

Abstract
Huntington's disease (HD) is a progressive polyglutamine disease that leads to a severe striatal and layer‐specific neuronal loss in the cerebral neo‐and allocortex. As some of the clinical symptoms (eg, oculomotor dysfunctions) suggested a degeneration of select brainstem nuclei, we performed a systematic investigation of the brainstem of eight clinically diagnosed and genetically confirmed HD patients. This post‐mortem investigation revealed a consistent neuronal loss in the substantia nigra, pontine nuclei, reticulotegmental nucleus of the pons, superior and inferior olives, in the area of the excitatory burst neurons for horizontal saccades, raphe interpositus nucleus and vestibular nuclei. Immunoreactive intranuclear neuronal inclusions were present in all degenerated and apparently spared brainstem nuclei and immunoreactive axonal inclusions were observed in all brainstem fiber tracts of the HD patients. Degeneration of brainstem nuclei can account for a number of less well‐understood clinical HD symptoms (ie, cerebellar, oculomotor and vestibular symptoms), while the formation of axonal aggregates may represent a crucial event in the cascades of pathological events leading to neurodegeneration in HD.
Keywords: axonal inclusions, brainstem, Huntington's disease, neurodegeneration, pathoanatomy, polyglutamine diseases
Introduction
Huntington's disease (HD) is an autosomal dominantly inherited, progressive and currently untreatable polyglutamine or CAG‐repeat disease 10, 16, 17, 24, 31, 46, 49, 56, 62, 72, 76, 77, 81, 82, 85, 86. Clinically, HD presents with somatomotor (ie, chorea, bradykinesia, dystonia, rigidity, dysarthria, dysphagia), oculomotor and psychiatric symptoms, cognitive decline and often with a severe weight loss 5, 16, 25, 26, 27, 34, 42, 44, 49, 51, 62, 63, 72, 76, 83, 85, 86. Macroscopic brain alterations in HD commonly include atrophy of all cerebral lobes, reduction of cerebral white matter, atrophy of the striatum and enlarged lateral ventricles 3, 5, 15, 25, 26, 27, 39, 40, 41, 62, 63, 72, 73, 81, 82, 83. Although the degenerative process of the HD brain is widespread, to date, the severe neuronal loss in the striatum and the layer‐specific neurodegeneration in distinct areas of the cerebral neo‐ and allocortex is considered as generally the hallmark of the neuropathological changes 3, 5, 15, 25, 26, 27, 31, 35, 39, 40, 41, 47, 52, 62, 63, 72, 73, 81, 82, 83. Based on the worsening and topographical spread of the degenerative process of the striatum, a neuropathological HD grading system has been developed 81, 82, 83. Some of the unexplained HD symptoms (eg, oculomotor dysfunctions) as well as the previous, but widely forgotten assumptions have suggested additional degeneration of select brainstem nuclei in HD and pointed to the necessity of detailed brainstem studies in HD 5, 32, 34, 35, 41, 42, 44, 45, 62, 64, 81, 82, 83.
Located on chromosome 4p16.3, the HD gene harbors meiotically unstable CAG‐repeats which encode the pathogenic mutated protein huntingtin 16, 17, 31, 46, 49, 51, 72, 73, 76, 78, 81, 85, 86. In symptomatic HD patients and asymptomatic gene carriers, these CAG‐repeats are expanded and lead to a polyglutamine expansion in mutant huntingtin. The normal physiological CAG‐repeat sequences comprise six to 35 CAG triplets, whereby sequences of 28 and more already behave instable during meiosis and may be prone to potentially symptomatic mutations. CAG‐repeat sequences longer than 35 are considered pathologically expanded, 36 to 40 CAG‐repeats lead to an incomplete penetrance and expansions of 41 or more CAG‐repeats to the fully developed clinical picture of HD 16, 31, 46, 49, 51, 66, 72, 76, 81, 85, 86.
The subcellular neuronal localization of huntingtin is complex and dynamic. It is normally associated with the Golgi complex and mitochondria, localizes to microtubules and synaptic vesicles, is believed to play an important role in embryogenic development, gene transcription, regulation of axonal transport mechanisms, anterograde and retrograde trafficking of vesicles and organelles, as well as in transport of signaling and trophic factors 1, 17, 21, 31, 46, 66, 79, 81. As with other polyglutamine disease proteins, mutant huntingtin tends to aggregate into insoluble neuronal intranuclear inclusions (NI), which also contain components of the intracellular machinery responsible for protein handling and degrading of misfolded proteins (eg, ubiquitin, proteasome subunits, chaperones) 2, 10, 14, 17, 19, 31, 36, 46, 47, 48, 65, 66, 73, 74, 77, 80, 81, 85, 86. Although these NI may be present in the cerebral cortex and striatum of HD patients, their pathogenic role in the demise of neurons still is uncertain 1, 2, 14, 17, 19, 22, 31, 36, 47, 48, 56, 72, 73, 74, 80, 81. Recently, we described the brain distribution, neuronal localization and composition of neuropil protein aggregates in another polyglutamine disease, the spinocerebellar ataxia type 3 (SCA3 or Machado‐Joseph disease) 69. Inspired by the enigmatic pathogenic role of NI in HD and our recent SCA3 findings, we performed a systematic immunohistochemical study of the brainstem of eight clinically diagnosed and genetically confirmed HD patients and analyzed the presence, distribution and localization of NI and neuropil aggregates in brainstem nuclei or fiber tracts. The results of these analyses were correlated with the pathoanatomical findings obtained upon investigation of unconventional thick tissue sections through the brainstem of these HD patients.
Patients and Methods
Patients
We investigated the brainstem of eight clinically diagnosed and genetically confirmed HD patients (age at death: 67.3 ± 11.3 years; Tables 1 and 2) and of 14 control individuals without histories of neurological or psychiatric diseases in their medical records and no diagnostic abnormalities recognized on neuropathological examination of their brain (eight females, six males; age at death: 61.6 ± 16.5 years) (Table 3). All of our HD patients suffered from involuntary choreatic movements and progressive cognitive decline (patients 1–8; Table 1). Seven showed a severe weight loss (patients 1, 2, 4–8; Table 1), five had dysphagia (patients 1, 2, 4, 5, 7; Tables 1 and 2) and four had dysarthria (patients 1, 4, 3, 7; Table 1). Oculomotor dysfunctions noticed in the medical records of the HD patients included slowed horizontal saccades (patient 1; Tables 1 and 2), saccadic smooth pursuits (patients 1 and 7; Tables 1 and 2) and inability to start horizontal saccades without an initial head thrust (patient 3; Tables 1 and 2). Symptoms indicating dysfunction of the cerebellum and/or the precerebellar brainstem nuclei included unsteady and broad‐based gait (patients 1, 2, 3, 7; Tables 1 and 2), dysarthria (patients 1, 3, 7; Table 1), slowed smooth pursuits (patients 1, 7; Tables 1 and 2), ataxia (patient 4, Tables 1 and 2), gait imbalance (patients 6 and 7; Tables 1 and 2) and impaired gait coordination (patient 6, Tables 1 and 2).
Table 1.
Overview of the Huntington's disease (HD) patients studied.Abbreviations: CAG = number of expanded CAG‐repeats in the mutated HD allele; F = female; HD = Huntington's disease; M = male; TI = time interval between the last neurological investigation and death
| HD Patient | Age | Gender | CAG | TI | Vonsattel grade | Onset of initial HD symptoms | Initial HD symtpoms | Duration of HD |
|---|---|---|---|---|---|---|---|---|
| 1 | 51 | M | 49 | 60 | 4 | 33 | Personality changes | 18 |
| 2 | 61 | F | 45 | 6 | 3 | 35 | Chorea | 26 |
| 3 | 64 | M | 41 | 3 | 2 | 55 | Chorea | 9 |
| 4 | 65 | M | 45 | 36 | 4 | 53 | Ataxia | 12 |
| 5 | 68 | F | 46 | 18 | 3 | 40 | Cognitive decline | 28 |
| 6 | 68 | M | 40 | 29 | 3 | 60 | Cognitive decline | 8 |
| 7 | 70 | F | 44 | 4 | 2 | 50 | Chorea | 20 |
| 8 | 91 | M | 42 | 72 | 2 | 55 | Chorea | 36 |
Patient number, age at death (years), gender, number of expanded CAG‐repeats in the mutated HD allele, time interval between the last neurological investigation and death (TI, months), Vonsattel grade of striatal atrophy, age at onset of initial HD symptoms (years), initial HD symptoms, duration of HD (years).
Table 2.
Disease symptoms of the Huntington's disease (HD) patients studied possibly related to brainstem damage
| Patient | Disease symptoms |
|---|---|
| 1 | Broad‐based gait, dysphagia, slowed horizontal saccades, slowed and saccadic smooth pursuits |
| 2 | Broad‐based gait, dysphagia |
| 3 | Broad‐based gait, inability to start horizontal saccades without initial head thrust |
| 4 | Ataxia, dysphagia |
| 5 | Dysphagia |
| 6 | Gait imbalance |
| 7 | Broad‐based gait, gait imbalance, dysphagia, slowed and saccadic smooth pursuits |
| 8 | — |
Patient number, disease symptoms possibly related to brainstem damage.
Table 3.
