

Abstract

Benzimidazole reverse amides were designed and synthesized as Pan RAF kinase inhibitors. Investigation of the structure–activity relationship of the compounds revealed that they were potent in vitro and exhibited desirable in vivo properties.
Keywords: MAP, ras-mitogen activated protein kinase, VEGF, vascular endothelial growth factor receptor, CSF1R, colony stimulating factor-1 receptor, RTKs, receptor tyrosine kinases, STKs, serine threonine kinases, TKs, tyrosine kinases
The RAS-RAF-MEK-ERK pathway is a key driver of oncogenic signals, and mutations in RAS and BRAF occur in approximately 30% of all human cancers.1,2 In normal tissues the RAS family of GTPases transmit signals from growth factor receptors on the cell membrane to trigger activation of the RAF-MEK-ERK kinase cascade leading to the promotion of proliferation and survival signals in the cytoplasm and nucleus. Disregulation of the pathway through mutation occurs most frequently at the level of RAS, where oncogenic mutations lead the stabilization of RAS in the active GTP-bound form resulting in constitutive activation of downstream effectors. Activating mutations in BRAF occur in approximately 8% of human tumors, most notably in 50% of human melanomas with the most frequent mutation resulting in a valine to glutamic acid mutation at codon 600 (BRAFV600E).2 BRAFV600E is constitutively active and functions completely independent of RAS.
Given the central role this pathway plays in cancer it has been the center of intense drug discovery efforts for over 20 years. While RAS has remained an elusive target for drug discovery, numerous inhibitors have entered human clinical trials targeting RAF, MEK, and, more recently, ERK. Most notably, the RAF inhibitors vemurafenib and dabrafenib as well as the MEK inhibitor trametinib have been approved for the treatment of human melanoma tumors expressing BRAFV600E. The significant therapeutic benefit provided by vemurafenib (PLX-4032) and dabrafenib confirms that RAF is an attractive target for drug development.3
Our group has previously disclosed a series of benzimidazole amides as potent RAF inhibitors.4,5 Out of several compounds synthesized, only compound 1 potently inhibited BRAFV600E (IC50 = 0.044 μM) and exhibited the best downstream target modulation pERK (EC50 = 0.3 μM, Figure 1) in the amide series.4,5 The poor correlation between the biochemical potency and cell-based activity in the benzimidazole amides prompted us to explore modifications on the pyridyl amide portion of the molecule, an area that had not been extensively investigated. The amide functionality was transposed to form the “reverse” amide compound 2, which showed better correlation between biochemical potency (BRAFV600E IC50 = 0.003 μM) and the cell based activity (EC50 = 0.044 μM). In this letter, the structure–activity relationship (SAR) of the reverse amide series and the in vitro and in vivo profiles of representative examples will be discussed.
Figure 1.

Design strategy of reverse amide series.
Synthesis of the reverse amide series was attained by the route outlined in Scheme 1. O-Arylation of chloropyridyl-t-butyl ester 4 with the commercially available nitroaminophenol 3 followed by N-methylation and hydrolysis yielded the corresponding acid 5. Curtius rearrangement of 5 yielded pyridyl t-butylcarbamate 6. Hydrolysis of the t-butyl ester in 6 afforded the key intermediate 7, which was acetylated to give 8. Hydrogenation of the nitro group in 8 gave the phenelene diamine, which was then converted to the thiourea and cyclized to afford analogues (9–18).
Scheme 1.

The SAR from our previous series was utilized to begin exploration of the SAR of the reverse amides.4 Lipophilic groups at the meta and para positions of ring A exhibited good biochemical potency (9–18). The tert-butyl (2 and 13) and the isopropyl groups (10 and 15) at the meta and para positions exhibited good biochemical and cellular potency. The same trend was observed in 18 with 2-F and 5-CF3 substitution on ring A. A trifluoromethoxy group at the meta (11) and para positions (16) on ring A were biochemically potent as 2, but the cell potencies were 2-fold less. On the basis of the p-ERK EC50 values, the trifluoromethyl group at the para position (17) on ring A proved superior than at the meta position (12) with an 8-fold improvement over 12. In general, the reverse amide series showed improved and consistent cellular activity compared to the amide series (Table 1).4
Table 1. SAR of the Benzimidazole Reverse Amide Series.
