

Abstract
Tafazzin, a mitochondrial acyltransferase, plays an important role in cardiolipin side chain remodeling. Previous studies have shown that dysfunction of tafazzin reduces cardiolipin content, impairs mitochondrial function, and causes dilated cardiomyopathy in Barth syndrome. Reactive oxygen species (ROS) have been implicated in the development of cardiomyopathy and are also the obligated byproducts of mitochondria. We hypothesized that tafazzin knockdown increases ROS production from mitochondria, and a mitochondria-targeted antioxidant prevents tafazzin knockdown induced mitochondrial and cardiac dysfunction. We employed cardiac myocytes transduced with an adenovirus containing tafazzin shRNA as a model to investigate the effects of the mitochondrial antioxidant, mito-Tempo. Knocking down tafazzin decreased steady state levels of cardiolipin and increased mitochondrial ROS. Treatment of cardiac myocytes with mito-Tempo normalized tafazzin knockdown enhanced mitochondrial ROS production and cellular ATP decline. Mito-Tempo also significantly abrogated tafazzin knockdown induced cardiac hypertrophy, contractile dysfunction, and cell death. We conclude that mitochondria-targeted antioxidant prevents cardiac dysfunction induced by tafazzin gene knockdown in cardiac myocytes and suggest mito-Tempo as a potential therapeutic for Barth syndrome and other dilated cardiomyopathies resulting from mitochondrial oxidative stress.
1. Introduction
Tafazzin is a mitochondrial phospholipid-lysophospholipid acyltransferase [1, 2]. It plays important role in cardiolipin side chains remodeling from their nascent forms to tetralinoleoyl cardiolipin, which is believed to be the functional species in adult mammal heart [3]. Cardiolipin, a mitochondrial signature phospholipid consisting of two phosphatidylglycerols, is essential for optimal mitochondrial function. Cardiolipin is initially synthesized as a premature form and becomes fully functional when its four fatty acid chains are remodeled by enzymes including tafazzin [1, 4]. Tafazzin is encoded by the G4.5 gene in humans [5]. Tafazzin mutation causes Barth syndrome, a rare and often fatal x-linked genetic disorder which is characterized by aciduria, neutropenia, dilated cardiomyopathy, and myocardial noncompaction [6, 7]. Heart failure and arrhythmias are the causes of death in the early childhood. Dysfunction of the tafazzin gene reduces cardiolipin and impairs mitochondrial structure and function in yeast and in patients with Barth syndrome [8–11]. Most of the cardiac abnormalities of Barth syndrome have been mimicked in animal models, for example, reduced locomotor activity in Drosophila [12], signs of Barth syndrome heart failure in zebrafish [13], and cardiomyopathies in mice [14, 15]. Decreased cardiolipin contents and impaired mitochondrial function due to tafazzin dysfunction have been demonstrated in several cell models including yeast [16], human lymphoblasts [11], neonatal cardiac fibroblasts, and cardiac myocytes [17, 18]. Tafazzin knockdown resulted in cardiac hypertrophy in neonatal cardiac myocytes [17]. Impaired mitochondrial function is the potential cause of cardiomyopathy seen in Barth syndrome since cardiomyopathy is also a common clinical presentation of mitochondrial disease [19–21].
Our previous study showed that tafazzin knockdown enhances mitochondrial reactive oxygen species (ROS) production in neonatal cardiac fibroblasts [18]. Tafazzin mutation causes oxidative stress in yeast as well [22]. Imbalanced ROS have been implicated in the pathogenesis of a variety of diseases, including diabetes [23], neurodegenerative diseases [24], ischemia-reperfusion injury [25], and heart failure [26]. Mitochondria occupy about 30% of the cardiac myocyte volume and are postulated to be the major cellular ROS source. ROS are the byproducts of the mitochondrial respiration chain complexes. Up to 2% of the oxygen consumed in the respiration chain was used to form superoxide in a quiescent condition [27]. ROS are also produced from mitochondrial enzymatic reactions catalyzed by aconitase, α-ketoglutarate dehydrogenase, pyruvate dehydrogenase, glycerol-3-phosphate dehydrogenase, dihydroorotate dehydrogenase, monoamine oxidase, and cytochrome b5 reductase [28]. Antioxidation defense systems exist in the mitochondria, including manganese superoxide dismutase (Mn-SOD), catalase, glutathione peroxidase, and thioredoxin peroxidase [28, 29]. Besides mitochondria, cellular ROS are also contributed by xanthine oxidase, NADPH oxidase, and uncoupled nitric oxide synthase [30]. The importance of mitochondrial ROS has demonstrated that mice null for Mn-SOD exhibit lethality due to cardiac dysfunction [31], heart/muscle-specific ablation of Mn-SOD produces progressive congestive heart failure [32], and overexpression of a mitochondrial ROS scavenger, peroxiredoxin3, prevents heart failure induced by myocardial infarction [33]. Clinical investigation showed that general antioxidant supplement has no beneficial effects on cardiovascular diseases [34]; however, the mitochondria-targeted antioxidant MitoQ10 proved effective for endothelial improvement and cardiac hypertrophy attenuation in stroke-prone spontaneously hypertensive rats [35].
Our current study was designed to demonstrate that mitochondrial ROS play critical roles in tafazzin knockdown induced cardiac and mitochondrial dysfunction in cultured cardiac myocytes. We found that tafazzin knockdown enhanced ROS production from the mitochondria, and a mitochondria-targeted antioxidant normalized tafazzin knockdown induced ATP decline, cardiac myocyte hypertrophy, contractile dysfunction, and cell death.
2. Materials and Methods
Animal protocols under IACUC #2011-0059 were approved by the Institutional Animal Care and Use Committee of the Sanford-Burnham Medical Research Institute, Orlando Diabetes and Obesity Research Center, Florida.
2.1. Supplies and Chemicals
Phosphatase and proteinase inhibitor cocktail tablets (PhosSTOP and Complete Mini) were obtained from Roche Applied Science (Indianapolis, IN). Primary antibodies against phospho-AMPKα (Thr172), phospho-Jak2 (Tyr107/108), cytochrome c, and β-actin and a horseradish peroxidase-conjugated secondary antibody against rabbit IgG were purchased from Cell Signaling Technology (Boston, MA). The antibody against tafazzin was from Santa Cruz (Santa Cruz, CA). Coomassie protein assay kit, MemCode reversible protein stain kit, mitochondria isolation kit for mammalian cells, SuperSignal West Pico chemiluminescent substrates, and Restore Plus Western blot stripping buffer were purchased from Thermo Scientific (Rockford, IL). Cell culture medium and supplements, precast tris-glycine polyacrylamide gels, polyvinylidene fluoride (PVDF) membranes, SYBR Green PCR Master Mix, MitoTracker Green, and the mitochondrial superoxide indicator MitoSOX Red were obtained from Life Technologies (San Diego, CA). 3H-leucine was purchased from PerkinElmer (Waltham, MA). Random primers and Omniscript reverse transcriptase were obtained from Qiagen (Valencia, CA). Custom primers were synthesized by TIB MolBiol (Adelphia, NJ). The tafazzin short hairpin RNA (shRNA) adenovirus (pSilencer adeno 1.0-CMV) was expanded and purified by the Gene Therapy Center Virus Vector Core Facility of the University of North Carolina at Chapel Hill. An ATP assay kit was purchased from BioVision (Milpitas, CA). The Tunel apoptosis detection kit was from EMD Millipore (Billerica, MA). Hematoxylin and eosin reagents were obtained from VWR (Atlanta, GA). Mito-Tempo and Tempol were obtained from ENZO Life Sciences (Farmingdale, NY). Other routine supplies and chemicals were purchased from Fisher and Sigma. A muscle derived cell line C2C12 was obtained from ATCC (Manassas, VA).