Overview of the control individuals studied
| Control individual | Age | Gender | Cause of death | Post‐mortem delay |
|---|---|---|---|---|
| 1 | 39 | F | Ulcerative colitis | 12 |
| 2 | 39 | F | Arrhythmia | 21 |
| 3 | 39 | F | Thrombotic embolism | 16 |
| 4 | 52 | F | Cachexia | 24 |
| 5 | 54 | M | Cardiac arrest | 24 |
| 6 | 57 | F | Arrhythmia | 8 |
| 7 | 59 | M | Arrhythmia | 12 |
| 8 | 63 | F | Lymphoproliferative disease | 18 |
| 9 | 65 | F | Cardiac arrest | 24 |
| 10 | 66 | M | Lymphoma, Sepsis | 20 |
| 11 | 76 | M | Urosepsis | 19 |
| 12 | 84 | M | Myocardial infarction | 20 |
| 13 | 84 | M | Myocardial infarction | 17 |
| 14 | 86 | F | Bronchopneumonia | 18 |
Number of control individual, age at death (years), gender (F = female; M = male), cause of death, post‐mortem delay (hours).
Genetical examination revealed the presence of pathologically expanded CAG‐repeats in the mutated HD allele of all HD patients (Table 1) 16, 49, 51, 76.
The primary causes of death of our HD patients included cachexia (patients 1, 2, 4, 8; Table 1), subdural hematoma (patient 3; Table 1), aspiration pneumonia (patients 5 and 7; Table 1), ruptured aortic aneurysm (patient 6; Table 1) and pneumonia (patient 7; Table 1).
Tissue preparation and treatment
The HD patients' time interval between the last neurological examination and death was 28.5 ± 26.2 months (Table 1). Autopsy of the HD patients was performed within 10.9 ± 5.2 h and that of the control individuals within 18.1 ± 4.8 h post‐mortem (Table 3). Neuropathological investigation of the brains of the HD patients and control individuals was performed by an experienced neuropathologist (WF den Dunnen). Along with macroscopical investigations, routine neuropathological examination also included assessment of thin brain tissue sections. After fixation in a 4% aqueous formaldehyde solution, the 22 brains (eight HD patients, 14 controls) were assessed macroscopically. A grade of neuropathological severity of striatal degeneration was assigned to the HD brains according to the Vonsattel grading system (Table 1) 81, 82, 83. Tissue blocks of the lower brainstem were embedded in polyethylene glycol (PEG 1000, Merck, Darmstadt, Germany) 75 and cut into seven equidistant series of 100 μm thick horizontal sections.
In each instance the first set of serial brainstem tissue sections was stained for lipofuscin pigment (aldehyde‐fuchsin) and Nissl material (Darrow red) 3, 4, 61, 62, 63, 64 and applied for neuroanatomical orientation and pathoanatomical assessment of nerve cell loss in the HD patients (Table 4).
Table 4.
Distribution and extent of neuronal loss in the lower brainstem of Huntington's disease (HD) patients.Abbreviations: EBR = area of the excitatory burst neurons for horizontal saccades; IO = inferior olive; LV = lateral vestibular nucleus; MV = medial vestibular nucleus; PCRT = parvocellular reticular nucleus; PN = pontine nuclei; RIP = raphe interpositus nucleus; RTTG = reticulotegmental nucleus of the pons; SNC = substantia nigra, compact part; SNR = substantia nigra, reticulate part; SO = superior olive; SUV = superior vestibular nucleus; VII = facial nucleus
| Patient | SNC | SNR | PN | RTTG | SUV | EBR | RIP | SO | VII | PCRT | LV | MV | IO |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | + | + | + | + | − | ++ | ++ | ++ | + | + | + | + | + |
| 2 | + | + | + | + | + | ++ | + | ++ | + | + | + | + | + |
| 3 | + | + | + | + | + | + | + | ++ | − | − | + | + | + |
| 4 | + | + | + | + | + | ++ | ++ | ++ | − | − | + | + | + |
| 5 | + | + | + | + | − | + | ++ | + | − | − | + | + | + |
| 6 | + | + | + | + | − | + | ++ | + | − | − | + | + | + |
| 7 | + | + | + | + | − | ++ | ++ | ++ | − | − | + | + | + |
| 8 | + | + | + | + | − | ++ | + | ++ | − | + | + | + | + |
The pathoanatomical approach complements the routine neuropathological examination of thin brain tissue sections in the field of basic neuropathological research. Its application to unconventionally thick serial brain tissue sections allows the recognition of subtle pathological changes and reveals more detailed information with respect to the extent and severity of the pathological processes of neurodegenerative diseases 57, 59, 62, 64. Pathoanatomical examination of neuronal loss in the HD brainstems was performed twice by U. Rüb and its extent was scored as not discernible (–), marked (+) or severe (++) (Table 4). The second assessment of neuronal loss was perfomed 6 months after the first one and blinded to its results.
In all HD patients and control individuals, the second set of serial brainstem tissue sections was immunolabeled with an antibody directed against the protein aggregation marker p62 (1:100, Santa Cruz, Dallas, TX, USA). The multifunctional protein p62 participates in the intracellular trafficking, aggregation and degradation of various neurodegenerative disease proteins and is known to bind to abnormal nuclear and/or cytoplasmic neuronal accumulations of the disease‐specific proteins of various neurodegenerative diseases (35, 36, 67–69). Pretreatment for antigen retrieval before the application of the p62 antibody was performed with 10 mM citrate buffer (pH 6.0) at 90°C for 30 minutes. Positive immunoreactions were visualized with a 3,3‐diaminobenzidine‐tetra‐HCl/H2O2 reaction (DAB, D5637 Sigma, Taufkirchen, Germany) resulting in a brown staining. The p62‐immunolabeled tissue sections were counterstained for lipofuscin pigment (aldehyde‐fuchsin) and Nissl material (Darrow red) 3, 4, 61, 62, 63, 64 and used for the detection of NI bodies. The evaluation of NI in the HD brainstem nuclei was performed twice by U. Rüb and scored as follows: NI not detectable and this after confirmative, sequential assessments of a given brainstem nucleus (−), rare (+) or many (++). The second assessment of the presence of NI was performed 6 months after the first investigation and blinded to the results of the first assessment.
In each instance the third set of serial brainstem tissue sections was immunostained for GFAP to reveal reactive astrocytes.
Double immunostaining procedures were performed to identify and examine the topographical and intra‐axonal localization, as well as composition of neuropil aggregates. The AT270 antibody (1:4000, Thermo Scientific, Rockford, IL; USA), directed against the neuronal cytoskeletal protein tau was used as axonal marker 18, 71 and the anti‐p62 antibody applied as marker for axonal protein aggregates 37, 38, 67, 68, 69. Pretreatment for antigen retrieval prior to the application of the p62 antibody was performed with 10 mM citrate buffer (pH 6.0) at 90°C for 30 minutes. Positive axonal immunostaining was visualized with SK4700 (Linaris, D‐97877 Wertheim, Germany), resulting in a blue‐grey staining, and the p62 immunolabeling with 3,3‐diaminobenzidine‐tetra‐HCl/H2O2 (DAB, D5637 Sigma) resulting in a brown staining. The presence of p62‐immunopositive axonal inclusions was semi‐quantitatively assessed by U. Rüb and M. Hentschel and categorized as follows: axonal inclusions not detectable even after a time period of 3 minutes of careful investigation of a given fiber tract (−), or rare, or detectable after a time period of 3 minutes of examination (+), many (++) (Table 5). The assessments of U. Rüb and M. Hentschel were performed independently and blinded to the results of each other.
Table 5.
