

Abstract
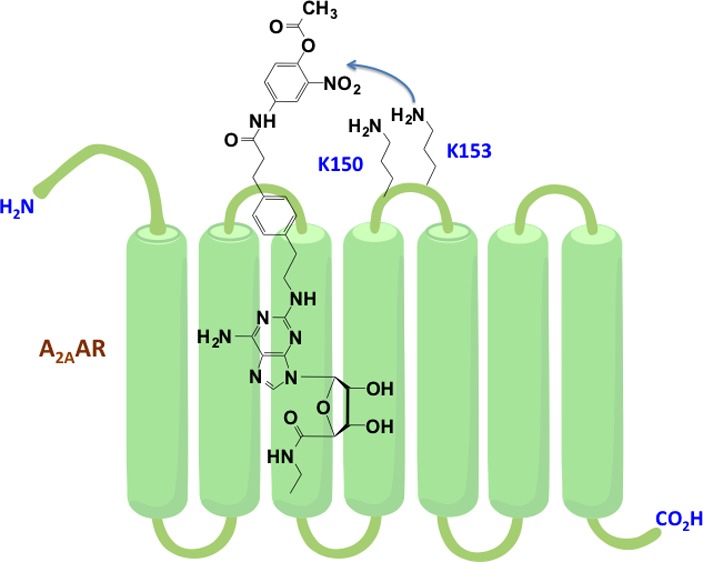
Adenosine receptors (ARs) are members of the G protein-coupled receptor (GPCR) superfamily and have shown much promise as therapeutic targets. We have used an agonist-bound A2AAR X-ray crystallographic structure to design a chemically reactive agonist for site-specific chemical modification of the receptor. To further explore and chemically engineer its binding cavity, a 2-nitrophenyl active ester was attached through an elongated chain at adenine C2 position. This general structure was designed for irreversible transfer of a terminal acyl group to a nucleophilic amino group on the A2AAR. Preincubation with several O-acyl derivatives prevented radioligand binding that was not regenerated upon extensive washing. In silico receptor docking suggested two lysine residues (second extracellular loop) as potential target sites for an O-acetyl derivative (MRS5854, 3a), and site-directed mutagenesis indicated that K153 but not K150 is essential. Similarly, a butyl azide for click reaction was incorporated in the active ester moiety (3b). These promising results indicate a stable, covalent modification of the receptor by several reactive adenosine derivatives, which could be chemical tools for future imaging, structural probing, and drug discovery. Thus, structure-based ligand design has guided the site-specific modification of a GPCR.
Keywords: G protein-coupled receptor, nucleoside, adenosine receptor, covalent modification, affinity labeling
The covalent modification of proteins is a useful research and diagnostic technique that benefits from a wide range of possible chemical attachments, including fluorescent and radiolabeled prosthetic groups to assist in both qualitative and quantitative characterization of a specific protein.1,2 Covalent modification of receptors and enzymes, such as formation of a disulfide bond with the P2Y12 receptor by thienopyridine drugs3 or acetylation of a specific serine on cyclooxygenase by acetylsalicylic acid,4 also has therapeutic utility. Previously, affinity labeling reagents for receptors were designed empirically,5 but recent advances in the structural elucidation of cell surface receptors make rational design a possible process.6,7
Adenosine receptors (ARs) are a 4-member subfamily of the large family of G protein-coupled receptors (GPCRs), which is of particular interest to biomedical research,8 consistent with the estimate that more than a third of all pharmaceuticals on the market target GPCRs.6,7 The A2AAR, which is the only AR subtype structurally elucidated with X-ray crystallography, has medical relevancy in various diseases including agonists for respiratory disorders and Huntington’s disease and antagonists for Parkinson’s disease, attention deficit disorder, cancer, and many others.9 Over the past several years, much work has been done toward structural characterization of the A2AAR, including the elucidation of its X-ray crystal structure in both agonist- and antagonist-bound conformations.10−12 Despite the large research effort devoted to understanding the structure and function of the A2AAR, the role of the flexible second extracellular loop (EL2), which makes contact with bound ligands, in recognition of orthosteric and allosteric ligands and in receptor activation, is still only partially understood.13−15 The EL2 of the A2AAR plays an important role in ligand recognition as indicated by the replacement of the EL2 of A2BAR with corresponding segment of the A2A-EL2.16 The chimeric receptor showed affinity for the prototypical selective A2AAR agonist 2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine (CGS21680, 1, Chart 1). Therefore, we focused on the A2AAR-EL2 and attempted to target specific nucleophilic amino acids on the receptor protein with high affinity ligands that incorporate chemically reactive, electrophilic moieties. We have used an agonist-bound A2AAR X-ray crystallographic structure12 to design chemically reactive agonists for site-specific chemical modification of the receptor. The covalent inhibition of GPCRs and enzymes is used therapeutically, for example, with the antithrombotic drug clopidogrel.3 Thus, the ability to modify the A2AAR protein effectively and permanently by targeting EL2 has implications for receptor characterization, imaging, and drug design.