2.2. Cell Culture
Neonatal ventricular myocytes (NVMs) were generated from 1-day-old Sprague-Dawley rat pups (Charles River Laboratories) as described previously [17]. Cells were cultured in DMEM containing 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM glutamine, and 10% fetal bovine serum in a humidified CO2 incubator at 37°C. Cells were treated in glucose-free DMEM after being serum-starved for 24 h.
Adult cardiac myocytes (ACMs) were isolated from 9-week old C57/B6 male mice as described previously [36] by retrograde perfusion of digestion buffer containing liberase (0.25 mg/mL) and trypsin (0.14 mg/mL). The cells were preplated in a MEM medium supplement with 5% fetal bovine serum and 10 mM 2,3-butanedione monoxime (BDM) to stop cell contraction for 1 h and then cultured in MEM containing 0.1 mg/mL bovine serum albumin, 10 mM BDM, insulin-transferrin-selenium, and antibiotics.
Adenoviral transduction of cardiac myocytes, protein extraction and Western blot, 3H-leucine incorporation, and ATP assay were performed as described previously [17].
2.3. Cardiolipin Analysis
Cells were scraped into PBS and the pellets kept at −80°C until just prior to processing. Lipids were extracted and cardiolipin analyzed by shotgun lipidomics [37].
2.4. Mitochondrial Reactive Oxygen Species (ROS)
Cells were incubated with 300 nM MitoTracker Green in serum-free medium at 37°C for 45 min. The cells were washed with HBSS (Hank's balanced salt solution) and incubated with 5 μM MitoSOX red at 37°C for 5 min in HBSS. The cells were kept in HBSS after washing. Five images were captured from each field using fluorescence microscopy with green and red filters and merged with Elements software. The red fluorescence intensity representing mitochondrial ROS was quantified using Image J and expressed as the corrected total cell fluorescence.
2.5. Mitochondrial Cytochrome C Assay
Mitochondria were isolated from NVMs with a kit from Thermo Fisher following the manufacturer's directions. The isolated mitochondria were dissolved in protein sample buffer by boiling. The mitochondrial protein samples were separated on 16% SDS polyacrylamide gel electrophoresis and transferred onto PVDF membranes. The major protein bands were captured for loading control after visualization using a MemCode reversible protein stain kit. After destaining, the PVDF membrane was subjected to Western blot for cytochrome c. The band intensity was quantified, corrected for the major protein bands, and expressed as a percentage of scrambled virus treated with vehicle.
2.6. Real-Time RT-PCR
Total RNA isolation from NVMs and real-time RT-PCR with the primers for tafazzin and β-actin in an Eppendorf RealPlex2 were performed as we described previously [17]. Target gene mRNA levels were determined using the ΔΔCt method [38] and expressed as relative to the control, which was NVMs infected with a scrambled adenovirus.
2.7. Cell Surface Area
NVMs were plated onto laminin-coated coverslips in 6-well plates at a density of 0.25 million cells per well. After treatments, cells were fixed with 3.7% formaldehyde for 20 min at room temperature. The fixed cells were stained with hematoxylin/eosin and mounted. Five images of each sample were acquired under an Olympus I × 71 inverted microscope. The cell surface area was analyzed with Image J.
2.8. Cell Count
Cells were trypsinized and fixed with 3.7% formaldehyde in PBS for 20 min at room temperature. Cells were counted with a hemocytometer.
2.9. Myocyte Contractility
ACMs were plated on laminin coated coverslips, incubated with tafazzin shRNA adenovirus for 24 h and treated with mito-Tempo overnight. They were stimulated at 1 Hz with a MyoPacer electric field in MEM culture medium containing 1.8 mM calcium at room temperature. Sarcomere length was measured with an IonWizars system from IonOptix. Myocyte contractility was represented by sarcomere shortening, for example, percent change in length.
2.10. Statistical Analysis
Data were expressed as mean ± SE and differences in mean values were analyzed using an unpaired 2-tailed t-test. P < 0.05 was considered statistically significant.
3. Results
3.1. Tafazzin Knockdown Decreased Cardiolipin
To see if tafazzin shRNA would knock down tafazzin expression, NVMs were transduced with the tafazzin shRNA adenovirus for 48 h and its expression measured by real-time RT-PCR and Western blot. We found that the tafazzin shRNA adenovirus significantly knocked down both tafazzin mRNA and protein compared with the scrambled adenovirus (Figures 1(a) and 1(b)). Since tafazzin plays an important role in cardiolipin remodeling, and since mutation of tafazzin decreases cardiolipin, we measured cardiolipin by shotgun lipidomics and found that tafazzin knockdown reduced cardiolipin to 86% while increasing both monolysocardiolipin (to 122%) and the ratio of monolysocardiolipin to cardiolipin (to 142%) compared control NVMs treated with scrambled virus (Figure 1(c)). Side chain analysis showed that cardiolipin species were deeply redistributed and shifted to the short chain. While most cardiolipin species tended to decrease (Figure 1(d)), monolysocardiolipin tended to increase (Figure 1(e)). Since there is no predominant species, we suggested that total cardiolipin plays more important roles than specific cardiolipin species in neonatal cardiac myocyte.
Figure 1.
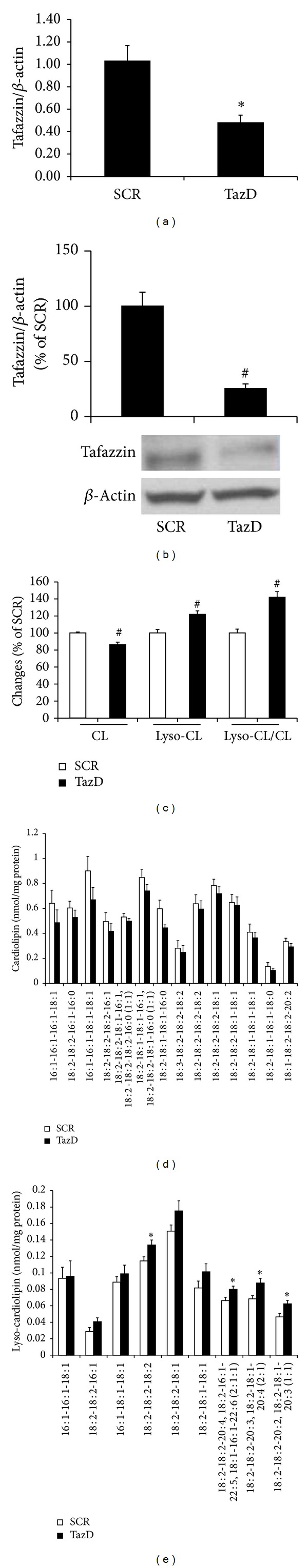
Tafazzin shRNA adenovirus knocked down tafazzin expression and cardiolipin. NVMs were transduced with the tafazzin (TazD) shRNA adenovirus for 48 h and tested for tafazzin expression ((a) and (b)) and cardiolipin ((c), (d), and (e)). (a) Tafazzin mRNA was measured by real-time RT-PCR using β-actin as a normalizer and expressed as relative level compared to the control (scrambled adenovirus or SCR). Data represent mean ± SE of 4 separate experiments. *P < 0.05 versus SCR. (b) Tafazzin protein was determined by Western blot and expressed as a percentage of SCR. Data represent mean ± SE of 3 separate experiments. # P < 0.01 versus SCR. (c) Cardiolipin (CL) and monolysocardiolipin (lyso-CL) were identified and quantified by mass spectrometry. Data represent mean ± SE of 9 separate experiments. # P < 0.01 versus SCR. The basal contents (including all detected species) of cardiolipin and monolysocardiolipin were 8.07 ± 0.36 and 0.92 ± 0.04 nmol/mg protein, respectively. Species of cardiolipin (d) and monolysocardiolipin (e) were identified and quantified by shotgun lipidomics. Data represent mean ± SE of 8 separate experiments. *P < 0.05 versus SCR.