Distribution of p62‐immunopositive protein aggregates in axons of brainstem fiber tracts of Huntington's (HD) patients.Abbreviations: CCT = central tegmental tract; CU = cuneate fascicle; DST = dorsal spinocerebellar tract; GR = gracile fascicle; ICP = inferior cerebellar peduncle; LL = lateral lemniscus; MCP = medial cerebellar peduncle; ML = medial lemniscus; MLF = medial longitudinal fascicle; NSP = nigrostriatal pathway; OCF = olivocerebellar fibers; PCF = pontocerebellar fibers; PYR = pyramidal tract; SCP = superior cerebellar peduncle; SOL = solitary tract; SPV = spinal trigeminal tract; TZ = trapezoid body; VST = ventral spinocerebellar tract; III = oculomotor nerve; IV = trochlear nerve; V = trigeminal nerve; VI = abducens nerve; VII = facial nerve; VIII = vestibulocochlear nerve; IX/X = vagal nerve; XII = hypoglossal nerve
| Patient | III | IV | NSP | MLF | LL | ML | SCP | V | MCP | PCF | VI | VII | TZ | CTT | VIII | ICP | PYR | SPV | IX/X | SOL | OCF | CU | GR | DST | VST | XII |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | + | + | ++ | + | + | + | ++ | + | + | + | + | + | + | + | ++ | + | + | + | + | x | + | + | + | ++ | ++ | + |
| 2 | + | + | ++ | + | + | + | ++ | + | + | + | + | + | + | ++ | ++ | + | + | + | + | + | + | + | + | + | + | + |
| 3 | + | + | ++ | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 4 | + | + | ++ | + | + | + | + | + | + | ++ | + | + | + | ++ | + | + | + | + | + | + | + | + | + | + | + | + |
| 5 | + | + | ++ | + | + | + | + | + | + | ++ | + | + | + | ++ | + | + | + | ++ | + | ++ | ++ | + | + | + | + | − |
| 6 | + | + | ++ | + | ++ | + | ++ | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | ++ | ++ | + |
| 7 | + | + | ++ | ++ | ++ | + | + | + | + | + | + | + | + | ++ | + | + | + | + | + | + | + | ++ | ++ | + | + | + |
| 8 | + | + | ++ | + | + | + | ++ | ++ | + | + | + | ++ | + | ++ | ++ | + | + | ++ | + | + | ++ | + | + | + | + | + |
Double immunofluorescence was also applied to investigate the colocalization of the p62 shuttle protein with (i) the pathologically altered disease protein huntingtin harboring an elongated polyglutamin tract or (ii) ubiquitin in axonal aggregates. We applied the 1C2 antibody (1:3000, Millipore, Billerica, CA, USA) to confirm the presence of huntingtin with an expanded polyglutamine sequence of more than 37 repeats in the axonal aggregates and used an ubiquitin antibody (1:1000, Cell Signaling, Danvers, MA, USA) to investigate their additional association with the small degradation‐related protein ubiquitin. Select brainstem tissue sections of all HD patients were (i) incubated with both the monoclonal mouse and polyclonal rabbit primary antibodies for 20 h at room temperature; (ii) followed by secondary incubation for 90 minutes with Alexa 488 (1:1000, Invitrogen, Carlsbad, CA, USA) and CY3 conjugated secondary antibodies (1:500, Jackson ImmunoResearch, West Grove, PA, USA); and (iii) treated for 10 minutes with 0.06% sudan‐black for quenching of autofluorescence. The primary and secondary antibodies were applied simultaneously 69, 78. Antigen retrieval for p62/ubiquitin double immunostainings was performed as described above. Retrieval for 1C2/p62 was performed with Tris/HCl pH 9 at 90–05°C for 30 minutes, followed by incubation in 100% formic acid for 3 minutes at room temperature.
We investigated the reproducibility of the examination of the presence of NI, occurrence of axonal p62 immunoreactive aggregates in brainstem fiber tracts, and of brainstem neurodegeneration in the HD patients by means of inter‐rater and intra‐rater reliabilities and calculated the weighted kappa coefficient Κw (BiAS for Windows, version 9.14, Epsilon, Darmstadt, Germany). In the case of disagreement of the investigators, the final assessment of neuronal loss, presence of NI and intra‐axonal aggregates was decided during a final consensus meeting. Kendall's rank correlation coefficient tau (τ) was applied to examine the correlation between neuronal loss in brainstem nuclei of the HD patients and the length of the CAG‐repeats in the mutated HD allele, age of HD onset, duration of HD, age at death and the Vonsattel grade of striatal atrophy (BiAS for Windows, version 9.14, Epsilon). The τ coefficient was also used to describe the correlation between the extent of neuronal loss in the brainstem nuclei of the HD patients and the presence of p62 immunoreactive axonal inclusions in associated brainstem fiber tracts (BiAS for Windows, version 9.14, Epsilon).
Examination of the HD and control brainstems was approved by the Ethical board of the Faculty of Medicine at the Goethe University of Frankfurt/Main.
Results
The lower brainstem of all HD patients along with a flattened pons showed no macroscopic signs of degeneration (Figure 1).
Figure 1.
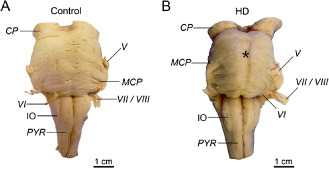
The lower brainstem in H untington's disease ( HD). A. Ventral aspect of the lower brainstem of a typical 76‐year‐old male control individual (control individual 11; Table 3). B. The lower brainstem with a flattened pons (asterisk) of a clinically diagnosed and genetically confirmed female HD patient (onset of HD symptoms: 40 years; age at death: 68 years; CAG‐repeats in the mutated HD allele: 46; Vonsattel grade of striatal atrophy: 3) (HD patient 5; Tables 1 and 2) 81, 82, 83.
Abbreviations: CP = cerebral peduncle; IO = inferior olive; MCP = medial cerebellar peduncle; PYR = pyramidal tract; V = trigeminal nerve; VI = abducens nerve; VII = facial nerve; VIII = Vestibulocochlear nerve.
In contrast to the routine neuropathological examination performed on thin tissue sections, our pathoanatomical study of thick tissue sections revealed neurodegeneration of select brainstem nuclei in all HD patients (Table 4).
A consistent neuronal loss was present in the dopaminergic compact part and GABAergic reticulate part of the substantia nigra, precerebellar pontine nuclei, reticulotegmental nucleus of the pons (Figure 2A,B), inferior olive (Figure 3E,F), in the premotor oculomotor area of the excitatory burst neurons for horizontal saccades (Figure 2C,D), raphe interpositus nucleus (Figure 2E,F), auditory superior olive (Figure 3C,D), lateral and medial vestibular nuclei (Figure 3A,B) Table 4). In addition, in a subset of our HD patients, a mild neuronal loss was also present in the superior vestibular nucleus, as well as in the ingestion‐related facial and parvocellular reticular nuclei (Table 4).
Figure 2.
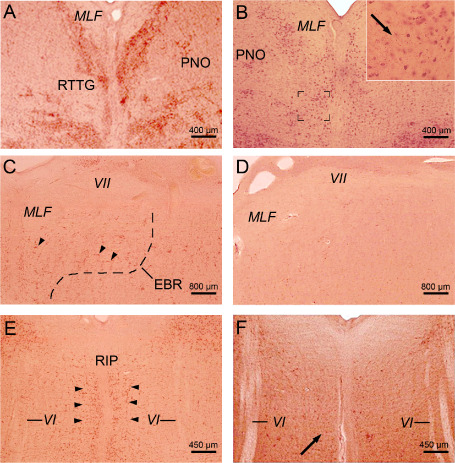
Neuronal loss in mid‐pontine and pontomedullary nuclei in H untington's disease ( HD ). A. Horizontal section through the mid‐pons of a representative 66‐year‐old male control individual depicting the oculomotor reticulotegmental nucleus of the pons (RTTG) (control individual 10; Table 3). B. Considerable neuronal loss in the RTTG of a 64‐year‐old male HD patient (CAG‐repeats in the mutated HD allele: 41; age at HD onset: 55 years; duration of HD: 9 years; Vonsattel grade of striatal atrophy: 2; patient 3; Tables 1, 2 and 4). Inset shows remaining RTTG nerve cells and lipofuscin deposits in the neuropil (arrow). C. Horizontal section through the caudal pons of a typical 54‐year‐old male control individual depicting the area of the excitatory burst neurons (arrowheads) for horizontal saccades (EBR) with its typical horizontally oriented nerve cells (control individual 5; Table 3). D. Complete neuronal loss in the EBR of a 51‐year‐old male HD patient (CAG‐repeats in the mutated HD allele: 49; age at HD onset: 33 years; duration of HD: 18 years; Vonsattel grade of striatal atrophy: 4; patient 1; Tables 1, 2 and 4). E. Horizontal section through the pontomedullary junction of a 39‐year‐old woman without any medical histories of neuropsychiatric diseases showing the RIP with its typical omnipause neurons for saccades in all directions (arrowheads) (control individual 3; Table 3). F. Severe loss of RIP omnipause neurons of a 70‐year‐old female HD patient. Arrow points to a surviving RIP nerve cell (CAG‐repeats in the mutated HD allele: 44; age at HD onset: 50 years; duration of HD: 20 years; Vonsattel grade of striatal atrophy: 2; patient 7; Tables 1, 2 and 4) (A–F: aldehyde‐fuchsin Darrow red staining; 100 μm PEG sections).
Abbreviations: MLF = medial longitudinal fascicle; PNO = pontine reticular nucleus, oral subnucleus; RIP = raphe interpositus nucleus; VI = abducens nerve; VII = facial nerve.
Figure 3.
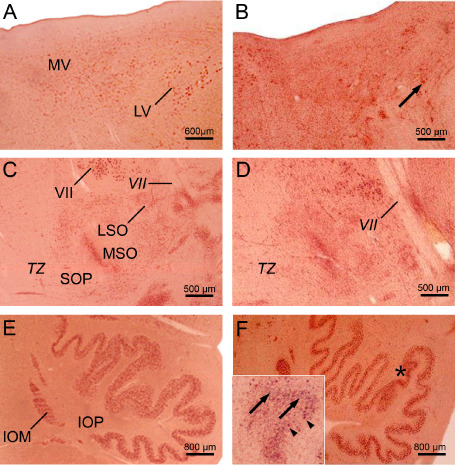
Neuronal loss in the pontomedullary junction and medulla oblongata in H untington's disease ( HD ). A. Horizontal section through the pontomedullary junction of a typical 54‐year‐old male control individual depicting the lateral (LV) and magnocellular portion of the medial (MV) vestibular nuclei (control individual 5; Table 3). B. Severe loss of LV and MV nerve cells of a 61‐year‐old female HD patient (CAG‐repeats in the mutated HD allele: 45; age at HD onset: 35 years; duration of HD: 26 years; Vonsattel grade of striatal atrophy: 3; patient 2; Tables 1, 2 and 4). Arrow points to a remaining giant LV nerve cell. C. Horizontal section through the mid‐pons of a representative 54‐year‐old male control individual depicting the auditory superior olive (SO) with its medial (MSO) and lateral subnuclei (LSO) as well as its periolivary group (SOP) (control individual 5; Table 3). D. SO neuronal loss of a 61‐year‐old female HD patient (CAG‐repeats in the mutated HD allele: 45; age at HD onset: 35 years; duration of HD: 26 years; Vonsattel grade of striatal atrophy: 3; patient 2; Tables 1, 2 and 4) predominantly affecting the LSO and SOP. E. Horizontal section through the medulla oblongata a 84‐year‐old male control individual without any medical histories of neuropsychiatric diseases showing the inferior olive (IO) with its principal (IOP) and medial accessory subnuclei (IOM) (control individual 13; Table 3). F. Circumscribed severe neuronal loss in the IOP (asterisk) of an 91‐year‐old male HD patient (CAG‐repeats in the mutated HD allele: 42; age at HD onset: 55 years; duration of HD: 36 years; Vonsattel grade of striatal atrophy: 2; patient 8; Tables 1, 2 and 4). Inset shows devastated area of the IOP (marked in F by an asterisk) with severe neuronal loss and the presence of extraneuronal lipofuscin deposits. Arrowheads point to remaining IOP nerve cells and arrows to extraneuronal lipofuscin deposits which can serve as reliable marker for neurodegeneration (A–F: aldehyde‐fuchsin Darrow red staining; 100 μm PEG sections).