Chart 1. Prototypical Competitive A2AAR Agonist (1) and Irreversibly Binding A2AAR Agonist (2)a.

a The target compounds for transfer of an acyl moiety to the receptor are active esters of the general formula 3. R2 is an alkyl group or reporter group.
Our experimental strategy aimed to covalently modify the A2AAR using reactive A2AAR selective agonist ligands (active esters 3, Chart 1) derived from 1. A set of high affinity acylating nucleosides was introduced with the aim of binding reversibly to the A2AAR and subsequently transferring an acyl group covalently to the receptor protein. There is a precedent for the design of acylating nucleoside ligands in our previous report of irreversible binding to the A2AAR of isothiocyanate derivatives,17 in which this reactive electrophilic group was attached through a long chain at the C2 position (R1). A potent ethylenediamine adduct of 1 was coupled to a single isothiocyanate group of a symmetric cross-linking reagent, p-phenylene diisothiocyanate, to form 2.17 This reactive nucleoside persisted in the binding site to produce long-lasting vasodilation of the coronary artery in the isolated guinea pig heart, which could not be reversed by the application of an AR antagonist, consistent with covalent reaction with the receptor. Additionally, various other derivatives containing reactive isothiocyanates at the extended C2 position were able to bind irreversibly to the rabbit A2AAR.5 Irreversible inactivation of the receptor was dependent on the presence of an aryl-NCS group.
In the design of acylating ligands applied here, unlike the isothiocyanate derivatives, these reactive nucleosides are intended to deliver a chemically functionalized handle to a nucleophilic amino group on the receptor. The nucleoside portion would serve as a leaving group, and the portion covalently bound to the A2AAR, i.e., detached from the nucleoside, would be used as a reporter group for receptor characterization.
Results and Discussion
Chemistry and Modeling
Our scheme for targeted covalent modification used as starting material a known selective A2AAR agonist 1 (Scheme 1), through modification of its terminal carboxylic acid. This functional group was condensed with a primary aniline 4 that has an o-nitrophenol group to serve as a good leaving group, consistent with the known reactivity of o-nitrophenyl esters.18 When acylated with a variety of acyl (RCO) species, this phenolic spacer group was converted into an electrophilic moiety, specifically an active nitrophenyl ester 3. Compound 3a was prepared by an alternate route, i.e., via 6a. This ester was intended to react with the nucleophilic groups in the vicinity of the terminal position of the C2 functionalized chain of the ligand (R1, Chart 1) when bound to the receptor. Thus, during acylation the nucleoside portion of the active ester would be detached along with the o-nitrophenol. In this manner, a R2CO group would be targeted for covalent transfer to a specific nucleophilic group on an EL region of the receptor, but the pharmacophore might not remain anchored to the receptor, unlike standard affinity labeling techniques. We varied R2 from the simplest acetyl group 3a to an alkyl azide 3b and biotin 3c.19 An alkyl azide is a particularly versatile group for site-specific attachment to GPCRs, due to its potential reactivity toward alkynes through a biocompatible Cu-free click reaction.20
Scheme 1. Synthesis of Reactive Agonist Ligands (3a−c) for Acyl Transfer to the A2AAR.

Reagents and yields: a. acyl anhydride, pyridine, 30-40%; b. EDC·HCl, DMF, 60%; c. RCO2H, DCC, DMAP, DMF, 30-50%, for 3b and 3c. d. PyBOP/DIEA, DMF, 70%, for 3a.
