicates activation of PPARγ-dependent transcription and also activation of secondary pathways. Evidence obtained from pharmacological activation of PPARγ by TZDs drugs suggests a possible therapeutic use against neurodegenerative diseases (Figure 2). Concomitantly, studies of PPARγ activation showed important effects against oxidative stress, mitochondrial dysfunction, and apoptosis in several cells models that resemble AD, HD, ALS, and SCI. Also, a large part of this evidence was corroborated in mice models for each of these neurological disorders, and additionally PPARγ activation improved cognitive decline observed in several neurodegenerative diseases. Moreover, the effects of PPARγ ligands on neuroinflammation in animal models suggest their possible use for treating human inflammatory pain and neuropathic pain.
Figure 2.
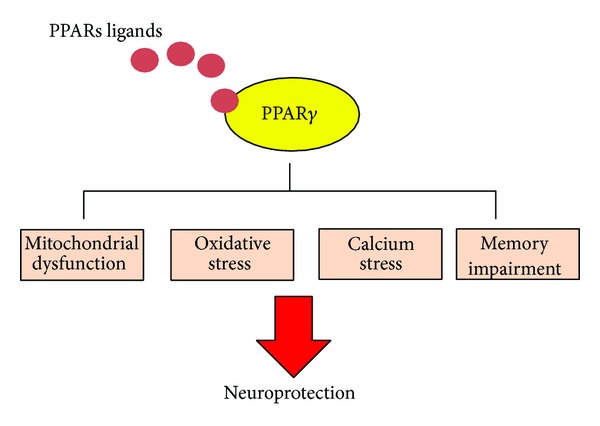
Activation of PPAR γ improves neuronal health. Several neurodegenerative diseases showed clear deficiencies in mitochondrial function, oxidative stress, and memory impairment. Activation of PPARγ by natural ligands or TZDs could prevent these neurodegenerative changes mainly improving mitochondrial function and increasing antioxidant capacity in neurons.
Altogether these observations suggest an important role for PPARγ in maintaining normal function of the brain and preventing neuronal damage induced by stressors and aging.
Acknowledgments
This work was supported by FONDECYT no. 1140968 (RAQ), grant of Vicerrectoría de Investigación, Pontificia Universidad Católica de Chile (VRI, PUC) (RAQ), PAI 79100009 (EU), and FONDECYT no. 11110136 (EU).
Conflict of Interests
The authors declare no conflict of interests.
References
- 1.Rosen ED, Spiegelman BM. PPARγ: a nuclear regulator of metabolism, differentiation, and cell growth. The Journal of Biological Chemistry. 2001;276(41):37731–37734. doi: 10.1074/jbc.R100034200. [DOI] [PubMed] [Google Scholar]
- 2.Kota BP, Huang TH, Roufogalis BD. An overview on biological mechanisms of PPARs. Pharmacological Research. 2005;51(2):85–94. doi: 10.1016/j.phrs.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 3.Ahmadian M, Suh JM, Hah N, et al. PPARγ signaling and metabolism: the good, the bad and the future. Nature Medicine. 2013;19(5):557–566. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forman BM, Chen J, Evans RM. The peroxisome proliferator-activated receptors: ligands and activators. Annals of the New York Academy of Sciences. 1996;804:266–275. doi: 10.1111/j.1749-6632.1996.tb18621.x. [DOI] [PubMed] [Google Scholar]
- 5.Coll T, Rodríguez-Calvo R, Barroso E, et al. Peroxisome proliferator-activated receptor (PPAR) β/δ: a new potential therapeutic target for the treatment of metabolic syndrome. Current Molecular Pharmacology. 2009;2(1):46–55. doi: 10.2174/1874467210902010046. [DOI] [PubMed] [Google Scholar]
- 6.Barak Y, Nelson MC, Ong ES, et al. PPARγ is required for placental, cardiac, and adipose tissue development. Molecular Cell. 1999;4(4):585–595. doi: 10.1016/s1097-2765(00)80209-9. [DOI] [PubMed] [Google Scholar]
- 7.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARγ . Annual Review of Biochemistry. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 8.Ryan KK, Li B, Grayson BE, Matter EK, Woods SC, Seeley RJ. A role for central nervous system PPAR-γ in the regulation of energy balance. Nature Medicine. 2011;17(5):623–626. doi: 10.1038/nm.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yki-Järvinen H. Thiazolidinediones. New England Journal of Medicine. 2004;351(11):1106–1158. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- 10.Feinstein DL, Galea E, Gavrilyuk V, et al. Peroxisome proliferator-activated receptor-γ agonists prevent experimental autoimmune encephalomyelitis. Annals of Neurology. 2002;51(6):694–702. doi: 10.1002/ana.10206. [DOI] [PubMed] [Google Scholar]