| compd | R′ | BRAFV600E IC50 (μM) | p-ERK EC50 (μM) |
|---|---|---|---|
| 2 | 3-t-Bu | 0.003 | 0.04 |
| 9 | 3-Et | <0.0004 | 0.1 |
| 10 | 3-iPr | <0.0004 | 0.05 |
| 11 | 3-OCHF3 | 0.0004 | 0.1 |
| 12 | 3-CF3 | 0.003 | 0.4 |
| 13 | 4-t-Bu | 0.009 | 0.05 |
| 14 | 4-Et | 0.001 | 0.08 |
| 15 | 4-iPr | 0.004 | 0.09 |
| 16 | 4-OCHF3 | 0.001 | 0.1 |
| 17 | 4-CF3 | 0.004 | 0.05 |
| 18 | 2-F, 5-CF3 | 0.001 | 0.09 |
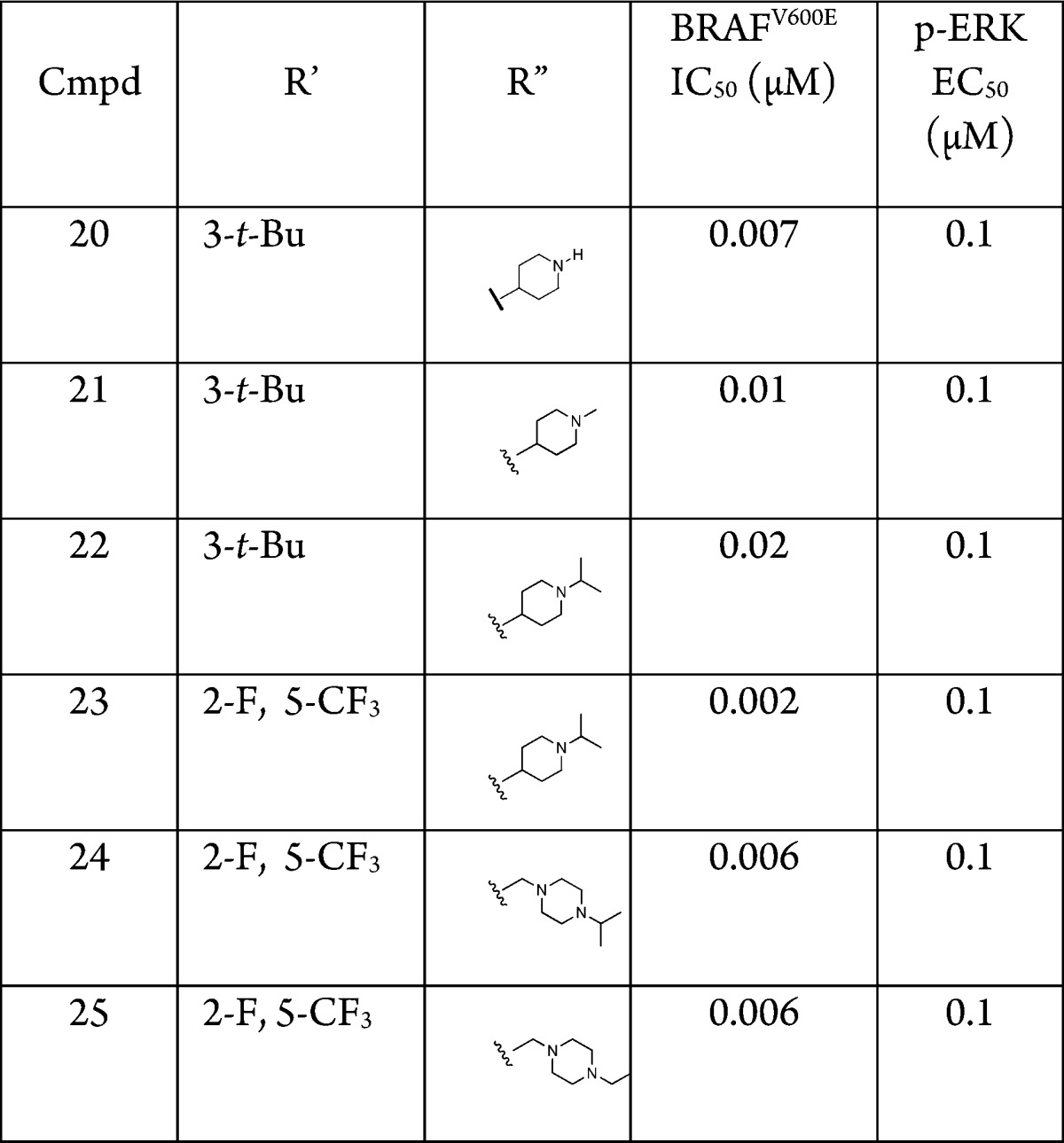
While the reverse amide series provided compounds that were highly potent in the both enzymatic and cellular assays, it was observed that 2 exhibited solubility of 0.28 μg/mL at pH 7 and inhibited CYP3A4 at 0.5 μM. Thus, in exploring the SAR, the focus was broadened to include improving physicochemical properties while retaining the biochemical and cellular potency. Incorporating basic amines onto the amide was found to significantlly impact the solubility of this series. After considerable work, the piperidinyl and methylpiperazinyl amide 20–25 (Table 2) were found to be superior in terms of potency and solubility as expected. However, because basic amines can cause an inhibition of hERG, we monitored this parameter during our optimization. The piperidinyl amides could be incorporated by coupling 7 with the corresponding carboxylic acids (Scheme 2).6 The N-methyl piperazinyl reverse amides were derived by the acylation of 7 with chloroacetyl chloride followed by nucleophilic substitution with the corresponding amine.6 The remainder of the syntheses were similar to Scheme 1 to give the desired products.
Table 2. SAR of the Reverse Amides.
Scheme 2.