From the X-ray structure of an agonist-bound A2AAR and subsequent molecular modeling (docking of various known adenosine ligands),11,21 it was evident that the distal portions of C2-extended chains in potent A2AAR agonists were in close proximity to EL2 and EL3.21 The only nucleophilic residues that were available for this interaction in the vicinity of the docked C2 chain were the flexible side chains of Lys150 and Lys153, being the only lysines present in the extracellular portion of the receptor. Thus, we predicted that a primary amine on one of the two lysine residues in EL2 (K150 or K153) would be in proximity to react with the active ester attachment. The same residues were predicted in our previous docking study of C2-extended A2AAR agonists to be proximal to the reactive isothiocyanate group of 2.17 Interestingly, hA1AR and hA3AR sequences lack lysine residues in this region of EL2.
We determined more precisely the docking of compound 3a in an agonist-bound structure of the A2AAR (Figure 1). This compound shows all the interactions important for agonist binding of A2AAR, highlighted by crystallographic complexes.11,12 In particular, the ribose moiety and the adenine core participate in H-bonds with key residues of the binding site, such as Thr88 (3.36), Glu169 (EL2), His250 (6.52), Asn253 (6.55), Ser277 (7.42), and His278 (7.43) (numbers in parentheses follow the Ballesteros–Weinstein notation).22 Moreover, the adenine core forms a π–π stacking interaction with Phe168 in EL2. The C2 substituent extends toward the extracellular environment in proximity of EL2 with the carbonyl of the amide group forming an H-bond with Lys153. Therefore, the active ester group, even though not in direct contact with the amino group of Lys153, is in close proximity to this residue making it the first possible site for covalent modification. In fact, even though during docking residues of the binding site were kept fixed, Lys153 can be expected to easily interact with the active ester moiety of compound 3a due to the flexibility of its side chain. Moreover, other low energy docking conformations (not shown) display the flexible C2 chain bending toward Lys150 and positioning the active ester favorably to interact with it as a possible second site for covalent modification. It is worth noting that in the A2AAR crystal structure both of these lysine residues are forming electrostatic interactions with Asp170 in EL2 (Figure 1). These interactions can be crucial in preserving the conformation of the loop, and consequently, acylation of the lysine residues could result in an altered loop conformation affecting ligand binding.
Figure 1.
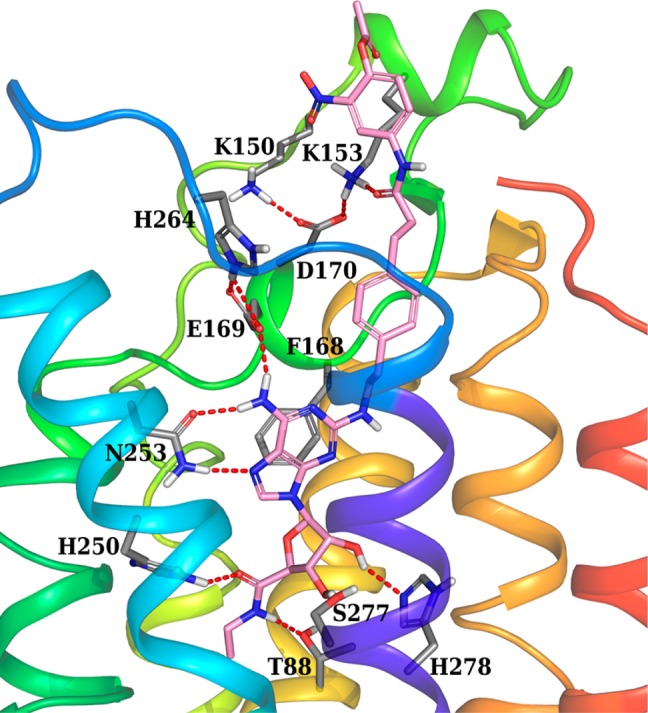
Docking pose of compound 3a (pink carbon sticks) at the hA2AAR (2YDV crystal structure12). Some residues important for ligand recognition are shown in sticks (gray carbons), and H-bonds are indicated by red dashed lines.