- 11.Allahtavakoli M, Moloudi R, Arababadi MK, Shamsizadeh A, Javanmardi K. Delayed post ischemic treatment with Rosiglitazone attenuates infarct volume, neurological deficits and neutrophilia after embolic stroke in rat. Brain Research. 2009;1271:121–127. doi: 10.1016/j.brainres.2009.03.040. [DOI] [PubMed] [Google Scholar]
- 12.McTigue DM. Potential therapeutic targets for PPARγ after spinal cord injury. PPAR Research. 2008;2008:7 pages. doi: 10.1155/2008/517162.517162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villapol S, Yaszemski AK, Logan TT, Sánchez-Lemus E, Saavedra JM, Symes AJ. Candesartan, an angiotensin II at 1-receptor blocker and PPAR-γ agonist, reduces lesion volume and improves motor and memory function after traumatic brain injury in mice. Neuropsychopharmacology. 2012;37(13):2817–2829. doi: 10.1038/npp.2012.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park S, Yi J, Miranpuri G, et al. Thiazolidinedione class of peroxisome proliferator-activated receptor γ agonists prevents neuronal damage, motor dysfunction, myelin loss, neuropathic pain, and inflammation after spinal cord injury in adult rats. The Journal of Pharmacology and Experimental Therapeutics. 2007;320(3):1002–1012. doi: 10.1124/jpet.106.113472. [DOI] [PubMed] [Google Scholar]
- 15.Kiaei M, Kipiani K, Chen J, Calingasan NY, Beal MF. Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Experimental Neurology. 2005;191(2):331–336. doi: 10.1016/j.expneurol.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 16.Shibata N, Kawaguchi-Niida M, Yamamoto T, Toi S, Hirano A, Kobayashi M. Effects of the PPARγ activator pioglitazone on p38 MAP kinase and IκBα in the spinal cord of a transgenic mouse model of amyotrophic lateral sclerosis. Neuropathology. 2008;28(4):387–398. doi: 10.1111/j.1440-1789.2008.00890.x. [DOI] [PubMed] [Google Scholar]
- 17.Benedusi V, Martorana F, Brambilla L, Maggi A, Rossi D. The peroxisome proliferator-activated receptor γ (PPARγ) controls natural protective mechanisms against lipid peroxidation in amyotrophic lateral sclerosis. Journal of Biological Chemistry. 2012;287(43):35899–35911. doi: 10.1074/jbc.M112.366419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heneka MT, Sastre M, Dumitrescu-Ozimek L, et al. Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1-42 levels in APPV717I transgenic mice. Brain. 2005;128(6):1442–1453. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- 19.Nicolakakis N, Aboulkassim T, Ongali B, et al. Complete rescue of cerebrovascular function in aged Alzheimer's disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor γ agonist. The Journal of Neuroscience. 2008;28(37):9287–9296. doi: 10.1523/JNEUROSCI.3348-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toledo EM, Inestrosa NC. Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1ΔE9 mouse model of Alzheimer’s disease. Molecular Psychiatry. 2010;15(3):272–285. doi: 10.1038/mp.2009.72. [DOI] [PubMed] [Google Scholar]
- 21.Denner LA, Rodriguez-Rivera J, Haidacher SJ, et al. Cognitive enhancement with rosiglitazone links the hippocampal PPARγ and ERK MAPK signaling pathways. The Journal of Neuroscience. 2012;32(47):16725–16735. doi: 10.1523/JNEUROSCI.2153-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mandrekar-Colucci S, Karlo JC, Landreth GE. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated Amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer's disease. The Journal of Neuroscience. 2012;32(30):10117–10128. doi: 10.1523/JNEUROSCI.5268-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nenov MN, Laezza F, Haidacher SJ, et al. Cognitive enhancing treatment with a PPARγ agonist normalizes dentate granule cell presynaptic function in Tg2576 APP mice. Journal of Neuroscience. 