The piperidinyl analogue of 2, compound 20, had a similar potency profile to 2 but greatly improved solubility (27 μg/mL at pH 7). It was also found that bulky groups on the basic nitrogen of the piperidine ring improved the CYP profile. For example, 22 was 6-fold less potent against CYP3A4 (3 μM) than 2, while also having improved solubility (581 μg/mL at pH 7). Since 22 met our criteria for potency, solubility, and CYP profile, a preliminary rat PK study of 22 was performed. Following a single dose 10 mg/kg oral administration 22 exhibited high clearance (90 mL/min/kg), volume of distribution (17784 mL/kg), adequate half-life (251 min), and poor oral bioavailability (15%). We hypothesized that the poor bioavailability could be due to the presence of an oxidative metabolism prone t-butyl group on ring A. Replacement of the t-butyl group with 2-fluoro-5-trifluoromethyl (23) maintained cell potency and solubility (561 μg/mL at pH 7). To confirm our hypothesis concerning oral bioavailability, we carried out a rat PK study with 23. Following a single dose 10 mg/kg oral administration, 23 proved superior to 22 with an oral bioavailability of 54%, improved CL (49 mL/min/kg), and similar half-life and volume of distribution. Upon further investigation, it was found that replacing the piperidinyl group in 23 with an isopropyl methylpiperazinyl (24) also gave a potent compound that exhibited solubility at 28 μg/mL at pH 7 and CYP3A4 inhibition at 14 μM. Further, replacing the isopropyl in 24 with an ethyl produced 25, which exhibited the best combination of cellular potency and clearance along with CYP3A4 inhibition >25 μM. Because of this superior profile, compound 25 was further evaluated.
The kinase profile of 25 was examined against 50 kinases including receptor tyrosine kinases (RTKs), serine/threonine kinases (STKs), tyrosine kinases (TKs), and AGC kinases. Compound 25, in addition to being a pan-Raf inhibitor, inhibited four RTKs: CSF1R kinase, VEGFR kinase, c-Kit, and PDGFR with IC50 values < 10 nM, respectively. Additionally, 25 inhibited one of the Src family of kinases, Lck with IC50 value of 240 nM.