The molecular modeling predictions also place the active ester 3c in a similar proximity to the two lysine residues on EL2 when this A2AAR-specific ligand is docked to the receptor. Probably due to the bulkiness of biotin active ester 3c, steric effects precluded its ability to bind unhindered to the A2AAR and irreversibly modify the receptor (see below), possibly through transfer of the reactive acyl group. The ability to effectively react with the receptor would be related to the residence time on the receptor,23 which was not evaluated here. Moreover, click reactions through the azide handle on the receptor protein, derived from 3b, could be used to tether many different kinds of groups, including reporter groups such as fluorescent labels, even in live cells and tissues. Other potential chemical reactions between 3a and Lys153 could include nucleophilic aromatic substitution or aminolysis of the arylamide, but both of these are much less likely than reaction at the highly activated o-nitrophenyl ester.
Pharmacology
Three active esters 3a–3c were initially tested in competitive radioligand binding assays at three human (h) AR subtypes (Table 1) and compared to nonreactive intermediates 1 and 5. Compound 3a, which contained the simple acetyl active ester moiety, had an 11-fold higher binding affinity at the A2AAR than compound 5, which contains the same o-nitrophenol spacer but lacks an active ester. Additionally, compound 3a displayed moderate selectivity for the A2AAR over the A1AR (22-fold) and A3AR (9-fold) that was not present in compound 5. Compound 3a also fully stimulated cAMP formation in CHO cells stably expressing the A2AAR with an EC50 value of 2.12 ± 0.81 nM, compared to 5.86 ± 1.29 nM for 1 (Supporting Information, Figure S1). Thus, highly potent full agonism is maintained in the acetyl active ester derivative.
Table 1. Competitive Binding Results for Nucleoside Derivatives at Three AR Subtypesa.
| compd | A1AR, % inhibition or Ki (nM) | A2AAR, Ki (nM) | A3AR, Ki (nM) |
|---|---|---|---|
| 1 | 380 | 70 | 570 |
| 3a | 500 ± 72 | 23.0 ± 6.7 | 207 ± 47 |
| 3b | (30 ± 5%) | 4360 ± 760 | 1810 ± 470 |
| 3c | (37 ± 5%) | 3280 ± 680 | 1960 ± 100 |
| 5 | 1890 ± 670 | 267 ± 66 | 171 ± 6 |
Binding in membranes of CHO or HEK293 (A2A only) cells stably expressing one of three hAR subtypes. The binding affinity for hA1, A2A, and A3ARs was expressed as Ki values (mean of 3–4 determinations ± SEM) using agonists [3H]N6-R-phenylisopropyladenosine 8 ([3H]R-PIA), [3H]2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine 1, or [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide 9 ([125I]I-AB-MECA), respectively. A percent in parentheses refers to inhibition of binding at 10 μM. Competition was performed at a concentration range of 1 nM to 10 μM against the selective radioligand agonist for each subtype. Nonspecific binding was determined using 10 μM 5′-N-ethylcarboxamidoadenosine 10. Values for 1 are from ref (21).
With the high potency and selectivity of compound 3a in mind, we tested the irreversibility of its A2AAR binding, which would be indicative of covalent receptor modification. Following preincubation with compound 3a at a 1 μM concentration for 30 min, membranes containing the A2AAR lost most of the ability to bind either agonist ([3H]1, 19% binding remaining) or antagonist ([3H]4-[2-[7-amino-2-(2-furyl)-1,2,4-triazolo[1,5-a][1,3,5]triazin-5-yl-amino]ethylphenol 7, 11% binding remaining) radioligand. This is in contrast to preincubation of the A2AAR-expressing membranes with reversible agonist ligands 1 or 5 (1 μM), in which binding function was largely restored following several washing steps (Figure 2). Furthermore, after repeated washing, the 3a-pretreated membranes were incubated with 50 μM theophylline for 1 h and again washed by cycles of centrifugation in an attempt to use this weak antagonist to regenerate binding ability. However, treatment with the antagonist failed to restore A2AAR binding of either [3H]1 or [3H]7, indicating that the modification was irreversible and not simply a function of slow dissociation kinetics. Additionally, binding saturation studies were performed with the antagonist radioligand [3H]7 following the same membrane preincubation procedure. The results showed similar Kd values for all compounds ranging from 3 to 5 nM and a 13-fold greater Bmax value for the control saturation curve at 9.90 ± 0.95 pmol/mg compared to membranes incubated with compound 3a at 0.79 ± 0.26 pmol/mg (Table 2A and Figure S2, Supporting Information). The preincubation step of this experiment was done alternatively with either membrane extracts or whole cells (Figure S3, Supporting Information), and nearly identical loss of binding was observed.