3.2. Tafazzin Knockdown Enhanced Mitochondrial ROS Production, which Is Abrogated by the Mitochondrial Antioxidant
Cardiolipin plays an important role in maintaining optimal function of the mitochondrial respiration chain, and dysfunction of mitochondria increases ROS production. We found that tafazzin knockdown obviously increased mitochondrial ROS production as assayed by MitoSOX red staining. The enhanced MitoSOX red stain was abolished by mito-Tempo, a mitochondria-specific antioxidant, which is antioxidant tempol covalently attached to lipophilic triphenylphosphonium cation. The alkyltriphenylphosphonium cations are preferably accumulated in mitochondria by mitochondrial membrane potential [39]. The MitoSOX red stain overlapped perfectly with MitoTracker green (Figure 2(a)). After quantification analysis, we found that the enhanced mitochondrial ROS production was blocked by the treatment with 25 μM mito-Tempo (Figure 2(b)).
Figure 2.
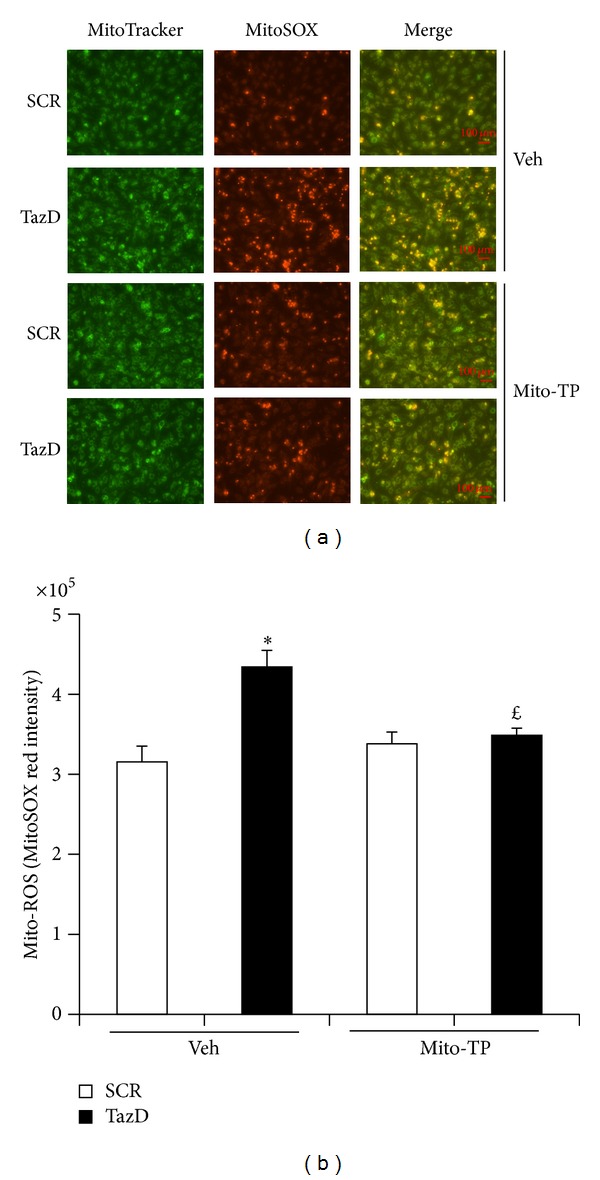
Mito-Tempo blocked the enhanced mitochondrial ROS production induced by tafazzin knockdown. NVMs were transduced with the shRNA adenovirus overnight, treated with mito-Tempo for 24 h and tested for ROS. (a) Representative picture of mitochondrial ROS. Each panel represents 15 images from 3 separate experiments. Scale bars, 100 μm. (b) Quantitative analysis of panel A MitoSOX red staining. Data represent mean ± SE of 3 separate experiments. *P < 0.05 versus SCR treated with vehicle. £ P < 0.05 versus Taz knockdown cells treated with vehicle.
3.3. Mitochondrial Antioxidant Normalized ATP Decline Induced by Tafazzin Knockdown
Our previous study showed that tafazzin knockdown decreases ATP production from the mitochondria [17], we next tested whether mito-Tempo affects intracellular ATP. The intracellular ATP was mainly from mitochondrial oxidative phosphorylation, since we used glucose-free DMEM. The only fuel molecule in our culture system is glutamine, which is used to produce ATP in the mitochondria. As showed in Figure 3(a), inhibition of mitochondrial respiration chain complex I with 5 nM rotenone dramatically decreased cellular ATP in C2C12 cells cultured in glucose-free medium but had no effect if the cells were cultured in glucose containing medium. Our results also showed that mito-Tempo normalized ATP decline induced by tafazzin knockdown compared with the vehicle treated cells in the glucose-free medium (Figure 3(b)).
Figure 3.
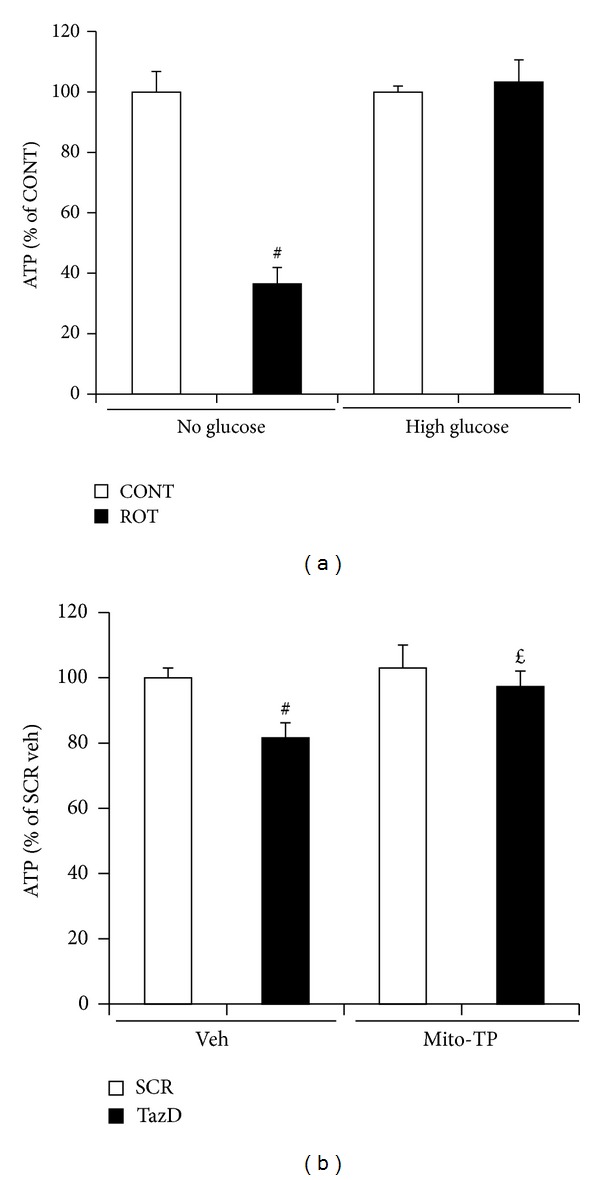
Mito-Tempo abolished ATP shortage induced by tafazzin knockdown. (a) C2C12 cells were cultured in high glucose DMEM to 70% confluence, serum starved, and treated with 5 nM rotenone (ROT) in glucose-free or high glucose DMED for 24 h. Data represent mean ± SE of 3 separate experiments. # P < 0.05 versus control cells which were treated with DMSO. (b). NVMs were transduced with the shRNA adenovirus for 48 h, treated with mito-Tempo in the last 24 h, and tested for ATP with a kit from BioVision. Data represent mean ± SE of 4 separate experiments. # P < 0.01 versus SCR treated with vehicle. £ P < 0.05 versus Taz knockdown cells treated with vehicle.
