

Abstract
Improving the gastrointestinal safety profile of nonsteroidal anti-inflammatory drugs (NSAIDs) is an important goal. Herein, we report two strategies, using the nonacidic propyphenazone structure, with potential to overcome the side effects of NSAIDs. Propyphenazone was employed to temporarily mask the free acid group of the widely used NSAIDs ibuprofen, diclofenac, and ketoprofen to develop three mutual prodrugs hypothesized to have minimal GI irritation. The three prodrugs exhibit in vivo anti-inflammatory and analgesic activities with improved potency over each parent drug when compared to a nonhydrolyzable control betahistine–propyphenazone (BET–MP). Additionally, ANT–MP formed by the irreversible coupling of propyphenazone and 4-aminoantipyrine, displayed exceptional COXII selectivity (COXII IC50 of 0.97 ± 0.04 μM, compared to no observed inhibition of COXI at 160 μM). Inhibition of COXII suppresses inflammatory diseases without affecting COXI-mediated GI tract events. ANT–MP exhibited maximal analgesic effect when tested in vivo in an abdominal writhing assay (100% protection) and its anti-inflammatory activity showed a peak at 2 h in a carrageenan-induced paw edema model. Its unique selectivity toward the COXII enzyme was investigated using molecular modeling techniques.
Keywords: Prodrugs, drug design, propyphenazone, cyclooxygenase-2 selective, NSAIDs
NSAIDs are widely prescribed for pain management and for the treatment of various inflammatory disorders. For example, NSAIDs annually account for 70 million prescriptions and 30 billion over-the-counter (OTC) medications sold in the United States alone. All of these drugs express their therapeutic actions by inhibiting the biosynthesis of prostaglandins (PGs) through inhibition of the cyclooxygenase isoforms cyclooxygenase-1 (COXI) and cyclooxygenase-2 (COXII).1 COXI is expressed constitutively and synthesized continuously. It is present in all tissues and cell types, whereas COXII is considered to be an inducible isoenzyme that plays an important role in acute pain and inflammatory processes.2
The use of NSAIDs has several limitations, including gastroduodenal, renal, and cardiovascular toxicities.3 The damage caused by NSAIDs to the gastroduodenal mucosa can be attributed to several mechanisms,4 including topical irritation of the epithelium that is predominantly associated with the free carboxylic acid residue of the NSAIDs.5,6 This residue causes uncoupling of the oxidative phosphorylation (mitochondrial respiration) process in the epithelial cells, which alters their ability to regulate normal functions, such as the maintenance of intracellular pH.7 Nonselective COX inhibitors suppress gastric prostaglandin synthesis due to the inhibition of COXI leading to severe gastric damage.8,9
Several strategies may be used to modulate the gastroduodenal toxicity caused by NSAIDs, such as the use of nitric oxide-donating NSAIDs (NO-NSAIDs), which are capable of generating gastroprotective NO.10,11 NO has been reported to raise mucosal blood flow,12 which increases the mucosal resistance to ulceration.13 It also affects the mucosal immune system14 and enhances the capability of ulcerated mucosal cells to undergo healing and repair.10 Other approaches include the use of a prodrug where the carboxylic acid moiety of NSAIDs is esterified to suppress their gastric toxicity without adversely affecting their anti-inflammatory activity. In addition, biotransformation of the prodrugs to the parent compounds at their target site or sites of activity may be used to achieve rate and time controlled drug delivery of the active entities.15 Several attempts have been made to develop prodrugs of NSAIDs.16,17 A third strategy is to identify inhibitors that selectively target the COXII enzyme to inhibit prostaglandin production at the site of inflammation, without affecting important prostaglandins present in normal tissues such as the stomach.2
The first generation of COXII inhibitors (coxibs) was introduced in 1995. The U.S. Food and Drug Administration (FDA) approved Celecoxib (by Monsanto) and Rofecoxib (by Merck) in 1999. In 2000, two large studies, CLASS (Celecoxib Long-Term Arthritis Safety Study) and VIGOR (Vioxx Gastrointestinal Outcomes Research) confirmed the improved gastrointestinal toxicity of these two drugs if compared to the nonselective NSAIDs, but the VIGOR study warned of possible cardiovascular side effects for Rofecoxib. Rofecoxib was voluntary withdrawn from the market by its manufacturer in September 2004. Since then several concerns have been raised against the use of COXII inhibitors.18,19 Given that Celecoxib is still prescribed without serious cardiovascular side effects, Zarraga et al.19 raised the question “Are all coxibs the same?” They reported that numerous studies indicate different degrees of cardiovascular risk associated with different coxibs, suggesting that COXII inhibitors can be used for the young, or low-risk individuals, but not for patients with coronary artery diseases (CAD) or multiple risk factors for CAD.