2014;34(3):1028–1036. doi: 10.1523/JNEUROSCI.3413-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamanaka M, Ishikawa T, Griep A, Axt D, Kummer MP, Heneka MT. PPARγ/RXRA-induced and CD36-mediated microglial amyloid-β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. Journal of Neuroscience. 2012;32(48):17321–17331. doi: 10.1523/JNEUROSCI.1569-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takahashi S, Ohshima T, Hirasawa M, et al. Conditional deletion of neuronal cyclin-dependent kinase 5 in developing forebrain results in microglial activation and neurodegeneration. The American Journal of Pathology. 2010;176(1):320–329. doi: 10.2353/ajpath.2010.081158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Utreras E, Hamada R, Prochazkova M, et al. Suppression of neuroinflammation in forebrain-specific Cdk5 conditional knockout mice by PPARγ agonist improves neuronal loss and early lethality. Journal of Neuroinflammation. 2014;11, article 28 doi: 10.1186/1742-2094-11-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dumont M, Stack C, Elipenahli C, et al. PGC-1α overexpression exacerbates β-amyloid and tau deposition in a transgenic mouse model of Alzheimer's disease. The FASEB Journal. 2014;28(4):1745–1755. doi: 10.1096/fj.13-236331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsunemi T, La Spada AR. PGC-1α at the intersection of bioenergetics regulation and neuron function: from Huntington's disease to Parkinson's disease and beyond. Progress in Neurobiology. 2012;97(2):142–151. doi: 10.1016/j.pneurobio.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mandrekar-Colucci S, Sauerbeck A, Popovich PG, McTigue DM. PPAR agonists as therapeutics for CNS trauma and neurological diseases. ASN Neuro. 2013;5(5) doi: 10.1042/AN20130030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Victor NA, Wanderi EW, Gamboa J, et al. Altered PPARγ expression and activation after transient focal ischemia in rats. European Journal of Neuroscience. 2006;24(6):1653–1663. doi: 10.1111/j.1460-9568.2006.05037.x. [DOI] [PubMed] [Google Scholar]
- 31.Sundararajan S, Gamboa JL, Victor NA, Wanderi EW, Lust WD, Landreth GE. Peroxisome proliferator-activated receptor-γ ligands reduce inflammation and infarction size in transient focal ischemia. Neuroscience. 2005;130(3):685–696. doi: 10.1016/j.neuroscience.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 32.Zhao X, Strong R, Zhang J, et al. Neuronal PPARγ deficiency increases susceptibility to brain damage after cerebral ischemia. The Journal of Neuroscience. 2009;29(19):6186–6195. doi: 10.1523/JNEUROSCI.5857-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meng Q, Liang X, Wang P, et al. Rosiglitazone enhances the proliferation of neural progenitor cells and inhibits inflammation response after spinal cord injury. Neuroscience Letters. 2011;503(3):191–195. doi: 10.1016/j.neulet.2011.08.033. [DOI] [PubMed] [Google Scholar]
- 34.Xu F, Li J, Ni W, Shen Y, Zhang X. Peroxisome proliferator-activated receptor-γ agonist 15d-prostaglandin J2 mediates neuronal autophagy after cerebral ischemia-reperfusion injury. PLoS ONE. 2013;8(1) doi: 10.1371/journal.pone.0055080.e55080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fuenzalida K, Quintanilla R, Ramos P, et al. Peroxisome proliferator-activated receptor γ up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. The Journal of Biological Chemistry. 2007;282(51):37006–37015. doi: 10.1074/jbc.M700447200. [DOI] [PubMed] [Google Scholar]