3.4. Mitochondrial Antioxidant Prevented Protein Kinases Activation Induced by Tafazzin Knockdown
Our previous study showed that tafazzin knockdown activates AMPK and is involved in mitochondrial biogenesis, which contributes to cardiac myocyte hypertrophy [17]. As shown in Figure 4, AMPK was significantly activated (via increases in its phosphorylation) by tafazzin knockdown and this activation was blocked by mito-Tempo (Figure 4(a)). ROS reportedly activate Janus kinase (JAK) involved in cardiac hypertrophy [40, 41]. We tested whether tafazzin knockdown activates JAK and the effects of mito-Tempo on JAK activation. We found that tafazzin knockdown dramatically activated JAK2 by increasing its phosphorylation. Mito-Tempo significantly abrogated tafazzin knockdown induced JAK2 phosphorylation (Figure 4(b)). A cell permeable antioxidant Tempol (25 μM) was failed to inhibit tafazzin knockdown induced phosphorylation of AMPK or JAK2 (Figures 4(c) and 4(d)).
Figure 4.
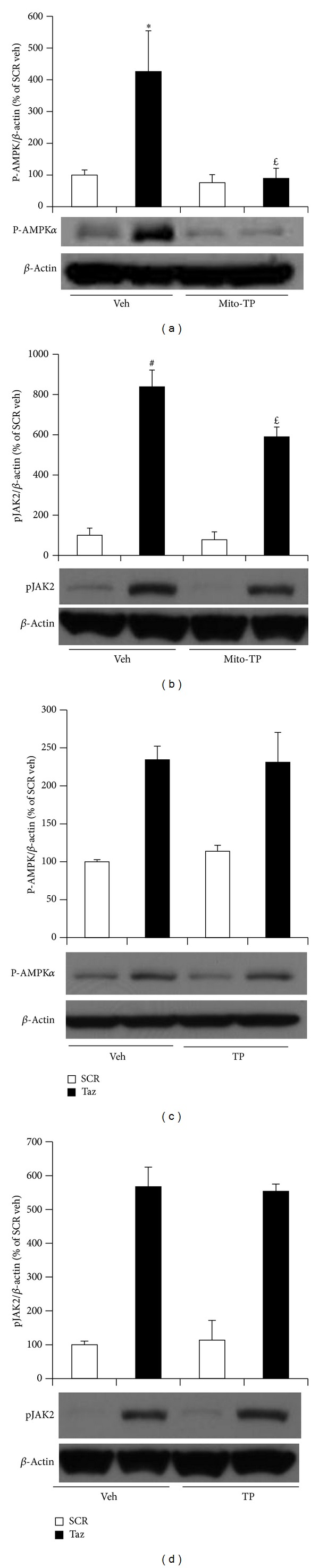
Mito-Tempo inhibited tafazzin knockdown induced protein kinases. NVMs were transduced with the shRNA adenovirus for 48 h, treated with 25 μM mito-Tempo (mito-TP) or Tempol (TP) for the last 24 h, and tested for phosphorylated AMPKα ((a) and (c)) and JAK2 ((b) and (d)). Data represent mean ± SE of 5 or 3 separate experiments for p-AMPKα with the treatments of mito-TP or TP and 4 separate experiments for JAK2 with the treatments mito-TP or TP. *P < 0.05 and # P < 0.01 versus SCR treated with vehicle. £ P < 0.05 versus Taz knockdown cells treated with vehicle.
3.5. Mitochondrial Antioxidant Prevented Cardiac Myocyte Hypertrophy Induced by Tafazzin Knockdown
Our previous study showed that tafazzin knockdown induces cardiac myocyte hypertrophy [17]. We questioned whether mito-Tempo affects hypertrophy. As showed in Figure 5(a), tafazzin knockdown increased cell surface area which is a marker of hypertrophy. This effect of tafazzin knockdown on cell surface area was blocked by the treatment of mito-Tempo. Further analysis revealed that mito-Tempo significantly attenuated not only cardiac myocyte surface area but also protein synthesis as assayed by 3H-leucine incorporation (Figure 5(b)). We conclude that mito-Tempo blocked cardiac hypertrophy induced by tafazzin knockdown.
Figure 5.
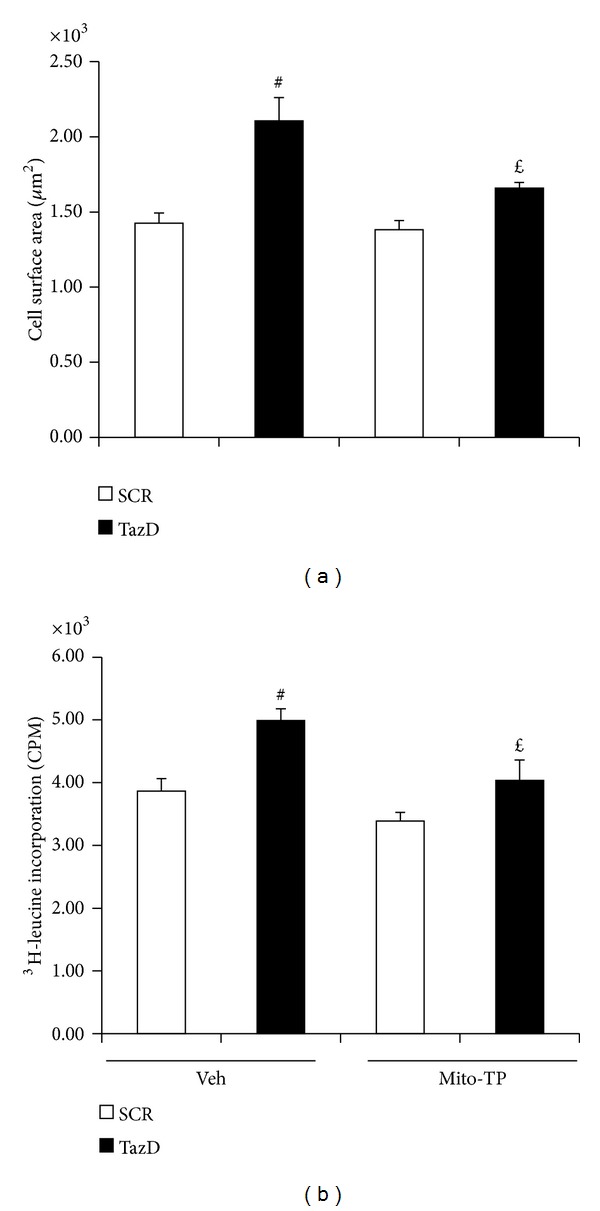
Mito-Tempo prevented hypertrophy induced by tafazzin knockdown. NVMs were transduced with the shRNA adenovirus for 48 h and assayed for hypertrophy. (a) Cell surface area. NVMs were fixed and stained for cell size. Data represent mean ± SE of 4 separate experiments. # P < 0.01 versus SCR treated with vehicle and £ P < 0.05 versus TazD treated with vehicle. (b). 3H-leucine incorporation. Data represent mean ± SE of 4 separate experiments. # P < 0.01 versus SCR treated with vehicle and £ P < 0.05 versus TazD treated with vehicle.