In this study, we selected propyphenazone as our lead NSAID for development into either a mutual prodrug (a composition of two drugs attached together in an inactive configuration)17 or a COXII selective inhibitor. Propyphenazone is a nonacidic pyrazole NSAID that has good analgesic activity with minimal anti-inflammatory activity.17,20,21 The coupling of propyphenazone with other widely used acidic NSAIDs such as ketoprofen, ibuprofen, and diclofenac produced mutual prodrugs with synergistic analgesic effects. Furthermore, its nonprodrug coupling to another nonacidic NSAID, 4-aminoantipyrine, highly enhanced its COXII selectivity. Recently, a few reports have emphasized the cardiovascular toxicities that appear in patients receiving propyphenazone.22 Administration of propyphenazone when irreversibly coupled to 4-aminoantipyrine is expected to minimize such toxicities.
Propyphenazone-Based Mutual Prodrugs
It has been reported that propyphenazone (PP) metabolism proceeds through the formation of 3-hydroxymethyl-1-propyphenazone (HPP in Figure 1A), which has been shown to be pharmacologically equivalent to the parent drug PP.17,23 We followed the strategy of Perez et al. and employed an esterase-triggered functional group to mask polar carboxylic acids.24 Herein, we used bromopropyphenazone (BMP) (Figure 1A) to mask the carboxylic group of three widely used acidic NSAIDs (ibuprofen, IB; diclofena, DIC; and ketoprofen, KET) (Figure 1C). The hydrolysis of each ester will result in pharmacologically active HPP and the corresponding acidic NSAID leading to both analgesic and anti-inflammatory effects (synergistic effect). As a negative control for the in vivo experiments, betahistine, BET, an antivertigo drug,25 was coupled with BMP through a nonhydrolyzable bond to produce a pharmacologically inactive compound (Figure 1C). The synthesis of the prodrugs was straightforward. The use of dioxan and methylene chloride as solvents afforded a good yield of BMP, with minimal dibrominated byproduct (Figure 1A). Esterification in dried acetone or methylene chloride followed by heating for 1 h at 90–100 °C yielded the products shown in Figure 1B. A higher temperature produced multiple degradation byproducts. A slight excess of the NSAIDs (1.01 mol) to 1 mol of BMP resulted in complete conversion.
Figure 1.

(A) Synthesis of bromopropyphenazone (BMP) and hydroxypropyphenazone (HPP). (B) Synthesis of ibuprofen–propyphenazone (IB–MP), diclofenac–propyphenazone (DIC–MP), ketoprofen–propyphenazone (KET–MP), betahistine–propyphenazone (BET–MP), and 4-aminoantipyrine–propyphenazone (ANT–MP) derivatives. (C) Chemical structures of the tested compounds.
COX Inhibition Studies
To evaluate the efficacy of the three mutual prodrugs (IB–MP, KET–MP, and DIC–MP) and to confirm that the protecting group will cleave effectively in the presence of esterases to generate active drugs, their ability to inhibit COXI and COXII enzymes was estimated in the absence and presence of porcine liver esterase (PLE) (Table 1 and Figure 2). Ovine COXI enzyme and human recombinant COXII were used in the assay. Before treatment with PLE the three prodrugs showed minimal inhibition of either COXI or COXII. Substrate-selective inhibition of cyclooxygenase has recently been reported by Windsor et al. and others.26 Such inhibitors (e.g., ketoprofen and ibuprofen) bind in one active site of cyclooxygenase and alter the structure of the second active site rendering it either fully or partially inhibited toward a particular substrate. For some substrates no inhibition is seen. As shown in Table 1 and Figure 2, ibuprofen–propyphenazone (IB–MP) did not inhibit COXI but minimally inhibited COXII with 15% maximum inhibition (IC50 = 1.8 ± 0.2 μM). Diclofenac–propyphenazone (DIC–MP) did not inhibit COXII but inhibited COXI with 29% maximum inhibition (IC50 = 7.1 ± 0.9 μM), while ketoprofen–propyphenazone (KET–MP) inhibited both COXI and COXII with 43% and 19% maximum inhibition (IC50 = 0.2 ± 0.01 and 0.04 ± 0.01 μM), respectively.