Figure 2.

Level of residual binding after preincubation of HEK-293 cell membranes expressing the hA2AAR with compounds 1, 3a, and 5 (1 μM) and 3b (10 μM), as well as controls in the absence of added nucleoside and with added 1. (A) Using an agonist radioligand, [3H]1 (10 nM). (B) Using an antagonist radioligand, [3H]7 (1.0 nM). The receptor was incubated with the compound indicated and then washed several times in order to restore binding to any A2AAR present that was intact and not chemically modified.
Table 2. Results of Antagonist ([3H]7) Radioligand Saturation Binding Experiments: (A) With HEK-293 Cells Stably Overexpressing the WT A2AAR Showing Kd and Bmax Values Following Preincubation with Various Nucleosides; or (B) with WT, K150A, and K153A A2AAR Mutant Receptors, Transiently Transfected in COS-7 Cells; Nonspecific Binding Was Determined Using 10 (10 μM).
| A | ||
|---|---|---|
| incubated with | Kd (nM) | Bmax (pmol/mg protein) |
| control | 5.28 ± 2.21 | 9.90 ± 0.95 |
| 3a | 5.04 ± 1.53 | 0.79 ± 0.26 |
| 1 | 4.36 ± 1.93 | 7.76 ± 1.38 |
| 3b | 3.15 ± 1.35 | 4.80 ± 0.60 |
| B | |||
|---|---|---|---|
| A2AAR construct | preincubated with 3a | Kd (nM) | Bmax (pmol/mg protein) |
| A2A–WT | – | 1.81 ± 0.76 | 2.21 ± 0.19 |
| A2A–WT | + | 2.24 ± 0.46 | 0.29 ± 0.08 |
| A2A–K150A | – | 1.99 ± 0.77 | 2.18 ± 0.16 |
| A2A–K150A | + | 3.17 ± 1.21 | 0.20 ± 0.07 |
| A2A–K153A | – | 1.47 ± 0.77 | 2.36 ± 0.13 |
| A2A–K153A | + | 1.03 ± 0.39 | 1.65 ± 0.18 |
To further probe the structural basis for the irreversible inhibition, we mutated the two target lysine residues in EL2 to determine if we were able to restore binding after treatment with compound 3a. Mutant A2AAR-K150A showed results similar to the A2AAR WT control in which binding was almost completely abolished. However, mutant A2AAR-K153A showed a restoration of binding after being treated with compound 3a of about 70% based on the respective Bmax values (Table 2B and Figure S4, Supporting Information). While the binding was not completely restored, these results indicate a covalent interaction between Lys153 and the active ester derivative.
Because of the ability of compound 3a to irreversibly modify the receptor, we attempted to further functionalize the acyl group of the o-nitrophenyl ester. A biotinylated derivative 3c showed poor binding affinity, selectivity, and an inability to permanently inhibit binding of the A2AAR. To overcome this limitation, a smaller (alkyl azide) group was attached through the acyl group in active ester 3b. The azide group, if covalently anchored to the receptor, would be subjected to a well-established method of Cu-free click reaction with a cyclooctyne-derivatized fluorophore or chromophore.24 Compound 3b displayed an A2AAR affinity similar to the biotinylated compound 3c; however, there was partial irreversible inhibition of binding. Following treatment with 3b and washing to remove all traces of the noncovalently bound ligand, approximately 50% of the initial receptor binding remained inhibited, as is demonstrated with the saturation curves that show a Bmax value of 4.80 ± 0.60 pmol/mg versus the control that showed a Bmax of 9.90 ± 0.95 pmol/mg (Table 2A and Figure S2, Supporting Information).
Initially, dibenzocyclooctyne (DIBO) with an AlexaFluor-488 fluorophore (Figure S5, Supporting Information) was tested for the ability to perform a Cu-free click reaction of the 3b-pretreated A2AAR.25,26 A high level of background fluorescence was observed in membrane extracts of control A2AAR-expressing cells not exposed to 3b, and specifically, post-3b labeling was not discernible. A similar experiment was performed using difluorooctyne (DIFO) with a rhodamine-based fluorophore (TAMRA), but there was no observable increase in fluorescence. Despite these results, there is still a potential for the site-specific functionalization of the A2AAR with the alkyl azide.