Abbreviations: IO = inferior olive; IOM = inferior olive, medial accessory subnucleus; IOP = inferior olive, principal subnucleus; LSO = lateral superior olive; LV = lateral vestibular nucleus; MSO = medial superior olive; MV = medial vestibular nucleus, magnocellular portion; SO = superior olive; SOP = superior olive, periolivary group; TZ = trapezoid body; VII = facial nerve.
Calculation of Kendall's rank correlation coefficient tau (τ) revealed no significant correlation between neuronal loss in brainstem nuclei and the length of the CAG‐repeats in the mutated HD allele, age of HD onset, duration of HD, age at death and Vonsattel grade of striatal atrophy (all P‐values > 0.10). Calculation of the weighted kappa coefficient revealed a nearly perfect intrarater reliability for the semi‐quantitative assessment of neuronal loss in the brainstem nuclei of the HD patients (Κw = 0.83; P < 0.0001).
No p62 immunoreactive nuclear inclusions (NI) were detected within the brainstem of control brains. However, NI were found within the brainstem of the eight HD patients. They involved both the brainstem nuclei with or without documented neuronal loss. The nucleus of a few neurons had two or even three NI (Figure 4). Calculation of the weighted kappa coefficient revealed a nearly perfect intrarater reliability for the semi‐quantitative assessment of p62 immunoreactive NI in brainstem nuclei of the HD patients (Κw = 0.90; P < 0.0001).
Figure 4.
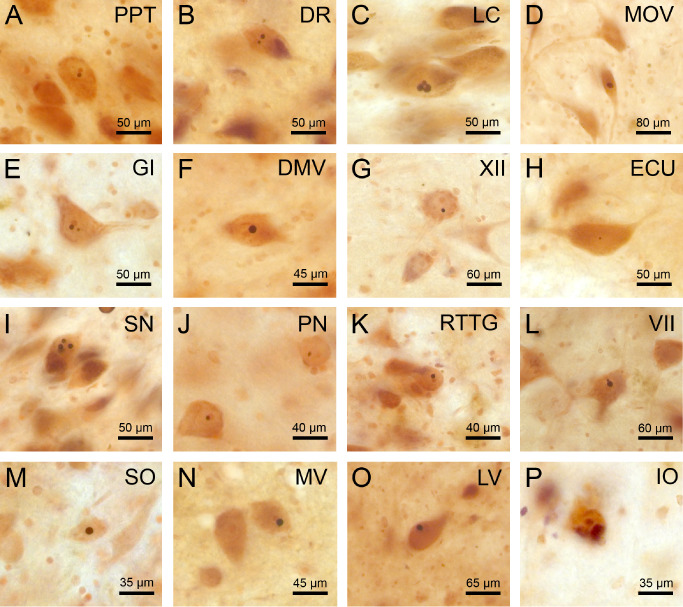
p62 immunoreactive neuronal intranuclear inclusions in spared and degenerated brainstem nuclei in Huntington's disease (HD). P62 immumopositive neuronal intranuclear inclusions (NI) in well‐preserved brainstem nuclei in HD: A. pedunculopontine nucleus (PPT), B. dorsal raphe nucleus (DR), C. locus coeruleus (LC), D. motor trigeminal nucleus (MOV), E. gigantocellular reticular nucleus (GI), F. dorsal motor vagal nucleus (DMV), G. hypoglossal nucleus (XII), H. external cuneate nucleus (ECU). NI in degenerated brainstem nuclei in HD: I. substantia nigra (SN), J. pontine nuclei (PN), K. reticulotegmental nucleus of the pons (RTTG), L. facial nucleus (VII), M. superior olive (SO), N . medial vestibular nucleus (MV), O. lateral vestibular nucleus (LV), P. inferior olive (IO) (A, B, F, L, N, O: HD patient 1—Table 1, CAG‐repeats in the mutated HD allele: 49; age at HD onset: 33 years; duration of HD: 18 years; Vonsattel grade of striatal atrophy: 4; C, G, H, M: HD patient 7—Table 1, CAG‐repeats in the mutated HD allele: 44; age at HD onset: 50 years; duration of HD: 20 years; Vonsattel grade of striatal atrophy 2: D, I: HD patient 4—Table 1, CAG‐repeats in the mutated HD allele: 45; age at HD onset: 53 years; duration of HD: 12 years; Vonsattel grade of striatal atrophy: 4; E, J, K, P: HD patient 6—Table 1, CAG‐repeats in the mutated HD allele: 40; age at HD onset: 60 years; duration of HD: 8 years; Vonsattel grade of striatal atrophy: 3) (A–P: p62 immunohistochemistry, counterstaining with aldehyde‐fuchsin Darrow red; 100 μm PEG sections).
Unlike in the control individuals studied, p62 immunoreactive axonal inclusions were ubiquitous throughout the brainstem of all HD patients (pyramidal tract, all cerebellar peduncles, medial and lateral lemnisci, medial longitudinal fascicle, trapezoid body, pontocerebellar and olivocerebellar fibers, pontine reticular formation, spinocerebellar tracts, cuneate and gracile fascicles) (Figure 5; Table 5). Axonal inclusions were present in the axonal hillock (Figure 5B) or in more distal parts of the axon (Figure 5A,C–I,K,L,O,P) and in some instances occurred at multiple sites of an affected axon (Figure 5J,M,N). Their shape varied from round (Figure 5H,O), ovoid (Figure 5A,F,P), club‐shaped (Figure 5B,E,K,L), irregular (Figure 5C,D,J,M), ramified (Figure 5N), to torpedo‐like (Figure 5G,I). Their diameter was either nearly identical to that of the affected axon (Figure 5F,J,N) or larger than the normal axonal diameter (Figure 5A–E,G–I,K–M,O,P). Calculation of weighted kappa revealed a substantial interrater reliability for the semi‐quantitative assessment of p62 immunoreactive axonal inclusions in brainstem fiber tracts (Κw = 0.74; P < 0.0001).
Figure 5.
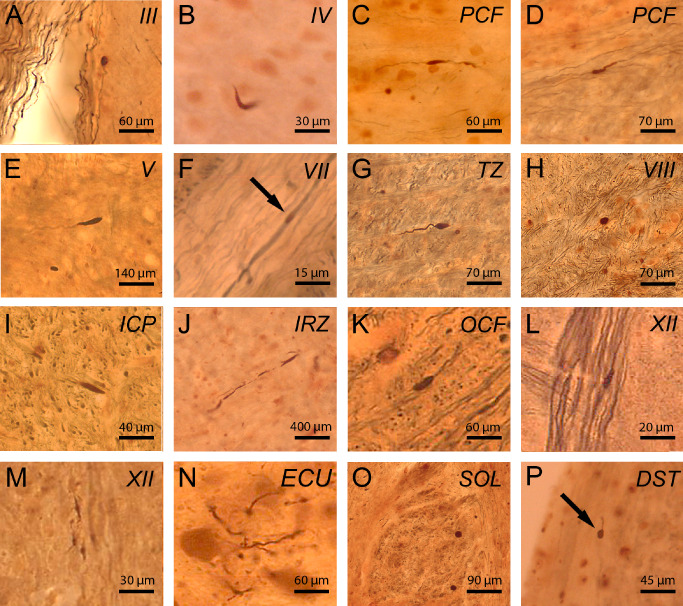
p62 immunoreactive axonal neuronal inclusions in brainstem fiber tracts in Huntington's disease (HD). P62 immumopositive axonal neuronal inclusions in HD: (A) oculomotor nerve (III), (B) trochlear nerve (IV), (C) pontocerebellar fibers (PCF), (D) PCF), (E) trigeminal nerve (V), (F) facial nerve (VII), (G) trapezoid body (TZ), (H ) vestibulocochlear nerve (VIII), (I) inferior cerebellar peduncle (ICP), (J) intermediate reticular zone (IRZ), (K) olivocerebellar fibers (OCF), (L) hypoglossal nerve (XII), (M) hypoglossal nerve (XII), (N) cuneate fascicle (ECU), (O) solitary tract (SOL), (P) dorsal spinocerebellar tract (DST). (A–P: p62/AT270 double immunohistochemistry; 100 μm PEG sections) (A, E, K, N: HD patient 4—Table 1, CAG‐repeats in the mutated HD allele: 45; age at HD onset: 53 years; duration of HD: 12 years; Vonsattel grade of striatal atrophy: 4; B, L, P: HD patient 8—Table 1, CAG‐repeats in the mutated HD allele: 42; age at HD onset: 55 years; duration of HD: 36 years; Vonsattel grade of striatal atrophy: 2; C, D, F, G, H: HD patient 2—Table 1, CAG‐repeats in the mutated HD allele: 45; age at HD onset: 35 years; duration of HD: 26 years; Vonsattel grade of striatal atrophy 3; I: HD patient 5—Table 1, CAG‐repeats in the mutated HD allele: 46; age at HD onset: 40 years; duration of HD: 28 years; Vonsattel grade of striatal atrophy 3; J, O: HD patient 6—Table 1, CAG‐repeats in the mutated HD allele: 40; age at HD onset: 60 years; duration of HD: 8 years; Vonsattel grade of striatal atrophy: 3; M: HD patient 1—Table 1, CAG‐repeats in the mutated HD allele: 49; age at HD onset: 33 years; duration of HD: 18 years; Vonsattel grade of striatal atrophy: 4).