Table 1. In Vitro COXI and COXII Inhibition by the Three Mutual Prodrugsa.
| |
+ porcine liver esterase |
||||
|---|---|---|---|---|---|
| compd | CoxI | CoxII | CoxI | CoxII | SIb |
| IB–MP | no inhibition | 1.8 ± 0.2 (15%) | 4.7 ± 0.3 (100%) | 1.3 ± 0.15 (100%) | 3.6 |
| IB | 10.1 ± 1.2 (100%) | 4.3 ± 0.46 (100%) | |||
| DIC–MP | 7.1 ± 0.9 (29%) | no inhibition | 0.25 ± 0.03 (100%) | 9.9 ± 0.22 (100%) | 0.03 |
| DIC | 0.32 ± 0.03 (100%) | 16.9 ± 1.8 (100%) | |||
| KET–MP | 0.2 ± 0.01 (43%) | 0.04 ± 0.01 (19%) | 0.48 ± 0.06 (100%) | 2.2 ± 0.21 (100%) | 0.22 |
| KET | 0.8 ± 0.06 (100%) | 3.8 ± 0.8 (100%) | |||
| BET–MP | 24.0 ± 2 (43%) | no inhibition | 24.0 ± 2 (43%) | no inhibition | <0.1 |
Values represent IC50 (μM) and % inhibition at saturation.
In vitro COXII selectivity index (CoxI IC50/CoxII IC50) calculated in the presence of PLE.
Figure 2.
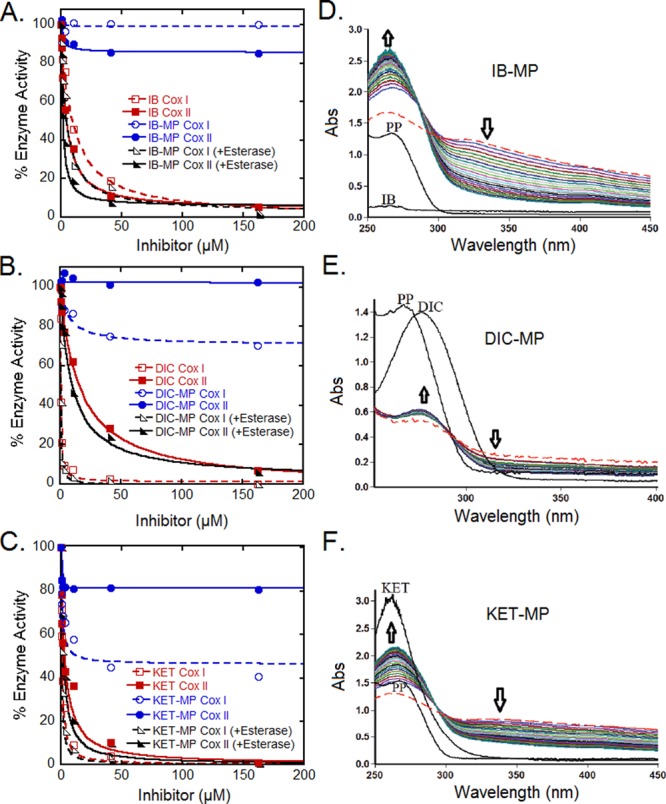
Effects of IB–MP (A), DIC–MP (B), and KET–MP (C) on the activity of ovine COXI and human COXII in the absence/presence of porcine liver esterase (PLE). Their activities were compared to the inhibitory effect of authentic samples of ibuprofen (IB) (A), diclofenac (DIC) (B), and ketoprofen (KET) (C). Absorption spectrum of each prodrug IB–MP (D), DIC–MP (E), and KET–MP (F) in the presence of porcine liver esterase (PLE) was monitored every 4 min for 100 min. The dashed line represents an initial absorbance spectrum before the addition of PLE. In each spectrum, one labeled line represents a spectrum of propyphenazone (PP), and the other labeled line depicts the absorption of an authentic sample of IB (D), DIC (E), and KET (F). The arrows indicate the direction of the spectral change over time.
Figure 2 shows the effect of PLE on the dose–response curve of the three prodrugs and compares them to the parent acidic NSAID. Upon the addition of PLE, the percentage inhibition of COXI and COXII by the three prodrugs increased to 100% indicating activation of the prodrugs by PLE.24 The potency and selectivity of each prodrug toward COXI and COXII after PLE treatment was comparable to the potency and selectivity of the released parent NSAID when assayed individually (Table 1). Noticeably, IB–MP showed more potent inhibition than its parent drug, for example, IB–MP inhibited COXI and COXII with IC50 of 4.7 ± 0.3 and 1.3 ± 0.15 μM, respectively, while ibuprofen (IB) alone inhibited them with an IC50 of 10 ± 1.2 and 4.3 ± 0.5 μM, respectively (Table 1). PLE addition to betahistine–propyphenazone (BET–MP) did not produce any enhancement of its activity against the COXI or COXII enzymes, validating its use as a negative control in in vivo experiments (Table 1).