- 36.Inestrosa NC, Godoy JA, Quintanilla RA, Koenig CS, Bronfman M. Peroxisome proliferator-activated receptor γ is expressed in hippocampal neurons and its activation prevents β-amyloid neurodegeneration: قole of Wnt signaling. Experimental Cell Research. 2005;304(1):91–104. doi: 10.1016/j.yexcr.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 37.Jope RS, Johnson GVW. The glamour and gloom of glycogen synthase kinase-3. Trends in Biochemical Sciences. 2004;29(2):95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 38.Inestrosa NC, Montecinos-Oliva C, Fuenzalida M. Wnt signaling: role in Alzheimer disease and schizophrenia. Journal of Neuroimmune Pharmacology. 2012;7(4):788–807. doi: 10.1007/s11481-012-9417-5. [DOI] [PubMed] [Google Scholar]
- 39.Santos MJ, Quintanilla RA, Toro A, et al. Peroxisomal proliferation protects from β-amyloid neurodegeneration. Journal of Biological Chemistry. 2005;280(49):41057–41068. doi: 10.1074/jbc.M505160200. [DOI] [PubMed] [Google Scholar]
- 40.Jung TW, Lee JY, Shim WS, et al. Rosiglitazone protects human neuroblastoma SH-SY5Y cells against acetaldehyde-induced cytotoxicity. Biochemical and Biophysical Research Communications. 2006;340(1):221–227. doi: 10.1016/j.bbrc.2005.11.177. [DOI] [PubMed] [Google Scholar]
- 41.Hernán MA, Logroscino G, García Rodríguez LA. A prospective study of alcoholism and the risk of Parkinson’s disease. Journal of Neurology. 2004;251(7):VII14–VII17. doi: 10.1007/s00415-004-1705-4. [DOI] [PubMed] [Google Scholar]
- 42.Jung TW, Lee JY, Shim WS, et al. Rosiglitazone protects human neuroblastoma SH-SY5Y cells against MPP+ induced cytotoxicity via inhibition of mitochondrial dysfunction and ROS production. Journal of the Neurological Sciences. 2007;253(1-2):53–60. doi: 10.1016/j.jns.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 43.Quintanilla RA, Jin YN, Fuenzalida K, Bronfman M, Johnson GVW. Rosiglitazone treatment prevents mitochondrial dysfunction in mutant huntingtin-expressing cells: possible role of peroxisome proliferator-activated receptor-γ (PPARγ) in the pathogenesis of huntington disease. The Journal of Biological Chemistry. 2008;283(37):25628–25637. doi: 10.1074/jbc.M804291200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quintanilla RA, Johnson GVW. Role of mitochondrial dysfunction in the pathogenesis of Huntington's disease. Brain Research Bulletin. 2009;80(4-5):242–247. doi: 10.1016/j.brainresbull.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Almad A, Lash AT, Wei P, Lovett-Racke AE, McTigue DM. The PPAR alpha agonist gemfibrozil is an ineffective treatment for spinal cord injured mice. Experimental Neurology. 2011;232(2):309–317. doi: 10.1016/j.expneurol.2011.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith SA, May FJ, Monteith GR, Roberts-Thomson SJ. Activation of the peroxisome proliferator-activated receptor-α enhances cell death in cultured cerebellar granule cells. Journal of Neuroscience Research. 2001;66(2):236–241. doi: 10.1002/jnr.1216. [DOI] [PubMed] [Google Scholar]
- 47.Gold M, Alderton C, Zvartau-Hind M, et al. Rosiglitazone monotherapy in mild-to-moderate alzheimer's disease: results from a randomized, double-blind, placebo-controlled phase III study. Dementia and Geriatric Cognitive Disorders. 2010;30(2):131–146. doi: 10.1159/000318845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Risner ME, Saunders AM, Altman JFB, et al. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer's disease. The Pharmacogenomics Journal. 2006;6(4):246–254. doi: 10.1038/sj.tpj.6500369. [DOI] [PubMed] [Google Scholar]
- 49.Dupuis L, Dengler R, Heneka MT, et al. A randomized, double blind, placebo-controlled trial of pioglitazone in combination with riluzole in amyotrophic lateral sclerosis. PLoS ONE. 2012;7(6) doi: 10.1371/journal.pone.0037885.e37885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nishijima C, Kimoto K, Arakawa Y. Survival activity of troglitazone in rat motoneurones. Journal of Neurochemistry. 2001;76(2):383–390. doi: 10.1046/j.1471-4159.2001.00039.x. [DOI] [PubMed] [Google Scholar]