In contrast to the control cases studied, GFAP immunopositive astroctyes were present in all degenerated and spared brainstem nuclei of all HD patients (data not shown).
Additional double immunostainings of brainstem tissue sections of our HD patients confirmed the presence of a protein with an expanded polyglutamine stretch (ie, detection of mutant huntingtin with the 1C2 antibody) in the p62‐immunopositive axonal protein aggregates (Figure 6) and also demonstrated their ubiquitination (Figure 7).
Figure 6.
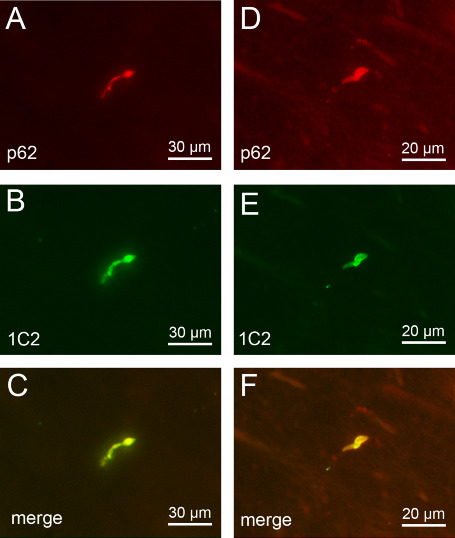
p62 and 1C2 double immunofluorescence of axonal inclusions in brainstem fiber tracts in Huntington's disease (HD). Double immunofluorescence of intra‐axonal neuronal inclusions in the nigrostriatal pathway (A–C) and (D–F) pontocerebellar fibers of a representative 51‐year‐old male HD patient (CAG‐repeats in the mutated HD allele: 49; age at HD onset: 33 years; duration of HD: 18 years; Vonsattel grade of striatal atrophy: 4; patient 1; Tables 1, 2 and 5). Immunolabeling with (A,D) an antibody against the proteasomal shuttle protein p62 (red) and (B,E) the anti‐polyglutamine marker 1C2 (green).
The positive immunoreactions for the anti‐polyglutamine marker 1C2 point to the recruitment of the pathologically altered form of the disease protein huntingtin with an elongated polyglutamine stretch into the p62 immunoreactive axonal inclusions (A, D: p62 immunostaining—Cy3; B, E: anti‐polyglutamine 1C2 immunostaining—Alexa 488; 100 μm PEG sections).
Figure 7.
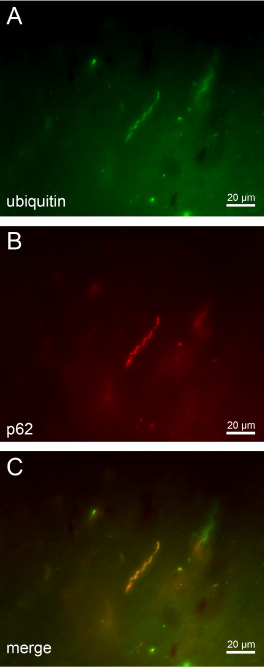
p62 and anti‐ubiquitin double immunofluorescence of axonal inclusions in brainstem fiber tracts in H untington's disease ( HD ). (A–C) Double immunofluorescence of intra‐axonal neuronal inclusions in the nigrostriatal pathway of a representative 64‐year‐old male HD patient (CAG‐repeats in the mutated HD allele: 41; age at HD onset: 55 years; duration of HD: 9 years; Vonsattel grade of striatal atrophy: 2; patient 3; Tables 1, 2 and 5) immunolabeled with (A) an antibody against the ubiquitin protein (green) and (B) the proteasomale shuttle protein p62 (red).
The presence of ubiquitin and p62 in intra‐axonal aggregates (i) suggests an involvement of the ubiquitin‐proteasome pathway in the HD pathogenesis and; (ii) points to defense mechanisms of affected nerve cells that attempt to reduce levels of mutant huntingtin; but (iii) may also reflect a decreased or compromised activity of the ubiquitin‐proteasome pathway or its overload in HD (A: p62 immunostaining—Alexa 488; B: anti‐ubiquitin immunostaining—Cy3; 100 μm PEG sections).
Discussion
Degeneration of the brainstem in HD
The degeneration of the brainstem and its possible clinical relevance in HD has been controversially discussed in neuropathological research for many decades 5, 32, 45, 52, 62, 82, 83. Neuropathological conjectures from years ago indicate that some brainstem nuclei may undergo neuronal loss in HD (ie, dopaminergic and GABAergic substantia nigra, auditory superior olive, lateral vestibular nucleus, precerebellar inferior olive), but these earlier findings have not been revisited or taken into account by current researchers. Thus, the involvement of the brainstem in HD is not among the established degenerative features of HD 5, 32, 41, 52, 82, 83. Our present pathoanatomical study, performed on serial thick tissue sections through the brainstem of eight clinically diagnosed and genetically confirmed HD patients, confirms and extends the earlier assumptions regarding brainstem involvement in HD. Based on our findings, damage to the brainstem in HD proved to be more widespread than documented so far in the published literature and included additional nuclei of the precerebellar, vestibular, oculomotor and ingestion‐related systems. As degeneration of brainstem nuclei did not correlate with the Vonsattel HD grade of striatal atrophy, it is likely that damage to these nuclei does not develop simultaneously with, but independently from the well‐known pathognomonic lesions in the striatum. Taken together, damage to the brainstem again shows that neurodegeneration during HD is not restricted to the striatum and cerebral neo‐ and allocortex. We conclude that HD is a multisystem neurodegenerative disease 3, 5, 15, 25, 26, 27, 39, 40, 41, 73, 81, 82, 83 characterized by a distribution of brain damage that shows more overlap with that of the polyglutamine ataxias (eg, spinocerebellar ataxias types 1, 2, 3 and 7) than has been believed 43, 54, 57, 58, 59, 60, 61, 70.
Clinicopathological correlation of brainstem degeneration in HD
The human brainstem is integrated into various brain neurotransmitter systems, as well as into circuits subserving auditory, precerebellar, ingestion‐related, and oculomotor functions, and regulation of posture and balance 7, 9, 28, 29, 33, 58, 60, 64, 84. Although a wide spectrum of oculomotor symptoms typically emerge in HD patients (ie, slowed and saccadic smooth pursuits, reduced gain of the vestibulo‐ocular reflex and optokinetic nystagmus, initiating deficits of saccades, slowing, restricted range or loss of horizontally and/or vertical saccades; impaired steady fixation) and may also occur in preclinical HD gene carriers 42, 44, 62, nearly all past attempts to decipher the morphological counterparts of these oculomotor dysfunctions have failed 32, 45, 62. Progress in the field of the functional neuroanatomy of the human oculomotor networks made over the last two decades led to the identification and characterization of important premotor oculomotor brainstem nuclei. As a result, a detailed and reliable basis for precise structure‐function relationships became available which enabled the establishment of clinicopathological correlations in various neurodegenerative diseases 7, 9, 29, 58, 60, 62, 64.
The raphe interpositus nucleus (RIP) (Figure 2E) was identified and described in non‐human primates and in humans 25 years ago. This small nucleus at the midline of the pontomedullary junction is located between the rootlets of the abducens nerve. The horizontally oriented nerve cells of the RIP are linearly arranged in a narrow band of only two or three nerve cells and represent omnipause neurons that trigger the initiation of vertical and horizontal saccades 7, 9, 29, 58, 62, 64. As the loss of omnipause neurons in the RIP has recently been shown to correlate with slowing of saccades 7, 9, 29, 58, 62, 64, the severe damage to the RIP of our HD patients most likely contributed to the slowing of horizontal saccades observed in our and other HD patients (Table 2) 42, 44, 45, 62, 64. The human premotor area of the excitatory burst neurons for horizontal saccades (EBR) (Figure 2C) was described only some years after the RIP. The EBR is only of about 2.5 mm in width and 2 mm in height and is located in the pontine reticular formation underneath the medial longitudinal fascicle and the internal genu of the facial nerve 7, 9, 29, 62, 64. The neurons of the EBR are essential for the initiation and generation of horizontal saccades 7, 9, 29, 62, 64. Accordingly, damage to the EBR most likely can account for the initiation deficits of horizontal saccades of our and other HD patients (Table 2) 42, 44, 45, 62.