Release Kinetics
To quantify the sensitivity of these prodrugs to esterase, we followed the procedure of Perez et al.24 by performing pseudo-first-order kinetic measurements using UV–vis absorption spectroscopy (Figures 2D–F and S1, Supporting Information). The absorbance of the reaction mixture was monitored for 100 min after the addition of PLE. The observed spectrum was recorded at different time points (Figure 2D–F) and the absorbance at λmax of 266 nm for IB–MB, 266 nm for DIC–MP, and 276 nm for KET–MP was plotted against time (in seconds) (Figure S1A–C, Supporting Information). The data presented in Figure S1A–C, Supporting Information, were fitted to pseudo-first-order kinetics (correlation coefficient ≥ 0.99). The rate constant computed at pH 7.5 were 0.0004, 0.001, and 0.0003 s–1 for IB–MP, DIC–MP, and KET–MP, respectively, which corresponds to half-lives of 28.8, 11.5, and 38.5 min for IB–MP, DIC–MP, and KET–MP, respectively (Figure S1D, Supporting Information). As expected, the addition of PLE to the negative control compound BET–MP did not affect its UV–vis spectrum after exhaustive incubation (Figure S1E, Supporting Information).
In Vivo Evaluation
Analgesic and anti-inflammatory activities of the three mutual prodrugs were tested and compared to equivalent doses of the parent drugs. For the analgesic activity, an abdominal writhing assay was employed.28,29 Intraperitoneal (i.p.) injection of freshly prepared acetic acid (1%, 10 mL/kg, i.p.) was used to induce writhing in mice. The frequency of the writhes induced by acetic acid was expressed as a nociceptive response. Mice were treated orally with each prodrug, parent drug, or vehicle then injected by acetic acid after 1 h of drug administration. The writhing response was recorded for 20 min (Tables 3 and 4). All three mutual prodrugs significantly reduced the writhing response compared to their parent drugs when taken individually. The protection from writhing caused by IB–MP was 95%, while IB or PP alone caused only 60 and 45% protection, respectively. Similarly, DIC–MP and KET–MP caused writhing protection of 87 and 73%, respectively (Table 3), while DIC and KET alone caused 63 and 66% protection, respectively. Interestingly, the negative control BET–MP caused a similar protection to PP when taken alone. The enhanced analgesic activity of the prodrugs evolves from their design as mutual prodrugs with synergistic analgesic effect.
Table 3. Analgesic Activity of the Mutual Prodrugsa.
| no. of writhings per 20 min ± SE | % protection | |
|---|---|---|
| control | 62 ± 0.6 | 0 |
| PP | 34 ± 0.9 | 45 |
| IB | 25 ± 0.6 | 60 |
| IB–MP | 3 ± 0.4 | 95 |
| DIC | 23 ± 5 | 63 |
| DIC–MP | 8 ± 2 | 87 |
| KET | 21 ± 4 | 66 |
| KET–MP | 17 ± 5 | 73 |
| BET–MP | 34 ± 0.9 | 45 |
No. of writhings values were obtained by averaging three independent trials; data represent the mean ± SE.
Table 4. Analgesic Activity of the New Cyclooxygenase-2 Selective Inhibitora.
| no. of writhings per 20 min ± SE | % protection | |
|---|---|---|
| ANT | 34 ± 0.8 | 45 |
| ANT+PP | 40 ± 0.8 | 35 |
| ANT–MP | 0 | 100 |
No. of writhings values were obtained by averaging three independent trials; data represent the mean ± SE and % protection values were calculated using the same control used in Table 3.
The effectiveness of these prodrugs for treatment of acute inflammation was estimated using a carrageenan-induced paw edema model in mice (Figure 3). Swelling of the injected paw was measured at 1, 2, 3, and 4 h using a mercury plethysmometer. Pretreatment with subcutaneous PP alone resulted in a limited reduction in swelling when compared to the parent drugs. IB, DIC, and KET showed significant reduction of swelling 1 h after injection with effects lasting for 4 h. The anti-inflammatory activity of the prodrugs improved with time with an onset of action, compared to control, appearing after 2 h. The anti-inflammatory activity of the three prodrugs was lower than their parent drugs in the first hour of the assay, consistent with the notion that the prodrugs themselves are devoid of anti-inflammatory activity and that the latent activity is due to the release of the parent drug (Figure S1D, Supporting Information). The negative control compound BET–MP showed similar anti-inflammatory activity to PP alone.
Figure 3.