3.6. Mitochondrial Antioxidant Prevented Cardiac Myocyte Contractile Dysfunction Induced by Tafazzin Knockdown
Tafazzin mutation causes impaired cardiac function in Barth syndrome patients [42] and tafazzin knockdown in mice reduces cardiac and skeletal muscle contractility [14, 43]. We questioned whether tafazzin knockdown impairs cardiac myocyte contractility and the effects of mito-Tempo treatment. As shown in Figure 6(a), tafazzin shRNA adenovirus significantly knocked down tafazzin protein in ACM. Tafazzin knockdown decreased cardiac myocyte contractility as measured by sarcomere shortening. The treatment of Mito-Tempo prevented tafazzin knockdown- induced contractile dysfunction (Figure 6(b)). Tafazzin knockdown did not affect sarcomere basal length at relaxation (Figure 6(c)).
Figure 6.
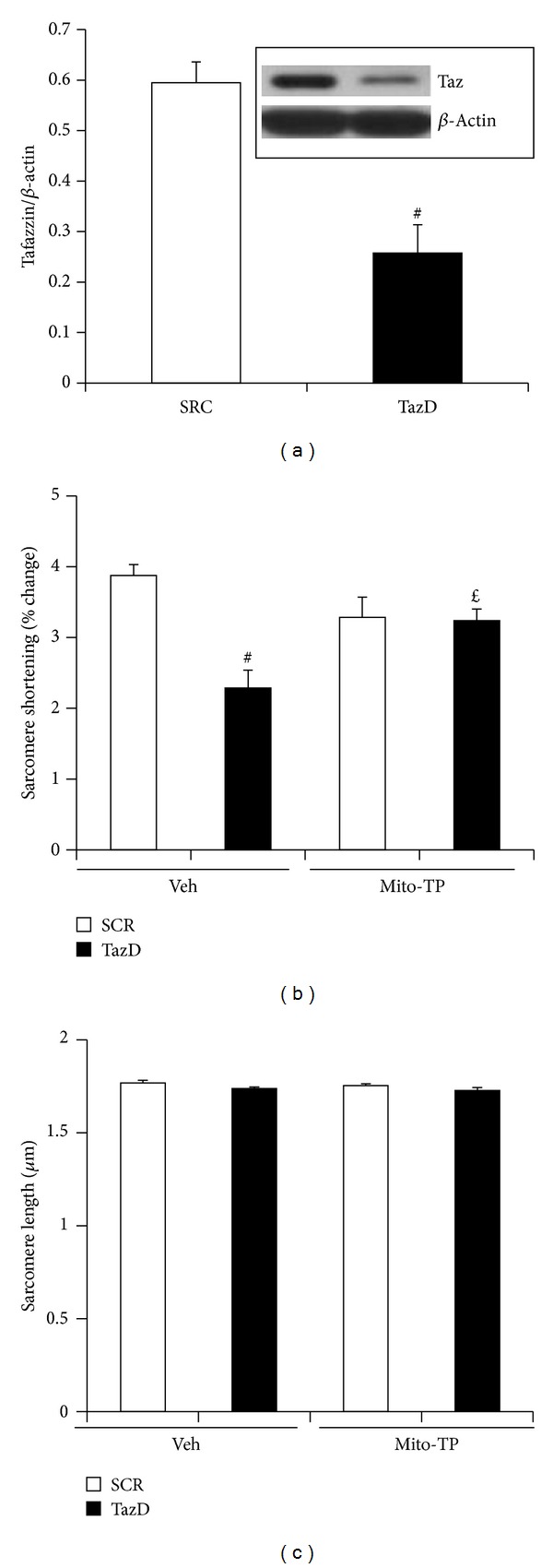
Mito-Tempo normalized contractile dysfunction induced by cardiolipin deficiency due to tafazzin knockdown. Cardiac myocytes isolated from adult mouse hearts were transduced with tafazzin shRNA adenovirus and treated with mito-Tempo overnight. (a) Tafazzin protein. Adenovirus containing tafazzin shRNA efficiently knocked down tafazzin protein. Data represent mean ± SE of 3 separate experiments. # P < 0.01 versus SCR. (b) Sarcomere shortening. Data represent mean ± SE of 4 separate experiments. # P < 0.01 versus SCR treated with vehicle and £ P < 0.05 versus TazD treated with vehicle. (c) Sarcomere length. Data represent mean ± SE of 4 separate experiments.
3.7. Mitochondrial Antioxidant Prevents Cytochrome C Release from the Mitochondria and Cell Death Induced by Tafazzin Knockdown
Since cytochrome c is attached to cardiolipin in the mitochondrial intermembrane space [44], cardiolipin deficiency would be expected to cause the release of cytochrome c into the cytoplasm. As shown in Figure 7(a), cardiolipin deficiency induced by tafazzin knockdown significantly decreased mitochondrial cytochrome c content and this decrease was blocked by mito-Tempo. Cytochrome c is released to the cytoplasm via mitochondrial outer membrane permeabilization [44] or opening of mitochondrial permeability transition pores (PTP) [45]. Cytochrome c release from mitochondria causes cell death via apoptosis. Cytochrome c release from mitochondria is also a sign of PTP opening leading to cell death via necrosis. We next checked the effect of tafazzin knockdown on cell survival and found that tafazzin knockdown decreased cell survival to about 75% compared to cells treated with a scrambled virus. Mito-Tempo normalized this decrease to 97% (Figure 7(b)). We did not find that tafazzin knockdown increased apoptosis by Tunel staining with a kit from EMD Millipore (data not shown). Most likely this is because the decreased cellular ATP in tafazzin knockdown cells since apoptosis is an ATP- dependent process [46].
Figure 7.
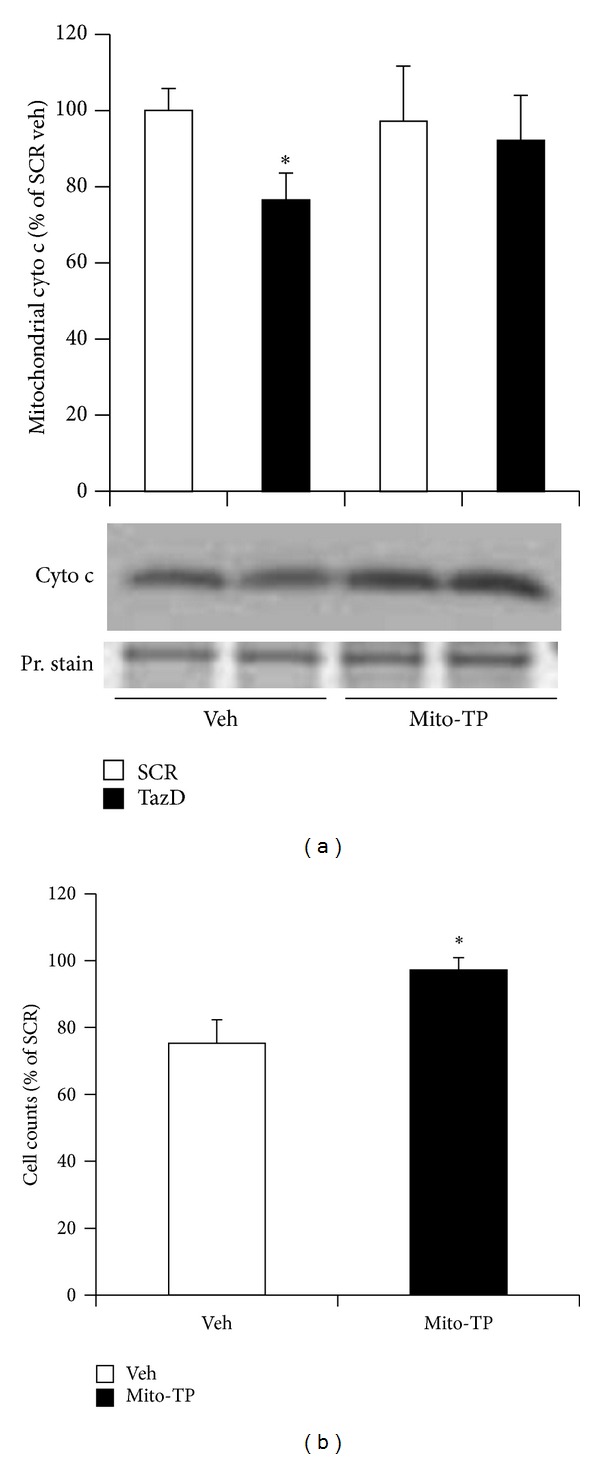
Mito-Tempo abrogated cytochrome c release and cell death induced by tafazzin knockdown. NVMs were transduced with the shRNA adenovirus for 48 h, mito-Tempo treated for the last 24 h, and tested for mitochondrial cytochrome c content and cell death. (a) Mitochondrial cytochrome c content. Data represent mean ± SE of 4 separate experiments. *P < 0.05 versus SCR treated with vehicle. (b) Cell count. Data represent mean ± SE of three separate experiments. *P < 0.05 versus vehicle.