The medial, lateral and superior vestibular nuclei (Figure 3A) via their descending spinal projections are crucial for the maintenance of truncal and postural stability 8, 60, 64. Their involvement in our HD patients may explain as to why our and other HD patients suffered from gait, truncal and postural instability with a tendency to fall (Table 2) 6, 20. Because these vestibular nuclei are important building blocks of the oculomotor circuits subserving the optokinetic nystagmus and vestibulo‐ocular reaction 8, 62, 64, damage to these nuclei contributes to the occurrence of this type of impaired eye movements in HD 42, 44, 45.
The reticulotegmental nucleus of the pons (RTTG) (Figure 2A) is a crucial component of the brain neural circuit engaged in the regulation and control of smooth pursuit eye movements 62, 63, 64. Thus, together with the lesions in the interconnected fastigial nucleus of the cerebellum 63, damage to the RTTG is well suited to explain why slowed and saccadic smooth pursuits occurred in our and other HD patients (Table 2) 42, 44, 45, 62.
The pontine nuclei and the inferior olive (Figure 3E) represent important building blocks in the precerebellar neuronal circuits and are intimately connected with the cerebellar hemispheres (neo‐ or pontocerebellum: pontine nuclei and inferior olive), vermal and paravermal regions of the cerebellum (vestibulo‐ or archicerebellum, spino‐ or paleocerebellum: inferior olive), as well as associated deep nuclei (ie, fastigial, globose, emboliform and dentate nuclei) 4, 84. Loss of Purkinje cells in the cerebellar cortex and degeneration of nerve cells in all four deep cerebellar nuclei of HD patients 63 in combination with neuronal loss in the precerebellar pontine nuclei and inferior olive are detrimental to the functions accomplished by the precerebellar and cerebellar neural circuits and most likely contributed substantially to the cerebellar disease symtoms observed in our and other HD patients (ie, impairments of rapid alternating movements and fine motor skills, dysarthria, hypotonia, postural instability and ataxia, impaired gait balance, broad‐based gait and stance) (Table 2) 34, 49, 63, 85, 86.
The superior olive is located in the caudal pontine tegmentum, embedded into the trapezoid body and involved in the performance of auditory functions, such as perception of the temporal patterns of sounds and analysis of the spatial attributes of sound sources (ie, location, distance and movement) (Figure 3C) 28, 50. Accordingly, the severe and consistent involvement of the superior olive in our HD patients suggests that impairment of these auditory functions may be among the disease symptoms of HD patients. In view of the striking discrepancy between the involvement of the superior olive in this study and missing clinical reports of auditory impairments in our and other HD patients, additional clinicopathological HD studies are required.
Degeneration of the dopaminergic compact part of the substantia nigra is a characteristic and unifying neuropathological hallmark of all clinical syndromes associated with parkinsonian features (eg, Parkinson's disease, progressive supranuclear palsy, multiple system atrophy, corticobasal degeneration) 13, 23. Although our study confirms a consistent loss of nerve cells of the compact and reticulate parts of substantia nigra in HD 39, 41, 52, the severity and extent of the neuronal loss in the compact part of the substantia nigra may not suffice to provoke clinically relevant parkinsonian features in adult HD patients 12, 53. This point of view is in agreement with the clinical experience that parkinsonian extrapyramidal motor features (eg, rigidity) only emerge in juvenile onset HD patients 82, 85, 86.
Neuronal intranuclear and axonal inclusions and their possible significance to the neurodegenerative process of HD
The aggregation of the disease protein huntingtin into NI bodies is a well‐known neuropathological feature of HD 3, 17, 19, 21, 22, 31, 36, 47, 48. NI are most frequent in the degenerated striatum and cerebral neo‐ and allocortex and may contain components of the cellular protein quality control machinery responsible for removing and degrading structurally abnormal, misfolded and ubiquitin‐tagged proteins (ie, ubiquitin, chaperones, proteasome subunits) 2, 17, 19, 21, 22, 30, 31, 47, 48, 55, 65, 74, 80. It is still an open question whether NI are toxic to or protective for affected nerve cells or merely represent pathognomonic morphological hallmarks of HD and currently no idea is in existence with respect to the possible mechanisms in which way they may contribute to damage of affected nerve cells or facilitate their survival 17, 19, 31, 36, 65, 74, 80. In this study, we observed NI in neurons of all brainstem nuclei of HD patients investigated, irrespective of whether they showed signs of degeneration or appeared unaffected, and failed to establish a correlation between the distribution of NI and the pattern of neuronal loss in the brainstem. Evidently, our correlative post‐mortem approach cannot provide a conclusive answer to the question whether the NI play a causal role in the cascades of pathological events leading to nerve cell loss. However, our study indicates that NI are to be considered as pathognomonic morphological hallmarks of HD.
In addition, NI neuropathological studies also described the occurrence of neuropil (ie, neuritic or axonal aggregates or swellings) in select brain gray and white matters of HD patients (eg, striatum, cerebral neocortex, internal capsule, cerebral peduncle) 17, 22, 31, 36, 47, 65, 74. Our study represents the first systematic analysis of axonal protein aggregates in fiber tracts of a coherent brain region of HD patients (ie, brainstem) and for the first time demonstrates that axonal protein aggregations are a consistent and widespread feature of brainstem pathology in HD. Accordingly, together with SCA3, HD has now been recognized as the second polyglutamine disease associated with the presence of axonal protein aggregates 69, 70.
The composition of the intra‐axonal inclusions observed here shares many biological similarities with that of the NI. Axonal inclusions in HD can be immunostained with the anti‐polyglutamine antibody 1C2 and along with the pathologically altered form of the disease protein huntingtin with elongated polyglutamin tract axonal inclusions in HD also contain the degradation‐related proteins p62 and ubiquitin. Together with ubiquitin, p62 plays a central role in the highly dynamic pathways that remove and degrade short‐lived or aberrantly structured proteins, thus preventing their aggregation 30, 55. Their presence in intra‐axonal aggregates of HD patients suggests an involvement of the ubiquitin‐proteasome pathway in the HD pathogenesis 10, 30, 84 and points to defense mechanisms of affected nerve cells that attempt to reduce levels of mutant and misfolded huntingtin 10, 17. The presence of ubiquitin and p62 in axonal aggregates also reflects a decreased or compromised activity of the ubiquitin‐proteasome pathway or its overload in HD causing aggregate formation (10, 30, 64, 80). Upregulation and enhancement of proteasome activity with drugs is considered as a promising therapeutical strategy to reduce the pathological levels of the polyglutamine protein huntingtin and the pathological consequences associated with this aggregation in affected nerve cells 10, 30, 55.
Several disease models have proposed the occurrence and a causative feature of axonal transport deficits in HD 11, 21. Although the pathological mechanisms leading to axonal transport deficits in HD remain unclear, it has been suggested that intra‐axonal transport deficits in HD may result from a physical block caused by intra‐axonal protein aggregation formation 11, 21. In the present study, we could indeed observe intra‐axonal protein aggregates in the brainstem fiber tracts of the HD patients studied, irrespective of the length, caliber or myelinization of the affected axons. In view of their size, diameter and insolubility, these intra‐axonal aggregations are likely to physically block and to pertubate anterograde and retrograde axonal transport processes 17, 21, 22, 31, 46, 65. The failure of anterograde transport mechanisms can impair movement of protein complexes, synaptic vesicles and neuronal organelles, can lead to structural changes in axons and accumulations of vesicles and organelles (eg, mitochondria), and can result in synaptic and mitochondrial dysfunction which are detrimental to ATP production and energy metabolism further impairing axonal transport processes 11, 17, 21, 31, 46, 63, 65. In view of the current uncertainties regarding the mechanisms of nerve cell death in HD and the possible consequences of intra‐axonal aggregates for neuronal viability or demise, we think it is justified and worthwhile to perform additional HD studies aimed to (i) reconstruct the spatial and temporal evolution of these axonal aggregates and their association with the development of neurodegeneration; and (ii) to investigate the presence of axonal accumulations of cellular organelles, vesicles and proteins in affected axons. These future studies may help to decipher the pathophysiological and pathobiochemical mechanisms of the neurodegenerative process of HD and represent a step forward on the road to a causative and disease modifying therapy for this severe and disabling human neuropsychiatric disease.
Acknowledgments
This study was supported by grants from the Dr. Senckenbergische Stiftung (Frankfurt/Main, Germany), the Deutsche Huntington Hilfe e.V. and NIH R01NS038712 to HLP. The skillful assistance of D. von Meltzer (secretary) is thankfully acknowledged.
Disclosure statement
All authors have no actual or potential conflicts of interest to disclose, including financial, personal or other relationships with other people or organizations, within 3 years of beginning the work submitted.