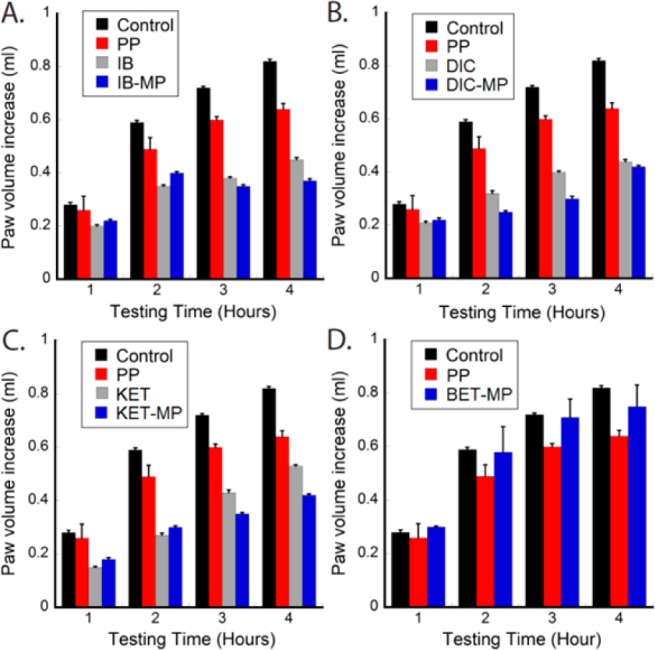
Acute inflammation mitigation by each prodrug and its corresponding individual NSAIDs in carrageenan model mice.
Propyphenazone-Based COXII-Selective Inhibitor
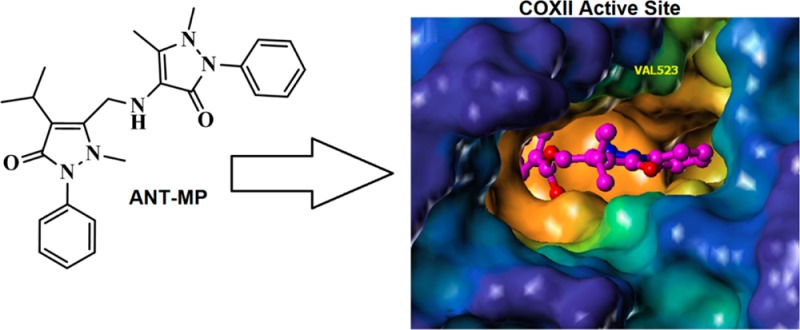
An important strategy to develop NSAIDs with minimal gastrointestinal toxicity is to target the COXII isoform with a selective inhibitor. When we tested the activity of the control compound BET–MP toward both COX isoforms, its partial inhibition of COXI (max inhibition 43%) and selectivity toward COXI was quite unexpected (Table 1). The observation suggested an ability to modulate propyphenazone COX selectivity when coupled to other NSAIDs in the same way as to BET. Luckily in our first trial where we coupled propophenazone to another nonacidic NSAID 4-aminoantipyrine (ANT) (Figure 1B), the coupled product 4-aminoantipyrine–propyphenazone (ANT–MP) (Figure 1C) showed absolute selectivity toward COXII, as it did not inhibit COXI at a concentration of 160 μM (Figure 4A), while it inhibited COXII with an IC50 of 0.97 ± 0.04 μM (100% maximum inhibition) (Table 2). ANT–MP showed an improved COXII selectivity index over the most widely used COXII selective inhibitor Celecoxib27 (Table 2). Noticeably, the coupling of propyphenazone to 4-aminoantipyrine dramatically enhanced its potency and improved its COXII selectivity. ANT alone inhibits both COX enzymes with 20–40-fold lower potency than ANT–MP (ANT IC50 for COXI is 26 ± 1.8 μM and for COXII is 42 ± 1.1 μM) (Figure 4A and Table 2). The stability of the compound toward PLE was tested following the same protocol used for the prodrugs. Neither the ANT–MP activity toward COX enzymes (Figure 4A and Table 2) nor its UV–vis spectrum (Figure S2, Supporting Information) was changed upon incubation with PLE confirming its expected stability toward the esterase.
Figure 4.
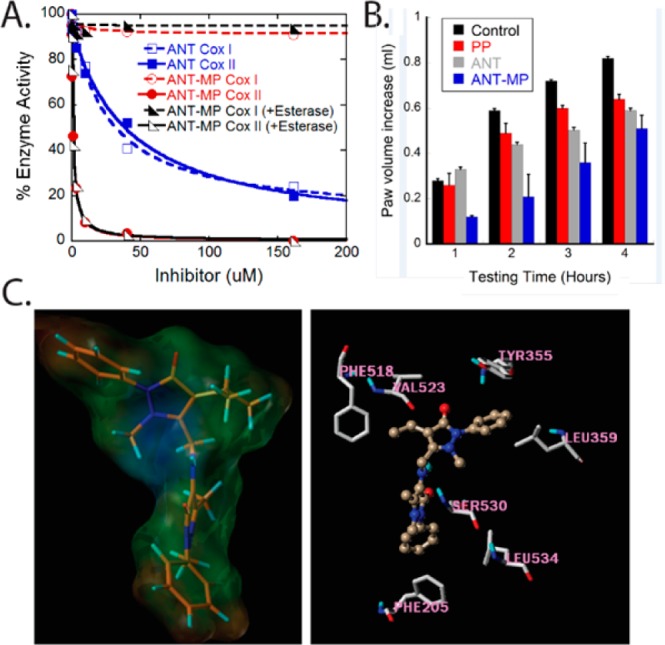
Characterization of ANT–MP as a specific COXII inhibitor. (A) Differential effect of ANT–MP and ANT on COXI and COXII activity in the absence and presence of porcine liver esterase (PLE). Unlike the nonselective ANT, ANT–MP showed 100% selectivity toward COX-II enzyme. (B) Acute inflammation mitigation by ANT–MP, ANT and PP in carrageenan model mice. (C) The 3-dimensional structure of ANT–MP covered by lipohilic potential water accessible surface. The brown portion of the molecule represents the most lipophilic potential, while the blue portion is the least lipophilic (left). Surflex docking of ANT–MP (golden brown) into COXII active site (right).