The irreversible inhibition of ligand binding was consistent with a covalent modification of an amino acid residue that is integral to the binding pocket of the A2AAR. A priori, we were uncertain if the modification of a residue on EL2 would impede subsequent radioligand binding. Evidently, the modification of the receptor by 3a completely blocks subsequent binding of reversible agonist or antagonist radioligand. We are uncertain if the residual nucleoside moiety of the affinity reagent is trapped in the binding site after covalent modification, or if it dissociates from the binding site. The nature of the covalently modified A2AAR and its functional pharmacological properties will be the subject of further investigations of this reagent family. We do not know if a prolonged activation of the receptor is present following 3a pretreatment, as was observed with isothiocyanate derivative 2 in the guinea pig heart.17
The question of why the chemically altered A2AAR loses its ability to bind radioligands remains. It is conceivable that blocking the entry of small molecule ligands to the pharmacophore binding site greatly impedes their high affinity binding. In principle, a reporter group could be directed toward the receptor and deposited there for detection by various methods.
Conclusions
In conclusion, a selective agonist ligand was modified as a selective agonist ligand containing an active ester intended for transferring various acyl moieties covalently to a GPCR protein. Covalent modification of the A2AAR was previously accomplished empirically, but this is the first structure-based, targeted covalent modification of the A2AAR. A strategically designed reactive O-acetyl derivative (3a) and a corresponding butyl azide for click reaction (3b) both caused irreversible loss of binding function. We have confirmed, using site-directed mutagenesis, our prediction from molecular modeling that a specific lysine residue (K153) on EL2 is the principal site of covalent modification. Determining if the receptor modification is by acyl transfer, as designed, and if the nucleoside portion of the agonist dissociates following covalent reaction will be topics for future study. Assuming a mechanism of transfer of a reactive acyl moiety, the agonist derivative was designed to contain a wide range of functionality for receptor characterization and quantification. Thus, structure-based ligand design of chemical tools has led to the site-specific modification of a GPCR, with the potential for future imaging, structural probing, and drug discovery.
Acknowledgments
We thank Dr. John Lloyd and Dr. Noel Whittaker (NIDDK) for mass spectral determinations.
Glossary
Abbreviations
- AR
adenosine receptor
- cAMP
adenosine 3′,5′-cyclic monophosphate
- CHO
Chinese hamster ovary
- EL
extracellular loop
- GPCR
G protein-coupled receptor
- HEK
human embryonic kidney
- I-AB-MECA
N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyl-uronamide
- HRMS
high resolution mass spectroscopy
- NMR
nuclear magnetic resonance
- WT
wild-type
Supporting Information Available
NMR and HRMS of nucleosides, structures of the cyclooctane reagents, and the coordinates of the model of the A2AAR complex with compound 3a. This material is available free of charge via the Internet at http://pubs.acs.org.
This research was supported by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Baslé E.; Joubert N.; Pucheault M. Protein chemical modification on endogenous amino acids. Chem. Biol. 2010, 17, 213–227. [DOI] [PubMed] [Google Scholar]
- Lang K.; Chin J. W. Bioorthogonal reactions for labeling proteins. ACS Chem. Biol. 2014, 9, 16–20. [DOI] [PubMed] [Google Scholar]
- Johnson D. S.; Weerapana E.; Cravatt B. F. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med. Chem. 2010, 2, 949–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N. P.; Dong L.; Yuan C.; Noon K. R.; Smith W. L. Asymmetric acetylation of the cyclooxygenase-2 homodimer by aspirin and its effects on the oxygenation of arachidonic, eicosapentaenoic, and docosahexaenoic acids. Mol. Pharmacol. 2010, 77, 979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K. A.; Stiles G. L.; Ji X.-D. Chemical modification and irreversible inhibition of striatal A2a-adenosine receptors. Mol. Pharmacol. 1992, 42, 123–133. [PMC free article] [PubMed] [Google Scholar]