- 51.Rohn TT, Wong SM, Cotman CW, Cribbs DH. 15-Deoxy-Δ12,14-prostaglandin J2, a specific ligand for peroxisome proliferator-activated receptor-γ, induces neuronal apoptosis. NeuroReport. 2001;12(4):839–843. doi: 10.1097/00001756-200103260-00043. [DOI] [PubMed] [Google Scholar]
- 52.Smith SA, Monteith GR, Holman NA, Robinson JA, May FJ, Roberts-Thomson SJ. Effects of peroxisome proliferator-activated receptor γ ligands ciglitazone and 15-deoxy-∆12,14-prostaglandin J2 on rat cultured cerebellar granule neuronal viability. Journal of Neuroscience Research. 2003;72(6):747–755. doi: 10.1002/jnr.10613. [DOI] [PubMed] [Google Scholar]
- 53.García-Giménez JL, Sanchis-Gomar F, Pallardó FV. Could thiazolidinediones increase the risk of heart failure in Friedreich's ataxia patients? Movement Disorders. 2011;26(5):769–771. doi: 10.1002/mds.23711. [DOI] [PubMed] [Google Scholar]
- 54.Geldmacher DS, Fritsch T, McClendon MJ, Landreth G. A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with alzheimer disease. Archives of Neurology. 2011;68(1):45–50. doi: 10.1001/archneurol.2010.229. [DOI] [PubMed] [Google Scholar]
- 55.Wada K, Nakajima A, Katayama K, et al. Peroxisome proliferator-activated receptor γ-mediated regulation of neural stem cell proliferation and differentiation. The Journal of Biological Chemistry. 2006;281(18):12673–12681. doi: 10.1074/jbc.M513786200. [DOI] [PubMed] [Google Scholar]
- 56.Yokoyama M, Nishi Y, Miyamoto Y, et al. Molecular cloning of a human neuroD from a neuroblastoma cell line specifically expressed in the fetal brain and adult cerebellum. Molecular Brain Research. 1996;42(1):135–139. doi: 10.1016/s0169-328x(96)00154-4. [DOI] [PubMed] [Google Scholar]
- 57.Esposito G, Scuderi C, Valenza M, et al. Cannabidiol reduces Aβ-induced neuroinflammation and promotes hippocampal neurogenesis through PPARγ involvement. PLoS ONE. 2011;6(12) doi: 10.1371/journal.pone.0028668.e28668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miglio G, Rattazzi L, Rosa AC, Fantozzi R. PPARγ stimulation promotes neurite outgrowth in SH-SY5Y human neuroblastoma cells. Neuroscience Letters. 2009;454(2):134–138. doi: 10.1016/j.neulet.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 59.Ghoochani A, Shabani K, Peymani M, et al. The influence of peroxisome proliferator-activated receptor γ 1 during differentiation of mouse embryonic stem cells to neural cells. Differentiation. 2012;83(1):60–67. doi: 10.1016/j.diff.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 60.Jung KM, Park KS, Oh JH, et al. Activation of p38 mitogen-activated protein kinase and activator protein-1 during the promotion of neurite extension of PC-12 cells by 15-deoxy-Δ12,14-prostaglandin J2. Molecular Pharmacology. 2003;63(3):607–616. doi: 10.1124/mol.63.3.607. [DOI] [PubMed] [Google Scholar]
- 61.Quintanilla RA, Godoy JA, Alfaro I, et al. Thiazolidinediones promote axonal growth through the activation of the JNK pathway. PLoS ONE. 2013;8(5) doi: 10.1371/journal.pone.0065140.e65140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu S, Levi L, Siegel R, Noy N. Retinoic acid induces neurogenesis by activating both retinoic acid receptors (RARs) and peroxisome proliferator-activated receptor β/δ (PPARβ/δ) The Journal of Biological Chemistry. 2012;287(50):42195–42205. doi: 10.1074/jbc.M112.410381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maden M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nature Reviews Neuroscience. 2007;8(10):755–765. doi: 10.1038/nrn2212. [DOI] [PubMed] [Google Scholar]