Table 2. In Vitro COXI and COXII Inhibition by the New Cyclooxygenase-2 Selective Inhibitora.
| |
+ porcine liver esterase |
||||
|---|---|---|---|---|---|
| compd | CoxI | CoxII | CoxI | CoxII | SIb |
| ANT–MP | no inhibition | 1.0 ± 0.04 (100%) | no inhibition | 1.0 ± 0.09 (100%) | Cox II selective |
| ANT | 26 ± 1.8 (100%) | 42 ± 1.1 (100%) | 0.6 | ||
| Celecoxib | 3027 | 0.0527 | 600 | ||
Values represent IC50 (μM) and % inhibition at saturation.
In vitro COXII selectivity index (CoxI IC50/CoxII IC50) calculated in the absence of PLE.
In Vivo Evaluation
The in vivo evaluation of ANT–MP suggests a highly improved analgesic activity for ANT–MP when compared to ANT alone. The percentage of protection from writhing reached 100% after administration of ANT–MP (no detected writhing,) while it was 45.2% for ANT alone (Table 4). To test if the analgesic effect of ANT–MP evolves from its in vivo hydrolysis to the original components ANT and PP, the analgesic effect of equimolar mixture of ANT and PP was investigated. The mixture produced 35% protection from writhing (Table 4), suggesting that the unique 100% protection caused by ANT–MP resulted from the effect of the intact compound. ANT–MP was also evaluated for its in vivo systemic anti-inflammatory activity using the carrageenan-induced paw edema model in mice (Figure 4B). ANT–MP showed a peak anti-inflammatory activity compared to control at 120 min, and the inhibitory activity on edema formation started to decline after that point (Figure 4B). The anti-inflammatory effect of ANT–MP showed an earlier onset than ANT or PP when used individually, and its anti-inflammatory effect was transient if compared to ANT or PP (Figure 4B).
Molecular Modeling
To rationalize the COXII selectivity of ANT–MP, the docking of ANT–MP in the active site of COXII was performed to investigate the nature of the interaction between ANT–MP and the active site residues. Several reports have determined the structural basis of the selective inhibition of COXII.30−32 To summarize, the free carboxylate moiety of NSAIDs orients toward the active site mouth of COXI where it is in a favorable position to interact with polar residues (Arg120 and Glu524).33 The COXII active site is 25% larger than COXI and is more flexible with a side pocket that is occluded in COXI.31 This infamous COXII side pocket is guarded by Val-523, which permits access to the pocket of the aromatic rings of ligands such as celecoxib.30 In COXI, a bulkier isoleucine (Ile-523) blocks access to this pocket. Several reports support the importance of hydrophobic interactions of COXII inhibitors with the hydrophobic channel of the active site.32 Docking of ANT–MP (Figure 4C, left) into the active site of COXII using the program Surflex suggested that the antipyrine side of ANT–MP accesses the active site of COXII where it is in a favorable position to interact with a group of hydrophobic amino acids composed of Phe-205, Phe-209, and Leu-534 (Figure 4C, right). The bulkier portion of the molecule (propyphenzone part) is predicted to bind the wide, more flexible mouth of the active site creating hydrophobic contacts with Phe-518 and Leu-359 and weaker interactions with Tyr-355. The docking of ANT–MP affords an explanation of its selectivity toward COXII over COXI.
Conclusions
Two strategies have been followed to prepare NSAIDs with minimal gastrointestinal toxicities based on the propyphenazone structure. Three mutual prodrugs have been evaluated both in vitro and in vivo, the release of the two pharmacologically active components of each prodrug has been studied using porcine liver esterase, and their release rate has been calculated. Interestingly, one selective COXII inhibitor was developed based on the irreversible coupling of propyphenazone and 4-aminoantipyrine. Its in vitro and in vivo activity was evaluated and its binding to COXII was investigated using molecular modeling techniques. ANT–MP, a new COXII selective lead, merits further investigation.