4. Discussion
Tafazzin plays an important role in cardiolipin remodeling in the mitochondria. Tafazzin knockdown leads to reduced cardiolipin and enhances both monolysocardiolipin and the ratio of lysocardiolipin to cardiolipin, which are consistent with previous studies involved in different model systems [9, 14, 47]. Our results from NVMs revealed that the ratio of monolysocardiolipin to cardiolipin is a more sensitive marker of tafazzin dysfunction than cardiolipin content. This notion has been proved in cultured human skin fibroblasts and bloodspots of Barth syndrome patients [47, 48]. However, the profiles of cardiolipin species are different from the adult heart where tetralinoleoyl cardiolipin (T4 18 : 2 CL) is the predominant species. T4 18 : 2 CL is one of several abundant species in NVMs. This profile of cardiolipin matched that from 1-day-old mice [49] and neonatal cardiac fibroblasts [18]. Our current cardiolipin results suggest that total cardiolipin other than specific species (e.g., T4 18 : 2 CL) is important for mitochondrial function in the early stages of the neonatal heart. This notion is also supported by a study showing that the functions of cardiolipin species are indistinguishable in yeast [50].
Cardiolipin is essential for proper function of the mitochondrial respiration chain, and dysfunction of the chain complexes leads to increased production of ROS [51]. ROS play an important role in the pathophysiology of cardiac remodeling [52]. Our previous study showed that tafazzin knockdown induced cardiomyocyte hypertrophy [17], which is most likely mediated by ROS. We have shown that tafazzin dysfunction increases ROS production in yeast and neonatal cardiac fibroblasts [22]. Our current data showing that tafazzin knockdown increased ROS production are consistent with this notion. Mitochondrial ROS have been postulated as the major resource in cardiac myocytes, since they are filled with mitochondria and ROS are byproducts of the respiration chain there. The enhanced mitochondrial ROS production directly damages mitochondrial protein, DNA, and lipids, leading to impaired mitochondrial function. As our data showed, tafazzin knockdown decreased mitochondrial ATP production and cytochrome c content. The mitochondria-targeted ROS scavenger, mito-Tempo, normalized these mitochondrial dysfunctions. Considering that tafazzin knockdown did not decrease cardiolipin dramatically though statistically significant, it is possible that tafazzin knockdown directly enhanced mitochondrial ROS production. This possibility warrants further investigation.
Our previous study showed that activation of AMPK leading to mitochondrial biogenesis involved in tafazzin knockdown induced cardiac hypertrophy in NVM since tafazzin knockdown causes mitochondrial dysfunction [17]. The increased dysfunctional mitochondria do not improve oxidative stress or cellular ATP decline. They even make the situation worsen leading to cardiac hypertrophy. Previous studies also showed that ROS activate Jak2 and thereafter STAT3 involved in cardiac hypertrophy [41, 53, 54]. Our current study showed that mito-Tempo abolished tafazzin knockdown induced AMPK activation and partial attenuated JAK2 activation implying that mitochondrial dysfunction is the major mechanism responsible for cardiac dysfunction induced by tafazzin knockdown and mitochondrial ROS triggered JAK2 signing is partially involved.
ROS mediate cardiac myocyte hypertrophy induced by mechanical stretch [55] and hypertrophic factors such as endothelin and phenylephrine [56]. Hydrogen peroxide directly induced cardiac hypertrophy [57], and antioxidants prevent hypertrophy induced by tumor necrosis factor-α and angiotensin II [58]. A recent clinical investigation showed that a supplemental antioxidant has no beneficial effects on cardiovascular diseases [59]; however, mitochondria-targeted antioxidant MitoQ10 proved effective for endothelial function improvement and cardiac hypertrophy attenuation in the stroke-prone spontaneously hypertensive rat [35]. Our current study showed that mito-Tempo blocked tafazzin knockdown induced cardiac hypertrophy suggesting that mitochondria-targeted antioxidants are promising targets for cardiac hypertrophy.
Mutation of tafazzin results in poor contractility in Barth syndrome patients [60, 61] and this phenotype has been reproduced in mice [43] and zebrafish [13]. Our data showed that tafazzin knockdown impaired contractility in cardiac myocytes and this impairment was blocked by mito-Tempo, suggesting that the enhanced mitochondrial ROS production mediates tafazzin knockdown induced contractile dysfunction.
Though cardiolipin deficiency directly results in the dissociation of cytochrome c from cardiolipin into the intermembrane space of mitochondria, extrusion of this protein still needs permeabilization of the outer mitochondrial membrane [44]. Release of mitochondrial contents including cytochrome c is a sign of PTP opening leading to necrotic cell death. Previous studies also showed that cytochrome c is released in a ROS-dependent fashion [62]. Tafazzin knockdown induced cell death but not by apoptosis. This agrees with a previous report showing that dysfunctional tafazzin does not lead to apoptosis [15, 63]. Our current study showing that mito-Tempo prevented tafazzin knockdown induced cytochrome c release and cell death demonstrates a means for mitochondrial stress induced cardiac myocyte death in cardiomyopathy.
5. Conclusion
In summary, our study demonstrated that tafazzin knockdown causes enhanced mitochondrial ROS production leading to mitochondrial and cardiac dysfunction, and a mitochondria-targeted antioxidant prevented the mitochondrial and cardiac dysfunction, including decreased mitochondrial ATP production, cardiac myocyte hypertrophy, contractile dysfunction, and cell death. Our results shed light on the development of mitochondria-targeted antioxidants for cardiomyopathies that resulted from tafazzin mutation and mitochondrial oxidative stress.
Acknowledgments
This work was supported by grants from the Barth Syndrome Foundation and the American Heart Association 11SDG7400019.