- 64.Brodbeck J, Balestra ME, Saunders AM, Roses AD, Mahley RW, Huang Y. Rosiglitazone increases dendritic spine density and rescues spine loss caused by apolipoprotein E4 in primary cortical neurons. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(4):1343–1346. doi: 10.1073/pnas.0709906104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chang S, Ma TR, Miranda RD, Balestra ME, Mahley RW, Huang Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(51):18694–18699. doi: 10.1073/pnas.0508254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brecht WJ, Harris FM, Chang S, et al. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. Journal of Neuroscience. 2004;24(10):2527–2534. doi: 10.1523/JNEUROSCI.4315-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maeda T, Kishioka S. PPAR and Pain. International Review of Neurobiology. 2009;85:165–177. doi: 10.1016/S0074-7742(09)85013-7. [DOI] [PubMed] [Google Scholar]
- 68.Xing G, Zhang L, Heynen T, et al. Rat PPARΔ contains a CGG triplet repeat and is prominently expressed in the thalamic nuclei. Biochemical and Biophysical Research Communications. 1995;217(3):1015–1025. doi: 10.1006/bbrc.1995.2871. [DOI] [PubMed] [Google Scholar]
- 69.Park S, Yi J, Miranpuri G, et al. Thiazolidinedione class of peroxisome proliferator-activated receptor γ agonists prevents neuronal damage, motor dysfunction, myelin loss, neuropathic pain, and inflammation after spinal cord injury in adult rats. Journal of Pharmacology and Experimental Therapeutics. 2007;320(3):1002–1012. doi: 10.1124/jpet.106.113472. [DOI] [PubMed] [Google Scholar]
- 70.Ambrosio ALB, Dias SMG, Polikarpov I, Zurier RB, Burstein SH, Garratt RC. Ajulemic acid, a synthetic nonpsychoactive cannabinoid acid, bound to the ligand binding domain of the human peroxisome proliferator-activated receptor γ . Journal of Biological Chemistry. 2007;282(25):18625–18633. doi: 10.1074/jbc.M702538200. [DOI] [PubMed] [Google Scholar]
- 71.Churi SB, Abdel-Aleem OS, Tumber KK, Scuderi-Porter H, Taylor BK. Intrathecal rosiglitazone acts at peroxisome proliferator-activated receptor-γ to rapidly inhibit neuropathic pain in rats. Journal of Pain. 2008;9(7):639–649. doi: 10.1016/j.jpain.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morgenweck J, Griggs RB, Donahue RR, Zadina JE, Taylor BK. PPARγ activation blocks development and reduces established neuropathic pain in rats. Neuropharmacology. 2013;70:236–246. doi: 10.1016/j.neuropharm.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maeda T, Kiguchi N, Kobayashi Y, Ozaki M, Kishioka S. Pioglitazone attenuates tactile allodynia and thermal hyperalgesia in mice subjected to peripheral nerve injury. Journal of Pharmacological Sciences. 2008;108(3):341–347. doi: 10.1254/jphs.08207fp. [DOI] [PubMed] [Google Scholar]
- 74.Morgenweck J, Abdel-aleem OS, McNamara KC, Donahue RR, Badr MZ, Taylor BK. Activation of peroxisome proliferator-activated receptor γ in brain inhibits inflammatory pain, dorsal horn expression of Fos, and local edema. Neuropharmacology. 2010;58(2):337–345. doi: 10.1016/j.neuropharm.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dill J, Patel AR, Yang X, Bachoo R, Powell CM, Li S. A molecular mechanism for ibuprofen-mediated RhoA inhibition in neurons. Journal of Neuroscience. 2010;30(3):963–972. doi: 10.1523/JNEUROSCI.5045-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hasegawa-Moriyama M, Ohnou T, Godai K, Kurimoto T, Nakama M, Kanmura Y. Peroxisome proliferator-activated receptor-gamma agonist rosiglitazone attenuates postincisional pain by regulating macrophage polarization. Biochemical and Biophysical Research Communications. 2012;426(1):76–82. doi: 10.1016/j.bbrc.2012.08.039. [DOI] [PubMed] [Google Scholar]
- 77.Hasegawa-Moriyama M, Kurimoto T, Nakama M, et al. Peroxisome proliferator-activated receptor-gamma agonist rosiglitazone attenuates inflammatory pain through the induction of heme oxygenase-1 in macrophages. Pain. 2013;154(8):1402–1412. doi: 10.1016/j.pain.2013.04.039. [DOI] [PubMed] [Google Scholar]