Glossary
ABBREVIATIONS
- CAD
coronary artery diseases
- CLASS
celecoxib long-term arthritis safety study
- COXI
cyclooxygenase-1
- COXII
cyclooxygenase-2
- NSAIDs
nonsteroidal anti-inflammatory drugs
- OTC
over-the-counter
- PGG2
prostaglandin g2
- PLE
porcine liver esterase
- SI
selectivity index
- UV–vis
ultraviolet–visible
- VIGOR
vioxx gastrointestinal outcomes research
Supporting Information Available
Pseudo-first-order kinetic measurements of the rates of conversion in the presence of porcine liver esterase (PLE) and detailed experimental methods. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ 107 West Dean Keaton, BME, The University of Texas at Austin, Austin, Texas 78712, United States.
Financial support was from grants from the National Institute of General Medical Sciences (R01GM059802), the Welch Foundation (F-1390), and Texas Institute for Drug & Diagnostic Development (H-F-0032) to K.N.D. T.S.K. acknowledges Cancer Prevention and Research Institute of Texas Postdoctoral Training Award (RP101501).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Green G. A. Understanding NSAIDs: from aspirin to COX-2. Clin. Cornerstone 2001, 3550–60. [DOI] [PubMed] [Google Scholar]
- Makhijia D. T.; Somani R. R. Improvement of GI tolerance of NSAIDs using oral prodrug approach. Der Pharm. Lett. 2010, 22300–309. [Google Scholar]
- Allison M. C.; Howatson A. G.; Torrance C. J.; Lee F. D.; Russell R. I. Gastrointestinal damage associated with the use of nonsteroidal antiinflammatory drugs. N. Engl. J. Med. 1992, 32711749–754. [DOI] [PubMed] [Google Scholar]
- Wallace J. L. How do NSAIDs cause ulcer disease?. Bailliere's Best Pract. Res., Clin. Anaesthesiol. 2000, 141147–159. [DOI] [PubMed] [Google Scholar]
- Fromm D. How do non-steroidal anti-inflammatory drugs affect gastric mucosal defenses?. Clin. Invest. Med. 1987, 103251–258. [PubMed] [Google Scholar]
- Somasundaram S.; Hayllar H.; Rafi S.; Wrigglesworth J. M.; Macpherson A. J.; Bjarnason I. The biochemical basis of non-steroidal anti-inflammatory drug-induced damage to the gastrointestinal tract: a review and a hypothesis. Scand. J. Gastroenterol. 1995, 304289–299. [DOI] [PubMed] [Google Scholar]
- Yamaoka S.; Urade R.; Kito M. Mitochondrial function in rats is affected by modification of membrane phospholipids with dietary sardine oil. J. Nutr. 1988, 1183290–296. [DOI] [PubMed] [Google Scholar]
- Rainsford K. D.; Willis C. Relationship of gastric mucosal damage induced in pigs by antiinflammatory drugs to their effects on prostaglandin production. Dig. Dis. Sci. 1982, 277624–635. [DOI] [PubMed] [Google Scholar]
- Whittle B. J. Temporal relationship between cyclooxygenase inhibition, as measured by prostacyclin biosynthesis, and the gastrointestinal damage induced by indomethacin in the rat. Gastroenterology 1981, 80194–98. [PubMed] [Google Scholar]
- Abuo-Rahma Gel D.; Abdel-Aziz M.; Beshr E. A.; Ali T. F. 1,2,4-Triazole/oxime hybrids as new strategy for nitric oxide donors: Synthesis, anti-inflammatory, ulceroginicity and antiproliferative activities. Eur. J. Med. Chem. 2014, 71, 185–198. [DOI] [PubMed] [Google Scholar]
- Abdel-Aziz M.; Abuo-Rahma Gel D.; Beshr E. A.; Ali T. F. New nitric oxide donating 1,2,4-triazole/oxime hybrids: synthesis, investigation of anti-inflammatory, ulceroginic liability and antiproliferative activities. Bioorg. Med. Chem. 2013, 21133839–3849. [DOI] [PubMed] [Google Scholar]
- Takeuchi K.; Yasuhiro T.; Asada Y.; Sugawa Y. Role of nitric oxide in pathogenesis of aspirin-induced gastric mucosal damage in rats. Digestion 1998, 594298–307. [DOI] [PubMed] [Google Scholar]
- Holzer P.; Pabst M. A.; Lippe I. T.; Peskar B. M.; Peskar B. A.; Livingston E. H.; Guth P. H. Afferent nerve-mediated protection against deep mucosal damage in the rat stomach. Gastroenterology 1990, 984838–848. [DOI] [PubMed] [Google Scholar]
- Wallace J. L.; Miller M. J. Nitric oxide in mucosal defense: a little goes a long way. Gastroenterology 2000, 1192512–520. [DOI] [PubMed] [Google Scholar]