The PK profile of 25 was examined in rat. Compound 25 exhibited acceptable clearance (32 mL/min/kg), volume of distribution (12737 mL/kg), half-life (340 min), and oral bioavailability (40%) after oral administartion of 10 mg/kg.
Because of its favorable PK profile, 25 was evaluated in a mouse tumor xenograft model. The SK-MEL-28 cell line could not be grown in vivo in nude mice so the HT29 (B-RAFV600E) human colorectal tumor xenograft model was used to test the effects of 25 on tumor growth in vivo.7 It demonstrated significant antitumor activity (Figure 2) at doses 10 mg/kg/day (ΔT/ΔC = 52%), 30 mg/kg/day (ΔT/ΔC = 72%), and 100 mg/kg/day (ΔT/ΔC = 75%) over the course of 16 days of treatment. It was very well tolerated at the 10 and 30 mg/kg doses; however, in the 100 mg/kg/day group there was an average body weight loss of 13%. The compound demonstrated time dependent inhibition of pERK consistent with inhibition of BRAFV600E in tumors (Figure 2B). These data demonstrate that the novel reverse amide 25 is efficacious in human colorectal xenografts.
Figure 2.
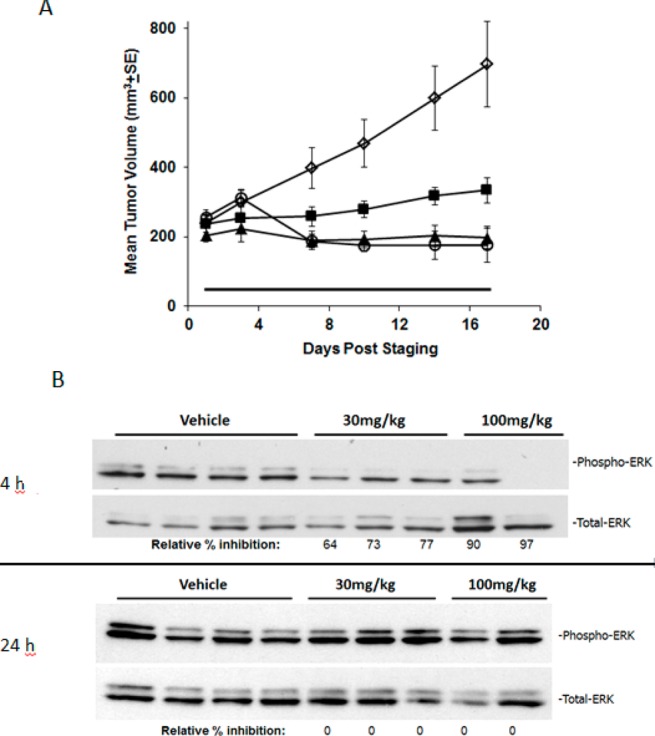
Efficacy and pathway inhibition of 25 in the HT29 (BRAFV600E) human colorectal carcinoma xenograft model: (A) vehicle (diamonds), 10 mg/kg/day (squares), 30 mg/kg/day (triangles), and 100 mg/kg (circles). (B) HT29 tumors from mice dosed with 25 at 30 and 100 mg/kg and tumors were harvested at 4 and 24 h after final dose, and lysates were analyzed by Western blot for pERK and total ERK.
Mouse plasma stability studies in vitro were conducted on the reverse acetamides, piperidinyl, and the methyl piperazinylamides. The stability of 17, 23, 24, and 25 were examined at 37 °C at an initial concentration of 1 μM. We observed that 100% of 17 and 52% of 23 remained after 45 min of incubation showing that the reverse acetamides and piperidinyl amides exhibited good to moderate plasma stability. However, only 1% of 24 and 25 remained after 45 min showing that methylpiperazinylamides possessed poor plasma stability. For other compounds in this series, it was found that the amide was being cleaved to the pyridyl amine. The pyridyl amine of 25 did not exhibit cell potency (pERK >10 μM) and hence might not have contributed to the tumor growth inhition. The increased plasma stability of the piperidinyl amides over the methyl piperazinyl amides could be attributed to the steric bulk on the carbon α to the amide carbonyl group. Further the hERG signal for 25 was 2.3 μM, which we considered suboptimal.