- Kobilka B. The structural basis of g-protein-coupled receptor signaling (Nobel Lecture). Angew. Chem., Int. Ed. 2013, 52, 6380–6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V.; Cherezov V.; Stevens R. C. Structure-function of the g protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 531–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K. A.; Gao Z. G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discovery 2006, 5, 247–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lera Ruiz M.; Lim Y. H.; Zheng J. Adenosine A2A receptor as a drug discovery target. J. Med. Chem. 2014, 57, 3623–3650. [DOI] [PubMed] [Google Scholar]
- Congreve M.; Langmead C. J.; Mason J. S.; Marshall F. H. Progress in structure based drug design for G protein-coupled receptors. J. Med. Chem. 2011, 54, 4283–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F.; Wu H.; Katritch V.; Han G. W.; Jacobson K. A.; Gao Z. G.; Cherezov V.; Stevens R. C. Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G.; Warne T.; Edwards P. C.; Bennett K.; Langmead C. J.; Leslie A. G. W.; Tate C. G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Jiang Q.; Glashofer M.; Yehle S.; Wess J.; Jacobson K. A. Glutamate residues in the second extracellular loop of the human A2a adenosine receptors are required for ligand recognition. Mol. Pharmacol. 1996, 49, 683–691. [PMC free article] [PubMed] [Google Scholar]
- Kennedy D. P.; McRobb F. M.; Leonhardt S. A.; Purdy M.; Figler H.; Marshall M. A.; Chordia M.; Figler R.; Linden J.; Abagyan R.; Yeager M. The second extracellular loop of the adenosine A1 receptor mediates activity of allosteric enhancers. Mol. Pharmacol. 2014, 85, 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K.; Zhang J.; Gao Z. G.; Paoletta S.; Zhang D.; Han G. W.; Li T.; Ma L.; Zhang W.; Müller C. E.; Yang H.; Jiang H.; Cherezov V.; Katritch V.; Jacobson K. A.; Stevens R. C.; Wu B.; Zhao Q. Agonist-bound structure of the human P2Y12R receptor. Nature 2014, 509, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibt B. F.; Schiedel A. C.; Thimm D.; Hinz S.; Sherbiny F. F.; Müller C. E. The second extracellular loop of GPCRs determines subtype-selectivity and controls efficacy as evidenced by loop exchange study at A2 adenosine receptors. Biochem. Pharmacol. 2013, 85, 1317–1329. [DOI] [PubMed] [Google Scholar]
- Niiya K.; Jacobson K. A.; Silvia S. K.; Olsson R. A. Covalent binding of a selective agonist irreversibly activates guinea pig coronary artery A2 adenosine receptors. Naunyn-Schmeideberg’s Arch. Pharmacol. 1993, 347, 521–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen B. J.; Kraus M. A.; Patchornik A. P. “Wolf and lamb” reactions: equilibrium and kinetic effects in multipolymer systems. J. Am. Chem. Soc. 1981, 103, 7620–1629. [Google Scholar]
- Lesch H. P.; Kaikkonen M. U.; Pikkarainen J. T.; Ylä-Herttuala S. Avidin-biotin technology in targeted therapy. Expert Opin. Drug Delivery 2010, 7, 551–564. [DOI] [PubMed] [Google Scholar]
- Baskin J. M.; Prescher J. A.; Laughlin S. T.; Agard N. J.; Chang P. V.; Miller I. A.; Lo A.; Codelli J. A.; Bertozzi C. R. Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 16793–16797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deflorian F.; Kumar T. S.; Phan K.; Gao Z. G.; Xu F.; Wu H.; Katritch V.; Stevens R. C.; Jacobson K. A. Evaluation of molecular modeling of agonist binding in light of the crystallographic structure of an agonist-bound A2A adenosine receptor. J. Med. Chem. 2012, 55, 538–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J.; Weinstein H. Integrated methods for the construction of three-dimensional models of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Guo D.; Mulder-Krieger T.; IJzerman A. P.; Heitman L. H. Functional efficacy of adenosine A2A receptor agonists is positively correlated to their receptor residence time. Br. J. Pharmacol. 2012, 166, 1846–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett J. C.; Bertozzi C. R. Cu-free click cycloaddition reactions in chemical biology. Chem. Soc. Rev. 2010, 39, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramil C. P.; Lin Q. Bioorthogonal chemistry: strategies and recent developments. Chem. Commun. 2013, 49, 11007–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E. J.; Kang D. W.; Leucke H. F.; Bond M. R.; Ghosh S.; Love D. C.; Ahn J. S.; Kang D. O.; Hanover J. A. Optimizing the selectivity of DIFO-based reagents for intracellular bioorthogonal applications. Carbohydr. Res. 2013, 377, 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.