- Halen P. K.; Murumkar P. R.; Giridhar R.; Yadav M. R. Prodrug designing of NSAIDs. Mini Rev. Med. Chem. 2009, 91124–139. [DOI] [PubMed] [Google Scholar]
- Calvi E.; Picciola G.; Carenini G.; Gentili P. Synthesis and pharmacologic study of new esters of naproxen with derivatives of N-oxyethylpiperazine. Il Farmaco; Edizione Scientifica 1985, 405334–346. [PubMed] [Google Scholar]
- Sheha M.; Khedr A.; Elsherief H. Biological and metabolic study of naproxen-propyphenazone mutual prodrug. Eur. J. Pharm. Sci. 2002, 173121–130. [DOI] [PubMed] [Google Scholar]
- Hsiao F. Y.; Tsai Y. W.; Huang W. F. Changes in physicians’ practice of prescribing cyclooxygenase-2 inhibitor after market withdrawal of rofecoxib: a retrospective study of physician-patient pairs in Taiwan. Clin. Ther. 2009, 31112618–2627. [DOI] [PubMed] [Google Scholar]
- Zarraga I. G.; Schwarz E. R. Coxibs and heart disease: what we have learned and what else we need to know. J. Am. Coll. Cardiol. 2007, 4911–14. [DOI] [PubMed] [Google Scholar]
- Boerlin V.; Maeglin B.; Hagler W.; Kuhn M.; Nuesch E. Analgesic activity of propyphenazone in patients with pain following oral surgery. Eur. J. Clin. Pharmacol. 1986, 312127–131. [DOI] [PubMed] [Google Scholar]
- Kraetsch H. G.; Hummel T.; Lotsch J.; Kussat R.; Kobal G. Analgesic effects of propyphenazone in comparison to its combination with caffeine. Eur. J. Clin. Pharmacol. 1996, 495377–382. [DOI] [PubMed] [Google Scholar]
- Akyel A.; Alsancak Y.; Yayla C.; Sahinarslan A.; Ozdemir M. Acute inferior myocardial infarction with low atrial rhythm due to propyphenazone: Kounis syndrome. Int. J. Cardiol. 2011, 1483352–353. [DOI] [PubMed] [Google Scholar]
- Goromaru T.; Matsuura H.; Furuta T.; Baba S. Identification of isopropylantipyrine metabolites in rat and man by using stable isotope tracer techniques. Chem. Pharm. Bull. 1984, 3283179–3186. [DOI] [PubMed] [Google Scholar]
- Perez C.; Daniel K. B.; Cohen S. M. Evaluating prodrug strategies for esterase-triggered release of alcohols. ChemMedChem 2013, 10.1002/cmdc.201300255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacour M. Betahistine treatment in managing vertigo and improving vestibular compensation: clarification. J. Vestibular Res. 2013, 233139–151. [DOI] [PubMed] [Google Scholar]
- Windsor M. A.; Hermanson D. J.; Kingsley P. J.; Xu S.; Crews B. C.; Ho W.; Keenan C. M.; Banerjee S.; Sharkey K. A.; Marnett L. J. Substrate-selective inhibition of cyclooxygenase-2: Development and evaluation of achiral profen probes. ACS Med. Chem. Lett. 2012, 39759–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H.; Yu R.; Tao Y.; Nikolic D.; van Breemen R. B. Measurement of cyclooxygenase inhibition using liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2011, 541230–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster R. M.; Anderson M.; Debeer E. J. Acetic acid for analgesic screening. Fed. Proc. 1959, 18, 412–416. [Google Scholar]
- Sawraj S.; Bhardawaj T.; Sharma P. Design, synthesis and evaluation of novel indomethacin-flavonoid mutual prodrugs as safer NSAIDs. Med. Chem. Res. 2011, 20, 687–694. [Google Scholar]
- Kurumbail R. G.; Stevens A. M.; Gierse J. K.; McDonald J. J.; Stegeman R. A.; Pak J. Y.; Gildehaus D.; Miyashiro J. M.; Penning T. D.; Seibert K.; Isakson P. C.; Stallings W. C. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996, 3846610644–648. [DOI] [PubMed] [Google Scholar]
- Luong C.; Miller A.; Barnett J.; Chow J.; Ramesha C.; Browner M. F. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat. Struct. Biol. 1996, 311927–933. [DOI] [PubMed] [Google Scholar]
- Vecchio A. J.; Orlando B. J.; Nandagiri R.; Malkowski M. G. Investigating substrate promiscuity in cyclooxygenase-2: The role of Arg-120 and residues lining the hydrophobic groove. J. Biol. Chem. 2012, 2872924619–24630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao P.; Knaus E. E. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): cyclooxygenase (COX) inhibition and beyond. J. Pharm. Pharm. Sci. 2008, 11281s–110s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.