In conclusion, a novel series of potent Raf inhibitors was developed.8 This series had significantly improved physicochemical properties compared to the earlier benzimidazole amide series.4 In addition, 25 demonstrated target inhibition in vivo with good antitumor activity. Further work on this scaffold to improve the inhibition of cellular proliferation, ERK phosphorylation, hERG, and plasma stability led us to identify compound RAF265, which has since entered human clinical trials.
Acknowledgments
The authors wish to acknowledge Paul Barsanti for mouse plasma stability study data and Dazhi Tang for HRMS analysis of these compounds.
Glossary
ABBREVIATIONS
- MAPK
ras-mitogen activated protein kinase
- VEGFR
vascular endothelial growth factor receptor
- CSFR1
colony stimulating factor-1 receptor
- RTKs
receptor tyrosine kinases
- STKs
serine threonine kinases
- TKs
tyrosine kinases
- KDR
kinase domain receptor
- SAR
structure–activity relationship
- DMAc
dimethylacetamide
Supporting Information Available
Experimental data and procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Bos J. L. Ras oncogenes in human cancer: A review. Cancer Res. 1989, 49, 4682–4689. [PubMed] [Google Scholar]
- Davies H.; Bignell G. R.; Cox C.; Stephens P.; Edkins S.; Clegg S.; Teague J.; Woffendin H.; Garnett M. J.; Bottomley W.; Davis N.; Dicks E.; Ewing R.; Floyd Y.; Gray K.; Hall S.; Hawes R.; Hughes J.; Kosmidou V.; Menzies A.; Mould C.; Parker A.; Stevens C.; Watt S.; Hooper S.; Wilson R.; Jayatilake H.; Gustereson B. A.; Cooper C.; Shipley J.; Hargrave D.; Pritchard-Jones K.; Maitland N.; Chenevix-Trench G.; Riggins G. J.; Bigner D. D.; Palmieri G.; Cossu A.; Flanagan A.; Nicholson A.; Ho J. W. C.; Leung S. Y.; Yuen S. T.; Weber B. L.; Seigler H. F.; Darrow T. L.; Paterson H.; Marais R.; Marshall C. J.; Wooster R.; Stratton M. R.; Futreal P. A. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [DOI] [PubMed] [Google Scholar]
- Flaherty K. T.; Puzanov I.; Kim K. B.; Ribas A.; McArthur G. A.; Sosman J. A.; O’Dwyer P. J.; Lee R. J.; Grippo J. F.; Nolop K.; Chapman P. B. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 3639809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramurthy S.; Subramanian S.; Aikawa M.; Payman A.; Costales A.; Dove J.; Fong S.; Jansen J. M.; Levine B.; Ma S.; McBride C. M.; Michaelian J.; Pick T.; Poon D. J.; Girish S.; Shafer C. M.; Stuart D..; Sung L.; Renhowe P. A. Design and synthesis of orally bioavailable benzimidazoles as Raf kinase inhibitors. J. Med. Chem. 2008, 51227049–52. [DOI] [PubMed] [Google Scholar]
- Ramurthy S.; Aikawa M.; Amiri P.; Costales A.; Hashash A.; Jansen J. M.; Lin S.; Ma S.; Renhowe P. A.; Shafer C. M.; Subramanian S..; Sung L. Design and synthesis of 5,6-fused heterocyclic amides as Raf Kinase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21113286–3289. [DOI] [PubMed] [Google Scholar]
- Synthetic methodology and characterization data for 25 are included in the Supporting Information.
- Tumors were established 10 days following subcutaneous implantation of 3 × 106 cells/mouse. Mice were randomized into 4 groups, and oral dosing commenced with vehicle (5 mM citrate) or 25 at 10, 30, and 100 mg/kg daily for 17 days.
- Ramurthy S.; Subramanian S.; Verhagen J.; Poon D. J.; Hansen T.; Shafer C.; McBride C.; Levine B. H.; Costales A.; Renhowe P.. Substituted benzazoles and use thereof as inhibitors of Raf kinase. Patent WO037273, 2005.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.