Abbreviations
- ROS:
Reactive oxygen species
- NVM:
Neonatal ventricular myocyte
- ACM:
Adult cardiac myocyte
- shRNA:
Short hairpin RNA
- Tempol:
4-Hydroxy-2,2,6,6-tetramethylpiperidinyloxy
- Mito-Tempo:
(2-(2,2,6,6-Tetramethylpiperidin-1-oxyl-4-ylamino)-2-oxoethyl) triphenylphosphonium chloride
- Mn-SOD:
Manganese superoxide dismutase
- BDM:
2,3-Butanedione monoxime
- PTP:
Permeability transition pores.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Schlame M. Cardiolipin remodeling and the function of tafazzin. Biochimica et Biophysica Acta. 2013;1831(3):582–588. doi: 10.1016/j.bbalip.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 2.Xu Y, Malhotra A, Ren M, Schlame M. The enzymatic function of tafazzin. The Journal of Biological Chemistry. 2006;281(51):39217–39224. doi: 10.1074/jbc.M606100200. [DOI] [PubMed] [Google Scholar]
- 3.Zachman DK, Chicco AJ, McCune SA, Murphy RC, Moore RL, Sparagna GC. The role of calcium-independent phospholipase A2 in cardiolipin remodeling in the spontaneously hypertensive heart failure rat heart. Journal of Lipid Research. 2010;51(3):525–534. doi: 10.1194/jlr.M000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He Q, Han X. Cardiolipin remodeling in diabetic heart. Chemistry and Physics of Lipids. 2013;179:75–81. doi: 10.1016/j.chemphyslip.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 5.Bione S, D'Adamo P, Maestrini E, Gedeon AK, Bolhuis PA, Toniolo D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nature Genetics. 1996;12(4):385–389. doi: 10.1038/ng0496-385. [DOI] [PubMed] [Google Scholar]
- 6.Barth PG, Valianpour F, Bowen VM, et al. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): an update. American Journal of Medical Genetics A. 2004;126(4):349–354. doi: 10.1002/ajmg.a.20660. [DOI] [PubMed] [Google Scholar]
- 7.Spencer CT, Bryant RM, Day J, et al. Cardiac and clinical phenotype in Barth syndrome. Pediatrics. 2006;118(2):e337–e346. doi: 10.1542/peds.2005-2667. [DOI] [PubMed] [Google Scholar]
- 8.Acehan D, Xu Y, Stokes DL, Schlame M. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Laboratory Investigation. 2007;87(1):40–48. doi: 10.1038/labinvest.3700480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gu Z, Valianpour F, Chen S, et al. Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Molecular Microbiology. 2004;51(1):149–158. doi: 10.1046/j.1365-2958.2003.03802.x. [DOI] [PubMed] [Google Scholar]
- 10.Ma L, Vaz FM, Gu Z, Wanders RJA, Greenberg ML. The human TAZ gene complements mitochondrial dysfunction in the yeast taz1delta mutant: implications for Barth syndrome. The Journal of Biological Chemistry. 2004;279(43):44394–44399. doi: 10.1074/jbc.M405479200. [DOI] [PubMed] [Google Scholar]
- 11.Xu Y, Sutachan JJ, Plesken H, Kelley RI, Schlame M. Characterization of lymphoblast mitochondria from patients with Barth syndrome. Laboratory Investigation. 2005;85(6):823–830. doi: 10.1038/labinvest.3700274. [DOI] [PubMed] [Google Scholar]
- 12.Xu Y, Condell M, Plesken H, et al. A Drosophila model of Barth syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(31):11584–11588. doi: 10.1073/pnas.0603242103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khuchua Z, Yue Z, Batts L, Strauss AW. A zebrafish model of human barth syndrome reveals the essential role of tafazzin in cardiac development and function. Circulation Research. 2006;99(2):201–208. doi: 10.1161/01.RES.0000233378.95325.ce. [DOI] [PubMed] [Google Scholar]
- 14.Acehan D, Vaz F, Houtkooper RH, et al. Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. The Journal of Biological Chemistry. 2011;286(2):899–908. doi: 10.1074/jbc.M110.171439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phoon CK, Acehan D, Schlame M, et al. Tafazzin knockdown in mice leads to a developmental cardiomyopathy with early diastolic dysfunction preceding myocardial noncompaction. Journal of the American Heart Association. 2012;1(2) doi: 10.1161/JAHA.111.000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang F, Ryan MT, Schlame M, et al. Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. The Journal of Biological Chemistry. 2000;275(29):22387–22394. doi: 10.1074/jbc.M909868199. [DOI] [PubMed] [Google Scholar]
- 17.He Q. Tafazzin knockdown causes hypertrophy of neonatal ventricular myocytes. The American Journal of Physiology—Heart and Circulatory Physiology. 2010;299(1):H210–H216. doi: 10.1152/ajpheart.00098.2010. [DOI] [PubMed] [Google Scholar]
- 18.He Q, Wang M, Harris N, Han X. Tafazzin knockdown interrupts cell cycle progression in cultured neonatal ventricular fibroblasts. The American Journal of Physiology—Heart and Circulatory Physiology. 2013;305(9):H1332–H1343. doi: 10.1152/ajpheart.00084.2013. [DOI] [PubMed] [Google Scholar]
- 19.Bunse M, Bit-Avragim N, Riefflin A, et al. Cardiac energetics correlates to myocardial hypertrophy in Friedreich's ataxia. Annals of Neurology. 2003;53(1):121–123. doi: 10.1002/ana.10419. [DOI] [PubMed] [Google Scholar]
- 20.Merante F, Tein I, Benson L, Robinson BH. Maternally inherited hypertrophic cardiomyopathy due to a novel T-to-C transition at nucleotide 9997 in the mitochondrial tRNA(glycine) gene. The American Journal of Human Genetics. 1994;55(3):437–446. [PMC free article] [PubMed] [Google Scholar]
- 21.Sproule DM, Kaufmann P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Annals of the New York Academy of Sciences. 2008;1142:133–158. doi: 10.1196/annals.1444.011. [DOI] [PubMed] [Google Scholar]
- 22.Chen S, He Q, Greenberg ML. Loss of tafazzin in yeast leads to increased oxidative stress during respiratory growth. Molecular Microbiology. 2008;68(4):1061–1072. doi: 10.1111/j.1365-2958.2008.06216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circulation Research. 2010;107(9):1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gandhi S, Abramov AY. Mechanism of oxidative stress in neurodegeneration. Oxidative Medicine and Cellular Longevity. 2012;2012:11 pages. doi: 10.1155/2012/428010.428010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perrelli MG, Pagliaro P, Penna C. Ischemia/reperfusion injury and cardioprotective mechanisms: role of mitochondria and reactive oxygen species. World Journal of Cardiology. 2011;3:186–200. doi: 10.4330/wjc.v3.i6.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seddon M, Looi YH, Shah AM. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart. 2007;93(8):903–907. doi: 10.1136/hrt.2005.068270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiological Reviews. 1979;59(3):527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 28.Addabbo F, Montagnani M, Goligorsky MS. Mitochondria and reactive oxygen species. Hypertension. 2009;53(6):885–892. doi: 10.1161/HYPERTENSIONAHA.109.130054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radical Biology and Medicine. 2009;47(4):333–343. doi: 10.1016/j.freeradbiomed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 30.Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension. 2007;49(2):241–248. doi: 10.1161/01.HYP.0000254415.31362.a7. [DOI] [PubMed] [Google Scholar]
- 31.Li Y, Huang T-T, Carlson EJ, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature Genetics. 1995;11(4):376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 32.Nojiri H, Shimizu T, Funakoshi M, et al. Oxidative stress causes heart failure with impaired mitochondrial respiration. The Journal of Biological Chemistry. 2006;281(44):33789–33801. doi: 10.1074/jbc.M602118200. [DOI] [PubMed] [Google Scholar]