References
- 1. Arrasate M, Finkbeiner S (2012) Protein aggregates in Huntington's disease. Exp Neurol 238:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Becher MW, Kotzuk JA, Sharp AH, Davies SW, Bates GP, Price DL, Ross CA (1998) Intranuclear neuronal inclusions in Huntington's disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol Dis 4:387–397. [DOI] [PubMed] [Google Scholar]
- 3. Braak H, Braak E (1992) Allocortical involvement in Huntington's disease. Neuropathol Appl Neurobiol 18:539–547. [DOI] [PubMed] [Google Scholar]
- 4. Braak H, Rüb U, Del Tredici K (2003) Involvement of precerebellar nuclei in multiple system atrophy. Neuropathol Appl Neurobiol 29:60–76. [DOI] [PubMed] [Google Scholar]
- 5. Bruyn GW, Bots GTAM, Dom R (1979) Huntington's chorea: current neuropathological status. Adv Neurol 23:83–93. [Google Scholar]
- 6. Busse ME, Wiles CM, Rosser AE (2009) Mobility and falls in people with Huntington's disease. J Neurol Neurosurg Psychiatry 80:88–90. [DOI] [PubMed] [Google Scholar]
- 7. Büttner‐Ennever JA (2006) The extraocular motor nuclei: organization and functional neuroanatomy. Prog Brain Res 151:95–125. [DOI] [PubMed] [Google Scholar]
- 8. Büttner‐Ennever JA, Gerrits NM (2004) Vestibular system. In: The Human Nervous System, 2nd edn. Paxinos G, Mai JK (eds), pp. 1213–1240. Elsevier: San Diego. [Google Scholar]
- 9. Büttner‐Ennever JA, Horn AKE (2004) Reticular formation: eye movements, gaze, and blinks. In: The Human Nervous System, 2nd edn. Paxinos G, Mai JK (eds), pp. 479–510. Elsevier: San Diego. [Google Scholar]
- 10. Davies JE, Sarkar S, Rubinsztein DC (2007) The ubiquitin proteasome system in Huntington's disease and the spinocerebellar ataxias. BMC Biochem 8(Suppl. 1):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. De Vos KJ, Grierson AJ, Ackerley S, Miller CC (2008) Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci 31:151–173. [DOI] [PubMed] [Google Scholar]
- 12. Den Dunnen WF (2013) Neuropathological diagnostic considerations in hyperkinetic movement disorders. Front Neurol 4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM et al (2009) Neuropathological assessment of Parkinson's disease: refining the diagnostic criteria. Lancet Neurol 8:1150–1157. [DOI] [PubMed] [Google Scholar]
- 14. Difiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277:1990–1993. [DOI] [PubMed] [Google Scholar]
- 15. Dom R, Malfroid M, Baro F (1976) Neuropathology of Huntington's chorea. Studies of the ventrobasal complex of the thalamus. Neurology 26:64–68. [DOI] [PubMed] [Google Scholar]
- 16. Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M et al (1993) Trinucleotide repeat length instability and age of onset in Huntington's disease. Nat Genet 4:387–392. [DOI] [PubMed] [Google Scholar]
- 17. Gil JM, Rego AC (2008) Mechanisms of neurodegeneration in Huntington's disease. Eur J Neurosci 27:2803–2820. [DOI] [PubMed] [Google Scholar]
- 18. Goedert M, Jakes R, Crowther RA, Cohen P, Vanmechelen E, Vandermeeren M, Cras P (1994) Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer's disease: identification of phosphorylation sites in tau protein. Biochem J 301(Pt 3):871–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gourfinkel‐An I, Cancel G, Duyckaerts C, Faucheux B, Hauw JJ, Trottier Y et al (1998) Neuronal distribution of intranuclear inclusions in Huntington's disease with adult onset. Neuroreport 9:1823–1826. [DOI] [PubMed] [Google Scholar]
- 20. Grimbergen YA, Knol MJ, Bloem BR, Kremer BP, Roos RA, Munneke M (2008) Falls and gait disturbances in Huntington's disease. Mov Disord 23:970–976. [DOI] [PubMed] [Google Scholar]
- 21. Gunawardena S, Goldstein LS (2005) Polyglutamine diseases and transport problems: deadly traffic jams on neuronal highways. Arch Neurol 62:46–51. [DOI] [PubMed] [Google Scholar]
- 22. Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R et al (1999) Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci 19:2522–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Halliday G (2004) Substantia nigra and locus coeruleus. In: The Human Nervous System, 2nd edn. Paxinos G, Mai JK (eds), pp. 449–463. Elsevier: San Diego. [Google Scholar]
- 24. Harper PS (1992) The epidemiology of Huntington's disease. Hum Genet 89:365–376. [DOI] [PubMed] [Google Scholar]
- 25. Heinsen H, Rüb U, Bauer M, Ulmar G, Bethke B, Schüler M et al (1999) Nerve cell loss in the thalamic mediodorsal nucleus in Huntington's disease. Acta Neuropathol 97:613–622. [DOI] [PubMed] [Google Scholar]
- 26. Heinsen H, Rüb U, Gangnus D, Jungkunz G, Bauer M, Ulmar G et al (1996) Nerve cell loss in the thalamic centromedian‐parafascicular complex in patients with Huntington's disease. Acta Neuropathol 91:161–168. [DOI] [PubMed] [Google Scholar]
- 27. Heinsen H, Strik M, Bauer M, Luther K, Ulmar G, Gangnus D et al (1994) Cortical and striatal neurone number in Huntington's disease. Acta Neuropathol 88:320–333. [DOI] [PubMed] [Google Scholar]
- 28. Hoche F, Seidel K, Brunt ER, Auburger G, Schöls L, Bürk K et al (2008) Involvement of the auditory brainstem system in spinocerebellar ataxia type 2 (SCA2), type 3 (SCA3) and type 7 (SCA7). Neuropathol Appl Neurobiol 34:479–491. [DOI] [PubMed] [Google Scholar]
- 29. Horn AK (2006) The reticular formation. Prog Brain Res 151:127–155. [DOI] [PubMed] [Google Scholar]
- 30. Huang Q, Figueiredo‐Pereira ME (2010) Ubiquitin/proteasome pathway impairment in neurodegeneration: therapeutic implications. Apoptosis 15:1292–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Imarisio S, Carmichael J, Korolchuk V, Chen CW, Saiki S, Rose C et al (2008) Huntington's disease: from pathology and genetics to potential therapies. Biochem J 412:191–209. [DOI] [PubMed] [Google Scholar]
- 32. Koeppen AH (1989) The nucleus pontis centralis caudalis in Huntington's disease. J Neurol Sci 91:129–141. [DOI] [PubMed] [Google Scholar]
- 33. Koutcherov Y, Huang XF, Halliday G, Paxinos G (2004) Organization of human brain stem nuclei. In: The Human Nervous System, 2nd edn. Paxinos G, Mai JK (eds), pp. 267–320. Elsevier: San Diego. [Google Scholar]
- 34. Kremer B, Weber B, Hayden MR (1992) New insights into the clinical features, pathogenesis and molecular genetics of Huntington disease. Brain Pathol 2:321–335. [DOI] [PubMed] [Google Scholar]
- 35. Kremer HP, Roos RA, Dingjan GM, Bots GT, Bruyn GW, Hofman MA (1991) The hypothalamic lateral tuberal nucleus and the characteristics of neuronal loss in Huntington's disease. Neurosci Lett 132:101–104. [DOI] [PubMed] [Google Scholar]
- 36. Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF et al (1999) Huntington aggregates may not predict neuronal death in Huntington's disease. Ann Neurol 46:842–849. [PubMed] [Google Scholar]
- 37. Kuusisto E, Kauppinen T, Alafuzoff I (2008) Use of p62/SQSTM1 antibodies for neuropathological diagnosis. Neuropathol Appl Neurobiol 34:169–180. [DOI] [PubMed] [Google Scholar]
- 38. Kuusisto E, Salminen A, Alafuzoff I (2001) Ubiquitin‐binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport 12:2085–2090. [DOI] [PubMed] [Google Scholar]
- 39. Lange H, Thorner G, Hopf A, Schroder KF (1976) Morphometric studies of the neuropathological changes in choreatic diseases. J Neurol Sci 28:401–425. [DOI] [PubMed] [Google Scholar]
- 40. Lange HW (1981) Quantitative changes of telencephalon, diencephalon, and mesencephalon in Huntington's chorea, postencephalitic, and idiopathic Parkinsonism. Verh Anat Ges 73:923–925. [Google Scholar]
- 41. Lange HW, Aulich A (1986) Die Hirnatrophie bei der Huntingtonschen Krankheit. In: Die Huntingtonsche Krankheit, 1st edn. Oepen H (ed.), pp. 25–41. Hippokrates Verlag: Stuttgart. [Google Scholar]
- 42. Lasker AG, Zee DS (1997) Ocular motor abnormalities in Huntington's disease. Vision Res 37:3639–3645. [DOI] [PubMed] [Google Scholar]
- 43. Lastres‐Becker I, Rüb U, Auburger G (2008) Spinocerebellar ataxia 2 (SCA2). Cerebellum 7:115–124. [DOI] [PubMed] [Google Scholar]
- 44. Leigh RJ, Newman SA, Folstein SE, Lasker AG, Jensen BA (1983) Abnormal ocular motor control in Huntington's disease. Neurology 33:1268–1275. [DOI] [PubMed] [Google Scholar]
- 45. Leigh RJ, Parhad IM, Clark AW, Buettner‐Ennever JA, Folstein SE (1985) Brainstem findings in Huntington's disease. Possible mechanisms for slow vertical saccades. J Neurol Sci 71:247–256. [DOI] [PubMed] [Google Scholar]
- 46. Li JY, Conforti L (2012) Axonopathy in Huntington's disease. Exp Neurol 246:62–72. [DOI] [PubMed] [Google Scholar]
- 47. Maat‐Schieman M, Roos R, Losekoot M, Dorsman J, Welling‐Graafland C, Hegeman‐Kleinn I et al (2007) Neuronal intranuclear and neuropil inclusions for pathological assessment of Huntington's disease. Brain Pathol 17:31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maat‐Schieman ML, Dorsman JC, Smoor MA, Siesling S, van Duinen SG, Verschuuren JJ et al (1999) Distribution of inclusions in neuronal nuclei and dystrophic neurites in Huntington disease brain. J Neuropathol Exp Neurol 58:129–137. [DOI] [PubMed] [Google Scholar]
- 49. Margolis RL, Ross CA (2003) Diagnosis of Huntington disease. Clin Chem 49:1726–1732. [DOI] [PubMed] [Google Scholar]
- 50. Moore JK, Linthicum FH (2004) Auditory system. In: The Human Nervous System, 2nd edn. Paxinos G, Mai JK (eds), pp. 1241–1279. Elsevier: San Diego. [Google Scholar]
- 51. Myers RH (2004) Huntington's disease genetics. NeuroRx 1:255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Oyanagi K, Takeda S, Takahashi H, Ohama E, Ikuta F (1989) A quantitative investigation of the substantia nigra in Huntington's disease. Ann Neurol 26:13–19. [DOI] [PubMed] [Google Scholar]
- 53. Paulus W, Jellinger K (1991) The neuropathologic basis of different clinical subgroups of Parkinson's disease. J Neuropathol Exp Neurol 50:743–755. [DOI] [PubMed] [Google Scholar]
- 54. Riess O, Rüb U, Pastore A, Bauer P, Schöls L (2008) SCA3: neurological features, pathogenesis and animal models. Cerebellum 7:125–137. [DOI] [PubMed] [Google Scholar]
- 55. Rubinsztein DC (2006) The roles of intracellular protein‐degradation pathways in neurodegeneration. Nature 443:780–786. [DOI] [PubMed] [Google Scholar]
- 56. Rubinsztein DC, Carmichael J (2003) Huntington's disease: molecular basis of neurodegeneration. Expert Rev Mol Med 5:1–21. [DOI] [PubMed] [Google Scholar]
- 57. Rüb U, Brunt ER, Deller T (2008) New insights into the pathoanatomy of spinocerebellar ataxia type 3 (Machado‐Joseph disease). Curr Opin Neurol 21:111–116. [DOI] [PubMed] [Google Scholar]
- 58. Rüb U, Brunt ER, Gierga K, Schultz C, Paulson H, de Vos RA, Braak H (2003) The nucleus raphe interpositus in spinocerebellar ataxia type 3 (Machado‐Joseph disease). J Chem Neuroanat 25:115–127. [DOI] [PubMed] [Google Scholar]
- 59. Rüb U, Brunt ER, Gierga K, Seidel K, Schultz C, Schöls L et al (2005) Spinocerebellar ataxia type 7 (SCA7): first report of a systematic neuropathological study of the brain of a patient with a very short expanded CAG‐repeat. Brain Pathol 15:287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rüb U, Brunt ER, de Vos RA, Del Turco D, Del Tredici K, Gierga K et al (2004) Degeneration of the central vestibular system in spinocerebellar ataxia type 3 (SCA3) patients and its possible clinical significance. Neuropathol Appl Neurobiol 30:402–414. [DOI] [PubMed] [Google Scholar]
- 61. Rüb U, Bürk K, Timmann D, den Dunnen W, Seidel K, Brunt E et al (2012) Spinocerebellar ataxia type 1 (SCA1): new pathoanatomical and clinico‐pathological insights. Neuropathol Appl Neurobiol 38:665–680. [DOI] [PubMed] [Google Scholar]
- 62. Rüb U, Heinsen H, Brunt ER, Landwehrmeyer B, Den Dunnen WF, Gierga K, Deller T (2009) The human premotor oculomotor brainstem system—can it help to understand oculomotor symptoms in Huntington's disease? Neuropathol Appl Neurobiol 35:4–15. [DOI] [PubMed] [Google Scholar]
- 63. Rüb U, Hoche F, Brunt ER, Heinsen H, Seidel K, Del Turco D et al (2013) Degeneration of the cerebellum in Huntington's Disease (HD): possible relevance for the clinical picture and potential gateway to pathological mechanisms of the disease process. Brain Pathol 23:165–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rüb U, Jen JC, Braak H, Deller T (2008) Functional neuroanatomy of the human premotor oculomotor brainstem nuclei: insights from postmortem and advanced in vivo imaging studies. Exp Brain Res 187:167–180. [DOI] [PubMed] [Google Scholar]
- 65. Sapp E, Penney J, Young A, Aronin N, Vonsattel JP, Difiglia M (1999) Axonal transport of N‐terminal huntingtin suggests early pathology of corticostriatal projections in Huntington disease. J Neuropathol Exp Neurol 58:165–173. [DOI] [PubMed] [Google Scholar]
- 66. Schulte J, Littleton JT (2011) The biological function of the Huntingtin protein and its relevance to Huntington's disease pathology. Curr Trends Neurol 5:65–78. [PMC free article] [PubMed] [Google Scholar]
- 67. Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW (2004) Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol 24:8055–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Seidel K, Brunt ER, de Vos RA, Dijk F, van der Want HJ, Rüb U, Den Dunnen WF (2009) The p62 antibody reveals various cytoplasmic protein aggregates in spinocerebellar ataxia type 6. Clin Neuropathol 28:344–349. [DOI] [PubMed] [Google Scholar]
- 69. Seidel K, Den Dunnen WF, Schultz C, Paulson H, Frank S, de Vos RA et al (2010) Axonal inclusions in spinocerebellar ataxia type 3. Acta Neuropathol 120:449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Seidel K, Siswanto S, Brunt ER, den Dunnen W, Korf HW, Rüb U (2012) Brain pathology of spinocerebellar ataxias. Acta Neuropathol 124:1–21. [DOI] [PubMed] [Google Scholar]
- 71. Shaw G, Osborn M, Weber K (1981) An immunofluorescence microscopical study of the neurofilament triplet proteins, vimentin and glial fibrillary acidic protein within the adult rat brain. Eur J Cell Biol 26:68–82. [PubMed] [Google Scholar]
- 72. Shoulson I, Young AB (2011) Milestones in Huntington disease. Mov Disord 26:1127–1133. [DOI] [PubMed] [Google Scholar]
- 73. Sieradzan KA, Mann DM (2001) The selective vulnerability of nerve cells in Huntington's disease. Neuropathol Appl Neurobiol 27:1–21. [DOI] [PubMed] [Google Scholar]
- 74. Sieradzan KA, Mechan AO, Jones L, Wanker EE, Nukina N, Mann DM (1999) Huntington's disease intranuclear inclusions contain truncated, ubiquitinated huntingtin protein. Exp Neurol 156:92–99. [DOI] [PubMed] [Google Scholar]
- 75. Smithson KG, MacVicar BA, Hatton GI (1983) Polyethylene glycol embedding: a technique compatible with immunocytochemistry, enzyme histochemistry, histofluorescence and intracellular staining. J Neurosci Methods 7:27–41. [DOI] [PubMed] [Google Scholar]
- 76. The Huntington's disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72:971–983. [DOI] [PubMed] [Google Scholar]
- 77. Trottier Y, Devys D, Imbert G, Saudou F, An I, Lutz Y et al (1995) Cellular localization of the Huntington's disease protein and discrimination of the normal and mutated form. Nat Genet 10:104–110. [DOI] [PubMed] [Google Scholar]
- 78. Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G et al (1995) Polyglutamine expansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature 378:403–406. [DOI] [PubMed] [Google Scholar]
- 79. Truant R, Atwal R, Burtnik A (2006) Hypothesis: Huntingtin may function in membrane association and vesicular trafficking. Biochem Cell Biol 84:912–917. [DOI] [PubMed] [Google Scholar]
- 80. Valera AG, Diaz‐Hernandez M, Hernandez F, Ortega Z, Lucas JJ (2005) The ubiquitin‐proteasome system in Huntington's disease. Neuroscientist 11:583–594. [DOI] [PubMed] [Google Scholar]
- 81. Vonsattel JP (2008) Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol 115:55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Vonsattel JP, Difiglia M (1998) Huntington disease. J Neuropathol Exp Neurol 57:369–384. [DOI] [PubMed] [Google Scholar]
- 83. Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr (1985) Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44:559–577. [DOI] [PubMed] [Google Scholar]
- 84. Voogd J (2004) Cerebellum and precerebellar nuclei. In: The Human Nervous System, 2nd edn. Paxinos G, Mai JK (eds), pp. 321–392. Elsevier: San Diego. [Google Scholar]
- 85. Walker FO (2007a) Huntington's disease. Semin Neurol 27:143–150. [DOI] [PubMed] [Google Scholar]
- 86. Walker FO (2007b) Huntington's disease. Lancet 369:218–228. [DOI] [PubMed] [Google Scholar]