- 33.Matsushima S, Ide T, Yamato M, et al. Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation. 2006;113(14):1779–1786. doi: 10.1161/CIRCULATIONAHA.105.582239. [DOI] [PubMed] [Google Scholar]
- 34.Myung S-K, Ju W, Cho B, et al. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: systematic review and meta-analysis of randomised controlled trials. British Medical Journal. 2013;346(7893, article f10) doi: 10.1136/bmj.f10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Graham D, Huynh NN, Hamilton CA, et al. Mitochondria-targeted antioxidant mitoq10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. 2009;54(2):322–328. doi: 10.1161/HYPERTENSIONAHA.109.130351. [DOI] [PubMed] [Google Scholar]
- 36.Wang F, He Q, Sun Y, Dai X, Yang X-P. Female adult mouse cardiomyocytes are protected against oxidative stress. Hypertension. 2010;55(5):1172–1178. doi: 10.1161/HYPERTENSIONAHA.110.150839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han X, Yang K, Yang J, Cheng H, Gross RW. Shotgun lipidomics of cardiolipin molecular species in lipid extracts of biological samples. Journal of Lipid Research. 2006;47(4):864–879. doi: 10.1194/jlr.D500044-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winer J, Jung CKS, Shackel I, Williams PM. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Analytical Biochemistry. 1999;270(1):41–49. doi: 10.1006/abio.1999.4085. [DOI] [PubMed] [Google Scholar]
- 39.Smith RAJ, Porteous CM, Gane AM, Murphy MP. Delivery of bioactive molecules to mitochondria in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(9):5407–5412. doi: 10.1073/pnas.0931245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu C, Cao F, Tang QZ, et al. Allicin protects against cardiac hypertrophy and fibrosis via attenuating reactive oxygen species-dependent signaling pathways. Journal of Nutritional Biochemistry. 2010;21(12):1238–1250. doi: 10.1016/j.jnutbio.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 41.Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. The American Journal of Physiology—Cell Physiology. 1998;275(6):C1640–C1652. doi: 10.1152/ajpcell.1998.275.6.C1640. [DOI] [PubMed] [Google Scholar]
- 42.Roberts AE, Nixon C, Steward CG, et al. The Barth Syndrome Registry: distinguishing disease characteristics and growth data from a longitudinal study. American Journal of Medical Genetics A. 2012;158(11):2726–2732. doi: 10.1002/ajmg.a.35609. [DOI] [PubMed] [Google Scholar]
- 43.Soustek MS, Falk DJ, Mah CS, et al. Characterization of a transgenic short hairpin RNA-induced murine model of tafazzin deficiency. Human Gene Therapy. 2011;22(7):865–871. doi: 10.1089/hum.2010.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(3):1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morin D, Pires F, Plin C, Tillement J-P. Role of the permeability transition pore in cytochrome C release from mitochondria during ischemia-reperfusion in rat liver. Biochemical Pharmacology. 2004;68(10):2065–2073. doi: 10.1016/j.bcp.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 46.Shimizu S. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Funkcialaj Ekvacioj. Serio Internacia. 1997;40(1):57–77. [PubMed] [Google Scholar]
- 47.van Werkhoven MA, Thorburn DR, Gedeon AK, Pitt JJ. Monolysocardiolipin in cultured fibroblasts is a sensitive and specific marker for Barth syndrome. Journal of Lipid Research. 2006;47(10):2346–2351. doi: 10.1194/jlr.D600024-JLR200. [DOI] [PubMed] [Google Scholar]
- 48.Kulik W, Van Lenthe H, Stet FS, et al. Bloodspot assay using HPLC-tandem mass spectrometry for detection of barth syndrome. Clinical Chemistry. 2008;54(2):371–378. doi: 10.1373/clinchem.2007.095711. [DOI] [PubMed] [Google Scholar]
- 49.Kiebish MA, Yang K, Sims HF, et al. Myocardial regulation of lipidomic flux by cardiolipin synthase: setting the beat for bioenergetic efficiency. The Journal of Biological Chemistry. 2012;287(30):25086–25097. doi: 10.1074/jbc.M112.340521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baile MG, Sathappa M, Lu YW, et al. Unremodeled and remodeled cardiolipin are functionally indistinguishable in yeast. The Journal of Biological Chemistry. 2014;289(3):1768–1778. doi: 10.1074/jbc.M113.525733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galati D, Srinivasan S, Raza H, et al. Role of nuclear-encoded subunit Vb in the assembly and stability of cytochrome c oxidase complex: implications in mitochondrial dysfunction and ROS production. The Biochemical Journal. 2009;420(3):439–449. doi: 10.1042/BJ20090214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. The American Journal of Physiology—Heart and Circulatory Physiology. 2011;301(6):H2181–H2190. doi: 10.1152/ajpheart.00554.2011. [DOI] [PubMed] [Google Scholar]
- 53.Modesti A, Bertolozzi I, Gamberi T, et al. Hyperglycemia activates JAK2 signaling pathway in human failing myocytes via angiotensin II-mediated oxidative stress. Diabetes. 2005;54(2):394–401. doi: 10.2337/diabetes.54.2.394. [DOI] [PubMed] [Google Scholar]
- 54.Wagner MA, Siddiqui MA. The JAK-STAT pathway in hypertrophic stress signaling and genomic stress response. JAKSTAT. 2012;1(2):131–141. doi: 10.4161/jkst.20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pimentel DR, Adachi T, Ido Y, et al. Strain-stimulated hypertrophy in cardiac myocytes is mediated by reactive oxygen species-dependent Ras S-glutathiolation. Journal of Molecular and Cellular Cardiology. 2006;41(4):613–622. doi: 10.1016/j.yjmcc.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 56.Tanaka K, Honda M, Takabatake T. Redox regulation of MAPK pathways and cardiac hypertrophy in adult rat cardiac myocyte. Journal of the American College of Cardiology. 2001;37:676–685. doi: 10.1016/s0735-1097(00)01123-2. [DOI] [PubMed] [Google Scholar]
- 57.Liu L, Li J, Liu J, et al. Involvement of Na+/K+-ATPase in hydrogen peroxide-induced hypertrophy in cardiac myocytes. Free Radical Biology and Medicine. 2006;41(10):1548–1556. doi: 10.1016/j.freeradbiomed.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 58.Nakamura K, Fushimi K, Kouchi H, et al. Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensin II. Circulation. 1998;98(8):794–799. doi: 10.1161/01.cir.98.8.794. [DOI] [PubMed] [Google Scholar]
- 59.Myung S-K, Ju W, Cho B, et al. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: systematic review and meta-analysis of randomised controlled trials. BMJ. 2013;346(7893) article f10 doi: 10.1136/bmj.f10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Momoi N, Chang B, Takeda I, Aoyagi Y, Endo K, Ichida F. Differing clinical courses and outcomes in two siblings with Barth syndrome and left ventricular noncompaction. European Journal of Pediatrics. 2012;171(3):515–520. doi: 10.1007/s00431-011-1597-0. [DOI] [PubMed] [Google Scholar]
- 61.Steward CG, Newbury-Ecob RA, Hastings R, et al. Barth syndrome: an X-linked cause of fetal cardiomyopathy and stillbirth. Prenatal Diagnosis. 2010;30(10):970–976. doi: 10.1002/pd.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Atlante A, Calissano P, Bobba A, Azzariti A, Marra E, Passarella S. Cytochrome c is released from mitochondria in a reactive oxygen species (ROS)-dependent fashion and can operate as a ROS scavenger and as a respiratory substrate in cerebellar neurons undergoing excitotoxic death. Journal of Biological Chemistry. 2000;275(47):37159–37166. doi: 10.1074/jbc.M002361200. [DOI] [PubMed] [Google Scholar]
- 63.Valianpour F, Mitsakos V, Schlemmer D, et al. Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis. Journal of Lipid Research. 2005;46(6):1182–1195. doi: 10.1194/jlr.M500056-JLR200. [DOI] [PubMed] [Google Scholar]